Synthesis, characterization and ethylene polymerization behavior of nickel dihalide complexes bearing bulky unsymmetrical α-diimine ligands†
Hao
Liu
ac,
Weizhen
Zhao
a,
Jiangang
Yu
a,
Wenhong
Yang
a,
Xiang
Hao
a,
Carl
Redshaw
b,
Langqiu
Chen
c and
Wen-Hua
Sun
*a
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100, 190, China. E-mail: whsun@iccas.ac.cn; Fax: +86 10 62618239; Tel: +86 10 62557955
bSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: carl.redshaw@uea.ac.uk; Fax: +44 (0)1603 592003; Tel: +44 (0)1603 593137
cKey Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education, College of Chemistry, Xiangtan University, 411105 Xiangtan, China
First published on 1st December 2011
Abstract
A series of nickel(II) dihalide complexes (C1–C10) bearing unsymmetrical α-diimine ligands of the type 2,4-dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-6-methylbenzenamine (L1–L5) were synthesized and fully characterized. Single-crystal X-ray diffraction revealed a distorted tetrahedral geometry around the nickel center in the complexes C3, C5 and C9. Upon activation with modified methylaluminoxane (MMAO), all nickel pro-catalysts performed with high activities in ethylene polymerization, producing highly branched polyethylene products.
1. Introduction
The use of late-transition metal complexes as catalysts for ethylene oligomerization and polymerization has undergone remarkable progress in the past two decades, especially since the milestone of the discovery of α-diimine nickel and palladium pro-catalysts by the Brookhart group.1 Significant influence on the catalytic activities of the metal complexes was achievable by the variation of the nature of the ancillary ligands present,2 and as a consequence the development of new pro-catalyst systems has relied heavily on the design of multi-dentate ligands such as those binding as N⁁N,3P⁁N,4O⁁N,5O⁁N⁁N,6N⁁P⁁N,7P⁁N⁁P,8P⁁O⁁P,9P⁁N⁁N10 and N⁁N⁁Nchelates.11 In general, α-diimine derivatives have been accessed via modification of the ligand frameworks 12 and in particular, via variation of the substituents on the arylimino groups.12,13 A unique feature of the resultant polyethylene obtained by using such nickel pro-catalysts is the high degree of branching in the resultant PEs,1a,13f,14 which is due to “chain-walking” on the active nickel species.14,15 Significant influences were observed through varying the substituents on the arylimino group by a number of researchers,16 and we have also noted that modification of a series of α-diimine nickel dihalides derived from the precursor 2,6-dibenzhydryl-6-methylaniline provided not only highly active pro-catalysts, but also highly branched PEs.13f However, the use of bulky anilines with benzhydryl substituents has not fully been explored. The limited studies to date have included the use of 2,6-dibenzhydryl-6-methylaniline as a precursor for preparing iron pro-catalysts,17 and, as mentioned previously, nickel pro-catalysts ligated by α-diimine ligands derived from 2,6-dibenzhydryl-6-methylaniline.13f There is thus scope for the fine-tuning of α-diimine ligands containing dibenzhydryl substituents with a view to further exploitation in nickel-based ethylene polymerization systems. Herein, the title nickel complexes are synthesized, characterized, and their performance in ethylene polymerization investigated.2. Results and discussion
2.1. Synthesis and characterization
According to our reported procedure,13f the stoichiometric condensation of acenaphthylene-1,2-dione with 2,4-dibenzhydryl-6-methylaniline produced the 2-(2,4-dibenzhydryl-6-methylphenylimino)acenaphthylenone, which was further reacted with a second aniline to afford the 2,4-dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-6-methylbenzenamine ligands (L1–L5) (Scheme 1). All the newly synthesized organic compounds were characterized by FT-IR and NMR spectroscopy, and the formulae were consistent with their elemental analyses.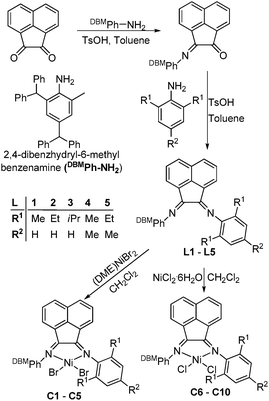 | ||
| Scheme 1 Synthesis of ligands (L1–L5) and nickel complexes (C1–C10). | ||
All 2,4-dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-6-methylbenzenamine ligands (L1–L5) acted as α-diimine type ligands and readily reacted with an equivalent of (DME)NiBr2 in dichloromethane to afford the corresponding nickel dibromide complexes (C1–C5), whilst on reaction with NiCl2·6H2O, the α-diimine ligands (L1–L5) produced the corresponding nickel chlorides (C6–C10) (see Scheme 1).
2.2. Crystal structures
The molecular structures are shown in Fig. 1–3, with the selected bond lengths and angles tabulated in Table 1. As shown in Fig. 1, the coordination geometry of C3 can be best described as a distorted tetrahedron, with the three atoms N1, N2 and Br1 forming the basal plane, and the Br2 atom occupying the apical position. The nickel atom is 0.818 Å out of the basal plane. Due to the bulky asymmetric nature of the ligand set, the plane composed of N1, N2 and Ni1 and the plane composed of Br1, Br2 and Ni1 form a dihedral angle of 82.26°. The dihedral angles between the aryl ring on N1 and the plane comprising N1, N2 and Ni1 is 86.54°, but the aryl ring on N2 and the plane composed of N1, N2 and Ni1 is 82.36°. There is a fused five-membered ring comprising the nickel and the ligand with an acute angle N1–Ni1–N2 of 83.14(15)°, and associated bond distances of Ni1–N1 2.028(4) Å and Ni1–N2 2.037(4) Å. The imino bond length N1–C12 is 1.285(6) Å, whereas N2–C1 is slightly longer at 1.295(6) Å.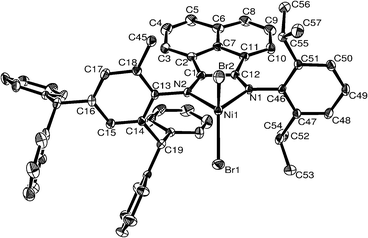 | ||
| Fig. 1 ORTEP structure of C3. Thermal ellipsoids are shown at 30% probability level. Hydrogen atoms have been omitted for clarity. | ||
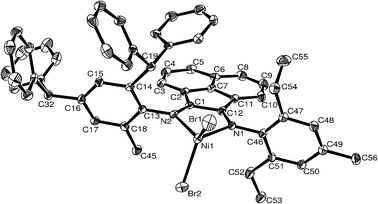 | ||
| Fig. 2 ORTEP structure of C5. Thermal ellipsoids are shown at 30% probability level. Hydrogen atoms have been omitted for clarity. | ||
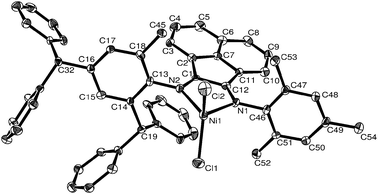 | ||
| Fig. 3 ORTEP structure of C9. Thermal ellipsoids are shown at 30% probability level. Hydrogen atoms have been omitted for clarity. | ||
| C3 | C5 | C9 | |
|---|---|---|---|
| Bond lengths (Å) | |||
| Ni(1)–N(1) | 2.028(4) | 2.018(4) | 2.018(5) |
| Ni(1)–N(2) | 2.037(4) | 2.037(4) | 2.046(5) |
| Ni(1)–Br(1) | 2.3188(10) | 2.3232(9) | 2.205(2) |
| Ni(1)–Br(2) | 2.3398(10) | 2.3265(11) | 2.204(2) |
| N(1)–C(12) | 1.285(6) | 1.287(6) | 1.308(7) |
| N(1)–C(46) | 1.442(5 | 1.440(6) | 1.459(7) |
| N(2)–C(1) | 1.295(6) | 1.283(6) | 1.284(7) |
| N(2)–C(13) | 1.435(5) | 1.444(5) | 1.442(7) |
| Bond angles (°) | |||
| N(2)–Ni(1)–N(1) | 83.14(15) | 82.50(15) | 82.34(19) |
| N(2)–Ni(1)–Br(1) | 114.60(12) | 124.26(10) | 120.88(15) |
| N(1)–Ni(1)–Br(1) | 118.56(11) | 109.68(11) | 109.14(15) |
| N(2)–Ni(1)–Br(2) | 107.48(12) | 102.21(10) | 101.48(15) |
| N(1)–Ni(1)–Br(2) | 102.40(11) | 112.71(11) | 110.63(15) |
| Br(1)–Ni(1)–Br(2) | 123.15(4) | 119.70(4) | 124.49(8) |
| C(12)–N(1)–C(46) | 119.1(4) | 119.4(4) | 120.5(5) |
| C(1)–N(2)–C(13) | 121.3(4) | 119.7(4) | 119.9(5) |
In the structure of C5, the nickel atom is coordinated by an N-(2-(2,4-dibenzhydryl-6-methylphenylimino)acenaphthylenylidene)-2,6-diethyl-4-methylbenzenamine ligand, and two terminal bromides. Similarly, the geometry of the four-coordinate complex can be described as a distorted tetrahedron, with the plane comprising the three atoms N1, N2 and Br2 as the basal plane and the Br1 atom occupying the apical position. The plane composed of N1, N2 and Ni1 and the plane composed of Br1, Br2 and Ni1 form a dihedral angle of 80.23°, slightly smaller than that observed for C3. Interestingly, the sterically bulky group [CH(Ph)2] is positioned above the square planar metal center, whereas for C3 the situation is reversed, and so these metal complexes can as being like a pair of ‘stereoisomers’.
As shown in Fig. 3, the structure of C9 more closely resembles that of C3. The plane composed of N1, N2 and Ni1 and the plane composed of Cl1, Cl2 and Ni1 form a dihedral angle of 81.87°. The dihedral angles between the aryl ring on N1 and the plane composed of N1, N2 and Ni1 is 83.46°, whereas the aryl ring on N2 and the plane composed of N1, N2 and Ni1 is 79.07°, both slightly smaller than observed in C3. There is a fused five-membered ring comprising nickel and the ligand with an acute angle N1–Ni1–N2 at 82.34(19)°. The imino bond lengths N1–C12, N2–C1 are 1.308(7) Å and 1.284(7) Å, both typical of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond character.
N double bond character.
2.3. Ethylene polymerization
The current focus of this study is the catalytic behavior of nickel complexes ligated by 2,4-dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-6-methylbenzenamine ligands. The two groups of nickel(II) bromide and chloride complexes were investigated separately, and evaluated to determine the optimum conditions for catalysis. Initially, the pro-catalyst C5 was employed with various alkylaluminum reagents as co-catalyst/activator at room temperature under 10 atm of ethylene. Treatment with either ethylaluminum sesquichloride (Et3Al2Cl3, EASC) or diethylaluminum chloride (Et2AlCl) afforded inactive systems (Table 2). However, in the presence of modified methylaluminoxane (MMAO), the pro-catalyst C5 exhibited outstanding catalytic activity, and as a consequence, modified methylaluminoxane was selected as the preferred co-catalyst for further investigation.| Entry | Catalyst | Co-catalyst | Al/Nib | Activityc | M w d , e | M w/Mnd | T m f (°C) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1.5 μmol; 20 °C; 30 min; 10 atm ethylene; 100 mL toluene. b Molar ratio of Al/Ni. c 103 kg mol−1(Ni) h−1. d Determined by GPC. e 104 g mol−1. f Determined by DSC. | |||||||
| 1 | C5 | MMAO | 1000 | 7.67 | 3.46 | 2.75 | 118.4 |
| 2 | C5 | Et2AlCl | 200 | Trace | |||
| 3 | C5 | EASC | 200 | Trace |
| Entry | Catalyst | Al/Ni | T b (°C) | Yield (g) | Activityc | M w d , e | M w/Mnd | T m f (°C) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: MMAO; 1.5 μmol; 30 min; 10 atm ethylene; 100 mL toluene. b Reaction temperature. c 103 kg mol−1(Ni) h−1. d Determined by GPC. e 104 g mol−1. f Determined by DSC. | ||||||||
| 1 | C5 | 1000 | 20 | 5.75 | 7.67 | 3.46 | 2.75 | 118.4 |
| 2 | C5 | 2000 | 20 | 5.97 | 7.96 | 4.97 | 2.42 | 117.3 |
| 3 | C5 | 3000 | 20 | 6.19 | 8.25 | 6.63 | 1.97 | 126.3 |
| 4 | C5 | 4000 | 20 | 5.39 | 7.19 | 5.46 | 2.01 | 122.6 |
| 5 | C5 | 3000 | 40 | 5.17 | 6.89 | 3.50 | 2.67 | 94.6 |
| 6 | C5 | 3000 | 60 | 4.60 | 6.12 | 1.92 | 2.78 | 70.2 |
| 7 | C5 | 3000 | 80 | 2.44 | 3.25 | 1.62 | 2.67 | 53.8 |
| 8 | C1 | 3000 | 20 | 3.16 | 4.21 | 5.07 | 2.47 | 127.6 |
| 9 | C2 | 3000 | 20 | 5.58 | 7.44 | 5.10 | 2.11 | 121.9 |
| 10 | C3 | 3000 | 20 | 6.71 | 8.95 | 6.03 | 1.91 | 124.3 |
| 11 | C4 | 3000 | 20 | 5.79 | 7.72 | 4.51 | 2.70 | 122.3 |
As for the other α-diimine catalytic systems, our new unsymmetrical bulky α-diimine catalyst yielded polyethylene with a high degree of branching, especially at higher temperature. As shown in Fig. 4, the number of branches was calculated according to the literature,18 and it was found that polyethylene with 166 branches/1000 carbons was obtained at 80 °C (entry 7 in Table 3), which was much lower than reported previously for polyethylenes obtained using analogous catalysts based on a series of unsymmetrical ligands using 2,6-dibenzhydryl-4-methylanilines.13f Meanwhile, polyethylene with 14 branches/1000 carbons was obtained at 20 °C (entry 3 in Table 3), consistent with the observation of obtaining higher branched polyethylenes on increasing the reaction temperature.1a Moreover, 13C NMR data showed that almost all of the branches present were methyl branches (Fig. 5). Indeed, a higher order of branches in the polyethylenes was achieved at the elevated reaction temperature (Fig. 4).
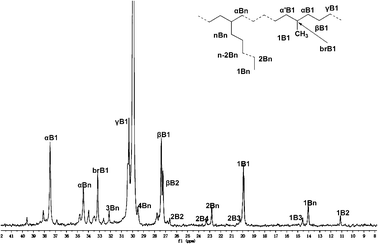 | ||
| Fig. 4 13C NMR spectrum of polyethylene prepared at 80 °C (entry 7 in Table 3). | ||
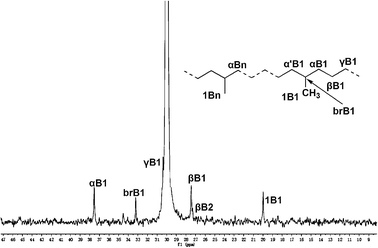 | ||
| Fig. 5 13C NMR spectrum of polyethylene prepared at 20 °C (entry 3 in Table 3). | ||
| Entry | Catalyst | Al/Ni | T (°C) | t b (min) | Yield (g) | Activityc | M w d , e | M w/Mnd | T m f (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: MMAO; 1.5 μmol; 30 min; 10 atm ethylene; 100 mL toluene. b Reaction time. c 103 kg mol−1(Ni) h−1. d Determined by GPC. e 104 g mol−1. f Determined by DSC. | |||||||||
| 1 | C9 | 1000 | 20 | 30 | 5.61 | 7.48 | 3.55 | 2.85 | 120.6 |
| 2 | C9 | 2000 | 20 | 30 | 6.51 | 8.68 | 3.76 | 2.75 | 124.3 |
| 3 | C9 | 3000 | 20 | 30 | 6.35 | 8.47 | 4.94 | 2.41 | 119.6 |
| 4 | C9 | 4000 | 20 | 30 | 5.15 | 6.87 | 4.28 | 2.58 | 117.3 |
| 5 | C9 | 2000 | 40 | 30 | 5.67 | 7.56 | 1.88 | 3.37 | 94.7 |
| 6 | C9 | 2000 | 60 | 30 | 4.15 | 5.53 | 1.31 | 2.58 | 93.2 |
| 7 | C9 | 2000 | 80 | 30 | 2.66 | 3.55 | 0.81 | 2.76 | 89.9 |
| 8 | C9 | 2000 | 20 | 5 | 1.83 | 14.6 | 4.70 | 2.31 | 118.4 |
| 9 | C9 | 2000 | 20 | 10 | 4.38 | 17.5 | 3.73 | 3.46 | 117.1 |
| 10 | C9 | 2000 | 20 | 60 | 8.72 | 5.81 | 4.20 | 2.85 | 116.5 |
| 11 | C6 | 2000 | 20 | 30 | 5.75 | 7.67 | 4.54 | 2.92 | 119.8 |
| 12 | C7 | 2000 | 20 | 30 | 6.58 | 8.77 | 5.38 | 2.14 | 118.8 |
| 13 | C8 | 2000 | 20 | 30 | 7.04 | 9.39 | 4.97 | 2.17 | 116.7 |
| 14 | C10 | 2000 | 20 | 30 | 7.12 | 9.49 | 5.20 | 2.20 | 117.4 |
Regarding the lifetime of the catalytic system, polymerizations using the C9/MMAO system were conducted over different time periods, namely 5, 10, 30 and 60 min (entries 2 and 8–10 in Table 4). On prolonging the reaction time from 5 to 30 min, the production of polyethylene followed an exponential increase, whereas on further prolonged reaction time, a distinct decrease of output was observed, suggesting that the active species suffered from severe deactivation at reaction times of over 30 min.
3. Conclusion
The synthesis and characterization of a new class of nickel-based ethylene polymerization pro-catalysts ligated by new bulky unsymmetrical α-diimines is described. Despite there been many reported α-diimine systems in the literature, the unsymmetrical α-diimine family presented herein represents a new avenue of research for α-diimine catalytic systems. On treatment with the co-catalyst MMAO, these complexes afforded good catalytic activity, and the polyethylenes obtained exhibited a high degree of branching; the degree of branching varied at different temperatures. The use of the ligand systems described herein with other metals will be reported separately.4. Experimental section
4.1. General considerations
All manipulations of air and/or moisture sensitive compounds were carried out under a nitrogen atmosphere using standard Schlenk techniques. Toluene was refluxed over sodium–benzophenone and distilled under nitrogen prior to use. Modified methylaluminoxane (MMAO, 1.93 M in heptane, 3A) was purchased from Akzo Nobel Corp. Diethylaluminum chloride (Et2AlCl, 0.79 M in toluene), and ethylaluminum sesquichloride (EASC, 0.87 M in toluene) were purchased from Acros Chemicals. High-purity ethylene was purchased from Beijing Yansan Petrochemical Co. and used as received. Other reagents were purchased from Aldrich, Acros, or local suppliers. NMR spectra were recorded on a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard. IR spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrometer. Elemental analysis was carried out using a Flash EA 1112 microanalyzer. Molecular weights and molecular weight distributions (MWD) of polyethylene were determined by a PL-GPC 220 at 150 °C, with 1,2,4-trichlorobenzene as the solvent. DSC trace and melting points of polyethylene were obtained from the second scanning run on a Perkin-Elmer DSC-7 at a heating rate of 10 °C min−1.4.2. Synthesis and characterization
2-(2,4-dibenzhydryl-6-methylphenylimino)acenaphthylenone . A mixture of 2,4-diphenylmethyl-6-methylaniline (1.30 g, 2.96 mmol), acenaphthylene-1,2-dione (0.53 g, 2.91 mmol) and a catalytic amount of p-toluenesulfonic acid in toluene (80 mL) was refluxed for 3 h. After solvent evaporation at reduced pressure, the crude product purified by column chromatography on silica with the eluent of petroleum ether/dichloromethane (V
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 10:1) to afford 0.84 g of the red product in 47% isolated yield. Mp: 98–99 °C. IR (KBr; cm−1): 3056.2(w), 3023.6(w), 1728.3(s), 1651.9(m), 1596.5(m), 1492.3(m), 1443.3(m), 1274.2(m), 1223.5(m), 1073.6(m), 1025.9(s), 909.5(m), 829.1(m), 805.0(s), 740.9(s), 696.3(vs). Anal. Calcd. for C45H33NO (603.75): C, 89.52; H, 5.51; N, 2.32; Found; C, 89.77; H, 5.61; N, 2.04. 1H NMR (400 MHz, CDCl3, TMS): δ 8.06 (t, J = 6.81, 2H), 7.85 (d, J = 8.34, 1H), 7.74 (t, J = 7.54, 1H), 7.32–7.17 (m, 7H), 7.13–7.07 (m, 7H), 6.95 (d, J = 7.42, 2H), 6.91 (s, 1H), 6.77 (d, J = 7.63, 2H), 6.68 (s, 1H), 6.41 (t, J = 6.36, 3H), 6.19 (t, J = 7.39, 1H), 5.54 (s, 1H), 5.50 (s, 1H), 1.98 (s, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 189.84, 161.59, 146.75, 144.28, 142.95, 142.69, 141.74, 139.68, 132.95, 131.94, 130.51, 130.44, 129.82, 129.49, 129.42, 129.09, 128.96, 128.31, 128.02, 127.54, 126.31, 126.08, 125.14, 124.59, 122.93, 121.77, 56.42, 52.57, 17.82.
:V = 10:1) to afford 0.84 g of the red product in 47% isolated yield. Mp: 98–99 °C. IR (KBr; cm−1): 3056.2(w), 3023.6(w), 1728.3(s), 1651.9(m), 1596.5(m), 1492.3(m), 1443.3(m), 1274.2(m), 1223.5(m), 1073.6(m), 1025.9(s), 909.5(m), 829.1(m), 805.0(s), 740.9(s), 696.3(vs). Anal. Calcd. for C45H33NO (603.75): C, 89.52; H, 5.51; N, 2.32; Found; C, 89.77; H, 5.61; N, 2.04. 1H NMR (400 MHz, CDCl3, TMS): δ 8.06 (t, J = 6.81, 2H), 7.85 (d, J = 8.34, 1H), 7.74 (t, J = 7.54, 1H), 7.32–7.17 (m, 7H), 7.13–7.07 (m, 7H), 6.95 (d, J = 7.42, 2H), 6.91 (s, 1H), 6.77 (d, J = 7.63, 2H), 6.68 (s, 1H), 6.41 (t, J = 6.36, 3H), 6.19 (t, J = 7.39, 1H), 5.54 (s, 1H), 5.50 (s, 1H), 1.98 (s, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 189.84, 161.59, 146.75, 144.28, 142.95, 142.69, 141.74, 139.68, 132.95, 131.94, 130.51, 130.44, 129.82, 129.49, 129.42, 129.09, 128.96, 128.31, 128.02, 127.54, 126.31, 126.08, 125.14, 124.59, 122.93, 121.77, 56.42, 52.57, 17.82.
Synthesis of 2,4-dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-6-methylbenzenamine (L1–L5).
2,4-dibenzhydryl-N-(2-(2,6-dimethylphenylimino)acenaphthylenylidene)-6-methylbenzenamine (L1). A solution of 2-(2,4-dibenzhydryl-6-methylphenylimino)acenaphthylenone (0.68 g, 1.13 mmol), 2,6-dimethylaniline (0.16 g, 1.32 mmol) and a catalytic amount of p-toluenesulfonic acid in toluene (50 mL) was mixed and refluxed for 8 h. The solution was evaporated at reduced pressure. The residual solids was further purified by silica column chromatography (V petroleum ether
:V ethyl acetate = 15:1) to afford L1 0.22 g (yellow, 28% yield). Mp: 147–148 °C. IR (KBr; cm−1): 3023.8(w), 2956.7(w), 1667.7(m), 1644.0(m), 1594.2 (m), 1492.6(m), 1442.1(m), 1277.6(w), 1232.0(m), 1203.7(m), 1077.2(m), 1034.4(m), 924.0(m), 830.9(m), 740.7(s), 696.4(vs). Anal. Calcd. for C53H42N2 (706.91): C, 90.05; H, 5.99; N, 3.96. Found; C, 90.33; H, 6.12; N, 3.55. 1H NMR (400 MHz, CDCl3, TMS): δ 7.78 (d, J = 8.27, 1H), 7.673 (d, J = 8.24, 1H), 7.32–7.27 (m, 5H), 7.24–7.18(m, 3H), 7.16–7.06(m, 10H), 6.99 (d, J = 7.12, 2H), 6.90 (d, J = 5.14, 2H, 6.88 (s, 1H), 6.70 (s, 1H), 6.58 (d, J = 7.05, 1H, 6.45 (t, J = 7.41, 2H , 6.38 (d, J = 7.06, 1H), 6.24 (t, J = 7.19, 1H), 5.73 (s, 1H), 5.51 (s, 1H), 2.28 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.58, 161.29, 149.34, 147.51, 144.51, 143.46, 141.83, 140.32, 139.01, 133.28, 130.45, 129.55, 129.48, 129.10, 128.83, 128.54, 128.45, 128.31, 127.99, 127.92, 127.48, 126.26, 126.00, 125.19, 125.07, 124.94, 124.83, 123.79, 123.07, 122.10, 56.45, 52.26, 18.30, 17.86, 17.77.
2,4-dibenzhydryl-N-(2-(2,6-diethylphenylimino)acenaph-thylenylidene)-6-methylbenzenamine (L2). Using the same procedure as for the synthesis of L1, L2 was obtained as a yellow powder in 32.8% (0.38 g) yield. Mp: 224–225 °C. IR (KBr; cm−1): 3023.0(w), 2964.0(w), 1675.6(m), 1650.0(m), 1596.6(m), 1492.5(s), 1438.7(s), 1278.6(w), 1235.1(w), 1192.4(m), 1079.3(m), 1031.8(m), 924.0(m), 832.9(m), 740.8(s), 697.1(vs). Anal. Calcd. for C55H46N2 (734.97): C, 89.88; H, 6.31; N, 3.81. Found; C, 90.21; H, 6.55; N, 3.59. 1H NMR (400 MHz, CDCl3, TMS): δ 7.76 (d, J = 8.24, 1H), 7.71 (d, J = 8.23, 1H), 7.36–7.28 (m, 5H), 7.27–7.22 (m, 3H), 7.20–7.10 (m, 10H), 6.99 (d, J = 7.20, 2H), 6.90 (d, 3H), 6.72 (s, 1H), 6.57 (d, J = 7.06, 1H), 6.47 (t, J = 7.32, 2H), 6.35 (d, J = 7.07, 1H), 6.25 (t, J = 7.23, 1H), 5.75 (s, 1H), 5.52 (s, 1H), 2.73 (m, 1H), 2.71–2.49 (m, 2H), 2.35 (m, 1H), 2.04 (s, 3H), 1.25 (t, J = 7.28, 3H), 1.05 (t, J = 7.44, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.55, 161.33, 148.56, 147.60, 144.49, 143.58, 141.85, 140.36, 139.02, 133.31, 131.00, 130.70, 130.45, 129.98, 129.56, 129.49, 129.31, 129.26, 129.13, 128.74, 128.52, 128.29, 127.98, 127.75, 127.54, 126.51, 126.40, 126.26, 125.99, 125.24, 124.92, 124.13, 123.07, 122.59, 56.47, 55.58, 24.89, 24.65, 17.73, 14.51, 13.83.
2,4-dibenzhydryl-N-(2-(2,6-diisopropylphenylimino)acenaphthylenylidene)-6-methylbenzenamine (L3). Using the same procedure as for the synthesis of L1, L3 was obtained as a yellow powder in 27% (0.34 g) yield. Mp: 243–244 °C. IR (KBr; cm−1): 3022.7(w), 2956.0(m), 1678.7(m), 1653.4(m), 1596.7(m), 1492.8(m), 1439.0 (m), 1277.67(w), 1248.1(m), 1187.5(w), 1078.7(w), 1032.1(m), 925.6(m), 833.1(m), 741.3(s), 6967.7(vs). Anal. Calcd. for C57H50N2 (763.02): C, 89.72; H, 6.60; N, 3.67. Found; C, 89.87; H, 6.96; N, 3.30. 1H NMR (400 MHz, CDCl3, TMS): δ 7.75 (d, J = 8.25, 1H), 7.70 (d, J = 8.23, 1H), 7.35–7.19 (m, 10H), 7.16–7.05 (m, 8H), 7.00 (d, J = 7.31, 2H), 6.92 (s, 1H), 6.89 (d, J = 7.52, 2H), 6.71 (s, 1H), 6.52 (d, J = 7.12, 1H), 6.46 (t, J = 7.44, 2H), 6.32 (d, J = 7.16, 1H), 6.24 (t, J = 7.31, 1H), 5.75 (s, 1H), 5.52 (s, 1H), 2.21 (m, 1H), 2.91 (m, 1H), 2.04 (s, 3H), 1.33 (d, J = 6.72, 3H), 1.20 (d, J = 6.76, 3H), 1.13 (d, J = 6.80, 3H), 0.86 (d, J = 6.76, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.62, 161.59, 147.63, 147.33, 144.52, 143.60, 141.79, 140.43, 139.00, 135.72, 135.54, 133.34, 130.45, 129.97, 129.56, 129.49, 129.11, 128.75, 128.51, 128.29, 127.97, 127,56, 127.50, 126.26, 125.98, 125.26, 124.92, 124.48, 123.70, 123.46, 123.08, 123.01, 56.46, 52.55, 28.70, 28.62, 23.91, 23.69, 23.50, 23.20, 17.70.
2,4-dibenzhydryl-N-(2-(2,4,6-trimethylphenylimino)acenaphthylenylidene)-6-methylbenzenamine (L4). Using the same procedure as for the synthesis of L1, L4 was obtained as a yellow powder in 32% (0.39 g) yield. Mp: 164–165 °C. IR (KBr; cm−1): 3023.5(w), 2967.6(w), 1667.4(m), 1640.8(m), 1596.7(m), 1493.2(m), 1441.8(m), 1278.4(w), 1232.7(m), 1207.3(mw), 1075.8(m), 1032.2(m), 922.4(m), 829.6(m), 738.4(s), 696.5(vs). Anal. Calcd. for C54H44N2 (720.94): C, 89.96; H, 6.15; N, 3.89. Found; C, 90.11; H, 6.54; N, 3.62. 1H NMR (400 MHz, CDCl3, TMS): δ 7.77 (d, J = 8.25, 1H), 7.71 (d, J = 8.24, 1H), 7.32–7.26 (m, 4H), 7.24–7.19 (m, 3H), 7.17–7.07 (m, 8H), 6.99 (d, J = 6.69, 3H), 6.95 (s, 1H), 6.89 (d, J = 8.46, 3H), 6.70 (s, 1H), 6.65 (d, J = 7.10, 1H), 6.45 (t, J = 7.41, 2H), 6.37 (d, J = 7.14, 1H), 6.23 (t, J = 7.28, 1H), 5.73 (s, 1H), 5.51 (s, 1H), 2.38 (s, 3H), 2.24 (s, 3H), 2.04 (s, 6H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.64, 161.46, 147.58, 146.85, 144.54, 143.50, 141.84, 140.31, 138.96, 133.30, 132.99, 130.46, 130.04, 129.56, 129.49, 129.33, 129.16, 129.10, 129.03, 128.71, 128.51, 128.30, 127.98, 127.91, 127.46, 126.26, 125.98, 125.18, 124.97, 124.84, 124.60, 123.02, 122.13, 56.46, 52.62, 21.05, 18.24, 17.77.
2,4-dibenzhydryl-N-(2-(2,6-diethyl-4-methylphenylimino)-acenaphthylenylidene)-6-methylbenzenamine (L5). Using the same procedure as for the synthesis of L1, L5 was obtained as a yellow powder in 39.5% (0.49 g) yield. Mp: 192–193 °C. IR (KBr; cm−1): 3022.7(w), 2967.5(w), 1674.4(m), 1649.3(m), 1597.4(m), 1492.5(m), 1442.3(m), 1277.9(w), 1233.1(w), 1205.1(w), 1076.3(m), 1030.4(m), 922.8(m), 833.3(m), 745.4(s), 697.6(vs). Anal. Calcd. for C56H48N2 (748.99): C, 89.80; H, 6.46; N, 3.74. Found; C, 89.67; H, 6.33; N, 3.69. 1H NMR (400 MHz, CDCl3, TMS): δ 7.75 (d, J = 8.17, 1H), 7.70 (d, J = 8.16, 1H), 7.30–7.26 (m, 5H), 7.24–7.20 (m, 2H), 7.18–7.09 (m, 8H), 7.04 (s, 1H), 7.00 (d, J = 6.43, 3H), 6.90 (d, J = 8.81, 3H), 6.71 (s, 1H), 6.63 (d, J = 6.99, 1H), 6.46 (t, J = 7.20, 2H), 6.34 (d, J = 7.01, 1H), 6.25 (t, J = 7.07, 1H), 5.75 (s, 1H), 5.51 (s, 1H), 2.70 (m, 1H), 2.58–2.45 (m, 2H), 2.42 (s, 3H), 2.32 (m, 1H), 2.03 (s, 3H), 1.23 (t, J = 7.40, 3H), 1.03 (t, J = 7.36, 3H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.67, 161.52, 147.67, 146.05, 144.54, 143.61, 141.84, 140.34, 138.96, 133.34, 133.28, 130.84, 130.54, 130.44, 130.00, 129.58, 129.50, 129.35, 129.11, 128.63, 128.49, 128.31, 127.98, 127.74, 127.54, 127.44, 127.31, 127.18, 126.27, 125.98, 125.23, 124.95, 123.02, 122.62, 56.48, 52.57, 24.90, 24.66, 21.34, 17.74, 14.70, 13.96.
Data for C2 Yield: 83.7%, red powder. IR (KBr; cm−1): 3023.9(w), 2969.5(w), 1646.0(w), 1620.2(m), 1580.2(m), 1492.7(m), 1444.3(m), 1294.6(m), 1184.5(w), 1030.9(w), 825.0(m), 774.9(s), 745.9(s), 699.5(vs). Anal. Calcd. for C55H46Br2N2Ni (953.47): C, 69.28; H, 4.86; N, 2.94. Found; C, 69.46; H, 4.93; N, 2.58.
Data for C3 Yield: 85.8%, red powder. IR (KBr; cm−1): 3023.0(w), 2963.9(w), 1651.0(w), 1622.4(m), 1581.3(s), 1493.7(s), 1442.0(s), 1293.2(m), 1181.0(m), 1030.7(m), 831.9(m), 776.2(s), 741.8(s), 696.5(vs). Anal. Calcd. for C57H50Br2N2Ni (981.52): C, 69.75; H, 5.13; N, 2.85. Found; C, 69.66; H, 5.21; N, 2.60.
Data for C4 Yield: 77.9%, red powder. IR (KBr; cm−1): 3023.3(w), 2968.6(w), 1644.7(w), 1619.7(m), 1579.4(s), 1492.8(s), 1446.4(s), 1294.1(m), 1200.9(m), 1030.7(m), 827.7(m), 773.5(s), 743.8(s), 699.8(vs). Anal. Calcd. for C54H44Br2N2Ni (939.44): C, 69.04; H, 4.72; N, 2.98. Found; C, 69.33; H, 4.87; N, 2.75.
Data for C5 Yield: 79.5%, red powder. IR (KBr; cm−1): 3024.1(w), 2968.1(w), 1644.7(m), 1620.2(m), 1580.0(s), 1492.6(s), 1451.4(s), 1293.4(m), 1200.5(m), 1030.9(m), 827.6(m), 774.7(s), 744.0(s), 699.0(vs). Anal. Calcd. for C56H48Br2N2Ni (967.5): C, 69.52; H, 5.00; N, 2.90. Found; C, 69.62; H, 5.20; N, 2.67.
The chloride complexes C6–C10 were synthesized by the reaction of NiCl2·6H2O with the corresponding ligands in dichloromethane. As a typical synthetic procedure complex C6 is described as follows: the solution of 0.080 g (0.113 mmol) ligand L1 and 0.03 g (0.126 mmol) of NiCl2·6H2O in 10 mL dichloromethane was stirred for 8 h at room temperature. The precipitate was washed with diethyl ether and dried in vacuo to obtain a brown solid in 0.078 g (82.8%) yield. IR (KBr; cm−1): 3024.6(w), 2964.8(w), 1657.6(w), 1626.5(m), 1578.2(s), 1493.4(s), 1444.0(m), 1289.9(m), 1190.3(m), 1032.9(s), 829.4(m), 772.7(s), 741.1(s), 697.6(vs). Anal. Calcd. for C53H42Cl2N2Ni (836.51): C, 76.10; H, 5.06; N, 3.35. Found; C, 76.27; H, 5.22; N, 3.19.
Data for C7 Yield: 86.0%, red powder. IR (KBr; cm−1): 3025.9(w), 2965.7(w), 1659.8(w), 1627.9(m), 1592.3(s), 1492.9(s), 1444.8(s), 1288.8(m), 1184.9(m), 1035.1(s), 827.1(m), 772.6(s), 740.4(s), 698.0(vs). Anal. Calcd. for C55H46Cl2N2Ni (864.57): C, 76.41; H, 5.36; N, 3.24. Found; C, 76.53; H, 5.59; N, 3.11.
Data for C8 Yield: 72.7%, red powder. IR (KBr; cm−1): 3025.1(w), 2963.4(w), 1653.7(w), 1624.0(m), 1585.4(s), 1493.4(s), 1442.5(s), 1289.8(m), 1182.8(m), 1032.3(s), 829.8(m), 777.3(s), 741.9(s), 697.4(vs). Anal. Calcd. for C57H50Cl2N2Ni (892.62): C, 76.70; H, 5.65; N, 3.14. Found; C, 76.88; H, 5.72; N, 3.01.
Data for C9 Yield: 80.3%, red powder. IR (KBr; cm−1): 3025.0(w), 2967.2(w), 1655.7(w), 1626.6(m), 1583.3(s), 1493.9(m), 1444.8(m), 1290.6 (m), 1193.4(w), 1031.8(m), 830.0(m), 775.0(s), 740.8(s), 698.7(vs). Anal. Calcd. for C54H44Cl2N2Ni (850.54): C, 76.25; H, 5.21; N, 3.29. Found; C, 76.44; H, 5.37; N, 2.91.
Data for C10 Yield: 77.8%, red powder. IR (KBr; cm−1): 3024.3(w), 2963.6(w), 1654.1(w), 1624.4(m), 1586.9(m), 1490.8(m), 1445.7(m), 1288.9(m), 1200.4(m), 1031.7(m), 829.6(m), 774.9(s), 740.4(s), 679.7(vs). Anal. Calcd. for C56H48Cl2N2Ni (878.59): C, 76.55; H, 5.51; N, 3.19. Found; C, 76.77; H, 5.87; N, 2.92.
4.3. General procedure for ethylene polymerization
Ethylene polymerization at 10 atm ethylene pressure was performed in a 0.3 L stainless steel autoclave equipped with a mechanical stirrer, a temperature controller and gas ballast through a solenoid clave for continuous feeding of ethylene at constant pressure. The pro-catalyst was dissolved in 30 mL toluene and injected into the autoclave; then the required co-catalyst toluene solution and additional toluene (to maintain the total volume of 100 mL) were added. When the required temperature was reached, the ethylene (10 atm pressure) was continuously introduced to initiate the polymerization. After the desired period of reaction, the autoclave was sealed and cooled in an ice–water bath for one hour. Then the resultant solution was quenched with 10% hydrochloric acid ethanol. The precipitated polyethylene was collected by filtration, and washed with ethanol and water, and then dried in a vacuum at 60 °C until of constant weight.4.4. X-Ray crystallographic studies
Single crystals of C3, C5 and C9 suitable for X-ray diffraction analysis were obtained by laying diethyl ether on their dichloromethane solutions at room temperature. With graphite-monochromatic MoKα radiation (λ = 0.71073 Å) at 173(2) K, cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least squares on F2. All hydrogen atoms were placed in calculated positions. Structure solution and refinement were performed by using the SHELXL-97 package.19 Details of the X-ray structure determinations and refinements are provided in Table 5.| C3 | C5 | C9 | |
|---|---|---|---|
| Formula | C57H50Br2N2Ni | C56H48Br2N2Ni | C54H44Cl2N2Ni |
| Formula weight | 981.52 | 967.49 | 850.52 |
| Temperature (K) | 173(2) | 173(2) | 173(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c | P21/c |
| a (Å) | 10.378(2) | 15.352(3) | 15.330(3) |
| b (Å) | 16.008(3) | 22.551(5) | 22.285(5) |
| c (Å) | 31.765(6) | 15.380(3) | 14.954(3) |
| α (°) | 90 | 90 | 90 |
| β (°) | 95.61(3) | 106.96(3) | 106.33(3) |
| γ (°) | 90 | 90 | 90 |
| Volume (Å3) | 5251.8(18) | 5093.0 | 4902.6(17) |
| Z | 4 | 4 | 4 |
| D calc (Mg m−3) | 1.241 | 1.262 | 1.152 |
| μ (mm−1) | 1.927 | 1.986 | 0.540 |
| F(000) | 2016 | 1984 | 1776 |
| Crystal size (mm) | 0.43 × 0.16 × 0.15 | 0.22 × 0.21 × 0.03 | 0.22 × 0.17 × 0.02 |
| θ range (°) | 1.29–26.34 | 1.39–25.05 | 1.38–27.47 |
| Limiting indices | −8 ≤ h ≤ 12, | −18 ≤ h ≤ 17, | −19 ≤ h ≤ 19, |
| −19 ≤ k ≤ 19, | −23 ≤ k ≤ 26, | −23 ≤ k ≤ 28, | |
| −36 ≤ l ≤ 37 | −18 ≤ l ≤ 18 | −17 ≤ l ≤ 19 | |
| No. of reflections collected | 36252 | 35074 | 39551 |
| No. of unique reflections | 10463 | 9011 | 11189 |
| R int | 0.0606 | 0.0898 | 0.0957 |
| Completeness to θ (%) | 97.8 (θ = 26.34°) | 99.8 (θ = 25.05°) | 99.7 (θ = 27.47°) |
| No. of parameters | 559 | 550 | 532 |
| Goodness-of-fit on F2 | 1.175 | 1.105 | 1.155 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0696 | R 1 = 0.0683 | R 1 = 0.1173 |
| wR 2 = 0.1778 | wR 2 = 0.1528 | wR 2 = 0.2780 | |
| R indices (all data) | R 1 = 0.0886 | R 1 = 0.0937 | R 1 = 0.1619 |
| wR 2 = 0.1894 | wR 2 = 0.1664 | wR 2 = 0.3059 |
Acknowledgements
This work was supported by MOST 863 program No. 2009AA033601. CR wishes to thank the EPSRC for an overseas travel grant (EP/H031855/1).References
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS; (b) C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664 CrossRef CAS; (c) B. L. Small, M. Brookhart and A. M. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS.
- (a) T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460 CrossRef CAS; (b) L. Wang, W.-H. Sun, L. Han, Z. Li, Y. Hu, C. He and C. Yan, J. Organomet. Chem., 2002, 650, 59–64 CrossRef CAS.
- (a) L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS; (b) S. A. Svejda and M. Brookhart, Organometallics, 1999, 18, 65 CrossRef CAS; (c) S. P. Meneghetti, P. J. Lutz and J. Kress, Organometallics, 1999, 18, 2734 CrossRef CAS; (d) T. V. Laine, U. Piironen, K. Lappalainen, M. Klinga, E. Aitola and M. Leskelä, J. Organomet. Chem., 2000, 606, 112 CrossRef CAS; (e) C. Shao, W.-H. Sun, Z. Li, Y. Hu and L. Han, Catal. Commun., 2002, 3, 405 CrossRef CAS; (f) S. Jie, D. Zhang, T. Zhang, W.-H. Sun, J. Chen, Q. Ren, D. Liu, G. Zheng and W. Chen, J. Organomet. Chem., 2005, 690, 1739 CrossRef CAS; (g) C. Zhang, W.-H. Sun and Z.-X. Wang, Eur. J. Inorg. Chem., 2006, 4895 CrossRef CAS; (h) R. Gao, L. Xiao, X. Hao, W.-H. Sun and F. Wang, Dalton Trans., 2008, 5645 RSC; (i) F. Yang, Y. Chen, Y. Lin, K. Yu, Y. Liu, Y. Wang, S. Liu and J.-T. Chen, Dalton Trans., 2009, 1243 RSC.
- (a) W. Keim, S. Killat, C. F. Nobile, G. P. Suranna, U. Englert, R. Wang, S. Mecking and D. L. Schröder, J. Organomet. Chem., 2002, 662, 150 CrossRef CAS; (b) W.-H. Sun, Z. Li, H. Hu, B. Wu, H. Yang, N. Zhu, X. Leng and H. Wang, New J. Chem., 2002, 26, 1474 RSC; (c) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Organometallics, 2004, 23, 2613 CrossRef CAS; (d) F. Speiser, P. Braunstein and L. Saussine, Organometallics, 2004, 23, 2625 CrossRef CAS; (e) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Inorg. Chem., 2004, 43, 1649 CrossRef CAS; (f) Z. Weng, S. Teo and T. S. A. Hor, Organometallics, 2006, 25, 4878 CrossRef CAS.
- (a) C. Wang, S. Friedrich, T. R. Younkin, R. T. Li, R. H. Grubbs, D. A. Bansleben and M. W. Day, Organometallics, 1998, 17, 3149 CrossRef CAS; (b) C. Carlini, M. Isola, V. Liuzzo, A. M. R. Galletti and G. Sbrana, Appl. Catal., A, 2002, 231, 307 CrossRef CAS; (c) S. Wu and S. Lu, Appl. Catal., A, 2003, 246, 295 CrossRef CAS; (d) F. Chang, D. Zhang, G. Xu, H. Yang, J. Li, H. Song and W.-H. Sun, J. Organomet. Chem., 2004, 689, 936 CrossRef CAS; (e) T. Hu, L.-M. Tang, X.-F. Li, Y.-S. Li and N.-H. Hu, Organometallics, 2005, 24, 2628 CrossRef CAS.
- (a) X. Tang, W.-H. Sun, T. Gao, J. Hou, J. Chen and W. Chen, J. Organomet. Chem., 2005, 690, 1570 CrossRef CAS; (b) Q.-Z. Yang, A. Kermagoret, M. Agostinho, O. Siri and P. Braunstein, Organometallics, 2006, 25, 5518 CrossRef CAS.
- F. Speiser, P. Braunstein and L. Saussine, Dalton Trans., 2004, 1539 RSC.
- P. Braunstein, Y. Chauvin, S. Mercier and L. Saussine, C. R. Chim., 2005, 8, 31 CrossRef CAS.
- P. Braunstein, Y. Chauvin, S. Mercier, L. Saussine, A. D. Cian and J. Fischer, J. Chem. Soc., Chem. Commun., 1994, 2203 RSC.
- J. Hou, W.-H. Sun, S. Zhang, H. Ma, Y. Deng and X. Lu, Organometallics, 2006, 25, 236 CrossRef CAS.
- (a) L. Wang, W.-H. Sun, L. Han, H. Yang, Y. Hu and X. Jin, J. Organomet. Chem., 2002, 658, 62 CrossRef CAS; (b) N. Ajellal, M. C. A. Kuhn, A. D. G. Boff, M. Hörner, C. M. Thomas, J.-F. Carpentier, Jr. and O. L. Casagrande, Organometallics, 2006, 25, 1213 CrossRef CAS; (c) S. Adewuyi, G. Li, S. Zhang, W. Wang, P. Hao, W.-H. Sun, N. Tang and J. Yi, J. Organomet. Chem., 2007, 692, 3532 CrossRef CAS; (d) M. Zhang, S. Zhang, P. Hao, S. Jie, W.-H. Sun, P. Li and X. Lu, Eur. J. Inorg. Chem., 2007, 3816 CrossRef CAS.
- (a) M. Helldörfer, J. Backhus, W. Milius and H. G. Alt, J. Mol. Catal. A: Chem., 2003, 193, 59 CrossRef; (b) M. M. Wegner, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 3624 CrossRef CAS; (c) F.-S. Liu, H.-B. Hu, Y. Xu, L.-H. Guo, S.-B. Zai, K.-M. Song, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789 CrossRef CAS.
- (a) R. J. Maldanis, J. S. Wood, A. Chandrasekaran, M. D. Rausch and J. C. W. Chien, J. Organomet. Chem., 2002, 645, 158 CrossRef CAS; (b) H.-R. Liu, P. T. Gomes, S. I. Costa, M. T. Duarte, R. Branquinho, A. C. Fernandes, J. C. W. Chien, R. P. Singh and M. M. Marques, J. Organomet. Chem., 2005, 690, 1314 CrossRef CAS; (c) C.-L. Song, L.-M. Tang, Y.-G. Li, X.-F. Li and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1964 CrossRef CAS; (d) B. K. Bahuleyan, G. W. Son, D.-W. Park, C.-S. Ha and Il. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1066 CrossRef CAS; (e) C. S. Popeney and Z. B. Guan, Macromolecules, 2010, 43, 4091 CrossRef CAS; (f) H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS.
- (a) D. P. Gates, S. A. Svejda, E. Onate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320 CrossRef CAS; (b) F. A. Kunrath, F. F. Mota, O. L. Casagrande, Jr., R. S. Mauler and R. F. de Souza, Macromol. Chem. Phys., 2002, 203, 2407 CrossRef CAS; (c) C. M. Killian, L. K. Johnson and M. Brookhart, Organometallics, 1997, 16, 2005 CrossRef CAS; (d) S. A. Svefda and M. Brookhart, Organometallics, 1999, 18, 65 CrossRef.
- J. Yu, H. Liu, W. Zhang, X. Hao and W.-H. Sun, Chem. Commun., 2011, 47, 3257 RSC.
- G. B. Galland, R. F. de. Mauler, R. S. Mauler and F. F. Nunes, Macromolecules, 1999, 32, 1620–1625 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC reference numbers 831465, 831466 and 831467 for crystallographic data of complexes C3, C5 and C9 respectively. |
| This journal is © The Royal Society of Chemistry 2012 |