Synthesis of dimethyl carbonate catalyzed by carboxylic functionalized imidazolium salt via transesterification reaction
Received
26th August 2011
, Accepted 7th November 2011
First published on 22nd December 2011
Abstract
We investigated the dependence of cations and anions of ionic liquids (ILs) on catalytic activity for the synthesis of dimethyl carbonate (DMC) via the transesterification of ethylene carbonate (EC) with methanol (CH3OH), and demonstrated that an easily prepared carboxylic functionalized imidazolium salt exhibited higher activity, 82% yield of DMC together with 99% selectivity was obtained under the metal-free and halogen-free conditions. The reaction mechanism was also proposed according to experimental and DFT studies. In addition, in order to simplify the catalyst separation and evaluate the catalyst stability, we also covalently anchored the functionalized imidazolium salt onto a highly cross-linked polystyrene resin (PS) as a heterogeneous catalyst for DMC synthesis, and continuously performed the reaction in a fixed bed reactor for 200 h without obvious loss of activity, which would have potential applications in industry. The process thus represented an environmentally friendly pathway for the synthesis of DMC via a transesterification reaction.
1. Introduction
Dimethyl carbonate has gained much attention as an environmentally friendly solvent or building block for the production of polycarbonate and other chemicals.1 Also, DMC has been mentioned as a potential additive to fuel oil due to its high oxygen content and octane number, good gasoline/water distribution coefficient, low toxicity, and rapid biodegradability.2 Additionally, it has been used as an electrolyte in lithium batteries due to its high dielectric constant.3 Therefore, the development of DMC production technology has become important for the oil and chemical industries.
Currently, there are three processes commercialized for the large-scale production of DMC,4 the phosgene process, the oxidative carbonylation of CH3OH process, and the two-step transesterification process (Scheme 1). Considerable effort has been devoted to the non-phosgene routes to DMC.5 In this context, the two-step transesterification process utilizing CO2 as a raw material is very attractive in view of the “sustainable society” and “green chemistry”.6 For the transesterification process, many homogeneous and heterogeneous catalysts have been successfully developed, such as tertiary amines,7 zirconium, titanium and tin salts or complexes,8 Group VBVIB complexes,9 Group V element-containing Lewis base and an epoxide or a cyclic amidine,10 alkali alcoholates or hydrates,11 alkali metal/or alkali metal compounds,12 polymers,13 and so on.14 Although significant progress has been made, unsatisfactory activities, instability or toxicity of metal-containing catalysts and harsh reaction conditions, are still the disadvantages needed to be overcome. So the development of more efficient, stable, easily prepared and environmentally friendly catalysts for DMC synthesis is still highly desired.
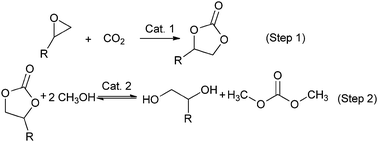 |
| | Scheme 1 Two-step process for DMC production. | |
Ionic liquids (ILs) have attracted significant attention from the scientific community from the viewpoint of “green chemistry” because of their special properties such as negligible vapor pressure, high thermal stability, environmental friendliness and easy designability.15 Various chemical reactions have been well performed in ILs.16 However, until recently, their potential use as catalysts for DMC synthesis through transesterification reactions have been reported.17
Park and coworkers first demonstrated that the transesterification of EC with CH3OH was successfully catalyzed by 1-alkyl-3-methylimidazolium ILs.17 He and coworkers ingeniously developed DABCO-derived (1,4-diazobicyclo[2. 2. 2]octane) ILs as effective catalysts for transesterification of EC with CH3OH.17d We have also initiated a program for environmentally friendly synthesis of DMC from epoxide, CO2 and CH3OH catalyzed by ILs through a two-step transesterification process.18 In this work, we systematically investigated the dependence of cations and anions of ILs on catalytic activity for synthesis of DMC via transesterification of EC with CH3OH, and demonstrated that an easily prepared carboxylic functionalized imidazolium salt exhibited higher activity, 82% yield of DMC together with 99% selectivity was obtained under the metal-free and halogen-free conditions. In addition, the reaction mechanism was proposed according to experimental and DFT studies. Another very interesting aspect of this work was that the corresponding PS supported imidazolium salt was also synthesized as a heterogeneous catalyst for DMC synthesis, and continuously performed in a fixed bed reactor for 200 h without obvious loss of activity, which could show much potential application in industry. This halogen-free and metal-free process thus represented an environmentally friendly pathway for synthesis of DMC.
2. Results and discussion
At the initial experimental study, the transesterification of EC with CH3OH was performed in the presence of a series of typical ILs to investigate the dependence of cations and anions on catalytic activity (Table 1, entries 1–9). The results revealed that anion activity increased in the following order: Cl− > PF6− > TFSI− > Br− > BF4− with the same cation (entries 1–3, 5–6). The 2-hydroxyethyl methyl carbonate (HEMC), being regarded as the intermediate for DMC formation, was detected and identified by 1HNMR and 13CNMR. Interestingly, the HEMC was preferentially formed in the presence of CnminTFSI (n = 4, 6, and 8). The EC conversion was decreasing as the alkyl length of the cation increased from C4 to C8 (entries 6–8), and the same trend was found for the BmimBF4 and OmimBF4 (entry 3 vs. 4), which indicated that a small bulk of cation was beneficial for improving catalytic activity. Additionally, other typical ILs (e.g. pyridinium salt, piperidinium salt, ammonium salt, phosphonium salt, choline chloride and guanidine salt) were also investigated (entries 10–18). The results showed these kinds of ILs demonstrated only moderate activity, and the selectivity of DMC was low for most of the ILs, being only 10% in the presence of HTAB (entry 15). In addition, the catalytic efficiency decreased in the order of Cl− > Br− >I− in the case of tetrabutylammonium salts (entries 12–14).
| Entry |
ILs |
EC Conv. (%)b |
DMC Yield (%)b |
HEMC Yield (%)b |
|
Reaction conditions: EC (1.76 g, 20 mmol), CH3OH (6.4 g, 200 mmol), ILs (0.2 mmol, 1 mol%), Temperature (110 °C), Time (80 min).
Conversion and yield were determined by GC using biphenyl as the internal standard.
DMIC (0.1 mmol, 0.5 mmol%). BminTFSI = (1-butyl-3-methylimidazoliumbis(trifluoromethylsulfonyl)imide, HminTFSI = (1-hexyl-3-methylimidazoliumbis(trifluoromthylsulfonyl)imide, OminTFSI = (1-octyl-3-methylimidazoliumbis(trifluoromethylsulfonyl)-imide, EmimOAc = 3-ethyl-1-methyl imidazolium acetate, BPBr = 1-n-butylpy-ridinium bromide, BMPBr = 1-butyl-1-methylpiperidinium bromide, TBAC = Tetrabutylammonium chloride, TBAB = Tetrabutylammonium bromide, HTAB = Hexadecyl trimethyl ammonium bromide, TTPC = Tetradecyltributylphosphonium chloride, CC = Choline chloride, GH = Guanidine hydrochloride, DMIC = 1,3-dimethylimidazolium-2-carboxylate.
|
| 1 |
BmimCl |
59 |
47 |
11 |
| 2 |
BmimBr |
39 |
25 |
13 |
| 3 |
BmimBF4 |
22 |
trace |
20 |
| 4 |
OmimBF4 |
17 |
trace |
16 |
| 5 |
BmimPF6 |
54 |
19 |
35 |
| 6 |
BmimTFSI |
43 |
trace |
42 |
| 7 |
HmimTFSI |
35 |
trace |
32 |
| 8 |
OmimTFSI |
23 |
trace |
22 |
| 9 |
EmimOAc |
50 |
31 |
19 |
| 10 |
BPBr |
54 |
26 |
27 |
| 11 |
BMPBr |
56 |
24 |
31 |
| 12 |
TBAC |
59 |
47 |
12 |
| 13 |
TBAB |
51 |
30 |
20 |
| 14 |
TBAI |
42 |
22 |
19 |
| 15 |
HTAB |
60 |
6 |
54 |
| 16 |
TTPC |
56 |
24 |
31 |
| 17 |
CC |
68 |
45 |
22 |
| 18 |
GH |
56 |
28 |
26 |
| 19 |
DMIC |
82 |
81 |
trace |
| 20c |
DMIC |
80 |
79 |
trace |
| 21 |
K2CO3 |
84 |
83 |
trace |
To sum up the above discussion, the catalytic activities of all of the typical ILs studied were unsatisfactory, and the DMC selectivity was also low due to the difficulty involving further conversion of HEMC. Subsequently, we then focused on the functionalized ILs. To be delighted, the carboxylic functionalized 1,3-dimethyl-imidazolium-2-carboxylate (DMIC), which could be easily prepared by imidazolium and DMC,19 observed high activity, 82% yield of DMC together with 99% selectivity was obtained, and the yield was even similar to conventional K2CO3 (entry 21). Furthermore, the transesterification reaction proceeded smoothly even at the low catalytic amount of 0.5% relative to EC (entry 20), which made the process more attractive. Interest in DMIC would presumably be thanks to facile preparation from commercially available and relatively inexpensive starting materials, without toxic metal or halogens and be environmentally benign. More importantly, CH3OH might be activated by its carboxylic group and special structure. Hence the DMIC was chosen as the model catalyst for further investigation.
2.2 Effect of other reaction parameters
With DMIC in hand, we investigated the influence of the reaction parameters on the transesterification EC with methanol for synthesis of DMC. The reaction temperature had a remarkable effect on the catalytic activity of this catalyst. The conversion of EC significantly increased with enhancement of the reaction temperature (Fig. 1). The conversion of EC was 5% at 50 °C, and increased to 82% at 110 °C. However, the selectivity of DMC was low due to the production of HEMC when the temperature was lower than 80 °C.
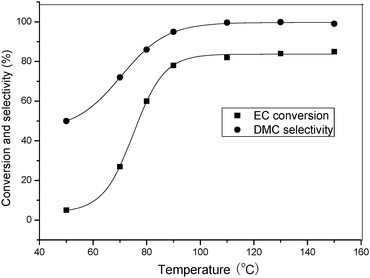 |
| | Fig. 1 Effect of reaction temperature on EC conversion and DMC selectivity. Reaction conditions: EC (1.76 g, 20 mmol), CH3OH (6.4 g, 200 mmol), DMIC (0.2 mmol, 1 mol%), Time (80 min). | |
Additionally, the influence of the CH3OH/EC feed molar ratio on the reaction was investigated under identical reaction conditions (Fig. 2). The results indicated that the CH3OH/EC feed molar ratio had a significant impact on the transesterification reaction. Both EC conversion and DMC selectivity increased gradually when the feed molar ratio of CH3OH/EC varied from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 20:1, which was presumably attributed to the reversible nature of the transesterification step.
:1 to 20:1, which was presumably attributed to the reversible nature of the transesterification step.
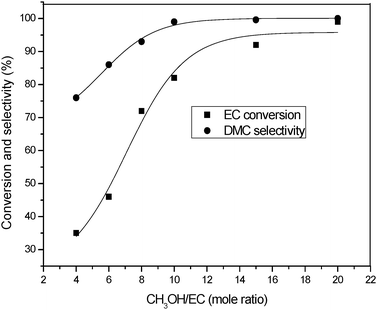 |
| | Fig. 2 The dependence of EC conversion and DMC yield on the molar ratio of CH3OH/EC. Reaction conditions: EC (1.76 g, 20 mmol), DMIC (0.2 mmol, 1 mol%), Temperature (110 °C), Time (80 min). | |
Subsequently, the dependence of DMC selectivity and EC conversion on reaction time was also evaluated. It was found that the reaction time had a remarkable influence on the reaction. As shown in Fig. 3, the reaction rate was faster in the initial stage and remained almost invariant after 80 min. This hinted that the transesterification reaction reached equilibrium after ca. 80 min under the reaction conditions. Conclusively, a reaction time of 80 min was appropriate for the synthesis of DMC in this study.
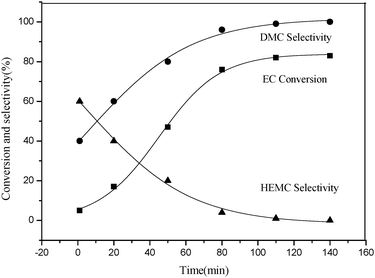 |
| | Fig. 3 Effect of reaction time on EC conversion, DMC and HEMC selectivity. Reaction conditions: EC (1.76 g, 20 mmol), CH3OH (6.4 g, 200 mmol), DMIC (0.2 mmol, 1 mol%), Temperature (110 °C). | |
2.3 Catalytic activity towards various substrates
Several substrates, alkyl alcohols including methanol, ethanol and 1-propanol, cyclic carbonates like EC and propylene carbonate (PC) were tested through the present process for synthesis of dialkyl carbonates (DAC) catalyzed by DMIC. As shown in Table 2, all the tested alcohols could be transesterified with EC or PC to the corresponding DAC. In addition, the EC conversion decreased with the increase of alkyl chain length of alcohols possibly because of the more serious equilibrium limitations (Table 2, entries 1, 3 and 5).20 Moreover, the results also demonstrated the activity of PC was much lower than EC under the same conditions, presumably due to its steric hindrance (entries 1 vs. 2 and 3 vs. 4, Table 2).
| Entry |
Alcohols |
Conv.(%)b |
DAC Selec. (%)b |
ME Selec. (%)b |
|
Reaction conditions: EC (1.76 g, 20 mmol), Alcohol (6.4 g, 200 mmol), DMIC (0.2 mmol, 1 mol%), Temperature (110 °C), Time (80 min).
Determined by GC.
PC (2.04 g, 20 mmol). ME = monoester.
|
| 1 |
CH3OH |
82 |
95 |
4 |
| 2c |
CH3OH |
38 |
92 |
5 |
| 3 |
C2H5OH |
46 |
86 |
10 |
| 4c |
C2H5OH |
3 |
99 |
1 |
| 5 |
C3H7OH |
20 |
76 |
20 |
2.4 A discussion of the reaction mechanism
Based on the published reports17d,21 and the results in this work, a probable catalytic cycle was proposed and depicted in Scheme 2. First, CH3OH was activated by DMIC through strong hydrogen bonding and more easily formed CH3O− (step 1), the DFT study disclosed the hydrogen bond length was 1.816 Å. Then, the CH3O− attacked the carbonyl carbon of EC and thus intermediate (I) was produced simultaneously (step 2). Subsequently, intermediate (II) (HEMC), which was a monoester product, was generated while the proton of CH3OH was captured by the intermediate (I) (step 3). Further nucleophilic substitution of intermediate (II) by CH3O− produced DMC and ethylene glycol anion (intermediate III) (step 4). Finally, EG was generated by proton transfer between intermediate (III) and CH3OH (step 5). Particularly, step 4 was proposed to be more difficult than step 2. Therefore it was reasonable that less active catalysts like the typical ILs especially with larger steric hindrance could give more monoester products, and low temperature also easily led to formation of HEMC.
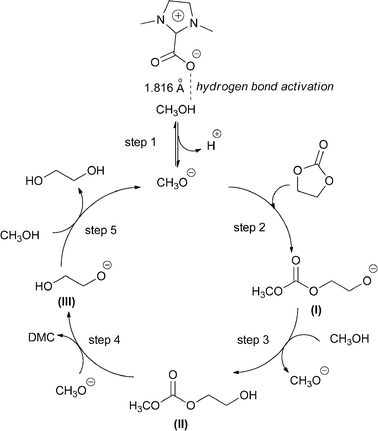 |
| | Scheme 2 Proposed mechanism for DMC formation. | |
2.5 Synthesis of PS supported DMIC and application in a fixed bed reactor
From the view of industrial application, heterogeneous catalysts are much more preferable than homogenous ones, and a further goal is the heterogenisation of DMIC. Being cheap, separable and easily surface-modified, PS were widely used in catalytic reactions.22 So we covalently anchored DMIC onto the PS as a heterogeneous catalyst for DMC synthesis (Scheme 4). The catalyst activity and stability were evaluated in a continuous flow fixed bed catalytic reactor. The continuous transesterification experiment was performed at 110 °C, 0.3 MPa and a molar ratio of CH3OH to EC of 4:1 with LHSV of 6.0 h−1, and the results were shown in Fig. 4. The EC conversion and DMC selectivity were around 67% and 95%, respectively, with about 5% unconverted HEMC. Furthermore, the PS-supported DMIC was continuously performed in the fixed bed reactor for 200 h without obvious loss of activity, which would show potential applications in industry.
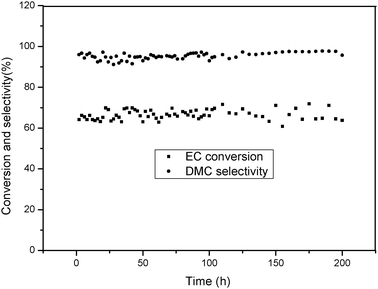 |
| | Fig. 4 Continuous transesterification experiment in a fixed bed reactor. Reaction conditions: molar ratio of CH3OH to EC of 4:1, 110 °C, 0.3 MPa, and catalyst total packed volume (4 mL), the LHSV (6.0 h−1). | |
3. Experimental
3.1 Materials
The ionic liquids were provided by Beijing zhongke anyin technology Co. Ltd. with a stated purity of > 99%, and dried in vacuo for 48 h before use. Highly cross-linked chloromethylated polystyrene (D301, 18–19% Cl content and 6% cross-linked with divinylbenzene) was received as a gift from Jiangsu Suqing Water Treatment Co., Ltd. (China). The alcohols, EC, imidazolium, DMC and PC were purchased from J&KCHEMICA without further purification, and other reagents were of analytical grade and used without further purification. The calculations were carried out by performing DFT by use of the B3PW91 functional with the 6-31++G(d,p) basis set as implemented in Gaussian 03 program package. GC-MS was measured on a Finnigan HP G1800 A, and GC analyses were performed on an Agilent GC-6820 equipped with a capillary column (DB-624, 30 m × 0.32 μm) using a flame ionization detector. NMR spectra were recorded on a Bruker 300 or Varian 400 spectrometer in CDCl3, and 1H and 13C NMR chemical shifts (δ) were given in ppm relative to TMS. 1H and 13C positive chemical shifts (δ) in ppm were downfield from tetramethylsilane (CDCl3: δC = 77.0 ppm; residual CHCl3 in CDCl3: δH = 7.26 ppm). Elemental analysis was carried out by using a Vario EL (Elementar Analysensysteme GmbH). Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Nicolet 380 spectrophotometer with anhydrous KBr as standard (Thermo Electron Co.).
3.2. Preparation of DMIC
The DMIC was prepared according to a previously reported procedure (Scheme 3),17 a mixture of DMC (1.5 mL) and 1-methylimidazole (1 mL) was heated in a 25 mL stainless-steel reactor equipped with a magnetic stirrer and automatic temperature control system for 24 h at 120 °C in a nitrogen atmosphere. After removing the solvent under reduced pressure, a yellow solid was obtained, then a white solid was further obtained by recrystallized from CH3OH. Yield: 86%, Anal: Found: C 51.07, H 5.72, N 19.66, calculated for C6H8N2O2: C 51.43, H 5.76, N 19.94. IR spectrum (NaBr): 3442, 2988, 1631, 1555, 1410, 1378, 1324, 1163, 916, 830, 783, 735, 631. 1H NMR (400 MHz, D2O): 3.84 (s, 6H, CH3), 7.24 (d, 2H, H-4 and H-5). 13C NMR (100.6 MHz, D2O): 37.16, 123.45, 140.16, 158.55.
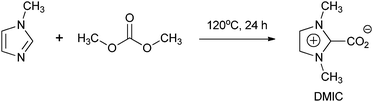 |
| | Scheme 3 Synthesis of DMIC. | |
3.3. Preparation of PS supported DMIC
The PS supported imidazole was prepared according to our previous procedure (Scheme 4, step 1).18d A mixture of D301 (10.0 g, 53.4 mmol), imidazole (53.5 mmol) and acetonitrile (70 mL) was heated at 80 °C for 24 h in a 125 mL three-necked flask. After being cooled to room temperature, the solid residue was collected by filtration and washed separately with ethyl acetate, 0.1 mol L−1 HCl, water and methanol. Then, the solid was dried under vacuum at 60 °C for 12 h to give PS supported imidazolium a 98% yield. The loading of imidazole attached on the resin was about 5.3 mmol g−1 determined by nitrogen content from elementary analysis.
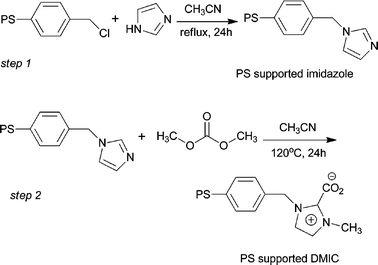 |
| | Scheme 4 Synthesis of PS supported DMIC. | |
The PS supported DMIC was prepared according to the following procedure (Scheme 4, step 2). A mixture of PS (10 g) supported imidazole and DMC (5 g) was heated in a 100 mL stainless-steel reactor equipped with a magnetic stirrer and automatic temperature control system for 24 h at 120 °C in a nitrogen atmosphere. After being cooled to room temperature, the solid residue was collected by filtration and washed with ethyl acetate. Then, the solid was dried under vacuum at 60 °C for 12 h to give the PS supported DMIC a 96% yield. The loading of imidazole attached on the resin was about 3.5 mmol g−1 determined by oxygen content from elementary analysis. The PS supported DMIC was further characterized by IR spectra: IR spectrum (NaBr): 3361, 3105, 2921, 1669, 1506, 1445, 1422, 1282, 1228, 1107, 1074, 1028, 740, 696, 661, 615.
3.4. Representative procedure for transesterification reaction
A mixture of EC (1.76 g, 20 mmol), CH3OH (6.4 g, 200 mmol) and ILs (0.2 mmol) was added to a 50 mL stainless-steel reactor equipped with a magnetic stirrer and automatic temperature control system. The reaction was carried out for 110 °C and 80 min. The vessel was then cooled to room temperature. The products were analyzed by GC using biphenyl as the internal standard and identified by GC-MS by comparing retention times and fragmentation patterns with authentic samples. The HEMC was further characterized by NMR spectra: 1H NMR (300 MHz, CDCl3): 2.43 (s, 1H, OH), 3.80 3.84 (s, 3H, CH3), (t, 2H, CH2OH), 4.26 (t, 2H, OCH2). 13C {1H} NMR (75 MHz, CDCl3): 54.85, 60.76, 69.36, 155.59.
3.5. Representative procedure for continuous transesterification experiment in fixed bed reactor
The continuous transesterification experiment was performed in a fixed bed reactor at 110 °C, 0.3 MPa (self-pressure control) and molar ratio of CH3OH to EC of 4:1. The catalyst total packed volume and the LHSV were 4 mL and 6.0 h−1 respectively. The products were analyzed by GC using biphenyl as the internal standard and identified by GC-MS by comparing retention times and fragmentation patterns with authentic samples.
Conclusions
We investigated the dependence of cations and anions of ILs on catalytic activity for synthesis of DMC via transesterification of EC with CH3OH, and found an easily prepared carboxylic functionalized imidazolium salt exhibited higher activity, 82% yield of DMC together with 99% selectivity was obtained under the metal-free and halogen-free conditions. In order to simply recycle the catalyst and evaluate the catalyst stability, we also covalently anchored the functionalized imidazolium salt onto the PS as a heterogeneous catalyst for DMC synthesis, and continuously performed in a fixed bed reactor for 200 h without obvious loss of activity, which could have potential applications in industry. The carboxylic functionalized imidazolium salt used in this study represented an easily synthesized, cheap, extremely robust, and environmentally benign catalyst, which can effectively activate methanol through hydrogen bond interactions. This halogen-free and metal-free process is thus an environmentally friendly pathway for synthesising DMC. Further structure optimizations of this functionalized imidazolium salt and also detailed mechanisms by DFT studies are currently under investigation in our laboratory.
Acknowledgements
Support of the grant from the National Basic Research Program of China (2009CB219901) and National Natural Sciences Foundation of China (No. 21003129, 21036007) is gratefully acknowledged. We thank Professor Liang-Nian He in Nankai University for useful discussions.
Notes and references
-
(a) F. Arico and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 Search PubMed;
(b) B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS;
(c) B. Schaffner, J. Holz, S. P. Verevkin and A. Borner, ChemSusChem, 2008, 1, 249–253 CrossRef;
(d) X. W. Miao, C. Fischmeister, C. Bruneau and P. H. Dixneuf, ChemSusChem, 2008, 1, 813–816 CrossRef CAS;
(e) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS;
(f) Y. Sato, T. Yamamoto and Y. Souma, Catal. Lett., 2000, 65, 123–126 CrossRef CAS;
(g) P. Tundo, Pure Appl. Chem., 2001, 73, 1117–1124 CrossRef CAS;
(h) S. Fujita, B. M. Bhanage, Y. Ikushima and M. Arai, Green Chem., 2001, 3, 87–91 RSC.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
-
(a) M. S. Park, G. X. Wang, Y. M. Kang, D. Wexler, S. X. Dou and H. Kiu, Angew. Chem., Int. Ed., 2007, 46, 750–753 CrossRef CAS;
(b) T. Wei, M. H. Wang, W. Wei, Y. H. San and B. Zhong, Green Chem., 2003, 5, 343–346 RSC.
-
(a) N. Keller, G. Rebmann and V. Keller, J. Mol. Catal. A: Chem., 2010, 317, 1–18 Search PubMed;
(b) R. X. Jiang, X. Q. Zhao and Y. J. Wang, Appl. Catal., A, 2003, 238, 131–139 CAS;
(c) Y. J. Wang, X. Q. Zhao and B. G. Yuan, Appl. Catal., A, 1998, 171, 255–260 CrossRef.
-
(a) A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CAS;
(b) K. Kohono, J. C. Choi, Y. Ohshima, H. Yasuda and T. Sakakura, ChemSusChem, 2008, 1, 186–188 CrossRef CAS;
(c) K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef;
(d) A.-A. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS;
(e) N. S. Isaacs and B. O'Sullivan, Tetrahedron, 1999, 55, 11949–11956 CrossRef CAS;
(f) S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konnog, Green Chem., 2003, 5, 497–507 RSC;
(g) J. C. Choi, L. N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230–234 RSC.
-
(a) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC;
(b) P. T. Anastas, Green Chem., 2003, 5, G29–G34 RSC;
(c) T. C. Williamson, M. Kirchhoff and P. T. Anastas, Green Chem., 2000, 2, G85–G96 RSC;
(d) P. T. Anastas and R. L. Lankey, Green Chem., 2000, 2, 289–295 RSC;
(e) B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 429–432 RSC;
(f) B. M. Bhanage, S. I. Fujita, Y. Ikushima, K. Torii and M. Arai, Green Chem., 2003, 5, 71–75 RSC;
(g) K. V. M. Selva, A. Perosa and M. Fabris, Green Chem., 2008, 10, 1068–1077 RSC.
-
(a)
U. Romano and U. Melis, US Patent, 4 062 884, 1977, to ANIC Search PubMed;
(b)
L. K. French and J. A. Gilpin, US Patent, 3 642 858, 1972, to Dow Chemical Co Search PubMed.
-
J. F. Knifton, US Patent, 4 661 609, 1987, to Texaco Inc Search PubMed.
-
J. F. Knifton, US Patent, 4 734 518, 1988, to Texaco Inc Search PubMed.
-
M. J. Green, US Patent, 4 559 180, 1985 , to BP Chemicals Limited Search PubMed.
-
H. J. B. Krefeld, A. K. Stolberg, R. L. Krefeld and F. J. M. Duesseldorf, US Patent, 523 112, 1993, to Bayer Aktiengesellschaft Search PubMed.
-
(a) M. Sankar, S. Satav and P. Manikandan, ChemSusChem, 2010, 3, 575–578 CrossRef CAS;
(b) G. Stoica, S. Abello and J. Perez-Ramirez, ChemSusChem, 2009, 2, 301–304 CrossRef CAS;
(c) Z. Z. Yang, X. Y. Dou, F. Wu and L. N. He, Can. J. Chem., 2011, 89, 544–548 Search PubMed;
(d) C. Murugan, H. C. Bajaj and R. V. Jasra, Catal. Lett., 2010, 137, 224–231 Search PubMed;
(e) G. Stoica, S. Abello and J. Perez-ramirez, Appl. Catal., A, 2009, 365, 252–260 Search PubMed.
-
(a) S. R. Jagtap, M. D. Bhor and B. M. Bhanage, Catal. Commun., 2008, 9, 1928–1931 Search PubMed;
(b) J. S. Tian, C. X. Miao, J. Q. Wang, F. Cai, Y. Du, Y. Zhao and L. N. He, Green Chem., 2007, 9, 566–571 RSC.
-
(a) B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259–266 CrossRef CAS;
(b) Y. H. Chang, T. Jiang, B. X. Han, Z. M. Liu, W. Z. Wu, L. Gao, J. C. Li, H. X. Gao, G. Y. Zhao and J. Huang, Appl. Catal., A, 2004, 263, 179–186 CrossRef CAS;
(c) Y. Li, X. Q. Zhao and Y. J. Wang, Appl. Catal., A, 2005, 279, 205–208 CrossRef CAS;
(d) Y. Kishimoto and I. Ogawa, Ind. Eng. Chem. Res., 2004, 43, 8155–8162 CrossRef CAS;
(e) M. Sankar, C. Madhavan Nair, G. K. Murty and P. Manikandan, Appl. Catal., A, 2006, 312, 108–114 CrossRef CAS;
(f) C. W. Hu, F. W. Chen, T. Dong, Y. N. Chi and Y. Q. Xu, Catal Today, 2010, 139, 38–41;
(g) B. M. Bhanage, S. R. Jagtap, V. P. Raje and S. D. Samant, J. Mol. Catal. A: Chem., 2007, 266, 69–74 CrossRef CAS;
(h) S. G. Davies, A. M. Fletcher, W. Kurosawa, J. A. Lee, G. Poce, P. M. Roberts, J. E. Thmson and D. M. Williamson, J. Org. Chem., 2010, 75, 7745–7756 CrossRef CAS.
-
(a) R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef;
(b) T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS;
(c) P. J. Scammels, J. L. Scott and R. D. Singer, Aust. J. Chem., 2005, 58, 155–169 CrossRef CAS;
(d) D. R. MacFarlane and R. K. Seddon, Aust. J. Chem., 2007, 60, 3–5 CrossRef CAS;
(e) T. L. Greaves and C. J. Drummond, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS;
(f)
An introduction to ionic liquids, ed. M. Freemantle, RSC Publishing, Cambridge, UK, 2009, p. 281 Search PubMed;
(g)
Ionic liquids, ed. B. Kirchner, Springer, Heidelberg, Germany, 2010, p. 345 Search PubMed.
-
(a) I. P. Vasile and H. Christopher, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS;
(b) V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS.
-
(a) H. Y. Ju, M. D. Manju and D. W. Park, React. Kinet. Catal. Lett., 2007, 90, 3–9 CrossRef CAS;
(b) D. W. Kim, C. H. Kim, J. C. Koh and D. W. Park, J. Ind. Eng. Chem., 2010, 16, 474–478 Search PubMed;
(c) M. M. Dharman, H. Y. Ju, H. L. Shim, M. K. Lee, K. H. Kim and D. W. Park, J. Mol. Catal. A: Chem., 2009, 303, 96–101 CrossRef CAS;
(d) Z. Z. Yang, L. N. He, X. Y. Dou and S. Chanfreau, Tetrahedron Lett., 2010, 51, 2931–2934 CrossRef CAS;
(e) D. W. Kim, D. O. Lim, D. H. Koh and D. W. Park, Catal. Today, 2011, 146, 556–560 Search PubMed.
-
(a) J. Q. Wang, J. Sun, C. Y. Shi, W. G. Cheng, X. P. Zhang and S. J. Zhang, Green Chem., 2011, 13, 3213 RSC;
(b) J. Sun, L. J. Han, W. G. Cheng, J. Q. Wang, X. P. Zhang and S. J. Zhang, ChemSusChem, 2011, 4, 502–507 CrossRef CAS;
(c) J. Sun, S. J. Zhang, W. G. Cheng and J. Y. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS;
(d) J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS;
(e) J. Sun, J. Y. Ren, S. J. Zhang and W. G. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasid and R. D. Rogers, Chem. Commun., 2003, 28–29 RSC.
-
(a) Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS;
(b) M. Honda, S. Kuno, N. Begum, K. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165–170 CrossRef CAS.
-
(a) Y. J. Fang and W. D. Xiao, Sep. Purif. Technol., 2004, 34, 255–263 CrossRef CAS;
(b) M. Fan and P. Zhang, Energy Fuels, 2007, 21, 633–635 Search PubMed.
- M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401–3429 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.