Organometallic hydrogen transfer and dehydrogenation catalysts for the conversion of bio-renewable alcohols
Andrew C.
Marr
School of Chemistry and Chemical Engineering, Queen's University, Belfast, David Keir Building, Belfast, UK BT9 5AG. E-mail: a.marr@qub.ac.uk; Fax: +44 (0)28 9097 6524; Tel: +44 (0)28 9097 4442
First published on 13th October 2011
Abstract
Organometallic hydrogen transfer and dehydrogenation provide straightforward atom efficient routes from alcohols to a variety of chemical products. The potential of these reactions to enable the conversion of biomass to value added chemicals is discussed, with reference to the products that can be prepared from aliphatic alcohols in good isolated yield.
 Andrew C. Marr | Dr Andrew Marr is green chemist. He completed his PhD training in homogeneous catalysis in 1998 (St Andrews) and then postdoctoral research in Nottingham (1998–2000) and Bristol (2000–2001), before being appointed to Queen's University Belfast, where he is a faculty member in inorganic chemistry. |
1. Introduction
As the cost of petroleum increases alternative feedstocks for chemical products will become more viable. This review examines the role of catalyzed hydrogen transfer reactions in utilizing alcohols that could be derived from biomass by fermentation and/or chemical transformation.1The alcoholic beverage industry stands as testament to the commercial potential of fermentation as a source of chemicals. Modern fermentation technologies, such as anaerobic fermentations,2 are becoming ever more diverse and a range of bulk chemical products can be obtained. Of note is the facile production of large quantities of alcohol products. As a result of well developed biocatalytic routes bio-ethanol and bio-butanol are being considered as potential petroleum replacements.3,4
The importance of fermentation as a source of bio-renewable chemicals was recognised recently with the award of 2011 EPA Presidential Green Chemistry Challenge Awards to Genomatica5 and BioAmber.6 Genomatica has developed fermentation technologies from sugars to 1,4-butanediol. BioAmber's core business is the production of bio-renewable succinnic acid and downstream products including 1,4-butanediol. The success of BioAmber shows that bio-renewable chemicals can be prepared at lower cost than fossil fuel equivalents.
During the fossil fuel era the energy and chemicals industries have had a close association. This has arisen due to the existence of high levels of carbon within the principle fuels of coal, petroleum and natural gas. Looking to the future, and the diversification of energy towards a more complex mixture of technologies, many energy-yielding technologies will be independent of carbon, and the chemical industries will not be able to rely on the energy industry for raw materials. In such circumstances, a potentially cheap source of organic functionality is biomass. The conversion of biomass into chemicals has been considered in a number of recent reviews.7–10 Of the bio-renewable feeds, biomass waste has particular merit due to its scale, availability, low cost and minimal impact on food production.
From the literature on the production of chemicals from biomass, it becomes clear that carboxylic acid and aliphatic alcohol functional groups are extremely common in the primary products. This review will concentrate on the chemical conversion of aliphatic alcohols of relevance to the biomass sector and the use of hydrogen transfer and dehydrogenation to activate aliphatic alcohols, and enable their incorporation into higher value chemical products. Products that can be derived from the coupling of amines and alcohols are emphasized, and in particular amines that may be prepared from aliphatic alcohols and ammonia.
2. Organometallic hydrogen transfer and dehydrogenation
In organometallic hydrogen transfer catalysis (a.k.a. hydrogen borrowing/autotransfer) a metal complex is used to increase the rate of a redox reaction which involves the transfer of hydrogen from one substrate, the donor, to another, the acceptor. It has been noted that this is particularly useful for the activation of alcohols11,12 which readily act as hydrogen donors and are oxidized to aldehydes or ketones.Early work on activating alcohols by hydrogen transfer was reviewed in 2007,13,14 and more recently dehydrogenation as a means of activating organic substrates, including alcohols, has been reviewed.15,16
Common catalysts for hydrogen transfer and dehydrogenation are based on Ir and Ru, although a few other catalysts, for example Fe catalysts,17,18 have been reported. The strength of the approach is that the primary product of alcohol oxidation, the aldehyde or ketone, can be reacted with an added reagent, producing a more complex product in one pot with no intermediate separation. The most common application of this cascade is the N-alkylation of amines by dehydrogenation of the alcohol, reaction to yield an imine and subsequent hydrogenation to yield an alkylated amine, the only by-product of the reaction is water (Scheme 1).
 | ||
| Scheme 1 N-Alkylation of an amine (amination of an alcohol) by hydrogen transfer. | ||
The synthesis of amines by organometallic homogeneous hydrogen transfer affords selectivities unattainable by other routes and catalytic reactions can be tuned to produce primary, secondary or tertiary amines with remarkable isolated yields.
A reaction has been developed in which dihydrogen rather than water is generated, leading to amides (Scheme 2).19

Catalytic hydrogen transfer is therefore a suitable strategy for the incorporation of aliphatic alcohols derived from biomass into amine and amide products.
This perspective focuses on the synthetic utility of these reactions, mechanistic aspects have been reviewed elsewhere.20,13–16
3. The incorporation of renewable alcohols into amines and amides
We can already predict the chemicals available from the fermentation and chemical modification of biomass.7,8 Saccharides are particularly suited to the production of aliphatic alcohols, and this has led to projected applications in energy, as noted above. In addition to mono-ol products, notable is the prevalence of diols amongst expected products, with examples including 1,3-propanediol via 3-hydroxy propanoic acid, 1,2-propanediol via lactic acid, 1,4-butanediol via succinic acid, 1,5-pentanediol via citric acid, amino diols via glutamic acid and methyl 1,4-diol via itaconic acid. Another rich source of propanediols is glycerol derived from fats and oils. Many interesting hydrogen transfer reactions have been reported employing aliphatic diols over the last decade and some exemplars are given; particular note is made of reactions that convert aliphatic alcohols to products in good isolated yield.3.1 Amination of Mono-ols
The hydrogen transfer of an alcohol in the presence of an amine will lead to the formation of a new N–C bond and ultimately an N-alkylated amine product.In 1981 Ron Grigg (whilst he was Professor of Organic Chemistry at Queen's Belfast) and co-workers reported the alkylation of amines by aliphatic alcohols catalyzed by [RhH(PPh3)4] under relatively mild conditions (e.g. methylation in methanol under reflux) affording high yields by GC-MS (Scheme 3).21 Applying 5 mol% catalyst shortened reaction times to a few hours in many cases. In the same paper the activities of Ru and Ir complexes were also demonstrated, these metals have come to dominate the field.
 | ||
| Scheme 3 RhH(PPh3)4 catalyzed methylation of aliphatic alcohols (*yield by GC-MS).21 | ||
Watanabe and co-workers introduced many of the most popular protocols for N-alkylation. In their early work they showed aniline could be successfully alkylated with primary aliphatic alcohols by employing Ru catalysts.22,23 [RuCl2(PPh3)3] catalyzed the reaction of ethanol, 1-propanol and 1-butanol with anilines at 180 °C to give a mixture of mono and di-alkylated anilines (Scheme 4, yields are quoted are isolated yields unless otherwise stated), no solvent was required. 0.5–1 mol% of catalyst with respect to substrate was used and this is a typical catalyst loading in modern reactions.
 | ||
| Scheme 4 N-alkylation of aniline by n-alcohols.22,23 | ||
[RuH2(PPh3)4] catalyses the coupling of aliphatic amines and simple aliphatic alcohols under similar conditions.24 More recently new catalysts have been discovered that enable coupling at lower temperatures. [CpRuCl(PPh3)2] (1 mol%) can be employed as a catalyst precursor to methylate a wide range of aliphatic amines with methanol at 100 °C (Scheme 5).25 In some cases total conversion was reported after a few hours. Kinetic studies on the reaction of primary and secondary amines with high concentrations of methanol in the presence of [CpRuCl(PPh3)2] (Cp = (η5-C5H5)) suggested that reaction rates increased as the basicity of the amine was increased.
![Aliphatic amine methylation catalyzed by [CpRuCl(PPh3)2].25](/image/article/2012/CY/c1cy00338k/c1cy00338k-s5.gif) | ||
| Scheme 5 Aliphatic amine methylation catalyzed by [CpRuCl(PPh3)2].25 | ||
Employing a Ru pincer complex at 0.1 mol% with respect to the alcohol, primary amines have been prepared from the reaction of primary amines and ammonia in organic solvents and water.26 This is a key transformation as it enables the more sustainable synthesis of simple amines, thus allowing the possibility of fully bio-renewable synthesis of primary, secondary and tertiary amines by employing hydrogen transfer mediated syntheses. Highly successful transformations are summarised in Scheme 6. Reaction times of 12–32 h were typical.
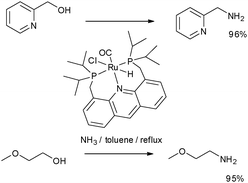 | ||
| Scheme 6 Primary amine synthesis by N-alkylation of alcohols.26 | ||
Vogt and co-workers have extended the reaction to the conversion of secondary alcohols to primary amines employing ammonia.27 Some good results were obtained by employing a catalyst based on Ru3(CO)12 (1 mol%) modified by phosphine ligands, an example is given in Scheme 7.
 | ||
| Scheme 7 The conversion of secondary alcohols to primary amines.27 | ||
A range of other catalysts for the N-alkylation of amines by alcohols have been reported based on RuCl3·nH2O,28,29 Ru3(CO)1230 and half sandwich complexes such as [RuCl2(p-cymene)]231 modified by a variety of ligands, typically phosphines e.g. n-tributylphosphine, tri-o-tolylphosphine, 1,1′-bis(diphenylphosphino)ferrocene) (dppf). Some combinations are suitable for the alkylation of amines by aliphatic alcohols.
The combination of [RuCl2(p-cymene)]2 and dppf or bis(2-diphenylphosphinophenyl)ether (DPEphos) could be used at 0.5 mol% in a wide range of N-alkylations in toluene, including alkylation of secondary amines.32 For example [RuCl2(p-cymene)]2 and DPEphos catalyzed the coupling of 1-octanol and dimethylamine in toluene heated to reflux for 24 h giving N,N-Dimethyloctylamine in 76% isolated yield, and morpholine was alkylated with pentan-2-ol using the same catalytic system heated in xylene at 150 °C for 24 h to give 4-(Pentan-2-yl)morpholine in 88% isolated yield (Scheme 8).
![N-alkylation of secondary alcohols catalyzed by [RuCl2(p-cymene)]2 promoted by phosphine.32](/image/article/2012/CY/c1cy00338k/c1cy00338k-s8.gif) | ||
| Scheme 8 N-alkylation of secondary alcohols catalyzed by [RuCl2(p-cymene)]2 promoted by phosphine.32 | ||
[Ru3(CO)12] promoted by N-phenyl-2-(dicyclohexylphosphanyl) pyrrole can also catalyse the amination of secondary amines.33
Urea has been coupled with a range of alcohols catalyzed by heterogenized ruthenium catalysts.16,34 The reaction could be effected using ruthenium hydroxide supported on titanium dioxide. This is one example from a range of heterogeneous catalysts for N-alkylation that are now being discovered. Beller and co-workers have examined some recent approaches to the synthesis of amines.35
Iridium centred homogeneous catalysts have been increasingly applied to amination reactions. Amongst the most successful is [Cp*IrCl2]2 (Cp* = η5-pentamethyl cyclopentadienyl), reported by Yamaguchi and co-workers.36 1-octanol has been reacted with aniline in the presence of 2 mol% [Cp*IrCl2]2 and NaHCO3 to yield the secondary amine in 97% isolated yield, the protocol was also successful for aliphatic secondary amines, and pyrrolidine could coupled with 1-octanol in 96% isolated yield (Scheme 9).
![N-alkylation catalyzed by [Cp*IrCl2]2.36](/image/article/2012/CY/c1cy00338k/c1cy00338k-s9.gif) | ||
| Scheme 9 N-alkylation catalyzed by [Cp*IrCl2]2.36 | ||
Recently Yamaguchi and co-workers have developed their system to render it recyclable, air-stable and water soluble.37 [Cp*Ir(NH3)3][X]2 catalyzed the N-alkylation of primary and secondary amines in water. For example the reaction of 1-hexylamine and 1-hexanol in water under air catalyzed by 1.0 mol% of [Cp*Ir(NH3)3][I]2 gave dihexylamine in 84% yield after 14 h. Under the same conditions dihexylamine and 1-hexanol coupled to give trihexylamine in 82% yield (Scheme 10).
 | ||
| Scheme 10 Coupling of aliphatic amines and aliphatic alcohols in water.37 | ||
Cp*Ir complexes ligated with N-heterocyclic carbenes (NHC) are powerful hydrogen transfer catalysts and their application to Oppenauer oxidation38 and chemoenzymic Dynamic Kinetic Resolution39 have been reported. Excellent results are also achieved in the N-alkylation of amines. Peris and co-workers40,41 tested a range of [Cp*IrCl2(NHC)] complexes as catalysts for the N-alkylation of alcohols, the bis-butyl imidazole carbene complex was found to catalyse a range of couplings. For example benzylamine was alkylated with an excess of 1-butanol in greater than 95% conversion (by NMR), giving a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of mono and di-alkylated products after 7 h at 110 °C using 5 mol% of catalyst (Scheme 11). Running the reaction for longer increased the proportion of the tertiary amine. Chloride containing catalyst precursors required the addition of AgOTf (OTf = trifluoromethanesulfonate) as an activator.
:1 ratio of mono and di-alkylated products after 7 h at 110 °C using 5 mol% of catalyst (Scheme 11). Running the reaction for longer increased the proportion of the tertiary amine. Chloride containing catalyst precursors required the addition of AgOTf (OTf = trifluoromethanesulfonate) as an activator.
![N-alkylation catalyzed by a [Cp*Ir(NHC)] complex, * yield by NMR.40](/image/article/2012/CY/c1cy00338k/c1cy00338k-s11.gif) | ||
| Scheme 11 N-alkylation catalyzed by a [Cp*Ir(NHC)] complex, * yield by NMR.40 | ||
Chelating ligands can be prepared by tethering a donor ligand to a cyclopentadienyl ring. Yamaguchi and co-workers demonstrated the application of amine functionalised cyclopentadienyl Ir NHC complexes to Oppenauer-type oxidation.42 The amine was appended in order to render the addition of a base unnecessary. We demonstrated the high transfer hydrogenation activity of highly chelated complexes of Rh by tethering Cp ligands to phosphines by dehydrofluorinative coupling.43 Pontes da Costa et al. developed the tethering strategy and prepared an Ir N-alkylation catalyst containing a Cp*-NHC ligand.41 Aniline could be monoalkylated by 1-butanol in 55% yield after 24 h.
Crabtree and co-workers have prepared air stable hydrogen transfer catalysts based on the Cp*Ir(NHC) motif.44 The NHC was modified by attaching a pyrimidine group to provide a chelating ligand. Benzyl amine was alkylated with 1-butanol in 56% yield (by NMR).
Although the majority of catalysts involve either Ru or Ir metal, the search for base metal catalysts was furthered recently with excellent isolated yields achieved for Fe based homogeneous catalysts.45 Catalysts were prepared in situ from Fe(III) halides (3 mol%), Cp*H and amino acids. The best system catalyzed the coupling of benzyl alcohol and aniline in 94% isolated yield after 24 h.
3.2 Amination of diols
The hydrogen transfer mediated coupling of primary amines and diols is an effective method for the formation of ring systems. Some of the earliest work was carried out by Watanabe and co-workers employing simple Ru phosphine complexes under fairly forcing conditions (150–180 °C).46 1,5-diols could be utilized for the preparation of piperidines, diethylene glycol for morpholines and diethanolamines for piperazines.47 Yields were highly variable but several good synthetic routes were identified. The reaction of aniline and ethylene glycol catalysed by [RuCl2(PPh3)3] gave a good yield of 1,4-diphenylpiperazine (78% isolated, Scheme 12).![Synthesis of piperazines catalyzed by [RuCl2(PPh3)3].47](/image/article/2012/CY/c1cy00338k/c1cy00338k-s12.gif) | ||
| Scheme 12 Synthesis of piperazines catalyzed by [RuCl2(PPh3)3].47 | ||
Marsella showed the coupling of secondary amines and ethylene glycol or 1,3-propanediol could be affected to yield mono-amines using [RuCl2(PPh3)3].48 In the same study RuCl3·nH2O and IrCl3·nH2O promoted by PPh3 were shown to promote the formation of diamines. Marsella later reported the [RuCl2(PPh3)3] catalyzed coupling of primary amines and ethylene glycol to form a mixture of amino alcohols, diamines and piperazines.49 Varying the substituent on the amine through methyl, iso-butyl, sec-butyl, neopentyl and tert-butyl, a trend in selectivity was observed, diamination was favoured by small alkyl groups.
RuCl3·nH2O with and without a phosphine promoter was used to couple ethylene glycol, 1,3-propanediol, 1,4-butanediol and 1,5-pentanediol to a range of aliphatic amines to prepare diamino compounds.28,50 For example 1,2-dipiperidinoethane could be prepared from piperidine and ethylene glycol in 79% isolated yield after 5 h in the presence of RuCl3·nH2O (1 mol% with respect to diol) at 180 °C in dioxane (Scheme 13).50

Imidazoles were prepared by the [RuCl2(PPh3)3] catalyzed reaction of N,N′-disubstituted ureas with vicinal-diols.51 A range of 1,2-diols were shown to be active, the most successful reaction reported was the coupling of N,N′-dimethylurea with propylene Glycol at 180 °C in diglyme for 12 h employing 2 mol% catalyst wrt diol (Scheme 14).
![Synthesis of imidazoles catalyzed by [RuCl2(PPh3)3], * yield by GLC.51](/image/article/2012/CY/c1cy00338k/c1cy00338k-s14.gif) | ||
| Scheme 14 Synthesis of imidazoles catalyzed by [RuCl2(PPh3)3], * yield by GLC.51 | ||
[RuCl2(PPh3)3] also catalyzed indole and quinoline formation at 180 °C.47 Indoles could be prepared via the hydrogen transfer activated coupling of aniline derivatives and 1,2-diols; similarly quinolines were prepared from aminoarenes and 1,3-propanediol. Substituted diols were employed to give a range of substituted indoles. The best yields obtained for indole formation were in the order of 50% employing ethylene glycol. The formation of quinolines was also catalyzed by the combination of RuCl3·nH2O and PBun3,52 the best conditions were found to be heating to reflux in diglyme and a P:Ru ration of 2:1 and 3 mol% catalyst wrt diol. Quinoline could be prepared from aniline and 1,3-propanediol in 59% isolated yield (Scheme 15).
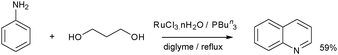 | ||
| Scheme 15 Synthesis of quinoline from aniline and 1,3-propanediol.52 | ||
Recently 1,2-diols and anilines have been coupled to yield indoles under neat conditions employing phosphine promoted RuCl3·nH2O systems.53 Ru (1 mol%) performed best when promoted by dimethylbisdiphenylphosphinoxanthene (xantphos) (Scheme 16).
 | ||
| Scheme 16 Synthesis of indoles catalyzed by RuCl3·nH2O promoted by phosphines.53 | ||
The addition of a co-catalyst can aid the hydrogen transfer mediated reaction between amines and alcohols.54 This has recently been demonstrated for the synthesis of quinolines from anilines and 1,3-diols.55 Madsen and co-workers added MgBr2·OEt2 to the catalytic system as a promoter. A wide variety of quinolines were prepared and isolated, with isolated yields ranging from 20–60%. The protocol was particularly successful at preparing 3-alkylquinolines, for example 3,6-dimethyl quinoline could be prepared in 61% isolated yield.
Amines can also be coupled with diols to form N-heterocycles in the presence of between 0.5 and 2.5 mol% [RuCl2(p-cymene)]2 promoted by DPEphos.32 Quantitative conversions and high isolated yields were achieved after 24 h in toluene heated to reflux. Enantiopure reactants could be employed without loss of chiral information (Scheme 17). The phosphine promoted [RuCl2(p-cymene)]2 system is also applicable to solvent free microwave conditions.56
![Phosphine promoted [RuCl2(p-cymene)]2 system for the formation of N-heterocycles.32](/image/article/2012/CY/c1cy00338k/c1cy00338k-s17.gif)
Cyclopentadienyl Iridium complexes have also been applied to the reaction of primary amines and diols to prepare ring systems.57,58 The application of [Cp*IrCl2]2 with a base enabled coupling to be achieved at lower temperatures with excellent isolated yields employing 1–5 mol% Ir. Most of the syntheses demonstrated employed benzylamine or aniline (Scheme 18) but the protocol also allowed the coupling of 1-octylamine and 1,4-butanediol and in 81% yield (by GC) and the asymmetric synthesis of 2-phenylpyridines in good yield and reasonable diastereomeric excess.57
![Examples of N-heterocyclic ring formation catalyzed by base-assisted [Cp*IrCl2]2.57](/image/article/2012/CY/c1cy00338k/c1cy00338k-s18.gif) | ||
| Scheme 18 Examples of N-heterocyclic ring formation catalyzed by base-assisted [Cp*IrCl2]2.57 | ||
Water soluble [Cp*Ir(NH3)3][I]2 catalyzed the preparation of cyclic amines from the reaction of benzyl amines with diols (Scheme 19).37 Saidi et al. have recently demonstrated highly efficient amine alkylation in water, in the absence of base, employing [Cp*IrCl2]2.59
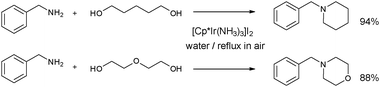
Tryptamine has been reacted with 1,4-butanediol or 1,5-pentanediol in the presence of 5 mol% [Ir(cod)Cl]2 (cod = 1,5-cyclooctadiene) and dppf at 110 °C for 24 h to yield the corresponding pyrrolidine in 83% isolated yield (Scheme 20), or piperidine in 72% isolated yield.60
 | ||
| Scheme 20 Pyrrolidine synthesis catalyzed by Ir/dppf.60 | ||
Piperazines have been prepared from diamines and 1,2-diols at 100–140 °C in toluene, in water or neat.61 Reactions were performed overnight. Excellent isolated yields were achieved, for example decahydro-2-methylquinoxaline could be prepared in 98% yield and high stereoselectively (Scheme 21). The reaction could be carried out with 0.5 mol% [Cp*IrCl2]2.
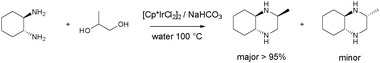 | ||
| Scheme 21 Piperazine synthesis from 1,2-diols and diamines.61 | ||
The synthesis of nitrogen heterocycles from napthylamines and 1,2 and 1,3-diols has been catalyzed by IrCl3·nH2O (5 mol%) promoted by 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP).62 Isolated yields as high as 96% were reported (Scheme 22). Ishii and co-workers have recently reviewed their contribution to this field.63

Indoles have been prepared by employing [Cp*IrCl2]2 (1 mol%) with an acid promoter,53 methanesulfonic acid was particularly promoting (Scheme 23). Reaction times were longer (2 days) than those required for RuCl3/phosphine based systems.
![Indole synthesis catalyzed by [Cp*IrCl2]2.53](/image/article/2012/CY/c1cy00338k/c1cy00338k-s23.gif) | ||
| Scheme 23 Indole synthesis catalyzed by [Cp*IrCl2]2.53 | ||
Catalysts for the hydrogen transfer mediated synthesis of nitrogen heterocycles have been reviewed recently.64
When heated at lower temperatures (115 °C) in the presence of [Cp*IrCl2(NHC)] catalysts (1 mol%), in toluene or Methyl-trioctylammonium bis(trifluoromethylsulfonyl)imide (N1,8,8,8NTf2) solution, we have found that aniline and 1,3-propanediol react to form three main products: the product of mono-amination, di-amination and the unexpected product of combined amination and dehydration: N-propylaniline (Scheme 24).65,66 The relative amount of amination and dehydration could be altered by altering the ratio of the reagents facilitating good selectivity of any of the three products.66 The catalyst was found to catalyze dehydration in the absence of the amine to form products of an ‘oxidative or dehydrogenative Guerbet’ reaction (see section 4).
 | ||
| Scheme 24 Reaction of aniline and 1,3-propanediol in N1,8,8,8NTf2.66 | ||
3.3 Dehydrogenative coupling and amidation
Early examples of the formation of amides by coupling of alcohols and amines include the formation of lactam rings from amino alcohols catalyzed by [RuH2(PPh3)4]67 or [Cp*RhCl2]2.68 More recently the intermolecular reaction has been reported.19Comparing amidation with N-alkylation (Schemes 1 and 2), the key difference is the retention of the alcohol oxygen in the final product; instead of H2O loss, leading to the formation of an amine, H2 is driven off to form the amide. These are therefore ‘dehydrogenative coupling’ reactions. Alternatively a hydrogen acceptor can be added enabling an oxidative coupling by hydrogen transfer. The application of chemicals derived from biomass to this reaction would be particularly advantageous, as the hydrogen evolved is a potential sustainable energy supply.
Milstein and co-workers coupled aliphatic alcohols and aromatic, benzyl and aliphatic primary amines.19 Mono and di-amidation was reported with only traces of secondary amine products detected and isolated yields of up to 99% using only 0.1 mol% catalyst (Scheme 25). The authors suggested that the mechanism of formation was via the formation and dehydrogenation of the hemiaminal.

Zeng and Guan have applied Milstein's catalyst to the synthesis of polyamides from diols and diamides (Scheme 26).69
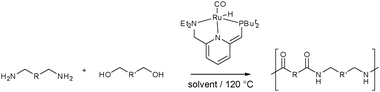 | ||
| Scheme 26 Synthesis of polyamides by hydrogen transfer mediated oxidative coupling of diols and diamines.69 | ||
Milstein and co-workers have also shown that changing the catalyst enables imines to be generated from the coupling of amines and alcohols, in this reaction one equivalent of hydrogen and one of water are eliminated.70 0.2 mol% of a Ru catalyst in xylene was able to catalyse the synthesis of imines with good isolated yields, (for an example see Scheme 27). Amides can also be formed from esters and amines.71

Madsen and co-workers72,73 achieved amidation by employing the combination of Ru(II) (2 mol%) with imidazolium and phosphonium promoters. Preformed Ru NHC catalysts were also found to be active catalysts. The best results were obtained for the coupling depicted in Scheme 28, affording isolated yields of 93 and 100%. Tricyclopentylphosphine (PCyp3) was added as the HBF4 salt along with 1,3-diisopropylimidazolium chloride (diPriimCl).
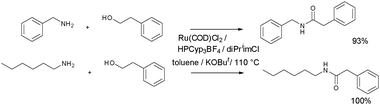 | ||
| Scheme 28 Amidations performed by the Madsen protocol.72,73 | ||
Williams and co-workers have applied their [RuCl2(p-cymene)]2 system to the formation of amides.74 The addition of bis(diphenylphosphino)butane (dppb) and a hydrogen acceptor, 3-methyl-2-butanone, enabled the coupling of a range of alcohols and benzyl amines to form amides. Using 2.5 mol% [RuCl2(p-cymene)]2 typical isolated yields of 65–75% were achieved after 24 h heating to reflux (Scheme 29). In the absence of a hydrogen acceptor a flow of nitrogen could be used to drive off H2.
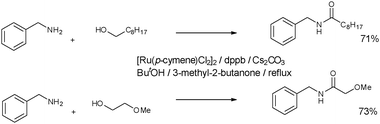 | ||
| Scheme 29 Amidations performed by the Williams protocol.74 | ||
Grützmacher and co-workers75 reported a versatile amidation method applied to primary and benzyl alcohols requiring 0.2 mol% catalyst. Perhaps most notable was the efficient synthesis of a primary amide in good yield by employing ammonia. Methyl methacrylate (MMA) was employed as the hydrogen acceptor at temperatures of −30 to 25 °C. Good yields were achieved in 4 h (Scheme 30). The catalyst [Rh(trop2N)(PPh3)] (trop2N = bis(5-H-dibenzocyclohepten-5-yl)-amide) could also be used for the conversion of alcohols to acids or esters using water or methanol in place of the amine reactant, in this case cyclohexanone was used as the hydrogen acceptor.
 | ||
| Scheme 30 Amidations performed by the Grützmacher protocol.75 | ||
New protocols are rapidly emerging, including important work by Hong and co-workers, who, amongst other avenues, applied aryl Ru NHC catalysts to this reaction.76 Their work thus far is summarised in a recent review.77 Aryl Ru NHC complexes have also been investigated by Prades et al.78 and Madsen and co-workers.73
4. Other reactions of synthetic potential
The N-alkylation of amines employing aliphatic alcohols over a homogeneous catalyst is now a well established synthetic protocol. As aliphatic alcohols are expected to be increasingly available and important in chemicals manufacture, other routes to value added chemicals must be developed. A variety of other synthetic strategies arising from the dehydrogenation of alcohols has recently been discussed.15 Of particular note for the activation of bio-renewable chemicals, are synthetic methods that involve dehydration and C–C bond formation.The products of biological processes are often highly oxygenated and diols, glycols and glycerol are common biological products. In order to transform these into chemicals that are synthetically flexible, some of the oxygen must be removed, dehydration is a suitable strategy for achieving this. In the presence of homogeneous [Rh2(CO)4Cl2] promoted by ammonium or imidazolium halides, simple aliphatic n-alcohols can be converted to alkenes by dehydration.79 Carrying out dehydration under conditions that favour hydrogen transfer, a cascade of reactions occurs that leads to new C–C bond formation. This is due to the formation of the highly reactive aldehyde, as observed in N-alkylation. In the absence of an added nucleophile (such as an amine), the aldehyde can react in an aldol condensation. In a common sequence of reactions the aldol product is dehydrated then reduced to form an aliphatic alcohol, this is the Guerbet reaction (auto β-alkylation of primary alcohols)80,81 (Scheme 31).
 | ||
| Scheme 31 Dehydration and dimerization of alcohols first observed by Guerbet.80,81 | ||
This reaction has a high potential in the sustainable chemical industry as simple alcohols, easily derived from biomass, can be built into longer chain functionalised molecules. Guerbet alcohols have applications in plasticizers, detergents, lubricants and plastics.15 The Guerbet reaction is most commonly catalyzed by heterogeneous catalysts but homogeneous systems are known. Early examples of homogeneous catalysts were reported by Gregorio et al.82 A range of phosphine promoted precious metal catalysts were tested for the dimerization of 1-butanol in the presence of sodium butoxide. The best systems tested were [Rh(CO)2Cl]2 or RhCl3·3H2O/PEt3, RuCl3·3H2O/PEt3 and [Ir(CO)Cl(PPh3)2]. All catalytic systems lost activity over time; the Rh and Ru systems were the most stable.
Burk et al. extended the study of Rh catalysts demonstrating that phosphine promoted rhodium precursors exhibited homogeneous and heterogeneous catalyst activity simultaneously.83 The heterogeneous component84 was shown to be more active than the homogeneous component.
More recently soluble Pd catalysts were evaluated against their heterogeneous counterparts.85 Isobutanol could be prepared by the condensation of methanol and 1-propanol, in the presence of a catalyst and MeONa. The heterogeneous catalyst Pd/C was found to leach. A range of Pd complexes could be used as alternative catalyst precursors, e.g. [PdCl2(dppe)] (dppe = 1,2-bis(diphenylphosphino)ethane). Both classes of catalyst exhibited a mixed homogeneous/heterogeneous speciation during catalysis. 1-butanol was reacted in the presence of a range of Pd catalysts and BunONa to form 2-ethyl-1-hexanol with good selectivity, but reactions were slow and high temperatures (typically 200–280 °C) were required.86 The system was not stable due to the hydrolysis of the base by the water formed.
Ir catalysts have been found to be highly active in the presence of alkene and base promoters.87 A range of aliphatic alcohols could be condensed in good yield and selectivity, for example 1-butanol was converted to 2-ethyl-1-hexanol in 70% isolated yield catalysed by 1 mol% [Cp*IrCl2]2 (Scheme 32), 40 mol% of base was required. Selectivity was assisted by the addition of the alkene, which acted as a hydrogen acceptor and was proposed to speed up the first step of the reaction.
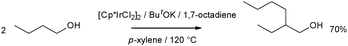
Initial results employing [Cp*IrCl2(NHC)] complexes suggest that Guerbet-like β-alkylation reactions can be catalyzed by these catalysts, and that it may be possible to suppress the hydrogenation step and introduce an ‘oxidative or dehydrogenative Guerbet reaction’ to yield an α,β-unsaturated aldehyde.66
The Guerbet reaction is not unique, many other hydrogen-transfer or dehydrogenation mediated alkylations employing alcohols have been demonstrated, for example the β-alkylation of ketones, and these are recorded in relevant literature reviews.13,15
5. Conclusion and outlook
The synthetic utility of hydrogen (auto)transfer/hydrogen borrowing and dehydrogenation chemistry is now without question. Analysis of the literature has revealed many examples of highly efficient reactions that convert simple aliphatic alcohol substrates into higher value products and these render the approach applicable to sustainable chemical synthesis. Alcohols derived from biomass can be converted to primary amines by N-alkylation of ammonia and these amines used in combination with alcohols to generate secondary and tertiary amines, amides or polyamides. Short chain alcohols can be converted to longer chain products by auto and cross β-alkylations such as the Guerbet reaction.The further development of catalysts for these reactions has unstoppable momentum, and a myriad of reactions are plausible, but the separation and purification processes necessary to utilize chemicals from biomass remain relatively unexplored. Sustainable (Green) chemistry requires consideration of the chemistry from origin to disposal, for petrochemicals this involves the processing of crude oils and for biomass this requires the processing of plant or animal matter, in whatever form. In order to design processes that convert bio-renewables to consumer products chemists and chemical engineers will have to increasingly engage in the crude, impure mixtures provided by nature and build the processing of biomass into their synthetic methods.1,65
Acknowledgements
Thanks to Dr Patricia Marr, Queen's University Belfast.Notes and references
- A. C. Marr and S. F. Liu, Trends Biotech., 2011, 29, 199–204 CrossRef CAS.
- R. A. Weusthuis, I. Lamot, J. van der Oost and J. P. M. Sanders, Trends Biotech., 2011, 29, 153–158 CrossRef CAS.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 RSC.
- S. Zinoviev, F. Muller-Langer, P. Das, N. Bertero, P. Fornasiero, M. Kaltschmitt, G. Centi and S. Miertus, ChemSusChem, 2010, 3, 1106–1133 CrossRef CAS.
- http://www.genomatica.com/ .
- http://www.bio-amber.com/succinic_acid.html .
- U.S. Department of Energy (2004) Top Value Added Chemicals from Biomass (Vol. 1) (T. Werpy and G. Petersen Eds), Pacific Northwest National Laboratory (PNNL), National Renewable Energy Laboratory (NREL),Office of Biomass Program (EERE). Available to download at http://www.eere.energy.gov/.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- P. Gallezot, Green Chem., 2007, 9, 295–302 RSC.
- P. Gallezot, Top. Catal., 2010, 53, 1209–1213 CrossRef CAS.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS.
- R. H. Crabtree, Organometallics, 2011, 30, 17–19 CrossRef CAS.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Angew. Chem. Int. Ed., 2007, 46, 2358–2364 CrossRef CAS.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS.
- S. Enthaler, B. Spilker, G. Erre, K. Junge, M. K. Tse and M. Beller, Tetrahedron, 2008, 64, 3867–3876 CrossRef CAS.
- V. V. K. M. Kandepi, J. M. S. Cardoso, E. Peris and B. Royo, Organometallics, 2010, 29, 2777–2782 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- Y. Watanabe, Y. Tsuji, H. Ige, Y. Ohsugi and T. Ohta, J. Org. Chem., 1984, 49, 3359–2670 CrossRef CAS.
- S.-I. Murahashi, K. Kondo and T. Hakata, Tetrahedron Lett., 1982, 23, 229–232 CrossRef CAS.
- A. Del Zotto, W. Baratta, M. Sandri, G. Verardo and P. Rigo, Eur. J. Inorg. Chem., 2004, 524–529 CrossRef.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS.
- D. Pingen, C. Müller and D. Vogt, Angew. Chem. Int. Ed., 2010, 49, 8130–8133 CrossRef CAS.
- G. Bitsi, E. Schleiffer, F. Antoni and G. Jenner, J. Organomet. Chem., 1989, 373, 343–352 CrossRef CAS.
- J. Luo, M. Wu, F. Xiao and G. Deng, Tetrahedron Lett., 2011, 52, 2706–2709 CrossRef CAS.
- A. Tillack, D. Hollman, D. Michalik and M. Beller, Tetrahedron Lett., 2006, 47, 8881–8885 CrossRef CAS.
- M. H. S. A. Hamid and J. M. J. Williams, Chem. Commun., 2007, 725–727 RSC.
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS.
- A. Tillack, D. Hollmann, K. Mevius, D. Michalik, S. Bähn and M. Beller, Eur. J. Org. Chem., 2008, 4745–4750 CrossRef CAS.
- J. He, J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem. Int. Ed., 2009, 48, 9888–9891 CrossRef CAS.
- K. Kruger, A. Tillack and M. Beller, ChemSusChem, 2009, 2, 715–717 CrossRef.
- K. Fujita, Y. Enoki and R. Yamaguchi, Tetrahedron, 2008, 64, 1943–1954 CrossRef CAS.
- R. Kawahara, K. Fujita and R. Yamaguchi, Adv. Synth. Catal., 2011, 353, 1161–1168 CrossRef CAS.
- F. Hanasaka, K. Fujita and R. Yamaguchi, Organometallics, 2004, 23, 1490–1492 CrossRef CAS.
- A. C. Marr, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 3283–3285 CrossRef CAS.
- A. Prades, R. Corberán, M. Poyatos and E. Peris, Chem. Eur. J., 2008, 14, 11474–11479 CrossRef CAS.
- A. Pontes da Costa, M. Viciano, M. Sanau′, S. Merino, J. Tejeda, E. Peris and B. Royo, Organometallics, 2008, 27, 1305–1309 CrossRef CAS.
- F. Hanasaka, K.-i. Fujita and R. Yamaguchi, Organometallics, 2006, 25, 4643–4647 CrossRef CAS.
- A. C. Marr, M. Nieuwenhuyzen, C. L. Pollock and G. C. Saunders, Organometallics, 2008, 27, 1954–1958 CrossRef CAS.
- D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321–325 CrossRef CAS.
- Y. Zhao, Siong W. Foo and S. Saito, Angew. Chem. Int. Ed., 2011, 50, 3006–3009 CrossRef CAS.
- Y. Tsuji, K.-T. Huh, Y. Ohsugi and Y. Watanabe, J. Org. Chem., 1985, 50, 1365–1370 CrossRef CAS.
- Y. Tsuji, K.-T. Huh and Y. Watanabe, Tetrahedron Lett., 1986, 27, 377–380 CrossRef CAS.
- J. A. Marsella, J. Org. Chem., 1987, 52, 467–468 CrossRef.
- J. A. Marsella, J. Organomet. Chem., 1991, 407, 97–105 CrossRef CAS.
- K.-T. Huh, S. C. Shim and C. H. Doh, Bull. Korean Chem. Soc., 1990, 11, 45–49 CAS.
- T. Kondo, S. Kotachi and Y. Watanabe, Chem. Commun., 1992, 1318–1319 RSC.
- Y. Tsuji, K.-T. Huh and Y. Watanabe, J. Org. Chem., 1987, 52, 1673–1680 CrossRef CAS.
- M. Tursky, L. L. R. Lorentz-Petersen, L. B. Olsen and R. Madsen, Org. Biomol. Chem., 2010, 8, 5576–5582 RSC.
- C. S. Cho, H. Y. Lim, S. C. Shim, T. J. Kim and H.-J. Choi, Chem. Commun., 1998, 995–996 RSC.
- R. N. Monrad and R. Madsen, Org. Biomol. Chem., 2011, 9, 610–615 RSC.
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, J. Org. Chem., 2011, 76, 2328–2331 CrossRef CAS.
- K. Fujita, T. Fujii and R. Yamaguchi, Org. Lett., 2004, 6, 3525–3528 CrossRef CAS.
- K. Fujita, Y. Enoki and R. Yamaguchi, Org. Synth., 2006, 83, 217–221 CAS.
- O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541–1543 RSC.
- G. Cami-Kobei, P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, J. Bioorg. Med. Chem. Lett., 2005, 15, 535–537 CrossRef CAS.
- L. U. Nordstrøm and R. Madsen, Chem. Commun., 2007, 5034–5036 RSC.
- H. Aramoto, Y. Obora and Y. Ishii, J. Org. Chem., 2009, 74, 628–633 CrossRef CAS.
- Y. Obora and Y. Ishii, Synlett, 2011, 30–51 CrossRef CAS.
- R. Yamaguchi, K. Fujita and M. W. Zhu, Heterocycles, 2010, 81, 1093–1140 CrossRef CAS.
- S. F. Liu, M. Rebros, G. Stephens and A. C. Marr, Chem. Commun., 2009, 2308–2310 RSC.
- S. D. Lacroix, A. Pennycook, S. F. Liu, T. T. Eisenhart and A. C. Marr, Catal. Sci. Technol. 10.1039/C1CY00339A.
- T. Naota and S.-I. Murahashi, Synlett, 1991, 693–694 CAS.
- K.-I. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785–2788 CrossRef CAS.
- H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159–1161 CrossRef CAS.
- B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem. Int. Ed., 2010, 49, 1468–1471 CAS.
- B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS.
- L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef CAS.
- J. H. Dam, G. Osztrovszky, L. U. Nordstrøm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820–6827 CrossRef CAS.
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667–2670 CrossRef CAS.
- T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS.
- Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374–1378 CrossRef CAS.
- C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20–26 RSC.
- A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162–1167 CrossRef CAS.
- G. R. M. Dowson, I. V. Shishkov and D. F. Wass, Organometallics, 2010, 29, 4001–4003 CrossRef CAS.
- M. Guerbet, C. R. Acad. Sci., 1899, 128, 1002–1004 Search PubMed.
- M. Guerbet, C. R. Acad. Sci., 1909, 149, 129–132 Search PubMed.
- G. Gregorio, G. F. Pregaglia and R. Ugo, J. Organomet. Chem., 1972, 37, 385–387 CrossRef CAS.
- P. L. Burk, R. L. Pruett and K. S. Campo, J. Mol. Catal., 1985, 33, 1–14 CrossRef CAS.
- P. L. Burk, R. L. Pruett and K. S. Campo, J. Mol. Catal., 1985, 33, 15–21 CrossRef CAS.
- C. Carlini, M. Di Girolamo, A. Macinai, M. Marchionna, M. Noviello, A. M. Raspolli Galletti and G. Sbrana, J. Mol. Catal. A: Chem., 2003, 204–205, 721–728 CrossRef CAS.
- C. Carlini, A. Macinai, A. M. Raspolli Galletti and G. Sbrana, J. Mol. Catal. A: Chem., 2004, 212, 65–70 CrossRef CAS.
- T. Matsu-ura, S. Sakaguchi, Y. Obora and Y. Ishii, J. Org. Chem., 2006, 71, 8306–8308 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |