Organocatalytic enantioselective methodologies using Morita–Baylis–Hillman carbonates and acetates
Ramon
Rios
*ab
aDepartment de Química Orgànica, Universitat de Barcelona, Martí i Franqués 1-11, 08028 Barcelona, Spain. E-mail: rios.ramon@icrea.cat; Web: http://www.runam.host22.com Tel: +34 934021257
bICREA, Passeig Lluis Companys 23, 08010 Barcelona, Spain
First published on 15th November 2011
Abstract
Lately, the use of Morita–Baylis–Hillman carbonates and acetates in organocatalysis has grown exponentially. Since the pioneering work of Kim and coworkers until the last cycloadditions reported by Barbas, a plethora of new methodologies have been developed. The use of these compounds opens a new gate for the synthesis of C–C or C-heteroatom bonds in an enantioselective fashion and under mild conditions giving access to highly functionalized structures. In this review, we aim to cover these exciting reactions, paying special attention on the nature of the MBH adduct.
1. Introduction
Since the advent of organic chemistry, most organic chemists have pursued the asymmetric synthesis of C–C or C-heteroatom bonds. Notable work on this field includes the work of Professors Sharpless, Noyori, and Knowles, who won the Nobel Prize in 2001 for their tremendous accomplishments in this field.During the past few decades, asymmetric allylic substitution (AAS) has emerged as one of the most powerful methodologies for the enantioselective synthesis of C–C bonds.1 In 1977, Trost and Strege reported the first example of an enantioselectively catalyzed allylic substitution with a stabilized nucleophile.2 Since this breakthrough, numerous new methodologies have been developed based on transition metal catalysts. The advancements in asymmetric allylic substitution make it one of the most commonly used methods for enantioselective bond formation.
Most of these methodologies involve the use of palladium as the metal catalyst; however, some also involve the use of other transition metal complexes such as Ir, Mo, Ru, Rh, and Cu with excellent results.3
The rediscovery of organocatalysis by List, Barbas, and Lerner through their seminal report on the proline-catalyzed intermolecular aldol reaction and the report on iminium catalysis by MacMillanet al. in 20004 have propelled the emergence of organocatalysis as one of the most active fields of research for the enantioselective construction of C–C or C-heteroatom bonds. In 2002, Kim and co-workers reported the use of Cinchona alkaloid derivatives for the hydrolysis of Morita–Baylis–Hillman (MBH) acetates with sodium bicarbonate; this led to the development of the organocatalytic AAS method (Scheme 1).5 Since then, the allylic alkylation of MBH adducts catalyzed by a metal-free organic Lewis base has attracted considerable attention from the organic chemistry community.
 | ||
| Scheme 1 Enantioselective allylic substitution catalyzed by Pd complexes reported by Trost. | ||
In this review, we aim to cover all the reported methodologies based on the AAS of MBH derivatives.
This review is organized to discuss the different types of AAS procedures. First, we present an overview of the AAS reaction mechanisms, followed by discussions on the enantioselective methodologies involving MBH acetates and the enantioselective methodologies involving MBH carbonates.
2. Mechanism of the asymmetric allylic substitution of Morita–Baylis–Hillman derivatives
MBH 6 adducts possess a high potential for use as synthetic intermediates; therefore, in recent years, several research groups have focused their efforts on the modification of the β-position (the alcohol moiety) of the MBH adducts. To achieve this transformation, the MBH reaction is performed in the absence of a chiral catalyst. Once the product is obtained, the alcohol moiety is transformed into a good leaving group, usually an acetate or a carbonate. These carbonates or acetates could react with a designed nucleophile via a tandem SN2′–SN2′ mechanism. In general, the first step, as shown in Fig. 1, is the attack of the catalyst (commonly, a nucleophilic chiral nitrogen or a phosphorous atom) on the vinylic moiety of the MBH adduct with subsequent ionization of the leaving group, affording intermediate 6. Next, the leaving group deprotonates the pronucleophile, which attacks intermediate 7via a SN2′ mechanism, leading to the product. | ||
| Fig. 1 General SN2–SN2′ mechanism for the AAS of MBH carbonates. | ||
Considering this mechanism, the first step is the kinetic resolution of the MBH acetate or carbonate, which affords the same intermediate 7. The formation rate of intermediate 7 is dependent on the interaction between each enantiomer of the MBH adduct and the chiral catalyst.
The nucleophilicity of the catalyst plays an important role in the mechanism of the reaction. Orena and co-workers7 demonstrated that when DBU (higher basicity, less nucleophilicity) was used instead of DABCO (less basicity, higher electrophilicity), a SN2′-decarboxylation mechanism results instead (11) of the normal SN2′–SN2′ pathway (12), as shown in Scheme 2.
 | ||
| Scheme 2 Influence of the catalyst on the mechanism. | ||
3. Morita–Baylis–Hillman acetates
The first report of AAS with MBH acetates was by Kim and co-workers in 2002.5 In this report, the first synthesis of enriched Baylis–Hillman alcoholsvia a combination of kinetic resolution of the MBH acetates and subsequent asymmetric induction during hydrolysis with NaHCO3 as a water surrogate has been reported. In this study, Kim and coworkers also reported the use of Cinchona alkaloids or Cinchona alkaloid dimers such as (DHQ)2PHAL (I) as a suitable catalyst for the reaction. As illustrated in Scheme 3, the reaction affords the final alcohols in moderate yields and with moderate to good enantioselectivities and enantioenriched starting material. In addition to reporting the hydrolysis of MBH acetates, Kim and co-workers reported that the addition of phenols, tosylamines, or mesilamines as nucleophiles rendered the final compounds in low to moderate yields and with moderate enantioselectivities (Scheme 3). | ||
| Scheme 3 Reaction reported by Kim. | ||
Two years later, Krische and co-workers reported a phosphine catalyzed dynamic kinetic resolution of MBH acetates.8a In this report, they showed a simple example of a dynamic kinetic resolution using (R)-Cl-MeO-BIPHEP (II) as a catalyst and phthalimide as the nucleophile, as shown in Scheme 4. After 62 h at 50 °C, the final compound was obtained in 80% yield and 56% enantiomeric excess.
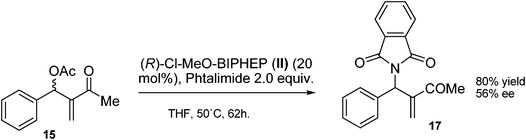 | ||
| Scheme 4 Dynamic Kinetic Resolution reported by Krische. | ||
Inspired by this work of Krische and co-workers and using the concept of the addition of γ-butenolides to MBH acetates,8b two research groups, Hou and co-workers and Shi and co-workers, developed a new asymmetric version of the dynamic kinetic resolution of MBH acetates.
In 2007, Hou and co-workers reported the allylic amination of MBH acetates catalyzed by planar-chiral [2,2]paracyclophane monophosphines (III).9 This reaction affords the aminated products with high regioselectivities and modest enantioselectivities with respect to limited substrates (Scheme 5).
 | ||
| Scheme 5 Allylic amination reported by Hou. | ||
In 2008, Shi and co-workers reported the phosphine catalyzed enantioselective construction of γ-butenolides via the addition of 2-trimethylsilyloxy furan (19) to MBH acetates.10 Multifunctional phosphines bearing a primary amide group were found to be the best catalysts for this reaction. A hydrogen bond interaction between the amide and the silyl compound was the key to achieving high stereoselectivities. Moreover, the use of water as an additive facilitated this interaction in aprotic solvents such as toluene, affording the final compounds in high yields and with high enantioselectivities, as shown in Scheme 6.
 | ||
| Scheme 6 Allylic alkylation reported by Shi. | ||
Around the same time, Cho and co-workers reported the first asymmetric intramolecular allylic substitution of MBH acetates bearing a nucleophilic nitrogen in the lateral chain (Scheme 7).11 This reaction was efficiently catalyzed by hydroquinidine 4-methyl-2-quinolyl ether (V) affording chiral 2-(α-methylene)-pyrrolidines (22) in good yields and up to 74% ee.
 | ||
| Scheme 7 Intramolecular allylic amination reported by Cho. | ||
In 2009, Shi and co-workers reported the use of bifunctional phosphine–proline catalysts for the allylic amination of MBH acetates.12 When phthalimide was made to react with MBH acetates in the presence of catalyst VI, the reaction afforded the final aminated products in good yields and with moderate to low enantioselectivities (Scheme 8).
 | ||
| Scheme 8 Allylic amination reported by Shi. | ||
Recently, Shi and co-workers improved the asymmetric allylic amination of MBH acetates described above, using a bifunctional phosphine–thiourea catalyst.13
Further, they suggested a catalytic double activation. In this reaction, the phosphine moiety undergoes direct addition to the MBH acetate bond generating the electrophilic leaving group while the thiourea moiety activates the ketone groupvia the hydrogen bond, as shown in Fig. 2.
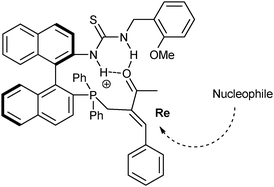 | ||
| Fig. 2 Proposed activation mode. | ||
The reaction afforded the allylic aminated product 17 in good yields and up to 90% ee when the reaction was carried out in 1,2-dichlorobenzene, at 10 °C (Scheme 9).
 | ||
| Scheme 9 Allylic amination catalyzed by bifunctional phosphines reported by Shi. | ||
4. Morita–Baylis–Hillman carbonates
Since the first report of asymmetric allylic substitution of MBH acetates, the research for obtaining a better leaving group has led to the use of MBH carbonates. These substrates are more reactive than the acetates mentioned above. Carbonates after decarboxylation of the carbamic acid render alcohols as waste unlike acetates that render AcOH as byproducts; thus, this allows the use of nucleophilic amines as catalysts without the risk of salt formation. Since 2004, new methodologies using MBH carbonates have increasingly been discovered. In this section, we review the different methodologies based on the nature of the nucleophiles.4.1 C-nucleophiles
The first example of the use of MBH carbonates with asymmetric synthesis was reported by Lu and co-workers in 2004.14 In this work, they reported the allylic substitution of MBH carbonates catalyzed by β-isocupreidine VIIIvia a SN2′–SN2′ mechanism, with different nucleophiles such as phenol, tosylamine, phthalimides, and malonates. The final products were obtained in good yields and moderate enantioselectivities, as shown in Scheme 10. | ||
| Scheme 10 Allylic substitution of MBH carbonates reported by Lu. | ||
Following this pioneering report, in 2007, Hiemstra and co-workers reported the enantioselective allylic substitution of MBH carbonates using α,α-cyanophenylacetate as the carbon nucleophile.15 This reaction presented several challenges such as the synthesis of adjacent quaternary and tertiary stereocenters. β-ICPD (VIII) was the best catalyst for this reaction, rendering the final compounds in good yields and with moderate to good diastereoselectivities and good enantioselectivities (Scheme 11).
 | ||
| Scheme 11 Allylic alkylation reported by Hiemstra. | ||
Chen and co-workers reported the addition of α,α-dicyanoalkenes to the MBH carbonates catalyzed by (DHQD)2AQN (IX).16 The mechanism of the reaction is as follows: first, the nucleophilic catalyst undergoes Michael addition to the MBH carbonate and generates cation 6 releasing CO2 and tBuO−. tBuO− reacts with α,α-dicyanoalkene deprotonating the γ position and, thus, generating an enolate. The enolate attacks cation 7, viaMichael addition, regenerating the double bond and releasing the catalyst, as shown in Scheme 12.
 | ||
| Scheme 12 Mechanism of the addition of α,α-dicyanoalkenes to MBH carbonates. | ||
This reaction afforded the final alkylated α,α-dicyanoalkenes 29 in good yields and with excellent diastereo- and enantioselectivities (Scheme 13).
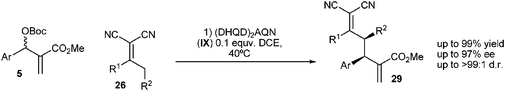 | ||
| Scheme 13 Allylic alkylation reported by Chen. | ||
Moreover, Chen and co-workers showed the utility of this reaction in the synthesis of different cyclic derivatives, as shown in Scheme 14.
 | ||
| Scheme 14 Synthesis of cyclic products developed by Chen. | ||
The same research group reported the use of oxindoles as a nucleophile.17N-Protected oxindoles (34) reacted with MBH-carbonates catalyzed by (DHQD)2AQN (IX), affording alkylated oxindoles in good yields and with excellent enantioselectivities.
The absolute configuration of the resulting adducts was ascertained using X-ray analysis and was easily derived via a [3+2] cycloaddition with in situ generated N-oxides (Scheme 15).
 | ||
| Scheme 15 Oxindole addition to MBH carbonates developed by Chen. | ||
Chen and co-workers completed their studies with the addition of α,α-cyano-olefin to MBH carbonates derived from oxindoles.18 The significance of this methodology is the synthesis of oxindoles with a C3-quaternary stereocenter. The above reaction was catalyzed by β-ICPD (VIII), resulting in highly functionalized compounds in good yields and with good stereoselectivities. Moreover, this reaction can result in spirocyclic oxindoles in only two steps by treating the alkylated product with DBU (Scheme 16).
 | ||
| Scheme 16 Oxindolic MBH carbonates addition to α,α-cyano-olefin developed by Chen. | ||
Chen and co-workers reported the use of butenolides as nucleophiles for the AAS of MBH carbonates.19 On the basis of the previous work done by Krische8 and Shi10 using MBH acetates, Chen and coworkers introduced the possibility of using MBH carbonates as well as directly using β,γ-butenolides 41 instead of using silyloxy furans. As compared to the precedent methodologies, the use of butenolides as nucleophiles has a clear advantage in terms of atom economy and reaction step reduction. The reaction was catalyzed by dimeric Cinchona alkaloid derivatives such as (DHQD)2PYR (XI) or (DHQD)2AQN (IX), rendering the final compounds in good yields and with good stereoselectivities (Scheme 17).
 | ||
| Scheme 17 Butenolide addition to MBH carbonates reported by Chen. | ||
Recently, Chen and co-workers reported a Lewis base assisted Brønsted base catalysis for the direct regioselective asymmetric vinylogous alkylation of allylic sulfones.20 They utilized the pKa of sulfone species possessing a simple functional group, such as allylphenylsulfone (pKa = 22.5 in dimethyl sulfoxide (DMSO)), for the deprotonation by the tert-butoxy anion (pKa tBuOH = 32.2 in DMSO) generated in situ in the catalytic cycle after the Michael addition of the catalyst to the MBH carbonate.
The reaction was efficiently catalyzed by Cinchona alkaloid derivatives such as (DHQD)2AQN (IX), affording the final compounds in good yields and with excellent enantioselectivities (Scheme 18).
 | ||
| Scheme 18 Asymmetric vinylogous alkylation of allylic sulfones reported by Chen. | ||
In 2011, Li and Cheng et al. reported the use of 3-substituted benzofuran-2(3H)-ones with MBH carbonates.21 The reaction was simply catalyzed by biscinchona alkaloids such as (DHQD)2AQN (IX), and rendered the final adducts, containing a quaternary center, in good yields and with good stereoselectivities (Scheme 19).
 | ||
| Scheme 19 Benzofuranone addition to MBH carbonates developed by Liu and Cheng. | ||
Huang, Jiang, and Tan reported the enantioselective addition of fluoro-bis(phenylsulfonyl)methane (FBSM, 47a) and bis(phenylsulfonyl)methane (BSM, 47b) to MBH carbonates catalyzed by Cinchona alkaloid derivatives.22a The reaction rendered the final compounds in good yields and with good enantioselectivities in both cases. Moreover, they reported that in the reaction where FBSM was used as a nucleophile, a simple filtration of the reaction mixture affords an enantiopure product (Scheme 20a). Almost at the same time, Shibata and coworkers reported a similar reaction.22b The main difference relies on the use of FeCl2 as a cocatalyst. The cooperative effect between Cinchona alkaloid and FeCl2 increases the yields and the enantioselectivities of the process (Scheme 20b).
 | ||
| Scheme 20 FBSM and BSM addition to MBH carbonates. | ||
In order to show the applicability of these methodologies, they described a short synthetic route to β-methyl-γ-fluoromethyl-substituted alcohols 51 in good yields and with excellent diastereoselectivities (Scheme 21).
 | ||
| Scheme 21 Synthesis of β-methyl-γ-fluoromethyl-substituted alcohols. | ||
Shi and co-workers reported an oxazolone addition to the MBH carbonates catalyzed by chiral phosphines bearing a thiourea moiety.23 Shi and co-workers used methyl vinyl ketone derivatives instead of acrylate derivatives. The reaction rendered the C4 regioisomer as the major product24 in good yields and with good stereoselectivities as illustrated in Scheme 22.
 | ||
| Scheme 22 Oxazolone addition to MBH carbonates. | ||
Shi and co-workers proposed that in the application of double activation, first, the phosphine attacks the MBH carbonatevia a Michael addition, forming the ion/pair, which is stabilized by intramolecular hydrogen bonding with a thiourea moiety; at the same time, the thiourea moiety activates and directs the oxazolone to attack the Re face of the MBH carbonate. The effectiveness of this methodology is demonstrated by the ring opening of oxazolones to afford the chiral quaternary amino acids in good yields.
Recently, Shibata and co-workers developed an extremely elegant trifluoromethylation of MBH carbonates using the Rupert–Prakash reagent.25 The reaction was catalyzed by Cinchona alkaloid derivatives such as (DHQD)2PHAL (I), affording the trifluoromethylated products in good yields and with good enantioselectivities (Scheme 23).
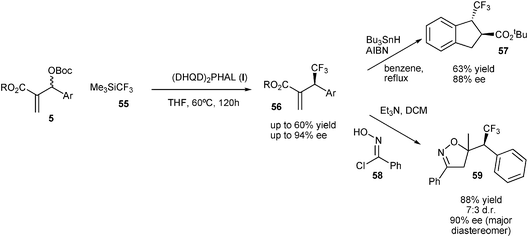 | ||
| Scheme 23 Trifluoromethylation of MBH carbonates. | ||
Very recently, Y.-C. Chen and coworkers have reported the reaction between indenes and MBH carbonates catalyzed by (DHQD)2AQN (IX). The reaction afforded the allylic indenes in good yields and excellent enantioselectivities.26
4.2 N-nucleophiles
In 2009, Chen and co-workers reported the allylic substitution of MBH carbonates using indoles as nucleophiles.27 The reaction was N-selective and was efficiently catalyzed by (DHQD)2PHAL (I), rendering the final amine derivatives in good yields and excellent enantioselectivities (Scheme 24). | ||
| Scheme 24 Asymmetric allylic amination of MBH carbonates. | ||
The same research group reported the use of a phthalimide derivative as a suitable N-nucleophile for the asymmetric allylic amination of MBH carbonates.28 The best catalyst for this reaction was (DHQD)2PYR (XI), affording the final compounds in excellent yields and with excellent enantioselectivities.
In 2011, Chen and co-workers also reported a highly enantioselective N-allylic alkylation of enamines with MBH carbonates.29 The chemoselective N-alkylation could be realized via deprotonation by the in situ generated tert-butoxy anion of the acidic proton of the enamide. The deprotonated enamide reacted with the MBH carbonatesvia a SN2′–SN2′ pathway catalyzed by (DHQD)2AQN (IX) resulting in multifunctional enamides in good yields and with good enantioselectivities (Scheme 25).
 | ||
| Scheme 25 N-Alkylation of enamines reported by Chen. | ||
4.3 O-nucleophiles
In 2009, Y.-C. Chen and co-workers reported the use of peroxides as suitable oxygen nucleophiles for the alkylation of MBH carbonates.30 The best catalyst for this reaction was (DHQD)2PHAL (I), which via a SN2′–SN2′ mechanism rendered the final peroxoethers in good yields and with excellent enantioselectivities (Scheme 26). Moreover, the products could be reduced to obtain alcohols in good yields by treating them with Zn powder in AcOH. | ||
| Scheme 26 O-Alkylation of peroxides reported by Chen. | ||
Jiang and co-workers were the first to use water as a nucleophile in the asymmetric allylic hydroxylation of MBH carbonates.31 The reaction was catalyzed by (DHQD)2AQN (IX) and rendered the final alcohols in good yields and enantioselectivities. Previously, Jiang and co-workers developed a highly enantioselective MBH reaction via a two-step procedure (Scheme 27).
 | ||
| Scheme 27 Allylic hydroxylation of MBH carbonates reported by Jiang. | ||
4.4 Other nucleophiles
In 2010, Rui Wang and co-workers were the first to report the addition of phosphine oxides to MBH carbonates catalyzed by Cinchona alkaloid derivatives.32 Allylic phosphine oxides are valuable intermediates for the synthesis of chiral tertiary allyl phosphines, and there is only one methodology, involving the asymmetric hydroarylation of diphenylphosphinylallenes, reported for the synthesis of allylic phosphine oxides. The reaction rendered the final phosphine oxide derivatives in good yields and with excellent enantioselectivities (Scheme 28); the absolute configuration of the reaction product was determined by X-ray analysis. | ||
| Scheme 28 Allylic phosphination of MBH carbonates reported by Jiang. | ||
4.5 [3+2] Cycloadditions
Barbas and co-workers reported a similar reaction using methyleneindolines as dipolarophiles.33 The best catalyst for this reaction was chiral diphosphines. Barbas and co-workers believe that the second diphosphine has a major impact on the stereoselectivity. The reaction was initiated by the displacement of the carbonate moiety by the phosphinevia a SN2′ mechanism, which was followed by deprotonation to generate the dipole 69. The second phosphine activated the methylene indoline that is attacked viaMichael addition, and subsequent ring closing via intermolecular conjugate addition afforded intermediate 72. Next, the elimination of phosphine completed the catalytic cycle and rendered the final compound (Scheme 29). | ||
| Scheme 29 Proposed mechanism. | ||
This reaction afforded the final spirocyclic products in good yields and with excellent stereoselectivities (Scheme 30). The clear limitation of this reaction is the low enantioselectivity obtained when alkyl MBH carbonates were used.
 | ||
| Scheme 30 Synthesis of spiro compounds reported by Barbas. | ||
In 2011, Shi and co-workers reported a [3+2] annulation between MBH carbonates and isatylidene malononitriles.34 As shown in Scheme 31, the reaction was catalyzed by phosphine catalysts.
 | ||
| Scheme 31 Synthesis of spiro compounds reported by Shi. | ||
Shi and co-workers only described a simple example of the enantioselective version of this reaction that used a chiral phosphine bearing a thiourea moiety as the catalyst and afforded the final product in good yields and with good stereoselectivity (Scheme 31).
5. Conclusions
The asymmetric allylic substitution by organocatalytic methods is considered a suitable option for the enantioselective formation of C–C or C-heteroatom bonds. Recently, several research groups have developed powerful methodologies for the direct asymmetric allylic substitution as well as for generating dipoles that can react via [3+2] cycloadditions. These new reaction modes suggest a brilliant future for this type of reagents. However, there are some limitations that are common to all methodologies, for instance, the use of aliphatic MBH carbonates is very narrow, probably due to the poor enantioselectivity seen in different examples. In terms of applicability, the use of Cinchona alkaloids has a clear advantage; however, restricted of the other enantiomer, thus, the use of commercial pseudo-enantiomers renders opposite asymmetric induction with lower enantioselectivities for a large number of reactions. It is clear that after this initial stage, more research is required in order to achieve beneficial asymmetric allylic substitution methods.MBH adducts represent an open and exciting avenue in the development of new procedures; their versatility and highly functionalized structures suggest their accelerated development in the near future.
Acknowledgements
R.R. thanks the Spanish Ministry of Science and Innovation (MICINN) for financial support (Project AYA2009-13920-C02-02).References
- For an excellent review on asymmetric transition metal-catalyzed allylic alkylations see: B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- B. M. Trost and P. E. Strege, J. Am. Chem. Soc., 1977, 99, 1650–1652 Search PubMed.
- For an exceptional review see: M. L. Crowley, Sci. Synth., 2011, 3, 403–441 Search PubMed.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- J. N. Kim, H. J. Lee and J. H. Gong, Tetrahedron Lett., 2002, 43, 9141–9146 CrossRef CAS.
- For excellent reviews on Morita–Baylis–Hillman reaction see: (a) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005–1018 CrossRef CAS; (b) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614–4628 CrossRef CAS; (c) G.-N. Ma, J.-J. Jiang, M. Shi and Y. Wei, Chem. Commun., 2009, 5496–5514 RSC; (d) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS.
- M. Ciclosi, C. Fava, R. Galeazzi, M. Orena and J. Sepulveda-Arques, Tetrahedron Lett., 2002, 43, 2199 CrossRef CAS.
- (a) C.-W. Cho, J.-R. Kong and M. J. Krische, Org. Lett., 2004, 6, 1337–1339 CrossRef CAS; (b) C.-W. Cho and M. J. Krische, Angew. Chem., Int. Ed., 2004, 43, 6689–6691 CrossRef CAS.
- T.-Z. Zhang, L.-X. Dai and X.-L. Hou, Tetrahedron: Asymmetry, 2007, 18, 1990–1994 CrossRef CAS.
- Y.-Q. Jiang, Y.-L. Shi and M. Shi, J. Am. Chem. Soc., 2008, 130, 7202–7203 CrossRef CAS.
- M.-Y. Kwak, S.-H. Kwon and C.-W. Cho, Bull. Korean Chem. Soc., 2009, 30, 2799–2802 CrossRef CAS.
- G.-N. Ma, S.-H. Cao and M. Shi, Tetrahedron: Asymmetry, 2009, 20, 1086–1092 CrossRef CAS.
- H.-P. Deng, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 1956–1960 CrossRef CAS.
- Y. Du, X. Han and X. Lu, Tetrahedron Lett., 2004, 45, 4967–4971 CrossRef CAS.
- S. D. J. V. C. van, T. Marcelli, M. Lutz, A. L. Spek, M. J. H. van and H. Hiemstra, Adv. Synth. Catal., 2007, 349, 281–286 CrossRef.
- H.-L. Cui, J. Peng, X. Feng, W. Du, K. Jiang and Y.-C. Chen, Chem.–Eur. J., 2009, 15, 1574–1577 CrossRef CAS.
- K. Jiang, J. Peng, H.-L. Cui and Y.-C. Chen, Chem. Commun., 2009, 3955–3957 RSC.
- J. Peng, X. Huang, H.-L. Cui and Y.-C. Chen, Org. Lett., 2010, 12, 4260–4263 CrossRef CAS.
- H.-L. Cui, J.-R. Huang, J. Lei, Z.-F. Wang, S. Chen, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 720–723 CrossRef CAS.
- L. Jiang, Q. Lei, X. Huang, H.-L. Cui, X. Zhou and Y.-C. Chen, Chem.–Eur. J., 2011, 17, 9489–9493 CrossRef CAS.
- C. Liu, B.-X. Tan, J.-L. Jin, Y.-Y. Zhang, N. Dong, X. Li and J.-P. Cheng, J. Org. Chem., 2011, 76, 5838–5845 CrossRef CAS.
- (a) W. Yang, X. Wei, Y. Pan, R. Lee, B. Zhu, H. Liu, L. Yan, K.-W. Huang, Z. Jiang and C.-H. Tan, Chem.–Eur. J., 2011, 17, 8066–8070 CrossRef CAS; (b) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 9684–9688 CrossRef CAS; (c) X. Companyó, G. Valero, V. Ceban, T. Calvet, M. Font-Bardia, A. Moyano and R. Rios, Org. Biomol. Chem., 2011 10.1039/c1ob06308a.
- Y.-L. Yang, C.-K. Pei and M. Shi, Org. Biomol. Chem., 2011, 9, 3349–3358 CAS.
- For a review of oxazolones in organocatalysis see: A.-N. R. Alba and R. Rios, Chem.–Asian J., 2011, 6, 720–734 CrossRef CAS.
- T. Furukawa, T. Nishimine, E. Tokunaga, K. Hasegawa, M. Shiro and N. Shibata, Org. Lett., 2011, 13, 3972–3975 CrossRef CAS.
- H.-L. Cui, X.-H. Sun, L. Jiang, L. Ding and Y.-C. Chen, Eur. J. Org. Chem., 2011 DOI:10.1002/ejoc.201101255.
- H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737–5740 CrossRef CAS.
- S.-J. Zhang, H.-L. Cui, K. Jiang, R. Li, Z.-Y. Ding and Y.-C. Chen, Eur. J. Org. Chem., 2009, 5804–5809 CrossRef CAS.
- J.-R. Huang, H.-L. Cui, J. Lei, X.-H. Sun and Y.-C. Chen, Chem. Commun., 2011, 47, 4784–4786 RSC.
- X. Feng, Y.-Q. Yuan, H.-L. Cui, K. Jiang and Y.-C. Chen, Org. Biomol. Chem., 2009, 7, 3660–3662 CAS.
- B. Zhu, L. Yan, Y. Pan, R. Lee, H. Liu, Z. Han, K.-W. Huang, C.-H. Tan and Z. Jiang, J. Org. Chem., 2011, 76, 6894–6900 CrossRef CAS.
- L. Hong, W. Sun, C. Liu, D. Zhao and R. Wang, Chem. Commun., 2010, 46, 2856–2858 RSC.
- B. Tan, N. R. Candeias and C. F. Barbas, J. Am. Chem. Soc., 2011, 133, 4672–4675 CrossRef CAS.
- H.-P. Deng, Y. Wei and M. Shi, Org. Lett., 2011, 13, 3348–3351 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |