CO2 reforming of methane over Mg-promoted Ni/SiO2 catalysts: the influence of Mg precursors and impregnation sequences†
Jianqiang
Zhu
,
Xiaoxi
Peng
,
Lu
Yao
,
Dongmei
Tong
and
Changwei
Hu
*
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: chwehu@mail.sc.cninfo.net; Fax: +86 28 85411105; Tel: +86 28 85411105
First published on 1st December 2011
Abstract
Mg-promoted Ni/SiO2 catalysts with different Mg precursors were prepared by different impregnation sequences and used in CO2 reforming of methane. The catalysts were characterized by XRD, TPR-H2, TPD-CO2, TG-DTA and SEM techniques. The use of Mg(CH3COO)2 as Mg precursor favored the performance of the catalyst. The impregnation of Mg(CH3COO)2 prior to Ni led to stronger interaction of nickel species with the support and the formation of stable Ni2SiO4 and Mg2SiO4 species, which inhibited the sintering of metallic Ni and then made the catalyst show better activity and stability. The catalyst thus prepared also exhibited enhanced capacity of CO2 adsorption, which accelerated the CO2 activation and the elimination of deposited carbon. No significant carbon deposition was observed on the surface of the catalysts, keeping the catalyst stable.
1. Introduction
Because of the increasing interest in reduction of CO2 emissions and effective utilization of natural gas, CO2 reforming of methane to produce synthesis gas has been paid much more attention from both academic and industrial standpoints.1–18,20–30 This process provides a good approach to convert the two greenhouse gases, CO2 and methane, into synthesis gas with a theoretical CO/H2 ratio of unity, which could be preferentially used for the production of oxygenated hydrocarbons. However, the industrialization of CO2 reforming of methane remains a great challenge due to the lack of an efficient and stable catalyst, since it suffers from severe catalyst deactivation by coke deposition and metal sintering.It is well known that noble metal-based catalysts exhibited high activity and good coke resistance.25–33 Nevertheless, by consideration of the availability and economy, the development of non-noble metal-based catalysts is a promising route for this process. Many researchers reported that Fe, Co and Ni-based catalysts possessed high activity, but deactivated with time on stream due to coke formation and/or metal sintering.5–7,13,14 Therefore, much more efforts were mainly focused on the enhancement of catalytic stability.
The catalytic performance of supported metallic catalysts for CO2 reforming of methane is mainly affected by the type of support, the nature of metal and corresponding precursor, as well as the strategy of catalyst preparation and the conditions of pretreatment.34–42 He et al.34 investigated the catalytic performance of Ni/SiO2 and Ni–Al2O3/SiO2 catalysts prepared by citrate and nitrate precursors for the combination reactions of CO2 reforming and partial oxidation of methane. They found that the catalytic activity significantly depended on the precursor of nickel and the interaction between NiO and support. Nickel citrate might be an excellent precursor for the preparation of Ni catalyst with high dispersion. The strong interaction between NiO and SiO2 resisted the catalyst sintering at high temperature. They also reported that Ni/SiO2 catalysts prepared with citrate precursor had excellent performances for their high nickel dispersion derived from the nickel silicate like species.35 Özkara-Aydınoğlu et al.39 studied the influence of cerium amount and the impregnation strategy on the activity and stability of Ce-promoted Pt/ZrO2 catalysts for CO2 reforming of methane. It was reported that the influence of the Ce promoter on catalytic activity depended on the cerium amount and impregnation strategy. The co-impregnation method resulted in extensive and strong Pt–Ce surface interaction and enhanced the Pt dispersion. Wang and Lu40 studied the effect of nickel precursors on the catalytic performance of Ni/Al2O3 catalysts for CO2 reforming of methane, and found that nickel nitrate was superior to nickel chloride and nickel acetylacetonate in terms of activity and stability. They also reported that Ni/Al2O3 catalysts prepared with different nickel precursors resulted in different carbon species formed on the catalyst surface, influencing the catalytic stability.
In our previous work, we investigated the effect of La, Mg, Co and Zn promoters on the catalytic performance of Ni/SiO2 with an emphasis on the activity and stability. We found that Ni–La/SiO2 catalysts exhibited excellent performance as compared to other catalysts. Ni–Mg/SiO2 and Ni–Co/SiO2 catalysts showed high initial activity, but poor stability. Meanwhile, Mg was reported to improve the catalytic performance due to its strong basicity, which enhanced the capacity of CO2 adsorption, altered the acid–base property of catalysts and improved the dispersion of active phase.11,16–18 Therefore, based on our previous investigation,18 the influence of Mg precursors and impregnation sequences on the performance of Mg-promoted Ni/SiO2 catalysts for CO2 reforming of methane was studied, aiming to develop a catalyst with enhanced activity and stability.
2. Experimental
2.1 Catalyst preparation
By modification of the catalyst preparation methods reported in previous literature,18,19 the catalysts used in the present work were prepared by an incipient wetness impregnation method. For all catalysts, the loadings of Ni and Mg were 10 wt% and 5 wt%, respectively.(1) The Ni–Mg/SiO2 catalysts with different Mg precursors were prepared by the following procedure: the SiO2 support (20–40 mesh, 320 m2 g−1) was impregnated with aqueous solution containing appropriate amounts of Ni(NO3)2 and Mg(CH3COO)2, or Mg(NO3)2, or MgCl2, or MgSO4 for 24 h, and dried at 110 °C for 4 h, then calcined in a muffle furnace at 800 °C for 5 h. These catalysts were denoted as Ni–Mg/SiO2–C, Ni–Mg/SiO2–N, Ni–Mg/SiO2–Cl and Ni–Mg/SiO2–S, respectively.
(2) The Ni/Mg/SiO2 catalysts with different Mg precursors were prepared by the following procedure: the SiO2 support (20–40 mesh, 320 m2 g−1) was firstly impregnated with aqueous solution containing an appropriate amount of Mg(CH3COO)2, or Mg(NO3)2, or MgCl2, or MgSO4 for 24 h, and dried at 110 °C for 4 h, then calcined in a muffle furnace at 800 °C for 5 h. The obtained samples were subsequently impregnated with aqueous solution containing an appropriate amount of Ni(NO3)2 for 24 h, and dried at 110 °C for 4 h, finally calcined in a muffle furnace at 800 °C for 5 h. These catalysts were denoted as Ni/Mg/SiO2–C, Ni/Mg/SiO2–N, Ni/Mg/SiO2–Cl and Ni/Mg/SiO2–S, respectively.
(3) The Mg/Ni/SiO2 catalysts with different Mg precursors were prepared by the following procedure: the SiO2 support (20–40 mesh, 320 m2 g−1) was firstly impregnated with aqueous solution containing an appropriate amount of Ni(NO3)2 for 24 h, and dried at 110 °C for 4 h, then calcined in a muffle furnace at 800 °C for 5 h. The obtained samples were subsequently impregnated with aqueous solution containing an appropriate amount of Mg(CH3COO)2, or Mg(NO3)2, or MgCl2, or MgSO4 for 24 h, and dried at 110 °C for 4 h, finally calcined in a muffle furnace at 800 °C for 5 h. These catalysts were denoted as Mg/Ni/SiO2–C, Mg/Ni/SiO2–N, Mg/Ni/SiO2–Cl and Mg/Ni/SiO2–S, respectively.
2.2 Catalytic activity test
The procedure of the activity test was reported in previous literature.18 The catalytic activity test was carried out in a fixed-bed flow under ambient atmospheric pressure. 0.25 g of catalyst was loaded in the middle of a micro-quartz-tube reactor. The catalyst was heated from room temperature to reaction temperature (800 °C) in argon flow. The catalyst was reduced in a H2/Ar atmosphere for 1 h (H2/Ar = 1, 60 ml min−1) prior to reaction. Then the reactants CH4 (99.99%) and CO2 (99.5%) without any diluents were co-fed with a total flow rate of 60 ml min−1 (CH4/CO2 = 1, F = 60 ml min−1). The flow rate of reactant gases was controlled by mass flow controllers (D07-11A/ZM made by Beijing Sevenstar Huachuang Electronic). The effluent after the removal of water was analyzed by an on-line gas chromatograph (GC) with a Plot-C2000 capillary column for separating the products and a TCD to determine the concentration of each component.In this work, the catalytic activity was calculated as mentioned in the literature.24 All the activity results discussed in this paper were obtained by at least three parallel tests and the deviation of activity was about ±0.2%.
2.3 Catalyst characterization
The actual amounts of Ni and Mg on the fresh catalysts were determined by atomic absorption spectroscopy (AAS). The fresh and used catalysts were comparatively characterized by an XRD technique. The X-ray diffraction patterns of the catalysts were recorded with a DX-1000 CSC diffractometer using Cu Kα monochromatic X-ray radiation (40 kV and 25 mA).The reduction behaviors of fresh catalysts were characterized by temperature-programmed reduction (TPR) with H2 according to our previous report.18 In short, 0.25 g of catalyst was pretreated in N2 flow at 150 °C for 30 min before reduction, and then cooled down to 80 °C. The TPR test was performed under a N2/H2 mixture (H2/N2 = 5/95 vol%) from 100 °C to about 830 °C with a heating rate of 8 °C min−1. The amount of consumed H2 was determined on-line by TCD. The conditions were selected to avoid the effect of mass transfer on the reduction behavior according to the literature.18,21,22
Temperature-programmed desorption (TPD) of CO2 was carried out on a conventional apparatus. 0.25 g of catalyst was pretreated in N2 flow at 300 °C for 60 min before the adsorption of CO2, and then cooled to below 50 °C. The samples were saturated by pure CO2 stream for 30 min, then the gas was changed to pure helium to purge the gas line and remove weakly adsorbed CO2. The TPD test was performed from room temperature to 800 °C with a heating rate of 8 °C min−1. The amount of CO2 desorbed was monitored on-line by TCD.
The morphologies of typical fresh and used catalysts were observed by scanning electron microscopy (SEM, FEI Inspect F). The samples were covered with a thin film of gold (ion sputtering) to improve the conductivity.
TG-DTA analysis was employed to determine the amount of carbon deposited on the surface of typical used catalysts. The TG-DTA measurements were carried out in air flow (70 ml min−1) with a NETZSCH 409 PC/PG analyzer using 15–30 mg and a heating rate of 10 °C min−1.
3. Results
3.1 Atomic absorption spectroscopy analysis
The actual amounts of Ni and Mg determined by AAS analysis are listed in Tables 1 and 2. It was found that the actual amounts are close to the controlled amounts with the exception of a relatively low Mg content for samples prepared with MgSO4 precursor. This illustrated that Ni and Mg were successfully impregnated on the SiO2 support during the preparation process.| Catalyst | Actual amountb (%) | Conversionc (%) | Yieldc (%) | CO/H2c | |||
|---|---|---|---|---|---|---|---|
| Ni | Mg | CH4 | CO2 | CO | H2 | ||
| a Conditions: 800 °C, F = 60 ml min−1, CH4/CO2 = 1/1, m = 0.25 g. b According to AAS results. c Activity obtained at 30 h on stream. | |||||||
| Ni/Mg/SiO2–C | 8.6 | 4.8 | 83.1 | 84.0 | 79.3 | 53.0 | 1.50 |
| Ni/Mg/SiO2–N | 8.4 | 4.5 | 78.5 | 80.6 | 74.6 | 49.5 | 1.51 |
| Ni/Mg/SiO2–Cl | 8.6 | 4.4 | 45.6 | 53.6 | 41.5 | 21.6 | 1.92 |
| Ni/Mg/SiO2–S | 7.9 | 2.0 | 44.6 | 52.4 | 40.0 | 20.7 | 1.93 |
| Ni–Mg/SiO2–C | 8.4 | 4.3 | 84.8 | 84.9 | 81.1 | 54.6 | 1.49 |
| Mg/Ni/SiO2–C | 8.3 | 4.5 | 50.0 | 52.8 | 43.1 | 27.9 | 1.54 |
| Catalyst | Actual amountb (%) | Conversionc (%) | Conversiond (%) | CO/H2d | |||
|---|---|---|---|---|---|---|---|
| Ni | Mg | CH4 | CO2 | CH4 | CO2 | ||
| a Conditions: 800 °C, F = 60 ml min−1, CH4/CO2 = 1/1, m = 0.25 g. b According to AAS results. c Activity obtained at 1 h on stream. d Activity obtained at 10 h on stream. | |||||||
| Ni–Mg/SiO2–N | 8.4 | 4.2 | 86.2 | 87.6 | 74.6 | 78.6 | 1.50 |
| Mg/Ni/SiO2–N | 8.2 | 4.6 | 33.9 | 36.4 | 46.9 | 52.1 | 1.66 |
| Ni–Mg/SiO2–Cl | 8.1 | 4.4 | 35.1 | 42.8 | 36.7 | 44.7 | 2.14 |
| Mg/Ni/SiO2–Cl | 8.4 | 4.6 | 34.4 | 42.4 | 11.2 | 12.7 | 1.62 |
| Ni–Mg/SiO2–S | 7.8 | 2.2 | 31.5 | 34.7 | 46.4 | 53.1 | 1.77 |
| Mg/Ni/SiO2–S | 7.5 | 1.9 | 15.7 | 20.7 | 37.0 | 47.5 | 1.76 |
3.2 XRD characterization
The X-ray patterns of several typical fresh catalysts are plotted in Fig. 1. By comparison of Ni/Mg/SiO2 catalysts with different Mg precursors, it was found that the XRD peaks attributed to NiO phase were detected on all the four catalysts. Especially, NiO was highly dispersed on Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2. Nevertheless, NiO species with particle sizes of 17 and 13 nm calculated from the Scherrer–Warren formula was observed on Ni/Mg/SiO2 prepared with MgCl2 and MgSO4 precursors. Moreover, as Mg(CH3COO)2 and Mg(NO3)2 were used, new phases of Ni2SiO4 and Mg2SiO4 appeared on Ni/Mg/SiO2–C and Ni/Mg/SiO2–N catalysts. This demonstrated that the interaction between Ni, Mg species and SiO2 support was enhanced on the two catalysts, leading to the formation of Ni2SiO4 and Mg2SiO4 species (it is hard to discriminate between Ni2SiO4 and Mg2SiO4 as the diffractions of these phases overlap partly). Ni2SiO4 and Mg2SiO4 species were formed due to the interaction of nickel oxide and magnesium oxide with SiO2 support as reported in other literature.43–47 However, other new peaks corresponding to MgSiO3 were detected on Ni/Mg/SiO2 catalysts prepared with MgCl2. For the series of Ni–Mg/SiO2 and Mg/Ni/SiO2 catalysts, NiO phase was found on all these catalysts. Ni2SiO4 and Mg2SiO4 species were only detected on Ni–Mg/SiO2–C, while MgSiO3 species was detected on Ni–Mg/SiO2–Cl and Mg/Ni/SiO2–Cl catalysts (see ESI†, Fig. S1).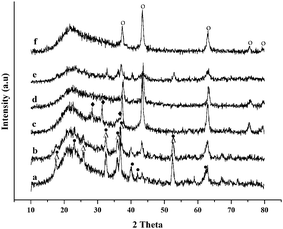 | ||
| Fig. 1 XRD patterns of the typical fresh catalysts. (a) Ni/Mg/SiO2–C; (b) Ni/Mg/SiO2–N; (c) Ni/Mg/SiO2–Cl; (d) Ni/Mg/SiO2–S; (e) Ni–Mg/SiO2–C; (f) Mg/Ni/SiO2–C; (○) NiO; (●) Ni2SiO4; (△) Mg2SiO4; (◆) MgSiO3. | ||
As Mg(CH3COO)2 was used, except for highly dispersed NiO species, Ni2SiO4 and Mg2SiO4 phases appeared on Mg-promoted Ni/SiO2 catalysts prepared by impregnating Mg prior to Ni and co-impregnation. It can be inferred that the two impregnation sequences were more beneficial to enhance the interaction with the support, leading to the formation of Ni2SiO4 and Mg2SiO4 species. However, only NiO species with a particle size of about 15 nm was detected on catalysts prepared by impregnating Ni prior to Mg. As Mg(NO3)2 was used, except for highly dispersed NiO phase, Ni2SiO4 and Mg2SiO4 appeared on the catalysts prepared by impregnating Mg prior to Ni, while only NiO phase was found on the samples prepared by other two impregnation sequences. Nevertheless, there was no significant influence of impregnation sequences on the structure of catalysts as MgCl2 and MgSO4 precursors were employed.
In brief, the structure of the catalysts was influenced significantly by the addition of different Mg precursors. Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2 possessed high NiO dispersion and stronger surface interaction, leading to the formation of Ni2SiO4 and Mg2SiO4 species. Meanwhile, the impregnation sequence influenced the structure of the catalysts. Especially, as Mg(CH3COO)2 and Mg(NO3)2 precursors were employed, the catalysts prepared by impregnating Mg prior to Ni exhibited higher NiO dispersion and enhanced interaction with the support, resulting in the formation of Ni2SiO4 and Mg2SiO4 species.
3.3 TPR results
The reduction behaviors of the catalysts were characterized by TPR experiments. The TPR-H2 profiles of typical samples are presented in Fig. 2. By comparison of Ni/Mg/SiO2 catalysts with different Mg precursors, it was found that Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2 precursors had only one reduction peak with a maximum temperature of higher than 800 °C, which was assigned to the reduction of nickel oxides bearing strong interaction with the support. Zhang et al.47 reported that the reduction of Ni2SiO4 needs a higher temperature of about 630–700 °C. Actually, the Ni2SiO4 and Mg2SiO4 species were detected by XRD characterization mentioned above. In addition, the low intensity of the reduction peak indicated that the amount of reducible species on Ni/Mg/SiO2–C and Ni/Mg/SiO2–N catalysts was low. It is well known that the compounds bearing strong interaction with the support provided higher metal dispersion and high resistance to sintering.15,16 On the other hand, except for the not ended peak at about 800 °C, a very broad peak starting from 435 to 730 °C was found on the TPR profile of Ni/Mg/SiO2 prepared with MgCl2. Meanwhile, three well-defined peaks with maxima at 440, 508, and 595 °C were detected on Ni/Mg/SiO2 catalysts prepared with MgSO4. This illustrated that nickel oxides bearing different interactions with the support existed on Ni/Mg/SiO2–Cl and Ni/Mg/SiO2–S catalysts. Moreover, the higher area of these peaks on the two catalysts demonstrated that the amount of reducible species was more than that on Ni/Mg/SiO2–C and Ni/Mg/SiO2–N catalysts. Similar results were obtained on the series of Ni–Mg/SiO2 and Mg/Ni/SiO2 catalysts. Especially, as MgCl2 and MgSO4 were used, the relatively lower reduction temperature and higher area of peaks were found on the profiles of TPR (see ESI†, Fig. S2).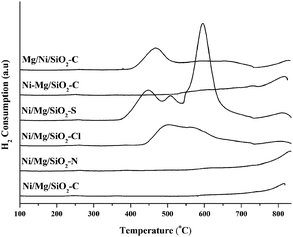 | ||
| Fig. 2 TPR-H2 profiles of the typical fresh catalysts. | ||
As Mg(CH3COO)2 was used, except for the peak located between 520–730 °C, another new obvious peak with maximum at 474 °C ascribed to “free state” nickel oxides bearing weak interaction with the support was detected on the catalyst prepared by impregnating Ni prior to Mg. The same peaks were reported in other literature.18,23,24 Nevertheless, only peaks with lower intensity located at higher than 800 °C were detected on catalysts prepared by co-impregnation and impregnating Mg prior to Ni. It can be inferred that the amount of reducible species was low and the metallic oxides bearing enhanced interaction with the support existed on the two catalysts, resulting in high reduction temperature. On the other hand, when the Mg(NO3)2 precursor was used, the catalyst prepared by impregnating Mg prior to Ni exhibited higher reduction temperature with lower peak area, as compared with other two impregnation sequences. Nevertheless, the impregnation sequence did not exert a significant effect on the reduction behavior of catalysts prepared with MgCl2 and MgSO4 precursors (see ESI†, Fig. S2).
In summary, Mg precursors and impregnation sequences influenced the reduction behavior of the catalysts. Totally speaking, Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2 exhibited high reduction temperature and enhanced interaction, leading to lower reducibility. Meanwhile, as Mg(CH3COO)2 and Mg(NO3)2 precursors were used, impregnating Mg prior to Ni increased the interaction of metallic oxide with SiO2 support much more significantly, keeping the catalysts stable.
3.4 TPD-CO2 results
Fig. 3 presents the CO2-TPD profiles of several typical fresh catalysts. Generally speaking, the temperature of CO2 desorption reflected the strength of basic sites, while the areas of desorption peaks reflected the amount of basic sites on the surface of catalysts. In analyzing the TPD-CO2 profiles of Ni/Mg/SiO2 with different Mg precursors, it was found that a CO2 desorption peak around 110 °C was detected on all the Ni/Mg/SiO2 catalysts, which was ascribed to the weak basic sites. A similar CO2 desorption peak was also reported on Ni-based catalysts in the literature.11,17,48,49 Moreover, the area of desorption peaks on the TPD profiles of Ni/Mg/SiO2–C and Ni/Mg/SiO2–S was much more than that on Ni/Mg/SiO2–N and Ni/Mg/SiO2–Cl catalysts. This illustrated that Ni/Mg/SiO2 prepared with Mg(CH3COO)2 and MgSO4 precursors had much more basic sites and enhanced ability of CO2 adsorption, which was much more beneficial to CO2 activation and the removal of deposited carbon.22,49 Especially, the higher intensity of a desorption peak around 110 °C on Ni/Mg/SiO2–C and Ni/Mg/SiO2–S demonstrated that the equilibrium of CO2 adsorption–desorption could take place much faster and easier at low temperature. However, except for the desorption peaks at 210 and 565 °C, which was attributed to moderate basic sites, another not ended peak with much higher intensity at about 800 °C was found on the TPD profile of Ni/Mg/SiO2–S. This illustrated that the amount of strong basic sites on Ni/Mg/SiO2–S was much higher and the CO2 desorption could take place at higher temperature. On the other hand, the high intensity of CO2 adsorption peaks at about 210 and 430 °C ascribed to moderate basic sites and a peak at 560–740 °C attributed to strong basic sites were detected on Ni/Mg/SiO2–C catalysts. Similar peaks were also found on the profiles of Ni/Mg/SiO2–Cl catalysts. Nevertheless, only a CO2 desorption peak at 430 °C with lower intensity and a not ended peak around 800 °C were detected on Ni/Mg/SiO2–S catalysts.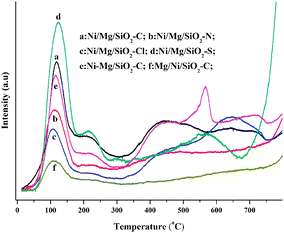 | ||
| Fig. 3 TPD-CO2 profiles of the typical fresh catalysts. | ||
When the Mg(CH3COO)2 precursor was used, by comparison with the TPD-CO2 desorption profiles of catalysts prepared by different impregnation sequences, it can be seen that the desorption area on Ni/Mg/SiO2–C and Ni–Mg/SiO2–C was higher than that on Mg/Ni/SiO2–C catalysts. This illustrated that the amount of basic sites on Mg-promoted Ni/SiO2 catalysts prepared by impregnating Mg prior to Ni and co-impregnation was more than on that prepared by impregnating Ni prior to Mg. The same desorption peak at a low temperature of about 110 °C was also detected on all the three Mg-promoted Ni/SiO2 catalysts prepared with Mg(CH3COO)2 precursor. Especially, the amount of weak basic sites on Ni/Mg/SiO2–C was much higher than that on Ni–Mg/SiO2–C and Mg/Ni/SiO2–C catalysts. This illustrated that the establishment of CO2 adsorption–desorption equilibrium was much easier on the former catalysts. Meanwhile, another CO2 desorption peak at 400–800 °C with low intensity was detected on Mg/Ni/SiO2. Several CO2 desorption peaks at 210, 430, 565, 720, 800 °C, and CO2 desorption peaks at 210, 430, 640, 720, 800 °C were found on the TPD-CO2 profiles of Ni–Mg/SiO2–C and Ni/Mg/SiO2–C catalysts, respectively. This indicated that the basic sites with different strengths existed on the surface of the catalysts.
In short, the basicity of Mg-promoted Ni/SiO2 catalysts was influenced by the addition of different Mg precursors and impregnation sequences. Three kinds of basic sites, named weak, moderate and strong basic sites existed on the surface of catalysts. The capacity of CO2 adsorption on the catalyst prepared with Mg(CH3COO)2 and MgSO4 was much stronger, especially the weak basic sites increased significantly, leading to the faster and easier establishment of CO2 adsorption–desorption equilibrium. Meanwhile, as Mg(CH3COO)2 was used, the basicity of catalysts prepared by impregnating Mg prior to Ni and co-impregnation was increased much more remarkably, which were more beneficial to the CO2 adsorption and the elimination of deposited carbon.
3.5 Catalytic activity and stability
The activity of the catalysts was monitored by means of the conversions of methane and CO2, the yields of CO and H2, as well as the molar ratio of CO/H2. The activity results of several typical catalysts at 30 h are listed in Table 1. Under the following conditions: 800 °C, F = 60 ml min−1, CH4/CO2 = 1/1, 0.25 g catalyst, by comparison with the activity of Ni/Mg/SiO2 catalysts with different Mg precursors, it was found that Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2 precursors possessed higher conversion of CH4 and CO2, as well as higher yield of CO and H2. However, Ni/Mg/SiO2 catalysts prepared with both MgCl2 and MgSO4 exhibited a relatively low activity. This indicated that Mg(CH3COO)2 and Mg(NO3)2 might be better candidates as Mg precursors for the preparation of Ni/Mg/SiO2 catalysts for use in CO2 reforming of methane as compared to other two Mg precursors. On the other hand, the catalytic performance was also influenced by the impregnation sequence remarkably. As seen in Table 1, it was found that Ni/Mg/SiO2–C and Ni–Mg/SiO2–C showed higher activity than Mg/Ni/SiO2–C catalysts. That is to say, as Mg(CH3COO)2 was used, impregnating Ni prior to Mg was not an effective method to promote the catalytic performance, while the other two impregnation methods improve the activity more significantly. The six typical catalysts were selected for further investigation with an emphasis on the influence of Mg precursors and impregnation sequences on the catalytic performance.Under the same reaction conditions mentioned above, the activity results of other six catalysts within 10 h are shown in Table 2. It was found that all the catalysts exhibited a relatively low activity and poor stability with the exception of Ni–Mg/SiO2–N and Mg/Ni/SiO2–Cl catalysts, which had a relatively higher initial activity but decreased with time on stream.
In all activity tests, the molar ratio of CO/H2 was more than unity due to the occurrence of reverse water-gas shift reaction. This is in accordance with other reported literature.4,7,18 In addition, higher conversions of CH4 and CO2 with lower CO/H2 molar ratio were obtained on these catalysts. Therefore, lower CO/H2 molar ratio was obtained on Ni/Mg/SiO2–C, Ni/Mg/SiO2–N and Ni–Mg/SiO2–C catalysts, which illustrated that the reverse water-gas shift reaction was inhibited much more significantly on these catalysts, leading to higher H2 yield.
Fig. 4 presents the tendency of CH4 conversion on selected catalysts with time on stream. It can be seen that the CH4 conversion on Ni/Mg/SiO2–C decreased slightly, which was similar to that on Ni/Mg/SiO2–N and Ni–Mg/SiO2–C catalysts. However, the other three catalysts showed low activity and poor stability. Similar results were found on the conversion of CO2 (see ESI†, Fig. S3).
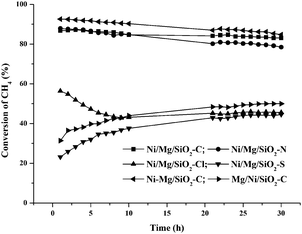 | ||
| Fig. 4 The tendency of CH4 conversion on selected catalysts within 30 h. | ||
3.6 The characterization of used catalysts
The XRD patterns of selected catalysts after 30 h reaction are comparatively plotted in Fig. 5. Typically, NiO species were totally reduced to metallic Ni. However, the peaks ascribed to carbon were not detected, which was different from our previous report.18 It might be inferred that low amount of carbon deposition and/or high dispersion of carbon occurred on all the used catalysts. On the other hand, as compared with the XRD profiles of fresh catalysts, the peaks attributed to Ni2SiO4 and Mg2SiO4 species still existed on used Ni/Mg/SiO2–C, Ni–Mg/SiO2–C and Ni/Mg/SiO2–N catalysts, even after long-time reaction of 30 h. This implied the excellent stability of Ni2SiO4 and Mg2SiO4 species, as reported in other literature.46,47 Nevertheless, the intensity of peaks corresponding to MgSiO3 species on Ni/Mg/SiO2–Cl catalysts weakened after 30 h reaction, indicating that MgSiO3 species exhibited a poor stability with time on stream. Świerczyński et al.44 reported that MgSiO3 species could be diminished in the presence of MgO and/or NiO to form Mg2SiO4 and/or NiMgSiO4 at high temperature. Mg2SiO4 and NiMgSiO4 species were not observed on the used Ni/Mg/SiO2–Cl catalyst in the present work, which might be due to their lower amount and/or their higher dispersion on the surface of the catalyst. By calculating the particle size of metallic Ni, it was found that Ni/Mg/SiO2–C, Ni–Mg/SiO2–C and Ni/Mg/SiO2–N catalysts had the particle size of about 11 nm, which was smaller than that on used catalysts of Mg/Ni/SiO2–C (18 nm), Ni/Mg/SiO2–Cl (21 nm) and Ni/Mg/SiO2–S (15 nm).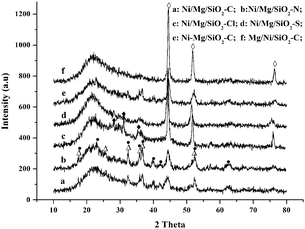 | ||
| Fig. 5 XRD patterns of the typical used catalysts after 30 h reaction. (⋄) Ni; (●) Ni2SiO4; (△) Mg2SiO4; (◆) MgSiO3. | ||
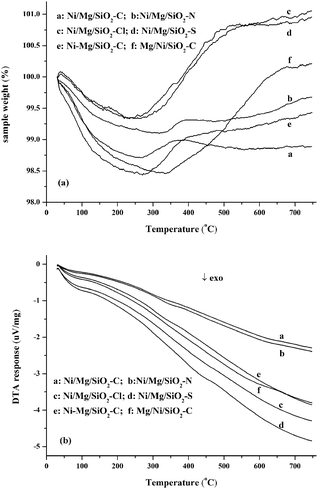
The carbon deposited on the catalyst surface after 30 h reaction was quantified by TG-DTA analysis in air flow. The TG-DTA profiles are plotted in Fig. 6. In analyzing the TG profiles, the initial step of weight loss occurred over the temperature range of 110–300 °C, which is attributed to the removal of moisture and easily oxidized carbonaceous species.18,41 Meanwhile, the oxidation of metallic Ni and carbonaceous species could take place simultaneously thereafter up to 500 °C. The net result depended on the relative amount of nickel and deposited carbon. An obvious weight increase was found on Ni/Mg/SiO2–Cl, Ni/Mg/SiO2–S and Mg/Ni/SiO2–C. Meanwhile, a slight weight decrease was found on Ni/Mg/SiO2–C, Ni/Mg/SiO2–N and Ni–Mg/SiO2–C catalysts. According to TG profiles, it might be deduced that the amount of carbon deposited on the catalysts was low, which could be also supported by the analysis of DTA profiles (Fig. 6b). No remarkable exothermic peaks were observed for the used catalysts in all temperature profiles, which indicated that the amount of carbon deposited on the catalyst surface might be low. This is in accordance with the TG results mentioned above.
The selected catalysts after 30 h reaction were characterized by a SEM technique and the results are presented in Fig. 7. The corresponding fresh catalysts were also characterized comparatively (see ESI†, Fig. S4). No significant difference in morphology was observed between the used and fresh catalysts.
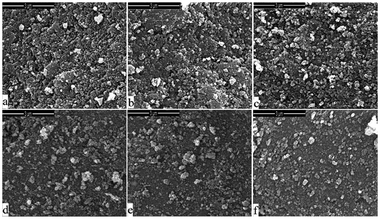 | ||
| Fig. 7 SEM images of the typical used catalysts after 30 h. (a) Ni/Mg/SiO2–C, (b): Ni/Mg/SiO2–N, (c) Ni/Mg/SiO2–Cl, (d) Ni/Mg/SiO2–S, (e) Ni–Mg/SiO2–C, (f) Mg/Ni/SiO2–C. | ||
Meanwhile, as shown in Fig. 7, by comparison of Ni–Mg/SiO2 catalysts prepared with Mg(NO3)2 precursor reported previously,18 the carbon whisker was not detected on all these Mg-promoted Ni/SiO2 catalysts. The IR spectra proved that no absorption peaks ascribed to carbon whisker were detected (see ESI†, Fig. S5). It might be deduced that the carbon whisker was eliminated quickly as formed for its low amount, or transferred into inactive carbon with time on stream, leading to the partial deactivation. However, by consideration of the TG-DTA results, it is reasonable to infer that the carbon deposited on these catalysts might be low, even on Ni/Mg/SiO2–C, Ni/Mg/SiO2–N and Ni–Mg/SiO2–C catalysts with higher catalytic activity.
4. Discussion
One of the major obstacles for the industrialization of CO2 reforming of methane into synthesis gas is the deactivation of catalysts caused by carbon deposition and metal sintering during reaction.5–7,13,14 Therefore, in recent decades, many researchers were devoted to develop nickel catalysts with improved activity and stability. They mainly focused on the preparation method of catalysts, the nature of the support and the addition of different promoters.1,8,36–42,504.1 The influence of Mg precursors
In the present work, four Mg precursors were employed to investigate the effect of Mg precursors on the structure, reducibility, basicity, as well as the catalytic activity and stability of the catalysts. Totally speaking, Mg(CH3COO)2 might be the better candidate as Mg precursor for the preparation of Mg-promoted Ni/SiO2 catalysts as compared to Mg(NO3)2, MgCl2 and MgSO4. For the series of Ni/Mg/SiO2 catalysts, except for NiO species observed on all the four catalysts, Ni2SiO4 and Mg2SiO4 species were formed on Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 and Mg(NO3)2 precursors, while MgSiO3 species was observed on Ni/Mg/SiO2 catalysts prepared with MgCl2. Moreover, the intensity of the peaks attributed to MgSiO3 species weakened, while Ni2SiO4 and Mg2SiO4 species still existed after long-time reaction. Meanwhile, higher reduction temperature observed on Ni/Mg/SiO2–C and Ni/Mg/SiO2–N catalysts illustrated that the interaction between metallic oxide and support was enhanced more significantly as Mg(CH3COO)2 and Mg(NO3)2 were employed, resulting in the decrease of their reducibility and the formation of Ni2SiO4 and Mg2SiO4 species on the two catalysts. Nevertheless, lower interaction of metallic oxide with support was found on Ni/Mg/SiO2 prepared with MgCl2 and MgSO4 deduced from their low reduction temperature. Bouarab et al.17 reported that Mg2SiO4 phase promoted the formation of well dispersed cobalt particles and inhibited the sintering at high temperature, and consequently improved the catalytic stability under severe dry reforming conditions. By consideration of the higher activity obtained on Ni/Mg/SiO2–C and Ni/Mg/SiO2–N catalysts, it can be inferred that the formation of stable Ni2SiO4 and Mg2SiO4 species on the two catalysts might be responsible for their better catalytic performance. However, the lower ability of CO2 adsorption on Ni/Mg/SiO2–N might lead to the slight decrease of its activity. In contrast, the basicity of Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 precursor was enhanced much more significantly, which promoted the ability of CO2 adsorption, resulting in a better CO2 conversion and removal of deposited carbon. On the other hand, the series of Ni–Mg/SiO2 and Mg/Ni/SiO2 catalysts prepared with Mg(CH3COO)2 precursor exhibited similar results, that is, they possessed better catalytic performance as compared to those prepared with other Mg precursors.In summary, the Mg precursors employed in this work influenced the catalytic performance significantly. In the preparation process, different precursors might influence the interaction between Mg precursors and the support. Moreover, the decomposition process of precursors and the producing gas atmosphere may affect the formation of active phases by altering the dispersion, structure, reducibility and basicity. For the series of Ni/Mg/SiO2 catalysts, the stable Ni2SiO4 and Mg2SiO4 species formed only when Mg(CH3COO)2 and Mg(NO3)2 precursors were used. Ni2SiO4 and Mg2SiO4 species might be beneficial to the formation of highly dispersed active phase and inhibit the sintering of metallic Ni. Consequently, the catalytic performance, especially the stability was improved.
4.2 The influence of impregnation sequences
In this section, the influence of impregnation sequences on the structure, reducibility, basicity as well as the catalytic activity and stability of the catalysts was discussed. Totally speaking, impregnating Mg prior to Ni might be more beneficial to improve the catalytic performance as compared to the methods of co-impregnation and impregnating Ni before Mg. As the Mg(CH3COO)2 precursor was employed, except for highly dispersed NiO, Ni2SiO4 and Mg2SiO4 species were detected on Mg promoted Ni/SiO2 catalysts prepared by impregnating Mg prior to Ni and co-impregnation. Meanwhile, the higher reduction temperature observed on Ni/Mg/SiO2–C and Ni–Mg/SiO2–C catalysts indicated that the interaction of metallic oxide with SiO2 support was enhanced more remarkably, correspondingly, resulting in the decrease of their reducibility and the formation of stable Ni2SiO4 and Mg2SiO4 species, which might prevent the sintering of catalysts. Moreover, the catalysts prepared by impregnating Mg prior to Ni and co-impregnation exhibited much more enhanced basicity, which was beneficial to CO2 adsorption and activation. Increasing the capacity of CO2 absorption was proposed to reduce carbon deposition and keep the catalyst stable.22,49 Additionally, the adsorption of CH4 and CO might be suppressed partially for the competitive adsorption of CO2 and could decrease the carbon deposition due to methane cracking and CO disproportionation reaction.49 In contrast, the catalyst prepared by impregnating Ni before Mg had a weak interaction of nickel oxide with the support and the relatively lower basicity, which might be related to its low activity and poor stability. As the Mg(NO3)2 precursor was used, although higher initial activity was obtained on Ni/Mg/SiO2–N and Ni–Mg/SiO2–N catalysts, the latter exhibited poor stability. Mg/Ni/SiO2–N catalysts showed only lower activity. In addition, the higher reduction temperature observed on Ni/Mg/SiO2–N illustrated its enhanced interaction of nickel oxide with the support. Therefore, impregnating Mg prior to Ni improved the catalytic performance more effectively as the Mg(NO3)2 precursor was used. On the other hand, as MgCl2 and MgSO4 precursors were employed, the structure, reduction behavior and the catalytic performance of the catalysts were not influenced significantly by the impregnation sequence. In a word, impregnating Mg prior to Ni was a more effective method for the preparation of Mg-promoted Ni/SiO2 catalysts used in CO2 reforming of methane.In summary, Mg-promoted Ni/SiO2 catalysts prepared by impregnating Mg prior to Ni with the Mg(CH3COO)2 precursor possessed stronger interaction between nickel species with the support, resulting in the formation of Ni2SiO4 and Mg2SiO4 species, which might be responsible for their high activity and good stability. Moreover, the catalyst simultaneously exhibited enhanced capacity of CO2 adsorption, which accelerated the removal of deposited carbon and kept the catalyst stable. Nevertheless, Ni/Mg/SiO2 catalysts prepared with Mg(NO3)2 exhibited higher interaction of nickel oxide with SiO2 support but relative lower ability of CO2 adsorption, while that prepared with MgSO4 had strong basicity but relative lower interaction between metallic oxide and support. Moreover, Ni/Mg/SiO2 catalysts prepared with MgCl2 only showed moderate interaction of nickel oxide with the support and a lower capacity of CO2 adsorption, resulting in their low activity and poor stability.
5. Conclusion
Mg-promoted Ni/SiO2 catalysts with different Mg precursors were prepared by different impregnation sequences and investigated in CO2 reforming of methane. Mg(CH3COO)2 might be the better candidate as Mg precursor as compared to Mg(NO3)2, MgCl2 and MgSO4. Moreover, impregnating Mg prior to Ni is more beneficial to improve the activity and stability than the other two impregnation sequences, especially as the Mg(CH3COO)2 precursor was employed.Ni/Mg/SiO2 catalysts prepared with Mg(CH3COO)2 exhibited lower reducibility and higher ability of CO2 adsorption. Meanwhile, when Mg(CH3COO)2 was used, the catalyst prepared by impregnating Mg prior to Ni possessed enhanced interaction of metallic oxide with the support and resulted in the formation of stable Ni2SiO4 and Mg2SiO4 species, which might prevent the catalyst sintering and be responsible for the better catalytic performance. Moreover, the catalyst prepared by this impregnation method simultaneously exhibited improved capacity of CO2 adsorption, which accelerated the activation of CO2 and the elimination of deposited carbon. Correspondingly, no whisker carbon was detected and the amount of inert carbon deposited on the surface of catalysts was low, which kept the catalyst stable.
Ni/Mg/SiO2 catalysts prepared with Mg(NO3)2 exhibited higher interaction of nickel species with SiO2 support but lower basicity, while that prepared with MgSO4 had enhanced capacity of CO2 adsorption but lower interaction of NiO with the support. Moreover, low activity and poor stability obtained on Ni/Mg/SiO2 catalysts prepared with MgCl2 resulted from lower ability of CO2 adsorption and weaker interaction of nickel oxide with the support.
Acknowledgements
The authors are grateful for financial support from the NNSFC (No. 20976109, 21021001), the Special Research Foundation of Doctoral Education of China (No. 20090181110046), and the characterization of the catalysts from Analytical and Testing Center of Sichuan University.Notes and references
- Y.-X. Pan, P. Y. Kuai, Y. Liu, Q. F. Ge and C.-J. Liu, Energy Environ. Sci., 2010, 3, 1322 CAS.
- F. Meshkani and M. Rezaei, Int. J. Hydrogen Energy, 2010, 35, 10295 CrossRef CAS.
- I. P. Silverwood, N. G. Hamilton, C. J. Laycock, J. Z. Staniforth, R. M. Ormerod, C. D. Frost, S. F. Parkerc and D. Lennon, Phys. Chem. Chem. Phys., 2010, 12, 3102 RSC.
- M. M. Barroso-Quiroga and A. E. Castro-Luna, Int. J. Hydrogen Energy, 2010, 35, 6052 CrossRef CAS.
- N. N. Sun, X. Wen, F. Wang, W. Wei and Y. H. Sun, Energy Environ. Sci., 2010, 3, 366 CAS.
- Ş. Özkara-Aydınoğlu and A. E. Aksoylu, Int. J. Hydrogen Energy, 2011, 36, 2950 CrossRef.
- Ş. Özkara-Aydınoğlu and A. E. Aksoylu, Catal. Commun., 2010, 11, 1165 CrossRef.
- X. S. Li, J.-S. Chang, M. Y. Tian and S.-E. Park, Appl. Organomet. Chem., 2001, 15, 109 CrossRef CAS.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, Appl. Catal., A, 2010, 377, 191 CrossRef.
- L. L. Xu, H. L. Song and L. J. Chou, Catal. Sci. Technol., 2011, 1, 1032 CAS.
- V. García, J. J. Fernández, W. Ruíz, F. Mondragón and A. Moreno, Catal. Commun., 2009, 11, 240 CrossRef.
- K. Nagaoka, K. Takanabe and K.-I. Aika, Chem. Commun., 2002, 1006 RSC.
- S. Therdthianwong, A. Therdthianwong, C. Siangchin and S. Yongprapat, Int. J. Hydrogen Energy, 2008, 33, 991 CrossRef CAS.
- D. P. Liu, W. N. E. Cheo, Y. W. Y. Lim, A. Borgna, R. Lau and Y. H. Yang, Catal. Today, 2010, 154, 229 CrossRef CAS.
- D. P. Liu, X. Y. Quek, W. N. E. Cheo, R. Lau, A. Borgna and Y. H. Yang, J. Catal., 2009, 266, 380 CrossRef CAS.
- Y. H. Wang, H. M. Liu and B. Q. Xu, J. Mol. Catal. A: Chem., 2009, 299, 44 CrossRef CAS.
- R. Bouarab, O. Akdim, A. Auroux, O. Cherifi and C. Mirodatos, Appl. Catal., A, 2004, 264, 161 CrossRef CAS.
- J. Q. Zhu, X. X. Peng, L. Yao, J. Shen, D. M. Tong and C. W. Hu, Int. J. Hydrogen Energy, 2011, 36, 7094 CrossRef CAS.
- J. Q. Zhu, S. Qin, S. T. Ren, X. X. Peng, D. M. Tong and C. W. Hu, Catal. Today, 2009, 148, 310 CrossRef CAS.
- R. Bouarab, O. Cherifi and A. Auroux, Green Chem., 2003, 5, 209 RSC.
- A. Venugopal, S. N. Kumar, J. Ashok, D. H. Prasad, V. D. Kumari, K. B. S. Prasad and M. Subrahmanyam, Int. J. Hydrogen Energy, 2007, 32, 1782 CrossRef CAS.
- Q. S. Jing, H. Lou, J. H. Fei, Z. Y. Hou and X. M. Zheng, Int. J. Hydrogen Energy, 2004, 29, 1245 CrossRef CAS.
- J. Gao, Z. Y. Hou, J. Z. Guo, Y. H. Zhu and X. M. Zheng, Catal. Today, 2008, 131, 278 CrossRef CAS.
- Y. X. Pan, C. J. Liu and P. Shi, J. Power Sources, 2008, 176, 46 CrossRef CAS.
- M. García-Diéguez, E. Finocchio, M. Á. Larrubia, L. J. Alemany and G. Busca, J. Catal., 2010, 274, 11 CrossRef.
- B. Nematollahi, M. Rezaei and M. Khajenoori, Int. J. Hydrogen Energy, 2011, 36, 2969 CrossRef CAS.
- N. E. McGuire, N. P. Sullivan, O. Deutschmann, H. Y. Zhu and R. J. Kee, Appl. Catal., A, 2011, 394, 257 CrossRef CAS.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, J. Catal., 2010, 270, 136 CrossRef.
- H. Arbag, S. Yasyerli, N. Yasyerli and G. Dogu, Int. J. Hydrogen Energy, 2010, 35, 2296 CrossRef CAS.
- G. K. Reddy, S. Loridant, A. Takahashi, P. Delichère and B. M. Reddy, Appl. Catal., A, 2010, 389, 92 CrossRef CAS.
- Z.-J. Wang, Y. Zhao, L. Cui, H. Y. Du, P. Yao and C.-J. Liu, Green Chem., 2007, 9, 554 RSC.
- S. Damyanova, B. Pawelec, K. Arishtirova, M. V. M. Huerta and J. L. G. Fierro, Appl. Catal., B, 2009, 89, 149 CrossRef CAS.
- O. Gamba, S. Moreno and R. Molina, Int. J. Hydrogen Energy, 2011, 36, 1540 CrossRef CAS.
- S. F. He, H. M. Wu, W. J. Yu, L. Y. Mo, H. Lou and X. M. Zheng, Int. J. Hydrogen Energy, 2009, 34, 839 CrossRef CAS.
- S. F. He, Q. S. Jing, W. J. Yu, L. Y. Mo, H. Lou and X. M. Zheng, Catal. Today, 2009, 148, 130 CrossRef CAS.
- S. Tang, L. Ji, J. Lin, H. C. Zeng, K. L. Tan and K. Li, J. Catal., 2000, 194, 424 CrossRef CAS.
- R. Pereñíguez, V. M. González-DelaCruz, J. P. Holgado and A. Caballero, Appl. Catal., B, 2010, 93, 346 CrossRef.
- C. E. Daza, S. Moreno and R. Molina, Int. J. Hydrogen Energy, 2011, 36, 3886 CrossRef CAS.
- Ş. Özkara-Aydınoğlu, E. Özensoy and A. E. Aksoylu, Int. J. Hydrogen Energy, 2009, 34, 9711 CrossRef.
- S. B. Wang and G. Q. Lu, Appl. Catal., A, 1998, 169, 271 CrossRef CAS.
- A. Nandini, K. K. Pant and S. C. Dhingra, Appl. Catal., A, 2005, 290, 166 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar, P. Bai, X. M. Liu and Z. F. Yan, Appl. Catal., B, 2008, 77, 346 CrossRef CAS.
- F. Pompeo, N. N. Nichio, M. G. González and M. Montes, Catal. Today, 2005, 107–108, 856 CrossRef CAS.
- D. Świerczyński, C. Courson, L. Bedel, A. Kiennemann and J. Guille, Chem. Mater., 2006, 18, 4025 CrossRef.
- Q. Zafar, T. Mattisson and B. Gevert, Energy Fuels, 2006, 20, 34 CrossRef CAS.
- Q. Zafar, T. Mattisson and B. Gevert, Ind. Eng. Chem. Res., 2005, 44, 3485 CrossRef CAS.
- L. F. Zhang, W. Li, J. Liu, C. L. Guo, Y. P. Wang and J. L. Zhang, Fuel, 2009, 88, 511 CrossRef CAS.
- H.-S. Roh, K.-W. Jun, W.-S. Dong, J.-S. Chang, S.-E. Park and Y.-I. Joe, J. Mol. Catal. A: Chem., 2002, 181, 137 CrossRef CAS.
- X. C. Li, M. Wu, Z. H. Lai and F. He, Appl. Catal., A, 2005, 290, 81 CrossRef CAS.
- P. Chen, Z.-Y. Hou and X.-M. Zheng, Chin. J. Chem., 2005, 23, 847 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: XRD patterns of several fresh catalysts; Fig. S2: TPR-H2 profiles of several fresh catalysts; Fig. S3: the tendency of CO2 conversion on selected catalysts within 30 h; Fig. S4: SEM images of the typical fresh catalysts; Fig. S5: IR spectra of the typical used catalysts after 30 h reaction. See DOI: 10.1039/c1cy00333j |
| This journal is © The Royal Society of Chemistry 2012 |