Silica nanoparticles efficiently catalyzed synthesis of quinolines and quinoxalines†
Alireza
Hasaninejad
*a,
Mohsen
Shekouhy
a and
Abdolkarim
Zare
*b
aDepartment of Chemistry, Faculty of Sciences, Persian Gulf University, Bushehr 75169, Iran. E-mail: ahassaninejad@yahoo.com; Fax: +98(771)4541494; Tel: +98(771)4222319
bChemistry Department, Payame Noor University, 19395-4697 Tehran, I. R. of Iran. E-mail: abdolkarimzare@yahoo.com; Fax: +98(771)5559489; Tel: +98(771)559486
First published on 27th October 2011
Abstract
In this work, highly efficient, green and inexpensive procedures for the preparation of quinoxaline and quinoline derivatives as attractive aza-polycyclic compounds are described. Silica nanoparticles-catalyzed condensation of 1,2-diamines with 1,2-diketones under solvent-free conditions at room temperature affords high yields of quinoxalines with short reaction times. Moreover, the microwave-assisted Friedländer hetero-annulation reaction between 2-aminoaryl ketones and carbonyl compounds in the presence of silica nanoparticles (NPs) as catalysts gives high yields of quinoline derivatives with short reaction times. These reactions have been done in neutral conditions. The NPs catalysts can be reused without lost of activity even after recycling fourteen times .
1. Introduction
Currently, the green synthesis of products without using volatile and toxic organic solvents is the subject of considerable interest in organic synthesis.1 In this regard, solvent-free conditions have received tremendous attention.1–4 Solvent-free conditions often lead to shorter reaction times, increased yields, easier workup, matching with the green chemistry protocols, and may enhance the regio- and stereoselectivity of reactions. Scale-up is also facilitated by the use of solvent-free techniques.1–4SiO2 is a very inexpensive, recyclable, and commercially available oxide which has been extensively used as a support, accompanied with a catalyst, in organic transformations.5–9 Nevertheless, the application of silica gel, solely, as a catalyst and a surface has been reported in few cases.10–14 Recent research has shown that remarkable improvement in the catalytic activity of catalysts can be achieved by manipulating their composition and/or structure at a level of a few nanometres.15–17 The development of new catalysts by nanoscale design has emerged as a fertile field for research and innovation.15–17 The ability of nanotechnology to enhance catalytic activity opens the potential to replace expensive catalysts with lower amounts of inexpensive nanocatalysts.15–17
Quinoxalines and quinolines are two important groups of aza-polycyclic compounds which have many biological and pharmaceutical properties. For example, quinoxalines have been applied as antimycobacterial,18 antibacterial,19 antifungal,19 anthelmintic,20antidepressant21 and antitumor agents.22Quinolines have been used as antimalarial, antiasthmatic, antihypertensive, antibacterial and tyrosine kinase inhibiting agents.23,24 The condensation of 1,2-diamines with 1,2-diketones has been used as a useful synthetic route toward quinoxalines. For this transformation some catalysts such as, Yb(OTf)3,25Zr(DS)4,26 (NH4)6Mo7O24·4H2O,27oxalic acid,28Zn[(L)proline],29iodine in DMSO,30 and sulfamic acid/MeOH31 have been reported. The Friedländer hetero-annulation reaction has been used as a straightforward method for the synthesis of quinoline derivatives.32,33 In this protocol, quinolines have been prepared by acid or base catalyzed condensation of 2-aminoaryl ketones with carbonyl compounds possessing a reactive α-methylene group followed by cyclodehydration.32–38 However, many of the reported protocols for the synthesis of quinoxalines and quinolines suffer from disadvantages such as, the use of expensive catalysts, the necessity for anhydrous conditions, prolonged reaction times, unsatisfactory yields, difficult experimental as well as workup procedures and the use of volatile organic solvents. Moreover, some methods need multi-steps procedures. Consequently, the development of efficient, inexpensive and simple methods for the preparation of quinoxaline and quinoline derivatives under solvent-free conditions is in demand.
Considering the above subjects and in continuation of our interest on the synthesis of aza heterocyclic compounds,39 herein we report highly efficient and simple methods for the synthesis of quinoxalines from 1,2-diamines and 1,2-diketone at room temperature as well as the synthesis of quinolines from 2- aminoaryl ketones and carbonyl compounds under microwave irradiation in the presence of silica nanoparticles (NPs) as catalysts under solvent-free conditions (Scheme 1). It is worth noting that our presented work has none of the above-mentioned disadvantages at all.
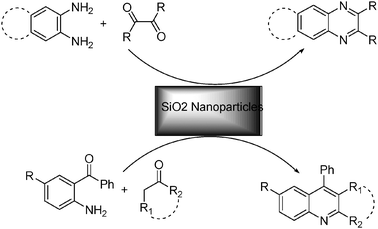 | ||
| Scheme 1 Silica nanoparticles-catalyzed synthesis of quinolines and quinoxalines. | ||
2. Experimental
All chemicals were purchased from Merck or Fluka chemical companies. Silica NPs were prepared according to the reported procedure.40 All known compounds were identified by comparison of their melting points and spectral data with those in the authentic samples. The silica nanoparticles sizes were measured using scanning electron microscopy (S-4160 SEM, HITACHI). A laboratory microwave oven (MicroSYNTH, Milestone Company, Italy) was used for the synthesis of quinolines. The 1H NMR (250 or 500 MHz) and 13C NMR (62.5 or 125 mhz) were run on a Bruker Avance DPX-250, FT-NMR spectrometer. Microanalysis was performed on a Perkin-Elmer 240-B microanalyzer. Melting points were recorded on a Stuart Scientific Apparatus SMP3 (UK) in open capillary tubes.2.1. General procedure for the synthesis of quinoxalines
A mixture of aryl 1,2-diamine (1 mmol), 1,2-diketone (1 mmol) and silica NPs (0.6 g) in a mortar was ground vigorously at room temperature. After completion of the reaction, as monitored by TLC, the reaction mixture was transferred to a 25 mL round-bottomed flask and hot ethanol (10 mL) was added to it followed by centrifugation for 20 min to separate the silica nanoparticles. The supernatant was concentrated to 5 mL, and allowed to stand at room temperature for 4–5 h. During this time, the product was precipitated and then collected on a sintered glass funnel and washed with ethanol and dried. After isolation of the product, hot ethanol was added to the recovered silica nanoparticles followed by centrifugation, the silica nanoparticles were then dried and successfully used for the next run under identical reaction conditions. For compounds 3u and 3v, ethyl acetate was used instead of ethanol during the catalyst separation.2.2. General procedure for the synthesis of quinolines
A well-ground mixture of 2-aminoaryl ketone (1 mmol), carbonyl compound (1.2 mmol) and silica NPs (0.5 g) in a test tube was irradiated in a microwave oven at 600 W (100 °C) for the times reported in Table 3. Afterward, the reaction mixture was cooled to room temperature and warm EtOAc (30 mL) was added to it. Then the reaction mixture was concentrated to 10 mL and centrifuged for 20 min to separate the silica nanoparticles. After evaporation of the solvent from the supernatant, the resulting solid was recrystallized from EtOH to give the pure product. EtOAc (10 mL) was added to the recycled silica nanoparticles and centrifuged (two times). Finally, the silica nanoparticles were dried and reused for the next run.2.3. Selected spectral data
3. Results and discussions
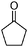
To optimize the reaction conditions for the synthesis of quinoxaline derivatives, the condensation of benzene-1,2-diamine (1) (1 mmol) with benzil (2) (1 mmol) was selected as a model reaction to provide quinoxaline 3a (Scheme 2). | ||
| Scheme 2 The condensation of benzene-1,2-diamine (1) with benzil (2). | ||
At first, the reaction was examined in the presence of some nontoxic, inexpensive, and easily available oxides (0.6 g) under solvent-free conditions at room temperature. All oxides (MgO, CaO, SiO2 (60–120 mesh) and neutral Al2O3) were purchased from Merck or Fluka chemical companies and SiO2 NPs were prepared according to the reported procedure.40
The results are summarized in Fig. 1. As Fig. 1 indicates, CaO and MgO gave low yields of compound 3a; Al2O3 afforded moderate yields of 3a; nevertheless, a good yield of the product was obtained in the presence of SiO2, and excellent results in order of yield and reaction time were observed when SiO2 nanoparticles were used as catalysts.
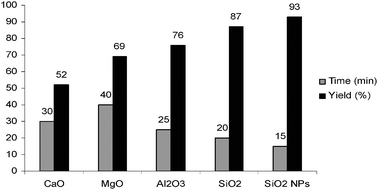 | ||
| Fig. 1 The solvent-free condensation of benzene-1,2-diamine with benzil in the presence of some oxides at room temperature. | ||
One of the most important aspects of the properties of oxides is their acid–base properties. Many oxides are basic or acidic anhydrides; that is they are compounds that are formed by the removal of water from a corresponding base or acid. Ionic oxides are usually basic anhydrides, whereas covalent oxides are usually acidic anhydrides. Oxides of the semimetals are amphoteric anhydrides, capable of acting as either an acid or base, depending on the circumstances. Note the greater degree of ionic character of the oxide, the more basic it is. If we list the oxides which were used as catalysts in this reaction, we find an orderly progression of their acid–base character. The acid strength increases with the acidity of the cation involved. CaO and MgO are basic oxides, Al2O3 is an amphoteric oxide and SiO2 is an acidic oxide. It is well-known that silica gel has many reactive hydroxyl groups on the surface and has been used as a catalyst in organic synthesis. Highly microporous solids such as silica NPs offer a wide range of active sites, and often can be regenerated if deactivated during reaction.40 On the other hand, reducing catalytic substances to nanometres in size greatly increases the surface area available per gram as well as the catalytic activity.15
In the next step, the reaction of benzene-1,2-diamine (1 mmol) with benzil (1 mmol) was tested using different amounts of silica NPs in the absence of solvent at room temperature (Fig. 2). As is clear from Fig. 2, the best amount of SiO2 nanoparticles per 1 mmol of each starting material was 0.6 g. This high catalyst loading to achieve a good yield of compound 3a is due to the numbers of silanol groups that catalyzed the reaction in comparison with the siloxan groups in the SiO2 NPs structure.
 | ||
| Fig. 2 Effect of different amounts of silica NPs on the reaction of benzene-1,2-diamine (1 mmol) with benzil (1 mmol) under solvent-free conditions at room temperature. | ||
To assess the generality and scope of the method, different aryl 1,2-diamines were reacted with structurally and electronically diverse 1,2-diketones; the respective results are displayed in Table 1.
| Entry | Product | Time (min) | Yielda (%) | M.p. (°C) | |
|---|---|---|---|---|---|
| Found | Reported | ||||
| a Isolated yield. | |||||
| 1 |

|
15 | 93 | 130–131 | 129–13026 |
| 2 |

|
15 | 92 | 188–190 | — |
| 3 |

|
10 | 90 | 133–135 | 132–13426 |
| 4 |

|
20 | 88 | 148–150 | 148–15028 |
| 5 |

|
20 | 87 | 126–128 | 127–12926 |
| 6 |

|
10 | 87 | 228–230 | — |
| 7 |

|
25 | 92 | 55–57 | 55–5726 |
| 8 |

|
5 | 92 | 222–224 | 218–21921 |
| 9 |

|
10 | 91 | 115–117 | 116–11827 |
| 10 |

|
10 | 89 | 164–166 | 163–16526 |
| 11 |

|
20 | 91 | 129–131 | 129–13128 |
| 12 |

|
20 | 92 | 139–140 | 138–13928 |
| 13 |

|
5 | 87 | 308–310 | — |
| 14 |

|
15 | 89 | 176–178 | 17231 |
| 15 |

|
5 | 93 | 302–304 | 302–30426 |
| 16 |

|
15 | 88 | 128–130 | — |
| 17 |
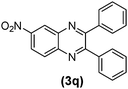
|
55 | 80 | 189–191 | 189–19127 |
| 18 |

|
50 | 85 | 169–171 | 174–17631 |
| 19 |

|
70 | 76 | 188–190 | 189–19127 |
| 20 |

|
100 | 83 | 133–135 | 136–13820 |
| 21 |
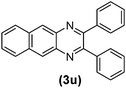
|
40 | 86 | 185–187 | 186–18828 |
| 22 |

|
50 | 82 | 196–198 | 196–19827 |
As can be seen in Table 1, the protocol was general and efficient; all reactions proceeded efficiently and the desired products were obtained with good to excellent yields in relatively short reaction times. In this study, the effect of electron-donating and electron-withdrawing substituents of aryl 1,2-diamines on the reaction was investigated. It was observed that electron-donating groups had no significant effect on the reaction results (Table 1, entries 9–16); however, electron-withdrawing substituents decreased the yields and increased the reaction times (Table 1, entries 17–19). When 1,2-diamines possessing electron-withdrawing substituents were utilized, the reaction was performed at 80 °C. The method was also efficient when pyridine-2,3-diamine (a deactivated 1,2-diamine) as well as naphthalene-2,3-diamine were condensed with 1,2-diketones (Table 1, entries 20–22). The results also showed that the structure of the aromatic ring of 1,2-diketones as well as their electronic properties had negligible influence on the reaction yields. Moreover, no effect of the aromatic ring substituents on the reaction rate was observed.
The other ecologically acceptable behavior of this method is the fact that silica NPs can be re-used after simple separation and purification. To check the recyclability of silica NPs (Fig. 3), the silica nanoparticles used in the condensation reaction of benzene-1,2-diamine with benzil, to 3a, after leaching out of the products, were thoroughly washed with ethyl acetate and ethanol and dried in air at 80 °C for 2 h. The dried silica nanoparticles were then reused directly in the next reaction, and an excellent yield of the expected product 3a was obtained. The same procedure was repeated for all further cycles. The recovered silica nanoparticles could be reused several times with only a slight loss of catalytic activity (see Fig. 3).
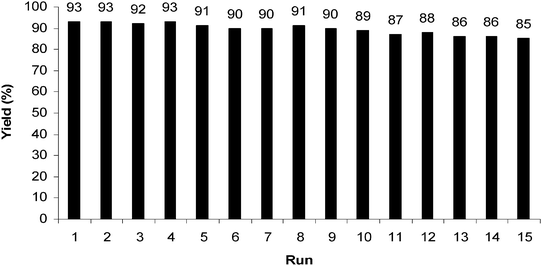 | ||
| Fig. 3 Recyclability of silica NPs in the condensation of benzene-1,2-diamine with benzil at room temperature (reaction time = 15 min). | ||
To highlight the catalytic activity of silica NPs with respect to silica gel, the reusability of silica gel for the condensation of benzene-1,2-diamine with benzil was examined. The catalytic activity of silica gel remarkably decreased after three recycling times (Fig. 4). These results confirmed the essential effect of nanoparticles on the reaction.
 | ||
| Fig. 4 Recyclability of silica gel in the condensation of benzene-1,2-diamine with benzil at room temperature (reaction time = 20 min). | ||
We believe that the presence of the reactive –OH groups on the surface of the silica NPs plays a major role in its catalytic activity. On the basis of the above results and by referring to the literature,41 we propose a plausible mechanism for the formation of quinoxalines (Scheme 3). First, the benzil is activated by the O–H group of silica NPs followed by the N-nucleophilic amine attacks on the carbonyl to form intermediate I and then to form intermediate II. Then dehydration occurs resulting in the generation of quinoxaline 3a. It may be speculated that the polar amphoteric surface hydroxyl groups of the silica NPs facilitate the interaction of absorbed weak acidic and basic components due to the stabilization of the corresponding transition states and intermediates by hydrogen bonding. This interaction with the neighboring silanol groups of the catalyst is shown in Scheme 3 for the first and second reaction steps. So, these surface hydroxyl groups also polarized the N–H bond of the benzene 1,2-diamines to act as nucleophiles. Participation of two proximate silanol groups (one as a hydrogen bond donor and another one as an acceptor) in the reaction mechanism also seems to be plausible.
 | ||
| Scheme 3 The proposed mechanism for the synthesis of quinoxalines using silica NPs. | ||
After the successful application of silica nanoparticles as catalysts in the synthesis of quinoxalines, we reasoned that silica NPs would be effective catalysts for the Friedländer synthesis of quinolines so, the ability of silica nanoparticles as catalysts for the synthesis of quinoline derivatives were studied (Scheme 4).
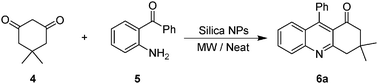 | ||
| Scheme 4 The reaction of dimedone with 2-aminobenzophenone in the presence of SiO2 NPs under microwave irradiation. | ||
For this purpose, a mixture of dimedone (4, 1.2 mmol) and 2-aminobenzophenone (5, 1 mmol) was stirred at 100 °C in the presence of silica NPs (0.5 g) under solvent-free conditions for 8 h to afford quinoline (6a) with a 57% yield (Scheme 4). Increasing the reaction time, temperature or the amount of the silica NPs did not significantly improve the reaction yield. Considering the usefulness of microwave irradiation to increase the reaction rates and yields,2 the condensation of dimedone with 2-aminobenzophenone was examined in the presence of various amounts of silica NPs under microwave irradiation (100–700 W, 100 °C) in the absence of solvent (Table 2).
As Table 2 shows, the product was obtained with a high yield and a short reaction time when the reaction was carried out in the presence of 0.5 g of silica NPs at 600 W of microwave power (100 °C). The model reaction was also tested in the absence of SiO2 nanoparticles; however, under these conditions, the reaction yield was very low (Table 2, entry 9).
To assess the efficiency of this method for the synthesis of quinolines, various α-methylene ketones were reacted with 2-aminoarylketones under microwave irradiation conditions (Scheme 1). The results are summarized in Table 3.
| Entry | Ketone | Product | Time (min) | Yielda (%) | M.p. (°C) | |
|---|---|---|---|---|---|---|
| Found | Reported | |||||
| a Isolated yield. | ||||||
| 1 |

|
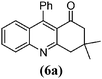
|
6 | 91 | 193–194 | 19234 |
| 2 |

|

|
8 | 92 | 153–154 | 151–15334 |
| 3 |

|
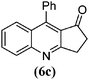
|
12 | 85 | 170–173 | 171–17535 |
| 4 |

|

|
7 | 90 | 109–110 | 105–10636 |
| 5 |
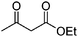
|
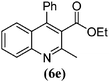
|
9 | 81 | 102–103 | 9934 |
| 6 |

|

|
8 | 89 | 103–105 | 10734 |
| 7 |

|

|
9 | 91 | 135–138 | 133–13437 |
| 8 |

|

|
12 | 89 | 140–142 | 13934 |
| 9 |
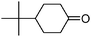
|
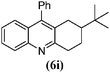
|
15 | 85 | 133–135 | 132–13336 |
| 10 |
|

|
15 | 83 | 132–133 | 140–14238 |
| 11 |

|

|
5 | 92 | 211–212 | 21134 |
| 12 |

|

|
7 | 90 | 187–188 | 18534 |
| 13 |

|

|
8 | 93 | 157–158 | 15734 |
| 14 |

|

|
8 | 88 | 99–100 | 10134 |
| 15 |

|
|
6 | 89 | 136–137 | 13534 |
| 16 |
|
|
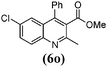
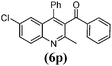
8 | 87 | 211–213 | 209–21137 |
| 17 |
|

|
9 | 84 | 165–166 | 16534 |
| 18 |

|
|
13 | 84 | 154–155 | — |
| 19 |

|

|
15 | 80 | 105–107 | 10537 |
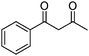
Various cyclic 1,3-diketones, acyclic 1,3-diketones as well as β-ketoesters were efficiently condensed with 2-aminoaryl ketones, and the desired quinolines were obtained with high yields and short reaction times. Interestingly, cyclic monofunctionalized ketones successfully reacted with 2-aminoaryl ketones to afford the respective tricyclic quinolines with high yields and short reaction times (Table 3, entries 8–10 and 17–19).
The important role of silica NPs in this interesting reaction may be attributed to its fitting acidity and ability to form H-bonds. So we supposed that the reaction was via a cascade procedure (Scheme 5): the aldol condensation between the protonated carbonyl group of 2-aminoarylketone and α-methylene group of carbonyl compound leads to intermediate A, and then this immediate converts to intermediate B by elimination of H2O and protonation of the carbonyl group of the carbonyl compound. Afterwards, intermediate B converts to Cvia intramolecular ring closure between the amine group and the protonated carbonyl group of B which is followed by water elimination of C to give the quinoline (Scheme 5). In fact, the polar skeleton of silica absorbs microwave irradiation extremely well and enables convenient heating of the reaction mixture.
 | ||
| Scheme 5 Proposed mechanism for the synthesis of quinolines using silica NPs. | ||
The prepared silica nanoparticles were characterized by scanning electron microscopy (SEM) (Fig. 5).

4. Conclusions
In conclusion, extremely efficient methods have been developed for the synthesis of quinoxaline and quinoline derivatives under solvent-free conditions using silica nanoparticles as catalysts. This method is bestowed with several unique merits, such as high conversions, simplicity in operation, cost efficiency and solvent-free conditions. Simple work-up, neutral reaction conditions, solvent-free conditions and high yields of the products make our methodologies valid contributions to the existing processes in the field of quinoxaline and quinoline synthesis.Acknowledgements
The authors thank the Persian Gulf University and Payame Noor University (PNU) Research Councils for the financial support of this work.References
- K. Tanaka, Solvent-free Organic Synthesis, Wiley-VCH, GmbH and KGaA, Weinheim, 2004 Search PubMed.
- A. Loupy, Microwaves in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- M. Góra, B. Kozik, K. Jamroży, M. K. Łuczyński, P. Brzuzan and M. Woźny, Green Chem., 2009, 11, 863–867 RSC.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- J. H. Clark and C. N. Rhodes, Clean Synthesis using Porous Inorganic Solid Catalysts and Supported Reagents, 1st edn, Royal Society of Chemistry, UK, 2000 Search PubMed.
- A. P. Kybett and D. C. Sherrington, Supported Catalysts and Their Applications, 1st edn, Royal Society of Chemistry, UK, 2001 Search PubMed.
- P. Salehi, M. A. Zolfigol, F. Shirini and M. Baghbanzadeh, Curr. Org. Chem., 2006, 10, 2171–2189 CrossRef CAS (Review).
- A. Hasaninejad, A. Zare, H. Sharghi, M. Shekouhy, R. Khalifeh, A. Salimi Beni and A. R. Moosavi Zare, Can. J. Chem., 2007, 85, 416–421 CrossRef CAS.
- A. Hasaninejad, A. Zare, H. Sharghi, K. Niknam and M. Shekouhy, ARKIVOC, 2007, xiv, 39–50 Search PubMed.
- B. Basu, S. Paul and A. K. Nanda, Green Chem., 2009, 11, 1115–1120 RSC.
- S. Balalaie, M. M. Hashemi and M. Akhbari, Tetrahedron Lett., 2003, 44, 1709–1712 CrossRef CAS.
- R. Ballini, D. Fiorini, M. V. Gil and A. Palmieri, Green Chem., 2003, 5, 475–476 RSC.
- S. Balalaie and A. Arabanian, Green Chem., 2000, 2, 274–276 RSC.
- L. You, S. Feng, R. An, X. Wang and D. Bai, Tetrahedron Lett., 2008, 49, 5147–5150 CrossRef CAS.
- S. Banerjee and G. Sereda, Tetrahedron Lett., 2009, 50, 6959–6962 CrossRef CAS.
- B. Zhou, S. Han, R. Raja and G. A. Somorjai, ed., Nanotechnology in Catalysis, 2007, XXII.
- U. Heiz and U. Landman, ed., Nanocatalysis, 2007, XVI, 503.
- L. E. Seitz, W. J. Suling and R. C. Reynolds, J. Med. Chem., 2002, 45, 5604–5606 CrossRef CAS.
- M. M. Badran, A. A. Moneer, H. M. Refaat and A. A. El-Malah, J. Chin. Chem. Soc., 2007, 54, 469–472 CAS.
- G. Sakata, K. Makino and Y. Karasawa, Heterocycles, 1988, 27, 2481–2515 CrossRef CAS.
- R. Sarges, H. R. Howard, R. C. Browne, L. A. Label and P. A. Seymour, J. Med. Chem., 1990, 33, 2240–2254 CrossRef CAS.
- S. T. Hazeldine, L. Polin, J. Kushner, K. White, N. M. Bouregeois, B. Crantz, E. Palomino, T. H. Corbett and J. P. Horwitz, J. Med. Chem., 2002, 45, 3130–3137 CrossRef CAS.
- G. Roma, M. D. Braccio, G. Grossi, F. Mattioli and H. Ghia, Eur. J. Med. Chem., 2000, 35, 1021–1035 CrossRef CAS.
- Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374–2377 CrossRef CAS.
- L. Wang, J. Liu, H. Tian and C. Qian, Synth. Commun., 2004, 34, 1349–1357 CrossRef CAS.
- A. Hasaninejad, A. Zare, M. A. Zolfigol and M. Shekouhy, Synth. Commun., 2009, 39, 569–579 CrossRef CAS.
- A. Hasaninejad, A. Zare, M. R. Mohammadizadeh and Z. Karami, J. Iran. Chem. Soc., 2009, 6, 153–158 CAS.
- A. Hasaninejad, A. Zare, M. R. Mohammadizadeh and M. Shekouhy, ARKIVOC, 2008, xiii, 28–35 Search PubMed.
- M. M. Heravi, M. H. Tehrani, K. Bakhtiari and H. A. Oskooie, Catal. Commun., 2007, 8, 1341–1344 CrossRef CAS.
- R. S. Bhosale, S. R. Sarda, S. S. Ardhapure, W. N. Jadhav, S. R. Bhusare and R. P. Pawar, Tetrahedron Lett., 2005, 46, 7183–7186 CrossRef CAS.
- H. R. Darabi, S. Mohandessi, K. Aghapoor and F. Mohsenzadeh, Catal. Commun., 2007, 8, 389–392 CrossRef CAS.
- P. Friedländer, Ber. Dtsch. Chem. Ges., 1882, 15, 2572–2575 CrossRef.
- R. Ghorbani-Vaghei and S. Akbari-Dadamahaleh, Tetrahedron Lett., 2009, 50, 1055–1058 CrossRef CAS and references cited therein.
- M. A. Zolfigol, P. Salehi, A. Ghaderi, M. Shiri and Z. Tanbakouchian, J. Mol. Catal. A: Chem., 2006, 259, 253–258 CrossRef CAS.
- S. S. Palimkar, S. A. Siddiqui, T. Daniel, R. J. Lahoti and K. V. Srinivasan, J. Org. Chem., 2003, 68, 9371–9378 CrossRef CAS.
- D. S. Bose and R. K. Kumar, Tetrahedron Lett., 2006, 47, 813–816 CrossRef CAS.
- S. Ghassamipour and A. R. Sardarian, Tetrahedron Lett., 2009, 50, 514–519 CrossRef CAS.
- S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 1647–1649 CrossRef CAS.
- (a) A. Hasaninejad, A. Zare, M. Shekouhy and J. Ameri Rad, J. Comb. Chem., 2010, 12, 844–849 CrossRef CAS; (b) A. Hasaninejad, A. Zare and Shekouhy, Tetrahedron, 2011, 67, 390–400 CrossRef CAS; (c) A. Hasaninejad, A. Zare, M. Shekouhy and J. Ameri Rad, Green Chem., 2011, 13, 958 RSC.
- K. S. Rao, K. El-Hami, T. Kodaki, K. Matsushige and K. Makino, J. Colloid Interface Sci., 2005, 289, 125–131 CrossRef CAS.
- Q. Ding, B. Cao, Z. Zong and Y. Peng, J. Comb. Chem., 2010, 12, 370–373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1cy00332a |
| This journal is © The Royal Society of Chemistry 2012 |