Covalently immobilized tris(triazolyl)methanol–Cu(I) complexes: highly active and recyclable catalysts for CuAAC reactions†
Erhan
Ozkal
a,
Salih
Özçubukçu
a,
Ciril
Jimeno
a and
Miquel A.
Pericàs
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. Fax: (+34)-977-920-222; E-mail: mapericas@iciq.es
bDepartament de Química Orgànica, Universitat de Barcelona, 08028 Barcelona, Spain
First published on 27th October 2011
Abstract
Tris(1-benzyl-1H-1,2,3-triazol-4-yl)methanol (3), a highly efficient ligand for CuAAC reactions, has been immobilized onto Merrifield resins through different strategies. The SN2-supported Cu complex (8) is stable in water and under air; it is active at low catalyst loadings (1 mol%) and at low concentration (down to 0.125 M) in both aqueous and purely organic media. Resin 8 can be repeatedly reused in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH–water for short reaction times (4 h) with the only precaution of Cu(I) reloading every five cycles.
:1 MeOH–water for short reaction times (4 h) with the only precaution of Cu(I) reloading every five cycles.
Introduction
The concept of click chemistry1 is closely linked to the development of the almost universally applicable copper-catalyzed alkyne–azide cycloaddition (CuAAC).2,3 Applications of this reaction span from materials science to biological systems, including medicinal chemistry and drug discovery.4,5 If a criticism can be made to the original CuAAC procedures, it would be related to the fact that fairly high amounts of copper catalyst must be used. This can lead to product contamination, which is unacceptable in electronics, or can lead to toxicity in biomedical applications.5 To overcome this problem, ligand stabilized Cu(I) species have been developed. In particular, tris(triazoles),6,7 tetramines,8 and N-heterocyclic carbenes9 have been successfully used under ambient conditions and at very low catalyst loadings.The covalent immobilization of such Cu(I) complexes onto solid supports adds further advantages, since catalyst separation can be achieved by simple filtration with an according reduction in copper contamination of reaction products, and (in principle) the catalyst can be recycled and reused with a subsequent improvement of the sustainable characteristics of the overall process.10
Up to now, however, the known covalently immobilized catalysts for CuAAC reactions have involved rather lengthy preparations,10a–e or have made use of commercial functional polymers involving polyamino appends. In these cases, catalytic use involves either over-stoichiometric amounts of catalytic resin (2 equiv.),10f or high copper loadings.10g We wish to report herein the development of a readily available and highly active polymer-supported catalyst for CuAAC reactions, whose immobilization takes advantage from the design of the corresponding homogeneous ligand.7,11
Results and discussion
Determination of the optimal supporting strategy
Complex 1 (Fig. 1), developed as a catalyst for CuAAC reactions,7 exhibits a notable stability even in the presence of free amino groups in the reacting molecules and has proved to be particularly efficient to catalyze CuAAC reactions on polymers.12 The free hydroxyl group present in its structure, arising from the Grignard approach used for its synthesis, offers a good opportunity for polymer supporting. Moreover, according to the topology of the system, the supporting process should not perturb the catalytic function of the assembly.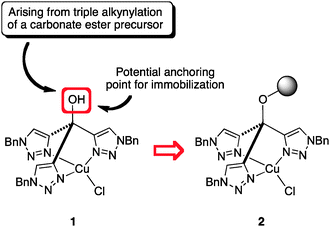 | ||
| Fig. 1 Immobilization strategy for complex 1, using a hydroxy group resulting from the synthetic design of the homogeneous ligand. | ||
In our initial approach, the CuAAC reaction was selected for the supporting process through the intermediacy of an O-propargyl derivative. Thus, treatment of tris(triazole) 3 with NaH in DMF at 0 °C and reaction with propargyl bromide (Scheme 1) afforded 4 in good yield (82%). For polymer supporting, the commercially available Merrifield resin (1% DVB; f = 1.1 mmol g−1) was first converted to azide 5 by treatment with sodium azide. The CuAAC reaction between 4 and 5 was performed in THF:DMF (1:1) at 40 °C for 48 h (IR monitoring) in the presence of 5 mol% of complex 1.7 The resulting functional resin 6 (f = 0.56 mmol g−1) was finally converted to its Cu(I) complex 7 by shaking with a stoichiometric amount of CuCl in THF at rt for 15 h.
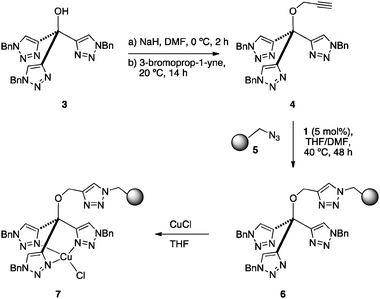 | ||
| Scheme 1 Synthesis of the immobilized tris(triazolyl)methanol copper(I) complex 7. | ||
To our delight, when the CuAAC reaction between benzyl azide and phenylacetylene was performed in the presence of 1 mol% of 7 in water at 40 °C, the desired 1,2,3-triazole was obtained in 94% yield after only 3 h. However, when the direct recycling of 7 was attempted, a significant decrease in its catalytic activity was observed. On the other hand, 7 could be recycled and reused for 5 times without any loss of activity with the simple precaution of a short re-conditioning with CuCl in THF after each cycle. This behavior was indicative of Cu leaching from resin 7, possibly arising from a less geometrically favourable coordination of copper (in addition to the one depicted in Scheme 1) involving the participation of the additional triazole unit present on the linker.
To circumvent this difficulty, a second-generation supported catalyst 8 lacking the triazole linker (Scheme 2) was prepared by alkylation of a Merrifield resin (1% DVB; f = 1.1 mmol g−1) with 3 and complexation with CuCl (1.05 eq). From the elemental analysis of the intermediate metal-free resin, a functionalization f = 0.46 mmol g−1 can be calculated for 8.
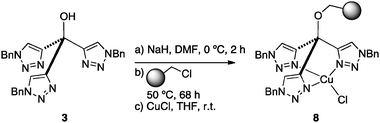 | ||
| Scheme 2 Synthesis of the copper(I) complex of polymer-supported tris(triazolyl)methanol 8. | ||
Optimization of reaction conditions for use in catalysis and recycling of resin 8
Using as a test the reaction of phenylacetylene with benzyl azide, a variety of solvents were screened both for exploring their suitability as reaction media and for determining the limits of recyclability of 8 (Table 1). Unless otherwise specified, the reactions were performed at 40 °C for 4 h.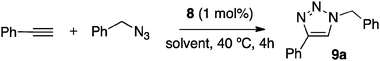
| Entry | Solvent | Yielda (%) | ||||
|---|---|---|---|---|---|---|
| Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 5 | ||
| a Isolated yield. b Yield determined by 1H NMR. c After resin re-conditioning with CuCl. d Complete conversion required 16 h at 40 °C. e Complete conversion required 16 h at rt. f All yields determined by average of two independent runs. g Conversion was complete in 6 h. Isolated yield was 95%. h Reactions performed at rt for 8 h. | ||||||
| 1 | H2O | 90 | 85 | 15b | 86c | 90 |
| 2 |
THF:H2O (1:1) |
85 | 7b | 79c | — | — |
| 3 | THF | 80 | 5b | 50c | — | — |
| 4 | Toluene d | 87 | — | — | — | — |
| 5 | CH2Cl2e | 86 | — | — | — | — |
| 6 | EtOAc d | 90 | — | — | — | — |
| 7f |
MeOH:H2O (1:1) |
99 | 99 | 96 | 95 | 79g |
| 8f,h |
MeOH:H2O (1:1) |
99 | 96 | 94 | 95 | 65 |
The catalytic resin 8 performed well in water, but became deactivated after the 2nd run (entry 1); as expected, catalytic activity was fully recovered when the polymer was reconditioned with CuCl (see cycles 4 and 5 in entry 1). A 1:1 THF–water mixture (entry 2) or THF alone (entry 3) gave promising results but, again, inter-cycle reconditioning was required. Other organic solvents were also tested (entries 4–6). In all these media, the cycloaddition took place at a lower reaction rate, requiring 16 h for complete conversion. Finally, a 1:1 methanol–water mixture was revealed to be the optimal solvent for the reaction (entries 7 and 8). Under these conditions, very high catalytic activity was recorded for four consecutive runs, whereas a slight decrease in yield was observed in the fifth one when the standard reaction time (4 h) was maintained. In fact, conversion for cycle #5 was complete in 6 h (95% yield). It is worth noting here that other covalently supported ligands for CuAAC for whom recycling data are available require, at identical catalyst loading (1%), reaction cycles of 2410a,10e or 48 h.10d In view of practical application, the achievement of complete conversion in short reaction times is very important. For this reason, we decided to introduce a reconditioning treatment after the fifth cycle. Thus, the sample of 8 used in the five cycles in entry 7 was treated with CuCl and used for five additional cycles, where a completely parallel activity pattern was observed (mean yield for cycles 6 to 10: 92%). After a second reconditioning with CuCl, the yield in cycle 11 was still 91%. Another set of experiments in 1:1 methanol–water was performed at room temperature, with 8 h as the reaction time for individual runs (entry 8). The behavior of 8 under these conditions almost exactly reproduced that recorded at 40 °C; slight de-activation became apparent after the fourth cycle, and reactivation with CuCl led to a complete recovery of catalytic activity.
To discard the possibility that the polymeric network could simply act as a copper chloride trap, and that catalysis was simply due to CuCl slowly released into solution, a control experiment was performed by treating a Merrifield resin (1% DVB; f = 1.1 mmol g−1) with a large excess (5.0 eq.) of CuCl and using this resin to promote the CuAAC reaction between phenylacetylene and benzyl azide. For comparison purposes, the amount of Merrifield resin/CuCl mixture employed in catalytic experiments corresponded to the weight of 8 used in experiments performed at the same scale. As anticipated, and in spite of its much higher copper content, the Merrifield resin/CuCl mixture behaved as a rather poor catalyst in comparison with 8. For reactions conducted at 40 °C for 4 h, conversion was 22% in the first cycle, and only 10% in the second one. When the Merrifield resin was mixed with 1.05 eq. CuCl, to mimic in a more realistic manner the copper content of 8, conversion in the model CuAAC reaction was <5% after 4 h at 40 °C. These results are a clear indication that polymer-bound complex 8 is the actual catalytic species in the process.
Scope of applicability of resin 8
The applicability of the supported catalyst 8 was next explored under the optimized conditions (MeOH:water 1:1, 40 °C, 1 mol% catalyst loading). The results have been summarized in Table 2.
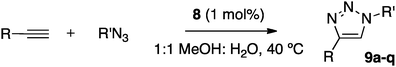
| Entry | Product | Time/h | Yieldb (%) |
|---|---|---|---|
|
a Samples of 8 with loadings of 0.34 mmol g−1 and 0.46 mmol g−1 were indistinctly used in these experiments.
b Isolated yield.
c In water.
d 3 mol% 8, rt.
e Microwave irradiation in n-BuOH:water 1:1 at 100 °C.
f
In situ formation of the azide from the corresponding bromide and sodium azide.
|
|||
| 1 |

|
4 | 99 |
| 2 |

|
4 | 98 |
| 3 |

|
4 | 98 |
| 4 |

|
5 | 99 |
| 5 |

|
6 | 99 (94)c |
| 6 |

|
5 | 99 |
| 7 |

|
18 | 92 |
| 8 |

|
5 | 93 (95)c |
| 9 |

|
6 | 91d |
| 10 |

|
4 | 96 |
| 11 |

|
16 | 97 |
| 12 |

|
5 | 99 |
| 13 |

|
16 | 99 |
| 14 |

|
1 | 60e,f |
| 15 |

|
5 | 95 |
| 16 |

|
5 | 94 |
| 17 |

|
8 | 99f |
Reaction of benzyl azide with several acetylenic alcohols yielded the expected products 9b–e in high yields (entries 2–5) and short reaction times. As indicated by the formation of 9f (entry 6), the corresponding esters exhibit a similar behavior. Even propiolic acid, a normally difficult substrate for CuAAC, leads to triazole formation in high yield, although a longer reaction time is required (9g, entry 7). With respect to acetylenic amines, N,N-dimethylpropargylamine afforded the triazole product 9h in very high yield (entry 8) and even substrates featuring free amino groups, like 4-aminophenylacetylene (entry 9) or a propargyloxypyrrolidine derivative (entry 10), that represent more stringent tests for the reaction given the known tendency of free amino groups to form Cu(I) complexes with deactivation of the catalyst, lead to the corresponding cycloadducts 9i and 9j in high yield. The CuAAC reaction of aliphatic alkynes (exemplified by 1-octyne) worked equally well, but required somewhat longer reaction times (16 h) for complete conversion (entries 11 and 13). The reaction with other alkyl azides also took place uneventfully, as seen in entries 12–14. As a particular case, the reaction leading to 9n (entry 14) was performed in a tandem manner from 1-bromooctane, sodium azide, and phenyl propargyl thioether under microwave irradiation at 100 °C. Under these conditions, triazole 9n could be isolated in 60% yield after one hour reaction time. Aryl azides were also tested (entries 15 and 16), the corresponding triazoles 9o and 9p being obtained in excellent yields after short reaction times. Finally, functional azides and functional alkynes could be combined for the preparation of difunctional triazoles, as in the example of entry 17. The formation of 9q, which involved the tandem assembly of the azide and the CuAAC reaction in essentially quantitative yield illustrates this possibility.
From a practical perspective, the use of 8 is simple and advantageous, allowing ample variation of reaction parameters. Reaction temperatures in the range 20–50 °C can be used at convenience, although in our hands 40 °C represents in most cases an optimal value. Reagent concentration can also be widely varied. While examples in Table 2 have been performed at ca. 0.5 M concentration, reactions at 0.125 M concentration worked equally well. As a general rule, examples in Table 2 showing complete conversion in <4 h13 require 5 h at 0.125 M concentration for completion of the reaction. From the point of view of product isolation, the separation of the triazole products (9) from 8 is simply performed by addition of ethyl acetate (to dissolve 9) and filtration (to recover 8). After solvent removal in vacuo, the triazole products are generally isolated in pure form.14
The high recyclability exhibited by 8 (see above) is indicative of very reduced leaching of Cu into aqueous methanol solutions. This fact, together with the low catalyst loading required for reaction, suggested that copper contamination in triazoles 9 should be very low. To confirm this, the amount of copper present in crude triazole 9a was repeatedly tested: several batches were analyzed by UV-Vis spectroscopy,15Cu concentration being in all cases below 250 ppm (mean value: 195 ppm). If required, the remaining Cu traces can be removed with Cu-scavenging resins.16
Conclusions
In summary, a tris(triazolyl)methanol ligand has been successfully immobilized in one step onto a polystyrene resin and converted into a Cu(I) complex. Probably due to the protecting cage formed around the metal center6 and to its particular functional groups arrangement, catalyst 8 behaves as a very active promoter of the CuAAC reaction, as it has been demonstrated with a broad variety of substrates (including free amines and thioethers), and exhibits excellent recycling characteristics.Experimental section
Synthesis of 3-[tris(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]propyne (4)
To a flame dried flask containing a suspension of NaH (80 mg, 60% in oil, 2.0 mmol) in DMF (2 mL), a solution of 3 (503 mg, 1.0 mmol) in DMF (2 mL) was added dropwise at 0 °C. After stirring for 2 h at room temperature, the suspension became a clean solution, which was cooled again to 0 °C. Then, a commercial solution of propargyl bromide in toluene (0.220 mL, 80% solution in toluene, 2.0 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and was stirred for an additional 14 h. Water (10 mL) was added to the reaction mixture, which was then extracted with dichloromethane (3 × 10 mL). The combined organic phase was dried over MgSO4 and concentrated under reduced pressure. Traces of DMF were removed by dissolving the crude product in an ethyl acetate: hexane (4:1) mixture (40 mL) and washing the solution with water (3 × 20 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure to afford crude 4, which was further purified by flash column chromatography using ethyl acetate as the eluent. The product was obtained as a thick orange oil (0.444 g, 82%). 1H NMR (400 MHz, CDCl3): δ = 7.85 (s, 3H), 7.34–7.24 (m, 15H), 5.47 (s, 6H), 4.11 (d, J = 2.4 Hz, 2H), 2.05 (t, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 148.4, 134.3, 129.1, 128.7, 128.2, 124.7, 80.0, 73.6, 73.1, 54.2, 53.0; IR(ATR): 3293, 3147, 3089, 2983, 2244, 1536, 1496, 1266, 1042 cm−1. HRMS calcd. for C31H27N9ONa: 564.2236. Found: 564.2253 ([M + Na]+).
Preparation of the first generation PS-supported catalyst 7
An azido-functionalized Merrifield resin (100 mg, f = 0.94 mmol g−1) was added to a solution of 4 (510 mg, 0.94 mmol) in a 1:1 mixture of DMF:THF (5 mL). A sample of 7 (5.6 mg, 0.0094 mmol) was added to the mixture, and the system was shaken for 48 h at 40 °C. The progress of the reaction was monitored by IR spectroscopy, and the process was interrupted after the disappearance of the azide band around 2100 cm−1. Solvents were separated by filtration for the recovery of excess 4. The resulting resin was washed with water, THF and methanol, respectively, and then dried at 50 °C overnight. Functionalization (f = 0.56 mmol g−1) was calculated at this point by elemental analysis of nitrogen (found: N 9.38%).16 The functional resin was then suspended in THF (5 mL) and shaken for 15 h with CuCl (9.7 mg, 0.1 mmol) at room temperature. The resulting light-green resin was washed with THF, dried, and stored for use.
Preparation of the second generation PS-supported catalyst 8
To a flame dried flask containing a suspension of NaH (16.8 mg, 95%, 0.7 mmol, stored and weighed in a glove box) in DMF (2 mL), a solution of 37 (252 mg, 0.5 mmol) in DMF (2 mL) was added dropwise at 0 °C under N2. After stirring for 2 h at room temperature, the suspension became a clean solution, which was cooled again to 0 °C and added via cannula to a pre-swollen sample (DMF, >30 min) of commercial Merrifield resin (1% DVB, 1.1 mmol g−1; 0.327 g, 0.36 mmol) in DMF (4 mL). The reaction mixture was allowed to warm to room temperature, then heated to 50 °C and shaken at that temperature for 96 h. After the reaction mixture cooled down to room temperature, the resin was separated by vacuum filtration and successively washed with DMF–H2O, H2O, THF, THF–MeOH (1:1), MeOH, and THF. The resulting polymer was dried in a vacuum drying oven at 40 °C overnight. The functionalization of the resin (f = 0.46 mmol g−1) was calculated from the results of elemental analysis of nitrogen (found: N 5.76%).17 A mixture of CuCl (8.4 mg, 0.084 mmol, 1.05 eq.) and the PS-supported tris(triazolyl)methoxy ligand (262 mg) in THF (10 mL) was shaken at room temperature for 16 h. The colour of the resin turned into green as the complexation proceeded. The CuCl-loaded resin was successively washed with THF (100 mL), H2O (100 mL), and THF (100 mL), then dried overnight under vacuum and stored for use.
General procedure for CuAAC reaction catalyzed by 8
Reconditioning of resin 8 in recycling experiments
Whenever a catalyst sample used in recycling experiments showed decreased activity (i.e.; complete conversion was not achieved under the standard reaction conditions for the particular combination of reagents), the employed catalyst was separated by vacuum filtration at the end of the reaction, transferred to a vial and shaken overnight with 1 eq. of CuCl in THF at room temperature. Then, the catalytic resin was washed with excess THF, H2O, and THF, and dried under vacuum at 40 °C. A full recovery of the original activity was observed in the next reaction cycle. For extended use of catalytic resin samples, it is convenient repeating the reconditioning process every five reaction cycles.Analysis of the Cu content in triazole adducts
A modified version of the UV-VIS spectroscopic procedure of Brenner and Harris was used for the analyses.14 For calibration, two solutions (A and B) have to be prepared. Solution A: a 100 mL aqueous solution of 120 μM bicinchoninic acid was prepared by adding bicinchoninic acid disodium salt hydrate (4.7 mg, 0.012 mmol) (BCA) and sodium ascorbate (39.6 mg , 0.2 mmol) (NaAsc) with 0.1 M sodium phosphate buffer at pH 7. Solution B: Solution A plus Cu(II)SO4·5H2O (1.5 mg, 0.006 mmol). The solutions were prepared separately. Different volumes of each solution A and B were mixed together to plot the calibration curve (see ESI†). For the determination of the Cu content in adducts 9, the following representative procedure was followed: to a 5.00 mg sample of the triazole product to be controlled (separated by filtration and only submitted to evaporation of volatiles) in a vial, 5 mL of solution A were added and the mixture was vigorously stirred for 15 minutes at RT. Then, the resulting almost colourless solution was filtered through a HPLC filter to a quartz UV cuvette, and its measured absorbance at 354.5 nm was used for the calculation of the copper content either graphically or with the calibration equation.Acknowledgements
We thank MICINN (grant CTQ2008-00947/BQU and Consolider Ingenio 2010 grant CSD2006-0003), DURSI (grant 2009SGR623), and the ICIQ Foundation for financial support. E. O. thanks MICINN for a predoctoral fellowship. We are grateful to Dr. Fernando Bravo (CSOL-ICIQ) for the scale-up of the preparation of 8.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- M. Meldal, C. Christensen and C. W. Tornøe, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- (a) Macromol . Rapid. Commun., 2008, 29, 943, special thematic issue; (b) QSAR Comb. Sci., 2007, 26, 1110, special thematic issue; (c) Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674 RSC; (d) E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (e) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS; (f) P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7 CAS; (g) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS; (h) C. O. Kappe and E. Van der Eicken, Chem. Soc. Rev., 2010, 39, 1280 RSC.
- Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley & Sons, Chichester (UK), 2009 Search PubMed.
- (a) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS; (b) W. G. Lewis, F. G. Magallon, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2004, 126, 9152 CrossRef CAS.
- S. Özçubukçu, E. Ozkal, C. Jimeno and M. A. Pericàs, Org. Lett., 2009, 11, 4680 CrossRef CAS.
- N. Candelon, D. Lastécouères, A. K. Diallo, J. Ruiz Aranzaes, D. Astruc and J.-M. Vincent, Chem. Commun., 2008, 741 RSC.
- (a) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558 CrossRef CAS; (b) S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881 CrossRef CAS.
- (a) T. R. Chan and V. V. Fokin, QSAR Comb. Sci., 2007, 26, 1274 CrossRef CAS; (b) G. M. Pawar, B. Bantu, J. Weckesser, S. Blechert, K. Wurst and M. J. Buchmeiser, Dalton Trans., 2009, 9043 RSC; (c) M. Lammens, J. Skey, S. Wallyn, R. O'Reilly and F. Du Prez, Chem. Commun., 2010, 8719 Search PubMed; (d) Y. Wang, J. Liu and C. Xia, Adv. Synth. Catal., 2011, 353, 1534 Search PubMed; (e) S. Wallyn, M. Lammens, R. K. O'Reilly and P. Du Prez, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2878 CrossRef; (f) A. Coelho, P. Diz, O. Caamaño and E. Sotelo, Adv. Synth. Catal., 2010, 352, 1179 CrossRef CAS; (g) C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689 CrossRef CAS.
- For the importance of ligand design in the success of immobilization strategies, see: (a) A. Vidal-Ferran, N. Bampos, A. Moyano, M. A. Pericàs, A. Riera and J. K. M. Sanders, J. Org. Chem., 1998, 63, 6309 CrossRef CAS; (b) M. A. Pericàs, D. Castellnou, I. Rodríguez, A. Riera and L. Solà, Adv. Synth. Catal., 2003, 345, 1305 CrossRef CAS; (c) D. Castellnou, L. Solà, C. Jimeno, J. M. Fraile, J. A. Mayoral, A. Riera and M. A. Pericàs, J. Org. Chem., 2005, 70, 433 CrossRef CAS.
- For examples of catalyst immobilization using CuAAC reactions, see: (a) D. Font, C. Jimeno and M. A. Pericàs, Org. Lett., 2006, 8, 4653 CrossRef CAS; (b) M. Tilliet, S. Lundgren, C. Moberg and V. Levacher, Adv. Synth. Catal., 2007, 349, 2079 CrossRef CAS; (c) E. Alza, C. Rodríguez-Escrich, S. Sayalero, A. Bastero and M. A. Pericàs, Chem.–Eur. J., 2009, 15, 10167 CrossRef CAS; (d) T. Chinnusami and O. Reiser, ChemSusChem, 2010, 3, 1040 Search PubMed; (e) P. Riente, C. Mendoza and M. A. Pericàs, J. Mater. Chem., 2011, 21, 7350 RSC; for the successful use of 1 as a catalyst for the CuAAC-immobilization of organocatalysts containing free amino groups, see: (f) E. Alza and M. A. Pericàs, Adv. Synth. Catal., 2009, 351, 3051 CrossRef CAS.
- For experiments in Table 2 performed at 40 °C for 4 h, reaction time for complete conversion was not optimized.
- In no case the presence in the reaction product of the regioisomeric 1,5-disubstituted 1,2,3-triazoles as minor components could be detected by NMR.
- Adapted from: (a) A. J. Brenner and E. D. Harris, Anal. Biochem., 1995, 226, 80 CrossRef CAS; (b) A. J. Brenner and E. D. Harris, Anal. Biochem., 1995, 230, 360 Search PubMed (erratum).
- (a) I. R. Baxendale, S. V. Ley, A. C. Mansfield and C. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 4017 CrossRef CAS; (b) C. D. Smith, I. R. Baxendale, S. Lanners, J. J. Hayward, S. C. Smith and S. V. Ley, Org. Biomol. Chem., 2007, 5, 1559 RSC.
- The degree of functionalization of a resin can be calculated from the results of elemental analysis with the formulas: fN = (0.714/nN)%N, where nN is the number of nitrogen atoms in the functional unit and %N is the percent of nitrogen provided by the elemental analysis. See: A. Bastero, D. Font and M. A. Pericàs, J. Org. Chem., 2007, 72, 2460 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00297j |
| This journal is © The Royal Society of Chemistry 2012 |