Low temperature hydrogenolysis of waxes to diesel range gasoline and light alkanes: Comparison of catalytic properties of group 4, 5 and 6 metal hydrides supported on silica–alumina
Sebastien
Norsic
a,
Cherif
Larabi
a,
Marco
Delgado
a,
Anthony
Garron
a,
Aimery
de Mallmann
a,
Catherine
Santini
a,
Kai C.
Szeto
*a,
Jean-Marie
Basset
*b and
Mostafa
Taoufik
*a
aLaboratoire de Chimie Organométallique de Surface, Université Lyon 1 – ICL - CNRS – C2P2 UMR 5265 – CPE Lyon, 43 Bld du 11 novembre 1918, F-69616 Villeurbanne, France. E-mail: kai.szeto@lcoms.cpe.fr; taoufik@cpe.fr
bKCC, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa
First published on 25th November 2011
Abstract
A series of metal hydrides (M = Zr, Hf, Ta, W) supported on silica–alumina were studied for the first time in hydrogenolysis of light alkanes in a continuous flow reactor. It was found that there is a difference in the reaction mechanism between d0metal hydrides of group 4 and d0 ↔ d2metal hydrides of group 5 and group 6. Furthermore, the potential application of these catalysts has been demonstrated by the transformation of Fischer–Tropsch wax in a reactive distillation set-up into typical gasoline and diesel molecules in high selectivity (up to 86 wt%). Current results show that the group 4 metal hydrides have a promising yield toward liquid fuels.
1 Introduction
Production of gasoline or diesel oil from Fischer–Tropsch waxes has gradually become an important process in the petroleum industry.1,2 One of the most established processes to fabricate liquid fuels apart from crude oil is the Fischer–Tropsch process.3 This reaction operates under high temperature and generates a significant amount of fairly pure paraffin wax as a side product with a relatively low economic value. Hence, upgrading wax to valuable products, especially liquid fuels, is preferable rather than dumping and can simultaneously supply the reservoir of the highly demanded fuels.A wide range of heterogeneous hydro-cracking catalysts based on metal particles supported on conventional supports or zeolites has been commercialized for years.4–6 However, the reported hydro-cracking catalysts are efficient only at high temperatures and suffer from deactivation by coke formation and poisoning of the active sites.7 Moreover, the catalytic cycles of those metal/acid bifunctional catalytic systems involve rather complex intermediates that are generally difficult to determine. Lack of fundamental understanding impedes rational design of a heterogeneous catalyst with optimal performance towards a given reaction. Therefore, it is interesting to develop new and efficient hydro-cracking catalysts running at low temperature and pressure and leading to an environmentally friendly process.
A more recent class of catalysts derived from isolated metal hydride or organometallic compounds (especially W-based catalysts) grafted on conventional support materials like silica, silica–alumina and alumina have shown outstanding activity and selectivity for a series of different reactions with saturated and unsaturated hydrocarbons under mild conditions (<200 °C).8–14 In particular, electron deficient supported d0metal hydrides of group 4 (Zr-H supported on silica–alumina, Hf-H supported on silica) are active in ethylene and propylene polymerization. These catalysts were also found to be able to depolymerize the corresponding polymer in the presence of hydrogen and yielded basically gaseous hydrocarbons (Ziegler–Natta depolymerisation). These preliminary studies carried out in a batch reactor could not provide the selectivity of the primary product since consecutive reactions can always occur in batch systems, giving light alkanes as the final products.15 Recently, we have shown that [Ti]s-H supported on silica and silica–alumina were active and selective to hydrogenolyze wax16 by combining the fixed-bed reaction with reactive distillation17 to typical gasoline and diesel compositions at low temperature in good yield. Previous studies also revealed that the acidity of silica–alumina has a promoting effect on this reaction compared to silica.16 Since the highly electrophilic d0[Ti]s-H center can readily convert wax to fuels, other electron deficient supported d0metal hydrides of group 4 (Zr, Hf), as well as d0 ↔ d2metal hydrides [M]s-(H)3 (M = Ta, W) supported on silica–alumina (simplified by [M]s-H, their structures are depicted in Scheme 1), have been studied under the same conditions and thereby affords a direct comparison between the group 4, 5 and 6 metal hydrides supported on silica–alumina. Furthermore, hydrogenolysis of ethane and butane in a fixed-bed reactor for the series of supported metal hydrides have been applied in order to obtain mechanistic insights since wax comprises molecules with different hydrocarbon chain lengths.
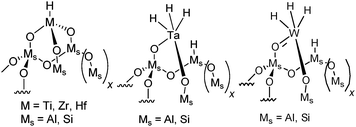 | ||
| Scheme 1 Metal hydrides supported on SiO2–Al2O3 of groups 4, 5 and 6. | ||
2 Experimental
Preparations of different compounds were performed under inert conditions (classical vacuum technique, glovebox and Schlenk line under Ar). Pentane was distilled from a reservoir withdrawn from a solvent purification system (Mbraun), degassed under vacuum and stored under NaK. Silica–alumina was obtained commercially (Akzo-Nobel, 25% Al). A batch of around 3 g of the support was first calcined at 500 °C for 20 hours followed by a dehydroxylation procedure involving heat treatment at 500 °C under high vacuum overnight. The surface organometallic precursors were prepared by impregnation of the corresponding metal neopentyl-based complexes (Zr[CH2C(CH3)3]4, Hf[CH2C(CH3)3]4, Ta[![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC(CH3)3][CH2C(CH3)3]3) dissolved in pentane onto the treated silica–alumina at room temperature.8,18,19W[
CHC(CH3)3][CH2C(CH3)3]3) dissolved in pentane onto the treated silica–alumina at room temperature.8,18,19W[![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC(CH3)3][CH2C(CH3)3]2 supported on silica–alumina was obtained through a solid reaction by stirring W[CC(CH3)3][CH2C(CH3)3]3 and the dehydroxylated support at 66 °C.20 In all cases, the excess of complex was washed off with pentane. The solids were then dried under high vacuum. The metal hydrides supported on silica–alumina were obtained after hydrogen treatment of the surface organometallic precursors at 150 °C for 16 hours. This procedure gives mainly the surface metal hydrides as depicted in Scheme 1. The metal loading was determined by elemental analysis (Zr: 3.9 wt%; Hf: 7.1 wt%; Ta: 7.8 wt%; W: 7.6 wt% through CNRS central analysis service in Solaize).
CC(CH3)3][CH2C(CH3)3]2 supported on silica–alumina was obtained through a solid reaction by stirring W[CC(CH3)3][CH2C(CH3)3]3 and the dehydroxylated support at 66 °C.20 In all cases, the excess of complex was washed off with pentane. The solids were then dried under high vacuum. The metal hydrides supported on silica–alumina were obtained after hydrogen treatment of the surface organometallic precursors at 150 °C for 16 hours. This procedure gives mainly the surface metal hydrides as depicted in Scheme 1. The metal loading was determined by elemental analysis (Zr: 3.9 wt%; Hf: 7.1 wt%; Ta: 7.8 wt%; W: 7.6 wt% through CNRS central analysis service in Solaize).
Catalytic reactions were carried out at 180 °C and 1 bar of H2 in a stainless steel fixed-bed reactor. The feed was purified by a column of activated molecular sieve and Cu2O/Al2O3. The catalysts were loaded in the glovebox. A mixture of 9 ml min−1 of hydrogen and 3 ml min−1 of ethane or butane was used for the hydrogenolysis reactions, respectively. About 70 mg of the catalyst was used (corresponds to 4 mol of hydrocarbon min−1 mol of [M]s-H−1). The reactor was coupled directly to a Varian CP 3800 GC for product determination. Low molecular-weight wax was purchased from Aldrich (ASTM D87, mp 70 °C, hydrocarbons range from C24–C42) and was dried at room temperature under high vacuum overnight before use. 400 mg of wax was placed on top of the catalyst bed when the hydrogen flow (20 ml min−1) was introduced from the bottom of the reactor.16 The heavier hydrocarbons were collected in a cold zone at 0 °C and analyzed in an external GC whereas the lighter hydrocarbons were quantified in the on-line HP 6890 GC.
3 Results and discussion
Catalytic C–C bond cleavage via hydrogenolysis can occur through two different mechanisms: (i) β-alkyl transfer corresponds to the reverse process of an olefin insertion into a metal–carbon bond (Scheme 2a);21 or (ii) carbene de-insertion corresponds to the reverse step known in organometallic chemistry postulated as the key step in Fischer–Tropsch synthesis (Scheme 2b).22,23 It is practically difficult to differentiate those mechanisms for long chain hydrocarbons such as wax. To obtain extensive information of the catalytic activity and mechanistic insights for the series of supported metal hydrides, hydrogenolysis of ethane and butane are studied. Ethane is selected since there is no alkyl group in the β-position of the metal-ethyl intermediate. Consequently, hydrogenolysis of ethane excludes the presence of β-alkyl transfer (Scheme 2a). The cleavage must then take place involving only one carbon atom at a time (according to Scheme 2b).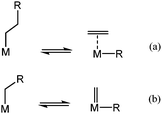 | ||
| Scheme 2 Mechanistic pathways for aliphatic C–C bond cleavage on metal-alkyl fragments: (a) β-alkyl transfer; (b) carbene de-insertion. | ||
C–C bond cleavage of butane by hydrogenolysis can in principle take place via β-alkyl and/or α-alkyl transfer and has hence been employed to compare the catalytic activity among the catalysts. Preliminary studies of similar systems (Zr-H and Ta-H supported on silica) carried out in a batch reactor provide an unreliable selectivity of the primary products since the formed components can undergo consecutive reactions.15,24 In the current work, all reactions were performed in a continuous flow reactor that gives a more representative view of the selectivity and allows a direct comparison among the supported metal hydrides of groups 4, 5 and 6.
3.1 Hydrogenolysis of ethane and butane
The results of ethane hydrogenolysis over different metal hydrides supported on silica–alumina are collected in Fig. 1. At 180 °C, 1 bar and under continuous flow of hydrogen and ethane (H2/Ethane = 3), [Zr]s-H and [Hf]s-H supported on silica–alumina are virtually inactive (conversion after 1000 minutes: 0.8% for [Zr]s-H; 0.07% for [Hf]s-H). On the other hand, ethane conversion stabilizes at 27% and 22% for [Ta]s-H and [W]s-H, respectively.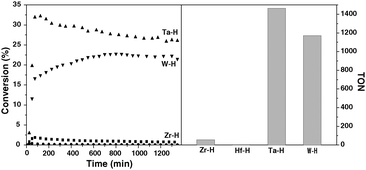 | ||
| Fig. 1 Comparison of the activity of the different metal hydrides (Zr: ■; Hf: ●; Ta: ▲; W: ▼) supported on silica–alumina in the hydrogenolysis of ethane. Left and right parts show the conversion with respect to time on stream and the cumulated turnover number after 1350 minutes, respectively. | ||
Evidently, a mixture of ethane and hydrogen behaves differently over [Zr]s-H and [Hf]s-H than [Ta]s-H and [W]s-H. It has been earlier shown that there is an equilibrium between d0 trishydrides [Ta]s-(H)3 and [W]s-(H)3 complexes with d2monohydride [Ta]s-H and [W]s-H complexes.12,19 As observed earlier for Ta-H supported on silica,24 the aliphatic C–C bond of ethane is broken through an α-alkyl transfer mechanism (Scheme 3). The current study also suggests that W-H cleaves the C–C bond via α-alkyl transfer on d2 W-H centers,25 and involves: (i) C–H activation of ethane on the d2 metal monohydride along with release of hydrogen; (ii) de-insertion of carbene, which results in a metal-methyl-methylidene d0 surface species; (iii) hydrogenolysis of the metal-methyl part and formation of the metal-methylidene-hydride moiety; (iv) insertion of hydride into metal-methylidene, giving a d2 metal-methyl fragment; (v) liberation of methane and regeneration of the metal-hydride site after hydrogen activation (Scheme 3). The inactivity of [Zr]s-H and [Hf]s-H precludes the presence of this mechanistic pathway. Note that the α-alkyl transfer requires d0 ↔ d2 transition of the metal to form the metal-methyl-methylidene intermediate. This elementary step is also suspected in the non-oxidative coupling of methane to ethane and hydrogen12,26 (the reverse reaction of ethane hydrogenolysis) by group 5 and group 6 metal hydrides while group 4 metal hydrides are inactive.27
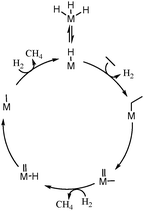 | ||
| Scheme 3 Proposed mechanism for the hydrogenolysis of ethane according to α-alkyl transfer (M = Ta, W). | ||
Fig. 2 summarizes the activity of the different metal hydrides supported on silica–alumina in the hydrogenolysis of butane. At 180 °C and atmospheric pressure, the maximum conversion is rapidly obtained. Clearly, [Zr]s-H supported on silica–alumina is the most active and stable catalyst among the series of metal hydrides. The activity decreases in the following order [Zr]s-H > [Hf]s-H > [Ta]s-H > [W]s-H. Period 6 metal hydrides show a slow deactivation at the beginning, but stabilize after 1000 minutes.
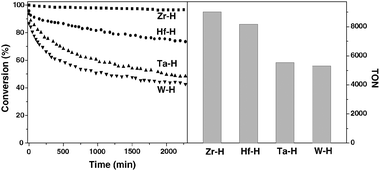 | ||
| Fig. 2 Comparison of activities of different metal hydrides (Zr: ■; Hf: ●; Ta: ▲; W: ▼) supported on silica–alumina in the hydrogenolysis of n-butane. Left and right parts show the conversion with respect to time on stream and the cumulated turnover number after 2300 minutes, respectively. | ||
For all catalysts, the hydrogenolysis of butane gives propane, ethane and methane as products. There is a notable difference in selectivity between the reactions carried out in a batch reactor15,24 and flow reactor. The major products obtained in the continuous flow reactor of group 4 metal hydrides are ethane and methane at steady-state as summarized in Table 1. While all products (methane, ethane and propane) are observed in the case of [Ta]s-H and [W]s-H (Table 1). Current observation is completely different with respect to a previous study in a batch reactor.24 No significant variation of the selectivity was observed at low conversion (between 5 and 20% conversion) by variation of contact time for the Zr and Ta based catalysts suggesting that the methane, ethane and propane are really primary products.
| Catalyst | [Zr]s-H | [Hf]s-H | [Ta]s-H | [W]s-H |
|---|---|---|---|---|
| Conversion (%) | 98 | 76 | 50 | 44 |
| C1 selectivity (mol%) | 44 | 30 | 36 | 47 |
| C2 selectivity (mol%) | 54 | 55 | 35 | 33 |
| C3 selectivity (mol%) | 2 | 15 | 29 | 20 |
According to the β-alkyl transfer mechanism in the case of the group 4, the initial step is C–H bond activation on the metal hydride, resulting in a metal(n-butyl) or metal(sec-butyl) surface species accompanied by release of hydrogen (Scheme 4). Then, the C–C bond cleavage of metal(n-butyl) via β-alkyl transfer occurs, giving a coordinated ethylene and a metal(ethyl) species (eqn (I), Scheme 4). Further activation of hydrogen on the metal center liberates ethane, leaving a coordinated ethylene and a metal hydride behind. The final products are formed after insertion of the hydride and hydrogenolysis of the metal ethyl giving the second molecule of ethane. In the case of metal(sec-butyl), the C–C bond cleavage by β-alkyl gives metal(methyl) and a coordinated propylene. The metal(propyl) intermediate can either produce propane or split to ethane and methane by another β-alkyl transfer process, as described by eqn (II) and (III) in Scheme 4. These competitive reactions explain the difference in selectivity of group 4 metal hydrides. The high selectivity in ethane (55%) is probably due to favorable activation on the primary C–H bonds rather than the secondary C–H bonds of butane (eqn (I) Scheme 4). It is quite possible that the initial C–H bond activation of butane is followed by rapid β-hydride elimination and alkyl reinsertion to favor eqn (I).28
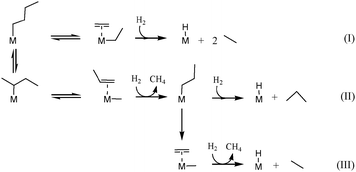 | ||
| Scheme 4 Basic equations illustrating the formation of different products via β-alkyl transfer originated from n-butane hydrogenolysis. | ||
There is also a pronounced difference in the product distribution between [Ta]s-H and [W]s-H. Tracing the product formation from different elementary reactions is inconvenient since both α-alkyl and β-alkyl transfer can occur. However, there are always two possible reaction pathways for the metal-alkyl surface species: (i) liberation of an alkanevia hydrogenolysis or (ii) successive alkyl cleavage (α-alkyl and β-alkyl transfer). A large amount of propane is released on the [Ta]s-H and [W]s-H catalysts compared to group 4, suggesting that the hydrogenolysis reaction of metal(propyl) is more dominant under the given conditions.
In summary, hydrogenolysis of butane shows that d0 supported metal hydrides of group 4 have an enhanced catalytic performance and they can only follow the β-alkyl transfer mechanism for the C–C bond breaking. The catalytic activities of group 5 and group 6 metal hydrides are lower and can undergo both α-alkyl and β-alkyl transfer.
3.2 Hydrogenolysis of wax
The differences among metal hydrides supported on silica–alumina in terms of activity and yield have been also investigated in the hydrogenolysis of wax to gasoline and diesel by combining the fixed-bed reaction with reactive distillation.As it can be seen in Table 2, the activity among the series of catalysts follows the same trend as observed for hydrogenolysis of butane and decreases in the following order: [Ti]s-H ≈ [Zr]s-H > [Hf]s-H > [Ta]s-H > [W]s-H. The products have been divided into three categories: gas (C1–C4), gasoline (C5–C9) and diesel (C10–C22). Table 2 presents the yields after 2000 minutes for the different catalysts. These results show that d0metal hydrides of group 4 supported on silica–alumina present in general higher activity and yield in gasoline and diesel than the other tested catalysts. But a striking feature is the propensity of W to form much higher amounts of methane than the other catalysts (see Fig. 3). Also of interest is the fact that [W]s-H and [Ta]s-H gave the highest selectivity for C2.
| Catalyst | [Ti]s-H16 | [Zr]s-H | [Hf]s-H | [Ta]s-H | [W]s-H |
|---|---|---|---|---|---|
| Conversion (%) | 100 | 100 | 66.1 | 64.4 | 38.0 |
| C1–C4 yield (wt%) | 13.7 | 23.4 | 8.3 | 7.1 | 5.2 |
| C5–C9 yield (wt%) | 24.3 | 25.3 | 16.9 | 16.3 | 9.2 |
| C10–C22 yield (wt%) | 62.0 | 51.3 | 40.9 | 41.0 | 23.6 |
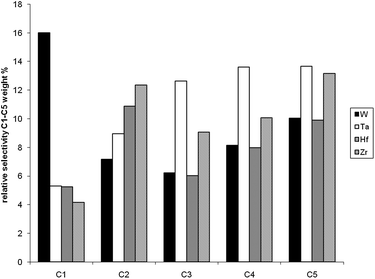 | ||
| Fig. 3 Relative selectivity of the various metal hydrides toward C1–C5 products (as the same conditions as in Table 2). | ||
4 Conclusions
Catalytic hydrogenolysis in a fixed-bed reactor of saturated hydrocarbons over [Zr]s-H, [Hf]s-H, [Ta]s-H and [W]s-H supported on silica-alumina have been exemplified by using ethane, butane and low molecular weight waxes as reactants. The activity decreases in the following order: [Zr]s-H > [Hf]s-H > [Ta]s-H > [W]s-H. Analysis of the selectivity using a continuous flow reactor offers a reliable product distribution of the reaction and illustrates that the C–C bond cleavage is in agreement with a β-alkyl transfer for [Zr]s-H and [Hf]s-H, while [Ta]s-H and [W]s-H can both undergo a β-alkyl and α-alkyl transfer. Wax has successfully been transformed into shorter hydrocarbon chains in typically gasoline or diesel range molecules. In particular, supported group 4 metal hydrides show promising activity and selectivity and thereby provide a potential application of these catalysts.Acknowledgements
We are grateful for the financial support from BP, 150 W Warrenville Road, IL 60563, Naperville, USA.References
- M. D. Hernandez, J. A. Reyes-Labarta and F. J. Valdes, Ind. Eng. Chem. Res., 2010, 49, 9068–9076 CrossRef CAS.
- Y. P. Huang and J. I. Chang, Renewable Energy, 2010, 35, 269–274 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- S. Hwang, J. Lee, J. G. Seo, D. R. Park, M. H. Youn, J. C. Jung, S.-B. Lee and I. K. Song, Catal. Lett., 2009, 132, 410–416 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- B. Louis, M. M. Pereira, F. M. Santos, P. M. Esteves and J. Sommer, Chem.–Eur. J., 2010, 16, 573–576 CrossRef CAS.
- M. Stocker, Microporous Mesoporous Mater., 2005, 82, 257–292 CrossRef.
- V. R. Dufaud and J. M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef CAS.
- F. Blanc, C. Coperet, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2006, 45, 6201–6203 CrossRef CAS.
- M. Taoufik, E. Le Roux, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2007, 46, 7202–7205 CrossRef CAS.
- M. Taoufik, E. Schwab, M. Schultz, D. Vanoppen, M. Walter, J. Thivolle-Cazat and J. M. Basset, Chem. Commun., 2004, 1434–1435 RSC.
- K. C. Szeto, S. Norsic, L. Hardou, E. Le Roux, S. Chakka, J. Thivolle-Cazat, A. Baudouin, C. Papaioannou, J. M. Basset and M. Taoufik, Chem. Commun., 2010, 46, 3985–3987 RSC.
- N. Merle, F. Stoffelbach, M. Taoufik, E. Le Roux, J. Thivolle-Cazat and J. M. Basset, Chem. Commun., 2009, 2523–2525 RSC.
- J. M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS.
- C. Lecuyer, F. Quignard, A. Choplin, D. Olivier and J. M. Basset, Angew. Chem., Int. Ed. Engl., 1991, 30, 1660–1661 CrossRef.
- C. Larabi, N. Merle, S. Norsic, M. Taoufik, A. Baudouin, C. Lucas, J. Thivolle-Cazat, A. de Mallmann and J. M. Basset, Organometallics, 2009, 28, 5647–5655 CrossRef CAS.
- S. Srinivas, S. M. Mahajani and R. K. Malik, Ind. Eng. Chem. Res., 2010, 49, 6350–6361 CrossRef CAS.
- G. Tosin, M. Delgado, A. Baudouin, C. C. Santini, F. Bayard and J. M. Basset, Organometallics, 2010, 29, 1312–1322 CrossRef CAS.
- S. Soignier, M. Taoufik, E. Le Roux, G. Saggio, C. Dablemont, A. Baudouin, F. Lefebvre, A. de Mallmann, J. Thivolle-Cazat, J. M. Basset, G. Sunley and B. M. Maunders, Organometallics, 2006, 25, 1569–1577 CrossRef CAS.
- E. Le Roux, M. Taoufik, A. Baudouin, C. Coperet, J. Thivolle-Cazat, J. M. Basset, B. M. Maunders and G. J. Sunley, Adv. Synth. Catal., 2007, 349, 231–237 CrossRef CAS.
- A. Motta, I. L. Fragala and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 16533–16546 CrossRef CAS.
- M. Leconte, A. Theolier and J. M. Basset, J. Mol. Catal., 1985, 28, 217–231 CrossRef CAS.
- G. Parkin, E. Bunel, B. J. Burger, M. S. Trimmer, A. Vanasselt and J. E. Bercaw, J. Mol. Catal., 1987, 41, 21–39 CrossRef CAS.
- M. Chabanas, V. Vidal, C. Coperet, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2000, 39, 1962–1965 CrossRef CAS.
- P. Sautet and F. Delbecq, Chem. Rev., 2010, 110, 1788–1806 CrossRef CAS.
- D. Soulivong, S. Norsic, M. Taoufik, C. Coperet, J. Thivolle-Cazat, S. Chakka and J. M. Basset, J. Am. Chem. Soc., 2008, 130, 5044–5045 CrossRef CAS.
- C. Thieuleux, C. Coperet, V. Dufaud, C. Marangelli, E. Kuntz and J. M. Basset, J. Mol. Catal. A: Chem., 2004, 213, 47–57 CrossRef CAS.
- G. P. Niccolai and J. M. Basset, Appl. Catal., A, 1996, 146, 145–156 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |