Aerobic oxidation of 5-hydroxylmethylfurfural with homogeneous and nanoparticulate catalysts†
Basudeb
Saha
*a,
Saikat
Dutta
a and
Mahdi M.
Abu-Omar
*b
aLaboratory of Catalysis, Department of Chemistry, University of Delhi, Delhi 110007, India. E-mail: bsaha@chemistry.du.ac.in; Fax: +91-11-2766-9974; Tel: +91-11-2766-6646
b5170 Brown Laboratory of Chemistry, Purdue University, West Lafayette, IN 47096, USA. E-mail: mabuomar@purdue.edu; Fax: +1-765-494-0239; Tel: +1-765-494-5302
First published on 23rd September 2011
Abstract
The aerobic oxidation of 5-hydroxymethylfurfural (HMF) with homogeneous Co(OAc)2/Zn(OAc)2/Br−, and heterogeneous Au–TiO2 and Au–CeO2 nanoparticulate catalysts was carried out in acetic acid with and without a trifluoroacetic acid (HTFA) additive. The selectivity of the oxidation products, 2,5-furandicarboxylic acid (FDCA) and 5-formyl-2-furancarboxylic acid (FFCA), improved in the presence of the HTFA (1 wt%) additive. The homogeneous oxidation catalyst gave higher selectivity for FDCA (60% yield) under 1 atm of O2 at 90 °C while Au–TiO2 afforded 80% FFCA under the same conditions.
In the last century, crude oil became the sole and essential feedstock for the production of bulk and fine chemicals, and polymeric materials.1 Depletion of fossil fuel and the awareness of climate change necessitate the utilization of bio-renewable resources on a large scale for chemicals and fuel production.2Terephthalic acid (PTA) is an important oil-based aromatic dicarboxylic acid for producing high purity polyethylene terephthalate (PET), a polymer commonly made into consumer goods for thousands of applications.3,4 The biomass-derived alternative 2,5-furandicarboxylic acid (FDCA) is a desirable replacement for oil-based terephthalic acid for polyester production. FDCA can also be used for the preparation of polyamides and polyurethane.5,6 Because of multiple applications, FDCA has been identified as one of the twelve biomass derived chemicals by the U.S. Department of Energy.1 The preferred route of FDCA synthesis is the catalytic oxidation of 5-hydroxymethylfurfural (HMF), which is derived from fructose, glucose and cellulose substrates.7,8
A thorough literature search reveals that the oxidation of HMF to FDCA has been reported for stoichiometric oxidants (e.g. KMnO4),9 highly polluting metal catalysts (e.g. Pb),10 and homogeneous metal salts comprising transition-metal acetates and a bromide source, Co(OAc)2/HBr/Mn(OAc)2, commonly known as the Amoco Mid-Century (MC) catalyst.11 The liquid phase oxidation of HMF over platinum on alumina catalysts produces 5-formyl-2-furancarboxylic acid (FFCA) as the main oxidation product, indicating that the hydroxymethyl group of HMF is oxidized first in the presence of the aldehyde group.12 Lilga et al. patented an oxidation method of HMF using Pt/ZrO2 catalyst. The patent claimed the formation of 2,5-diformylfuran (DFF) and FFCA with 68% and 32% selectivity, respectively.13 Contrary to some reports of using homogeneous and heterogeneous catalysts for the aerobic oxidation of HMF to FDCA, the application of nanocatalysts for this reaction is limited. Christensen et al. used titanium dioxide-supported gold nanocatalyst for the oxidation of HMF in methanol and reported the formation of 2,5-furandimethylcarboxylate (FDMC) in excellent yield (98%) at 130 °C and 4 bar O2 pressure in the presence of sodium methoxide as a base.14 In a subsequent communication, the effect of oxygen pressure and concentrations of hydroxide on FDCA yield and selectivity were addressed.15 The findings showed 99% HMF conversion with 84% yield of FDCA when the reaction was conducted in water using ceria (CeO2) and titania (TiO2) nanoparticulate materials supported gold catalysts, Au–CeO2 and Au–TiO2, in the presence of NaOH. Without base, the authors claimed catalyst deactivation by the initially formed acids, and therefore, FDCA selectivity was poor. In a recent publication, Gupta et al. demonstrated an excellent yield of FDCA (99%) from HMF using hydrotalcite-supported gold nanoparticles in water at 95 °C under atmospheric oxygen pressure without addition of base.16 By comparison, the analogous MC homogenous catalysts for para-xylene oxidation are activated by acid additives.17,18 However, the catalytic activities of both homogeneous and heterogeneous catalysts were not tested in the presence of acid additives for HMF oxidation. In this paper, we report the effect of an acid additive, trifluoroacetic acid (HTFA), on the aerobic oxidation of HMF with Co(OAc)2/HBr/Mn(OAc)2, Au/TiO2 and Au/CeO2 catalysts. HTFA, a simple and stable fluorinated acetic acid, was chosen as an acid additive because of its prior use in methylarenes oxidation18 and that it does not form insoluble metal complexes with catalyst metals that are usually formed with mineral acids.18
A preliminary reaction for HMF oxidation was carried out with 240 mM HMF, 40 mM Co(OAc)2, 3 mM Mn(OAc)2 and 40 mM NaBr in a glass reactor using acetic acid (HOAc) as a solvent at 90 °C under atmospheric pressure of O2. Caution: The use of highly oxidizing metal salts under a pure oxygen atmosphere at 90 °C could be potentially explosive and dangerous. These transformations should be performed only with adequate barriers for protection. Complete oxidation of HMF was realized in 4.5 h; however, the reaction did not progress beyond the formation of DFF (96%). The same reaction in the presence of 5 wt% HTFA enabled further oxidation beyond DFF to give 37% FDCA and 31% FFCA (Table 1). A new peak was detected in the GC chromatogram corresponding to the mass number of the methoxyketal of DFF. The formation of a methoxyketal derivative of DFF might occur by the reaction of DFF and methanol (used for diluting the aliquot sample) at the injector temperature of the GC. The concentration of DFF in the form of methoxyketal was 32%.
| Entry # | HTFA | Time | Conv. (%) | Product (%) | ||
|---|---|---|---|---|---|---|
| DFF | FFCA | FDCA | ||||
| 1 | 0 | 4.5 h | 100 | 96 | — | — |
| 2 | 5 wt% | 3 h | 100 | 32 | 31 | 37 |
The beneficial effect of HTFA has also been reported for the aerobic oxidation of para-xylene to terephthalic acid,17,18 and the enhancement attributed to the formation of Mn(TFA)3 salt, which is a stronger oxidizing agent than Mn(OAc)3.19 The improved oxidation of HMF in the presence of the HTFA additive is an indication of a radical chain redox mechanism similar to that reported for para-xylene oxidation.20–22
To further test the beneficial effect of HTFA, the oxidation of HMF was carried out with the Co(OAc)2/Zn(OAc)2/Br−catalyst in HOAc in the presence and absence of HTFA. Zn(OAc)2 is known to co-catalyze the oxidation of para-xylene and minimize the loss of the active bromide promoter to the form of inactive benzyl bromide.17 The concentrations of inorganic bromide and Zn(OAc)2 co-catalyst were varied in the range of 2–44 mM and 2.5–11 mM, respectively. The corresponding results are summarized in Table 2.
| Expt # | HTFA, wt% | Zn(OAc)2/mM | NaBr/mM | Time/h | Conv. (%) | Product (%) | ||
|---|---|---|---|---|---|---|---|---|
| DFF | FFCA | FDCA | ||||||
| 1 | 0 | 11 | 44 | 2.2 | 100 | 88 | — | — |
| 2 | 1 | 11 | 44 | 3 | 90 | — | 29 | 60 |
| 3 | 1 | 12 | 10 | 4.2 | 89 | — | 33 | 55 |
| 4 | 1 | 11 | 5.3 | 4 | 82 | — | 28 | 41 |
| 5 | 1 | 11 | 2 | 4 | 80 | 7.6 | 32 | 30 |
| 6 | 1 | 4.5 | 3 | 4 | 67 | 17 | 22 | 26 |
| 7 | 1 | 2.5 | 3 | 3 | 70 | 22 | 22 | 23 |
| 8 | 0 | 2.5 | 3 | 4 | 91 | 90 | — | — |
Experiments 1 and 8 in Table 2 were performed without the HTFA additive. In both cases, only DFF was formed as the oxidation product. In the presence of 1 wt% HTFA (experiments 2–7), HMF oxidation progressed beyond the formation of DFF, and the reaction produced a maximum of 60% FDCA under the molar ratio of total metal acetates to NaBr of 1.2. The conversion of HMF and the formation of FDCA decreased with an increase in molar ratio of total metal catalysts to NaBr.
The effect of the HTFA additive was also investigated for the autoxidation of HMF with Au–TiO2 and Au–CeO2 nanocatalysts. These reactions were carried out with and without the HTFA additive under 8–10 bar oxygen pressure at 130 °C in HOAc as a solvent. The results show the formation of FFCA as the major oxidation product. Without the HTFA additive, HMF conversions with Au–TiO2 and Au–CeO2 catalysts were 62% and 29% with corresponding FFCA yields of 45% and 17%, respectively.
The beneficial effect of HTFA was immediately realized when both reactions were repeated in the presence of 1 wt% HTFA; HMF conversions for the Au–TiO2 and Au–CeO2 catalyzed oxidations improved to 84% and 83% with corresponding FFCA yields of 79% and 71%, respectively. FDCA was not formed during our reaction time of 3 h. The results are shown in Fig. 1.
![(a) The reaction profile for HMF oxidation with Co(OAc)2/Zn(OAc)2/Br−catalyst under the conditions of [Co(OAc)2] = 40 mM, [Zn(OAc)2] = 12 mM, [Br−] = 10 mM, HMF = 240 mM, HTFA = 1 wt% and T = 90 °C. (b) The effect of HTFA additive on HMF conversion and FFCA yields for HMF oxidation with Au/TiO2 and Au/CeO2 catalysts under the reaction conditions of HMF/catalyst = 18–21, pO2 = 10 bar, T = 130 °C and reaction time = 3 h.](/image/article/2012/CY/c1cy00321f/c1cy00321f-f1.gif) | ||
| Fig. 1 (a) The reaction profile for HMF oxidation with Co(OAc)2/Zn(OAc)2/Br−catalyst under the conditions of [Co(OAc)2] = 40 mM, [Zn(OAc)2] = 12 mM, [Br−] = 10 mM, HMF = 240 mM, HTFA = 1 wt% and T = 90 °C. (b) The effect of HTFA additive on HMF conversion and FFCA yields for HMF oxidation with Au/TiO2 and Au/CeO2 catalysts under the reaction conditions of HMF/catalyst = 18–21, pO2 = 10 bar, T = 130 °C and reaction time = 3 h. | ||
Fig. 1(b) shows that the Au–TiO2 catalyst is more effective than the Au–CeO2 catalyst, which could be due to the (i) higher Au content in the Au–TiO2 catalyst (Au = 1.9%) than that in the Au–CeO2 catalyst (Au = 1.4%) and (ii) higher BET surface area of the TiO2 support (285 m2 g−1) than that of the CeO2 support (72 m2 g−1). Corma23 and Riisager15groups have investigated the catalytic effectiveness of both Au–TiO2 and Au–CeO2 nanomaterials for HMF oxidation in aqueous medium. However, a base additive was needed for realizing good FDCA yield. It was suggested that the reaction suffered from incomplete oxidation in the absence of a base additive, primarily due to the deactivation of the catalysts with the initially formed acids.15,23 The reasons for the deactivation of the catalysts by the initially formed acid products were not discussed. In contrast, hydrotalcite supported gold nanoparticle catalyzed HMF oxidation to give 99% FDCA without using a base additive. We found an improvement in catalytic activity of Au–TiO2 and Au–CeO2 materials in the presence of HTFA over their catalytic activity without the HTFA additive.
The exact pathway of HMF oxidation to FDCA is a matter of debate. According to Corma23 and Riisager15groups, the –CHO group of HMF is oxidized first to –COOH and then hydroxymethyl is oxidized in a slower step. Partenheimer and Grushin11 described both possibilities, with preference to the hydroxymethyl group being oxidized first. In our work, we found DFF to be the only oxidation product without the HTFA additive, suggesting that the hydroxymethyl group is oxidized first. Based on this understanding, a sequence of the HMF oxidation process is shown in Scheme 1.
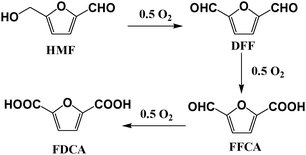 | ||
| Scheme 1 Sequence of HMF oxidation with Co(OAc)2/Zn(OAc)2/Br−catalyst. | ||
In conclusion, the effectiveness of Co(OAc)2/Zn(OAc)2/Br−, Au–TiO2 and Au–CeO2 catalysts were investigated for HMF oxidation in HOAc with and without HTFA. Without HTFA, Co(OAc)2/Zn(OAc)2/Br− catalyzed oxidation of HMF produced DFF as the only oxidation product, suggesting that the hydroxymethyl group of HMF is oxidized first. In the presence of HTFA, HMF oxidation proceeded to the desired product, FDCA. The selectivity of FDCA decreased with an increase in total metal to bromide ratio. In the case of Au–TiO2 and Au–CeO2 catalyzed oxidation, FFCA was formed without using the HTFA additive. In the presence of HTFA, HMF conversion and FFCA selectivity improved. Further studies on the interaction of the HTFA additive with Au–TiO2 and Au–CeO2 nanocatalysts are underway.
MMA-O is grateful to the US Department of Energy, Office of Basic Energy Sciences for financial support (Grant DE-FG02-06ER15794). BS acknowledges the financial support by the University Grant Commission (UGC), India, and the University of Delhi. Authors acknowledge Dr. Trenton Parsell for his help with operation of the Parr reactor and Mr. Sudipta De for assistance in characterization of Au-supported catalysts.
Notes and references
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, Pacific Northwest National Laboratory, vol. 1, 2004, p. 27 Search PubMed.
- A. Boisen, T. B. Christensen, W. Fu, Y. Y. Gorbanev, J. S. Jensen, S. K. Klitgaard, S. Pedersen, A. Riisager, T. Stahlberg and J. M. Woodley, Chem. Eng. Res. Des., 2009, 87, 1318 CrossRef CAS.
- D. Raju Burri, K.-W. Jun, J. S. Yoo, C. W. Lee and S.-E. Park, Catal. Lett., 2002, 81, 169 CrossRef CAS.
- N. C. Wyeth and R. N. Roseveare, US Pat., 3733309, 1973 Search PubMed.
- A. Gandini and M. N. Belgacem, J. Polym. Environ., 2002, 10, 105 CrossRef CAS.
- C. Moreaua, M. N. Belgacemb and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754 RSC.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1 CrossRef CAS.
- M. Toshinari, K. Hirokazu, K. Takenobu and M. Hirohide, US Pat., 232815, 2007 Search PubMed.
- P. Verdeguer, N. Merat and A. Gaset, J. Mol. Catal. A: Chem., 1993, 85, 327 CAS.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102 CrossRef CAS.
- P. Vinke, H. E. van Dam and H. van Bekkum, Stud. Surf. Sci. Catal., 1990, 55, 147 CrossRef CAS.
- M. A. Lilga, R. T. Hallen, J. Hu, J. F. White and M. J. Gray, US Pat., 0103318, 2008 Search PubMed.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75 CrossRef CAS.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672 CrossRef CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824 RSC.
- B. Saha and J. H. Espenson, J. Mol. Catal. A: Chem., 2007, 271, 1 CrossRef CAS.
- B. Saha and J. H. Espenson, J. Mol. Catal. A: Chem., 2005, 241, 33 CrossRef CAS.
- J. Hanotier, M. Hanotier-Bridoux and P. De Radzitzky, J. Chem. Soc., Perkin Trans. 2, 1973, 381 RSC.
- I. V. Zakharov, Y. V. Geletii and V. A. Adamian, Kinet. Katal., 1991, 32, 31 Search PubMed.
- B. Saha, N. Koshino and J. H. Espenson, J. Phys. Chem. A, 2004, 108, 425 CrossRef CAS.
- B. Saha and J. H. Espenson, J. Mol. Catal. A: Chem., 2004, 207, 123 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, materials, Au–TiO2 and Au–CeO2, preparation and characterization. See DOI: 10.1039/c1cy00321f |
| This journal is © The Royal Society of Chemistry 2012 |