Theoretical investigation on copper hydrides catalyzed hydrosilylation reaction of 3-methylcyclohex-2-enone: mechanism and ligands' effect†
Liang
Dong
,
Song
Qin
,
Huaqing
Yang
,
Zhishan
Su
and
Changwei
Hu
*
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan, 610064, China. E-mail: chwehu@mail.sc.cninfo.net; gchem@scu.edu.cn; Fax: +86-28-85411105; Tel: +86-28-85411105
First published on 8th December 2011
Abstract
The mechanism of the hydrosilylation reactions of 3-methylcyclohex-2-enone with tetramethyldisiloxane (TMDS) catalyzed by (Ph3P)CuH and (IPr)CuH has been investigated by DFT. The catalytic cycle is composed of two steps: the addition of the copper hydrides to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the substrate, and the regeneration of the copper hydrides assisted by TMDS. The calculations indicate that the catalyst recovery step is the rate-determining step. The assistances of IPr and Ph3P ligands to the CuH catalysts make the transition state structures compact and stable. The steric bulk of the ligands could help to stabilize the central Cu atom and promote the coordination of the central Cu atom with the substrate. The higher nucleophilicity of the catalysts and the stronger interaction of the ligands with the central Cu atom make the catalysts interact more easily with the substrate. The hydrosilylation reaction proceeds more favorably when catalyzed by (IPr)CuH as compared to (Ph3P)CuH.
C bond in the substrate, and the regeneration of the copper hydrides assisted by TMDS. The calculations indicate that the catalyst recovery step is the rate-determining step. The assistances of IPr and Ph3P ligands to the CuH catalysts make the transition state structures compact and stable. The steric bulk of the ligands could help to stabilize the central Cu atom and promote the coordination of the central Cu atom with the substrate. The higher nucleophilicity of the catalysts and the stronger interaction of the ligands with the central Cu atom make the catalysts interact more easily with the substrate. The hydrosilylation reaction proceeds more favorably when catalyzed by (IPr)CuH as compared to (Ph3P)CuH.
Introduction
The reduction of α,β-unsaturated carbonyl compounds by hydrogenation or hydrosilylation is an important transformation in organic synthesis.1 Hydrogenation reactions could obtain good yields, but only under high pressure or temperature. In contrast, hydrosilylation reactions are often achieved by the use of silanes under mild conditions and offer better effects. As silanes, the earlier noted tetramethyldisiloxane1e,g (TMDS) and polymethylhydrosiloxane2 (PMHS) are most frequently used. These silanes are easier to handle than hydrogen gas at high pressures, on the other hand, they could also essentially avoid the over-reduction. Gratifyingly, the high tolerance toward functional groups, the mildness of the reaction conditions, and especially the chemoselectivity are not affected.3 Therefore, conjugate hydrosilylation is an important reduction method for carbonyl compounds from both practical and economic points of view.1g,h,4Transition-metal hydride, in particular copper hydride, has been successfully applied for the reduction of many α,β-unsaturated carbonyl compounds.5 In general, the hydrosilylation process proceeded via the asymmetric 1,4-addition catalytic cycle. The proposed mechanism1g,5d,6 for the hydrosilylation reaction of α,β-unsaturated ketone catalyzed by copper hydride is shown in Scheme 1. The entire catalytic cycle includes two steps: first, the copper hydride A coordinates to the CC bond of the substrate B followed by a hydrogen transfer to the β carbon atom. The resulting copper enolate D then undergoes heterolytic σ-bond metathesis with silane X–H, thereby liberating the product F and regenerating the catalytically active species A. The postulated catalytically active catalyst A, a copper hydride species CuH, can be easily formed by σ-bond metathesis between a copper alkoxide and a silane, and this step is supported by experiments.7
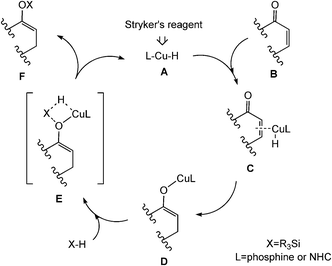 | ||
| Scheme 1 Proposed mechanism of the hydrosilylation catalyzed by CuH species. | ||
“Cu–H”, as one of the earliest metal hydrides reported in the literature, was too unstable to have potential applications as a reagent in organic chemistry.1g,5d,8 Several groups have made great contributions involving Cu–H-based conjugate reduction.1d,3,6,7b,c,9 The first report on the use of the stabilized form of copper hydride is the hexameric [(Ph3P)CuH]6, in the conjugate reduction of enones by Stryker and coworkers.10 That is why the copper hydride is commonly referred to as “Stryker's reagent”. The most common and widely used Stryker's reagent is the phosphine-stabilized copper hydride [(Ph3P)CuH]6.1d,g,5d,6,11 (Ph3P)CuH is considered as the active component to catalyze the hydrosilylation of the substrate.1a,6,12 A major disadvantage of this reagent is its sensitivity to air. The quality of the commercially available [(Ph3P)CuH]6 is quite variable and special precautions are needed for its storage. Lipshutz et al.12 achieved the catalytic hydrosilylation of 3-methylcyclohex-2-enone with 5 mol% of 1/6[(Ph3P)CuH]6 and stoichiometric amounts of TMDS. The hydrosilylation reaction could obtain its silyl ether product in 85% yield in toluene (room temperature, 24 h) (Scheme 2, eqn (1)).
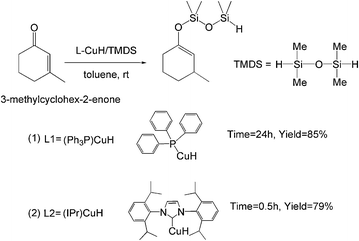 | ||
| Scheme 2 Copper hydrides catalyzed hydrosilylation reactions. | ||
During the past decade, N-heterocyclic carbenes (NHCs) have emerged as a new family of ligands for homogeneous catalysis.9a,d,13 Compared to tertiary phosphines, the NHC ligands have a stronger interaction with the metal center, thereby minimizing ligand dissociation. In addition, their significant steric bulk results in metal–NHC complexes having unique and distinct catalytic behavior compared to their phosphine-based analogues.9d,14 In previous studies, it was found that metal–NHC complexes acted as an efficient and convenient precatalyst for hydrosilylation, and could be a good alternative to the widely-used Stryker's reagent.9a,c Yun et al.15 reported an easily-handled and highly effective copper(II) precatalyst (IPr)CuII(OAc)2. This well-defined precatalyst can be effectively activated by silanes to generate the catalytically active species NHC–CuIH (namely (IPr)CuH), which performs higher activity than traditional Stryker's reagent in the hydrosilylation reaction. The conjugate reduction of 3-methylcyclohex-2-enone was completed at faster reaction rates (0.5 h, 79% yield) with lower catalyst loading (1 mol%) than that of using [(Ph3P)CuH]6 (Scheme 2, eqn (2)).
Theoretical calculation concerning the hydrosilylation reaction of α,β-unsaturated carbonyl compounds catalyzed by copper hydride is rare in the literature. To the best of our knowledge, only Gathy et al.16 reported a theoretical mechanistic study concerning the hydrosilylation of formaldehyde catalyzed by Cu(I) hydride. They proposed the hydrosilylation mechanism of formaldehyde and the rate-determining step (RDS) is identified as the first step concerning the formation of a copper alkoxide. Although great efforts have been made experimentally to explain the mechanism of the hydrosilylation reaction, the detailed information at the molecular level over copper hydride catalyst is still lacking. Furthermore, as compared to (Ph3P)CuH, NHC–CuH exhibits better catalytic performance for hydrosilylation reaction. In an attempt to gain a better understanding of the hydrosilylation reaction, and to explore the reason of the higher reactivity of the NHC-based copper hydride as compared to the Ph3P-based copper hydride, here, we present our theoretical results concerning the reaction mechanism of 3-methylcyclohex-2-enone with TMDS (as the silane source) catalyzed by the active species (Ph3P)CuH (L1–CuH) and (IPr)CuH (L2–CuH), respectively (Scheme 2). Furthermore, we clarify the impact of the ligands of copper hydrides for the hydrosilylation in this paper.
Computational details
Among the correlated functional theories, B3LYP performs well for the metal catalyzed reactions.17 Previous investigations also indicated that the B3LYP method can give reasonable structures and energies for copper-catalyzed reactions.16,18 All calculations were performed at the DFT level, by means of the hybrid B3LYP functional method,19 as implemented in Gaussian 03 program.20 Cu atom was described using an effective core potential (LANL2DZ)21 for the inner electrons, and its associated double-ζ basis set for the outer ones. The 6-31G(d, p) basis set22 was used for the C, H, O, N, P, and Si atoms. Zero-point vibrational energies (ZPVE) were also applied in relative Gibbs free energies. Vibrational frequencies were also obtained at the B3LYP/[6-31G(d, p), LANL2DZ] level, and the species were characterized as a minimum (no imaginary frequency) or a transition state (unique imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were performed to further confirm that the optimized transition state correctly connects the relevant reactant and product.23 Considering the effect of the solvent, single-point B3LYP (PCM/toluene)/[6-311++G(d, p), SDD] calculations24 (the SDD basis set for Cu atom and the 6-311++G(d, p) basis set for all other atoms) were performed on gas phase optimized structures at 298 K, with RADIIUAHF. The Gibbs free energies G298 K in toluene including the entropy and nonelectrostatic solvation energies were obtained from these single-point energies combined with the Gibbs free energy corrections at the B3LYP/6-31G(d, p) level in the gas phase. All energies discussed in the following refer to G298 K. To obtain a further insight into the electronic properties along the reaction, natural bond orbital (NBO)25 analysis at the B3LYP/[6-31G(d, p), LANL2DZ] level was also performed. A DFT analysis based on the electrophilicity index ω and the nucleophilicity index N was performed.26 The Potential Energy Surface Scanning was carried out to search for the most stable conformation of the catalyst.
Results and discussion
Mechanism
Since the postulated CuH species1g,5d,8 is the widely accepted catalytically active species for the conjugate reduction of ketone to its silyl ether in the hydrosilylation reaction, we continue the previous proposal16 considering CuH species as the most probable effective catalytic species in this reaction. Our calculations start from the simulation of the complex (L–COM) which the copper hydrides (L1–CuH and L2–CuH) coordinate to the substrate 3-methylcyclohex-2-enone. The optimized structures along the L1–CuH and L2–CuH catalyzed reaction paths are given in Fig. 1.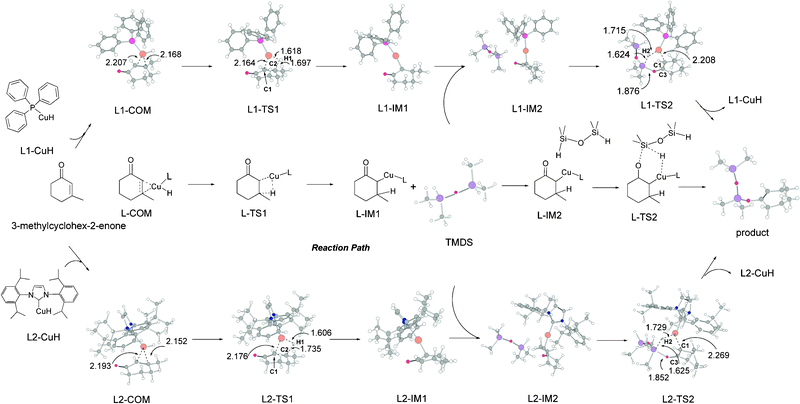 | ||
| Fig. 1 Optimized structures of the species in the hydrosilylation reactions along (Ph3P)CuH and (IPr)CuH catalyzed reaction paths (bond length in Å). | ||
(Ph3P)CuH catalyzed hydrosilylation reaction
Fig. 2 shows the calculated potential energy surface (PES) of the (Ph3P)CuH catalyzed hydrosilylation reaction in toluene at the B3LYP/[6-311++G(d, p), SDD] level. The relative Gibbs free energy in toluene is presented in Fig. 2.![Relative Gibbs free energy profile for the (Ph3P)CuH catalyzed hydrosilylation reaction path in toluene at the B3LYP/[6-311++G(d,p), SDD] level.](/image/article/2012/CY/c1cy00320h/c1cy00320h-f2.gif) | ||
| Fig. 2 Relative Gibbs free energy profile for the (Ph3P)CuH catalyzed hydrosilylation reaction path in toluene at the B3LYP/[6-311++G(d,p), SDD] level. | ||
As shown in Fig. 1, our calculations indicate that the hydrosilylation reaction catalyzed by (Ph3P)CuH (L1–CuH) is composed of two steps: (1) the addition of the catalyst L1–CuH to the CC bond on the substrate, and (2) the regeneration of the catalyst L1–CuH.
As shown in Fig. 1, the catalyst L1–CuH first coordinates to the CC bond of the substrate 3-methylcyclohex-2-enone with the formation of the precursor complex L1–COM. Then, the hydrogen of L1–CuH migrates from L1–CuH onto the β carbon (C2) of the substrate via a four center transition state L1–TS1 (involving C1, Cu, H1 and C2 atoms). The bond lengths of the Cu–H1 and H1–C2 bond are 1.618 and 1.697 Å in L1–TS1. Next, the corresponding intermediate L1–IM1 is formed. Subsequently the silane TMDS enters the catalytic cycle, and coordinates to L1–IM1 with the formation of L1–IM2. The hydrosilylation occurs between the silane moiety and the copper-substrate moiety. The hydrogen of TMDS migrates from TMDS to the Cu atom via a six center transition state L1–TS2 (involving C1, C3, O, Si, H2, and Cu atoms). Finally the silyl ether product is generated with the recovery of the L1–CuH catalyst. The bond lengths of the Si–H2 and H2–Cu bond in L1–TS2 are 1.624 and 1.715 Å. In summary, L1–CuH catalyzed the hydrosilylation of 3-methylcyclohex-2-enone along with TMDS which occurs through 1,4-reduction.
From Fig. 2, it is found that the relative Gibbs free energy of L1–TS2 is calculated to be 45.0 kJ mol−1. The relative Gibbs free energy of L1–TS1 is 38.3 kJ mol−1, ca. 7 kJ mol−1 lower than that of L1–TS2. Since L1–TS2 bears the largest energy barrier (72.8 kJ mol−1) and is the energy top on the PES, the RDS can be identified as the regeneration of the L1–CuH catalyst via L1–TS2.
(IPr)CuH catalyzed hydrosilylation reaction
The path concerning the hydrosilylation reaction catalyzed by (IPr)CuH is similar to that along the hydrosilylation catalyzed by (Ph3P)CuH. Fig. 3 shows the calculated PES of the (IPr)CuH catalyzed hydrosilylation reaction in toluene at the B3LYP/[6-311++G(d,p), SDD] level. The relative Gibbs free energy in toluene is presented in Fig. 3.![Relative Gibbs free energy profile for the (IPr)CuH catalyzed hydrosilylation reaction path in toluene at the B3LYP/[6-311++G(d,p), SDD] level.](/image/article/2012/CY/c1cy00320h/c1cy00320h-f3.gif) | ||
| Fig. 3 Relative Gibbs free energy profile for the (IPr)CuH catalyzed hydrosilylation reaction path in toluene at the B3LYP/[6-311++G(d,p), SDD] level. | ||
As shown in Fig. 1, the proposed mechanism for the hydrosilylation reaction of 3-methylcyclohex-2-enone catalyzed by L2–CuH also occurs through a two step cycle. The first step concerns the coordination of the L2–CuH to the CC bond of the substrate 3-methylcyclohex-2-enone with the formation of the complex L2–COM followed by a hydrogen migration from L2–CuH to the substrate via a four center transition state L2–TS1 to form L2–IM1. The bond lengths of the Cu–H1 and H1–C2 bond in L2–TS1 are 1.606 and 1.735 Å, respectively. In the second step, L2–IM1 reacts with TMDS to yield the corresponding silyl ether product via a six center transition state L2–TS2 (involving C1, C3, O, Si, H2 and Cu atoms) with the regeneration of the L2–CuH catalyst. The bond lengths of the Si–H2 and H2–Cu bond in L2–TS2 are 1.625 and 1.729 Å, respectively. The entire catalytic cycle along the (IPr)CuH catalyzed hydrosilylation reaction path also occurs through 1,4-reduction.
As shown in Fig. 3, the relative Gibbs free energy of L2–TS2 is calculated to be 40.3 kJ mol−1. Since L2–TS2 bears the largest energy barrier (65.3 kJ mol−1) and is the energy top on the PES, the regeneration step of the L2–CuH catalyst is identified as the RDS of the entire catalytic cycle. This result is consistent with that in the (Ph3P)CuH catalyzed hydrosilylation reaction system. Furthermore, the energy barrier via L2–TS2 is 65.3 kJ mol−1 in the (IPr)CuH catalyzed process, ca. 7.5 kJ mol−1 lower than that via L1–TS2 in the (Ph3P)CuH catalyzed process. This result indicates that the hydrosilylation reaction is more kinetically favored by using the (IPr)CuH catalyst as compared to the (Ph3P)CuH catalyst. This is compatible with the experimental observation where (IPr)CuH exhibits higher reactivity and better catalytic performance than (Ph3P)CuH.15 On the other hand, it is noted that since L2–IM1 is 19.8 kJ mol−1 lower than L1–IM1 in the relative Gibbs free energy, reaction from L2–IM1 to L2–TS2 requires more energy than from L1–IM1 to L1–TS2. However, the relative Gibbs free energies of IM1 and IM2 are relatively low, the reaction proceeds easily from IM1 to IM2. While TS2 is the energy top on the PES, and the relative Gibbs free energy of L1–TS2 is 45.0 kJ mol−1, ca. 5 kJ mol−1 higher than that of L2–TS2. Besides, the RDS of the catalytic cycle is the regeneration step of the catalyst via TS2, and the energy barrier via L1–TS2 is 72.8 kJ mol−1, ca. 7.5 kJ mol−1 higher than that via L2–TS2. In summary, the hydrosilylation reaction proceeds more favorably when catalyzed by L2–CuH as compared to L1–CuH.
The impact of the ligand for the hydrosilylation reaction
Ligands are obviously of great importance for determining the activity of the catalysts in general. Considering the differences of the reaction rate resulted by the association of the different ligands with CuH, it is essential to clarify the effect of the ligand assistance on the catalytic process.Structural analysis
Numerous investigations suggested that the steric bulk and the electronic properties of the ligands are closely related to the properties of the ligands.27 To expound further the reason why NHC–CuH exhibits better catalytic performance than (Ph3P)CuH, we focus our attention on the structures of the catalysts. Additionally, the Potential Energy Surface Scanning was carried out by rotating the isopropyl group in (IPr)CuH to search for the most stable conformation. The optimized structures of the catalysts (Ph3P)CuH and (IPr)CuH are presented in Fig. 4.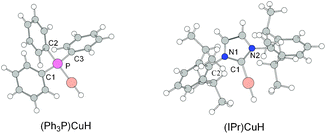 | ||
| Fig. 4 Optimized structures of (Ph3P)CuH and (IPr)CuH. | ||
According to the Potential Energy Surface Scanning, the isopropyl group rotates on the N1–C2 bond by σ-bond metathesis. The PES scanning finds the minimum at which the surface of the phenyl of the isopropyl group positions almost parallel to the surface of the six-membered ring of the substrate. It is obvious that this conformation has relatively small steric hindrance.
As shown in Fig. 4, it is obvious that the steric bulk of the IPr ligand is much more significant than that of the Ph3P ligand. Since necessary steric bulk could help stabilizing the central metal atom and facilitate the reaction,28 (IPr)CuH might be more stable than (Ph3P)CuH. Furthermore, the isopropyl groups are located at both sides of the imidazole ring like the wings of the aeroplane, and this conformation could decrease the steric bulk of the isopropyl groups. Hence the steric hindrance of the isopropyl groups of IPr ligand would not affect the central Cu atom accessing to the substrate thereby reducing the catalyst reactivity.
Electronic property analysis
We wish to clarify the impact of the ligand for the hydrosilylation from more micro aspects, therefore NBO analysis was carried out for the studied system. NBO analysis shows that the natural charge on the Cu atom is 0.30 e and 0.33 e in (Ph3P)CuH and (IPr)CuH, respectively. The increase of charge on the Cu atom indicates that the interaction between Cu atom with the IPr ligand is stronger than that between Cu atom with the Ph3P ligand and the structure of the (IPr)CuH becomes more compact than that of the (Ph3P)CuH. NBO calculations show that the Wiberg bond index between Cu atom and P atom in (Ph3P)CuH is 0.284, and that between Cu atom and C1 atom in (IPr)CuH is 0.330. As shown in Fig. 4, the σ orbitals of P–C1, P–C2 and P–C3 in Ph3P simultaneously interact with the σ* orbital of CuH in (Ph3P)CuH. NBO calculations give the corresponding stabilization interaction energy to be 0.56 kcal mol−1. In (IPr)CuH, the σ orbitals of C1–N1 and C1–N2 in NHC simultaneously interact with the σ* orbital of CuH. NBO calculations give the corresponding stabilization interaction energy to be 2.46 kcal mol−1. The above results suggest that the NHC ligand has a stronger interaction with the central Cu atom, as compared to Ph3P ligand.The electrophilicity and nucleophilicity indices defined within the framework of DFT are powerful tools for correctly explaining the electronic activation.26Table 1 shows the electronic chemical potential μ, chemical hardness η, electrophilicity index ω and global nucleophilicity N of the TS2s along the (Ph3P)CuH and (IPr)CuH catalyzed reaction paths. According to the electrophilicity index ω and the global nucleophilicity index N, it is clear that from L1–TS2 to L2–TS2, the electrophilicity decreases, and the nucleophilicity increases, suggesting that the catalyst moiety is more nucleophilic. This means that (IPr)CuH might react more easily than (Ph3P)CuH with the substrate, and the interaction between the substrate and (IPr)CuH is stronger than that between the substrate and (Ph3P)CuH. Therefore the structure of L2–TS2 becomes more compact and stable than that of L1–TS2, compatible with the lowering reaction barriers.
| Species | Chemical potential μ (a.u.) | Chemical hardness η (a.u.) | Electrophilicity index ω/eV | Global nucleophilicity N/eV |
|---|---|---|---|---|
| L1–TS2 | −0.1159 | 0.1537 | 1.19 | 4.35 |
| L2–TS2 | −0.1012 | 0.1523 | 0.91 | 4.60 |
In summary, according to the structural analysis and the electronic property analysis, we can conclude that the introduction of the ligand makes the transition state structure compact and stable. The steric bulk of the ligands of the catalysts could help to stabilize the central metal atom and promote the coordination of the central metal atom with the substrate. The higher nucleophilicity of the catalysts and the stronger interaction between the catalysts and the ligands make the catalysts interact more easily with the substrate. The hydrosilylation reaction proceeds more favorably when catalyzed by (IPr)CuH as compared to (Ph3P)CuH, and this is consistent with the experimental observation.
Conclusions
The mechanism of the hydrosilylation reactions of 3-methylcyclohex-2-enone catalyzed by (Ph3P)CuH and (IPr)CuH has been investigated theoretically. The impacts of the ligands of copper hydrides for the hydrosilylation are also interpreted. The major conclusions are listed as follows:The hydrosilylation of 3-methylcyclohex-2-enone with TMDS catalyzed by copper hydrides occurs through a 1,4-addition catalytic cycle. The entire catalytic cycle may include two steps: the addition of L–CuH to the CC bond on the substrate, and the regeneration of the L–CuH catalyst assisted by TMDS. The calculations indicate that the RDS is the regeneration step of the catalyst.
The assistances of IPr and Ph3P ligands to the copper hydride catalysts make the transition state structures compact and stable. While the steric bulk of the ligands of the catalysts could help to stabilize the central Cu atom and promote the coordination of the central Cu atom with the substrate. The higher nucleophilicity of the catalysts and the stronger interaction between the catalysts and the ligands make the catalysts interact more easily with the substrate. The hydrosilylation reaction proceeds more favorably when catalyzed by (IPr)CuH as compared to (Ph3P)CuH, and this is consistent with the experimental observation.
Acknowledgements
The authors are grateful for the financial support provided by NNSF (No. 21021001, 20732003 and 20772085) and PCSIRT (No. IRT0846) of China, Specialized Research Fund for the Doctoral Program of Higher Education (No. 200806101007).Notes and references
- (a) W. S. Mahoney and J. M. Stryker, J. Am. Chem. Soc., 1989, 111, 8818 CrossRef CAS; (b) J. M. Grosselin, C. Mercier, G. Allmang and F. Grass, Organometallics, 1991, 10, 2126 CrossRef CAS; (c) O. Vassylyev, A. Panarello and J. G. Khinast, Molecules, 2005, 10, 587 CrossRef CAS; (d) B. H. Lipshutz, J. Keith, P. Papa and R. Vivian, Tetrahedron Lett., 1998, 39, 4627 CrossRef CAS; (e) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349 CrossRef; (f) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (g) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS; (h) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729 CrossRef CAS.
- (a) V. M. Mastranzo, L. Quintero, C. A. de Parrodi, E. Juaristi and P. J. Walsh, Tetrahedron, 2004, 60, 1781 CrossRef CAS; (b) V. Bette, A. Mortreux, D. Savoia and J. Carpentier, Tetrahedron, 2004, 60, 2837 CrossRef CAS; (c) H. Mimoun, J. Y. de S. Laumer, L. Giannini, R. Scopelliti and C. Floriani, J. Am. Chem. Soc., 1999, 121, 6158 CrossRef CAS.
- (a) J. Yun and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 5640 CrossRef CAS; (b) S. Sirol, J. Courmarcel, N. Mostefai and O. Riant, Org. Lett., 2001, 3, 4111 CrossRef CAS; (c) J. Issenhuth, S. Dagorne and S. Bellemin-Laponnaz, C. R. Chim., 2010, 13, 353 CrossRef CAS.
- O. Pàmies, C. Claver and M. Diéguez, J. Mol. Catal. A: Chem., 2006, 249, 207 CrossRef.
- (a) B. H. Lipshutz, K. Noson, W. Chrisman and A. Lower, J. Am. Chem. Soc., 2003, 125, 8779 CrossRef CAS; (b) B. H. Lipshutz, A. Lower, R. J. Kucejko and K. Noson, Org. Lett., 2006, 8, 2969 CrossRef CAS; (c) C. Lee and B. H. Lipshutz, Org. Lett., 2008, 10, 4187 CrossRef CAS; (d) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498 CrossRef CAS; (e) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS; (f) J. Christoffers, G. Koripelly, A. Rosiak and M. Röessle, Synthesis, 2007, 1279 CrossRef CAS.
- B. H. Lipshutz, W. Chrisman and K. Noson, J. Organomet. Chem., 2001, 624, 367 CrossRef CAS.
- (a) H. Ito, T. Ishizuka, K. Arimoto, K. Miura and A. Hosomi, Tetrahedron Lett., 1997, 38, 8887 CrossRef CAS; (b) S. Díez-González, N. M. Scott and S. P. Nolan, Organometallics, 2006, 25, 2355 CrossRef; (c) D. Lee and J. Yun, Tetrahedron Lett., 2004, 45, 5415 CrossRef CAS.
- B. H. Lipshutz and J. M. Servesko, Angew. Chem., Int. Ed., 2003, 42, 4789 CrossRef CAS.
- (a) V. Jurkauskas, J. P. Sadighi and S. L. Buchwald, Org. Lett., 2003, 5, 2417 CrossRef CAS; (b) S. Díez-González, E. D. Stevens, N. M. Scott, J. L. Petersen and S. P. Nolan, Chem.–Eur. J., 2008, 14, 158 CrossRef; (c) H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157 CrossRef CAS; (d) S. Díez-González and S. P. Nolan, Synlett, 2007, 2158 CrossRef; (e) R. Moser, Ž. V. Bošković, C. S. Crowe and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 7852 CrossRef CAS.
- D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749 CrossRef CAS.
- J. Chen, J. F. Daeuble and J. M. Stryker, Tetrahedron, 2000, 56, 2789 CrossRef CAS.
- B. H. Lipshutz, W. Chrisman, K. Noson, P. Papa, J. A. Sclafani, R. W. Vivian and J. M. Keith, Tetrahedron, 2000, 56, 2779 CrossRef CAS.
- (a) C. Song, C. Ma, Y. Ma, W. Feng, S. Ma, Q. Chai and M. B. Andrus, Tetrahedron Lett., 2005, 46, 3241 CrossRef CAS; (b) A. S. Veige, Polyhedron, 2008, 27, 3177 CrossRef CAS; (c) A. Bittermann, P. Härter, E. Herdtweck, S. D. Homann and W. A. Herrmann, J. Organomet. Chem., 2008, 693, 2079 CrossRef CAS.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (b) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS.
- J. Yun, D. Kim and H. Yun, Chem. Commun., 2005, 5181 RSC.
- T. Gathy, D. Peeters and T. Leyssens, J. Organomet. Chem., 2009, 694, 3943 CrossRef CAS.
- (a) L. W. Chung, H. G. Lee, Z. Lin and Y. Wu, J. Org. Chem., 2006, 71, 6000 CrossRef CAS; (b) T. Tuttle, D. Wang, W. Thiel, J. Köhler, M. Hofmann and J. Weis, J. Organomet. Chem., 2007, 692, 2282 CrossRef CAS; (c) Y. Yang, M. N. Weaver and K. M. Merz, Jr., J. Phys. Chem. A, 2009, 113, 9843 CrossRef CAS; (d) M. N. Glukhovtsev, R. D. Bach and C. J. Nagel, J. Phys. Chem. A, 1997, 101, 316 CrossRef CAS.
- (a) T. Tüğsüz and F. Sevin, THEOCHEM, 2006, 775, 29 CrossRef; (b) L. Rulíšek and J. Šponer, J. Phys. Chem. B, 2003, 107, 1913 CrossRef; (c) M. Mitani, M. Inoue and Y. Yoshioka, Chem. Phys. Lett., 2007, 440, 296 CrossRef CAS; (d) G. Wu, Y. Li, H. Xiang, Y. Xu, Y. Sun and H. Jiao, THEOCHEM, 2003, 637, 101 CrossRef CAS; (e) L. A. Goj, E. D. Blue, S. A. Delp, T. B. Gunnoe, T. R. Cundari, A. W. Pierpont, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2006, 45, 9032 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Jr. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, B05, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS.
- (a) J. P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS ; the concept of electrophilicity index ω has been employed to gain a further interpretation of the entire reaction process.
- (a) C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS; (b) A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem.–Eur. J., 2010, 16, 14348 CrossRef CAS; (c) L. Cavallo and M. Solá, J. Am. Chem. Soc., 2001, 123, 12294 CrossRef CAS; (d) R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485 CrossRef CAS; (e) H. Y. Liu, L. L. Koh, K. Eriks, W. P. Giering and A. Prock, Acta Crystallogr., Sect. C, 1990, 46, 51 CrossRef; (f) S. J. Dossett, A. Gillon, A. G. Orpen, J. S. Fleming, P. G. Pringle, D. F. Wass and M. D. Jones, Chem. Commun., 2001, 699 RSC; (g) E. Daura-Oller, J. M. Poblet and C. Bo, Dalton Trans., 2003, 92 RSC; (h) D. Benitez, E. Tkatchouk, A. Z. Gonzalez, W. A. Goddard III and F. D. Toste, Org. Lett., 2009, 11, 4798 CrossRef CAS; (i) J. Mathew and C. H. Suresh, Organometallics, 2011, 30, 3106 CrossRef CAS.
- B. H. Lipshutz, K. Noson and W. Chrisman, J. Am. Chem. Soc., 2001, 123, 12917 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, geometries and energies available. See DOI: 10.1039/c1cy00320h |
| This journal is © The Royal Society of Chemistry 2012 |