Highly efficient direct large-scale stoichiometric aldol reactions catalyzed by a new diamine salt in the presence of water†
Jing
Huang
,
Guodong
Chen
,
Xiangkai
Fu
*,
Chao
Li
,
Chuanlong
Wu
and
Qiang
Miao
College of Chemistry and Chemical Engineering Southwest University, Research Institute of Applied Chemistry Southwest University, The Key Laboratory of Applied Chemistry of Chongqing Municipality, The Key Laboratory of Eco-environments in Three Gorges Reservoir Region Ministry of Education, Chongqing 400715, China. E-mail: hj41012@163.com; fxk@swu.edu.cn; Fax: +86 2368254000; Tel: +86 2368253704
First published on 5th December 2011
Abstract
A new type of primary–tertiary diamine salt prepared can be used as a catalyst for direct aldol reaction and can display high yields, diastereoselectivities (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and enantiomeric excesses (up to 98%), by using stoichiometric amounts of cyclic ketones and various aryl aldehydes in the presence of water. This new catalyst can also be efficiently applied in large-scale reactions with the enantioselectivity being maintained at the same level, which indicates the potential for applications in industry.
:1), and enantiomeric excesses (up to 98%), by using stoichiometric amounts of cyclic ketones and various aryl aldehydes in the presence of water. This new catalyst can also be efficiently applied in large-scale reactions with the enantioselectivity being maintained at the same level, which indicates the potential for applications in industry.
1. Introduction
Asymmetric synthesis is one of the most potent and useful methods for introduction of chirality into target molecules.1 For decades, the field of asymmetric catalysis, previously dominated by biocatalysis and metal catalysis, has been complemented by organocatalysis using small organic molecules as a third class of powerful asymmetric catalysts. The advent of organocatalysis brought the prospect of a complementary mode of catalysis, with the potentiality for savings in cost, time and energy, an easier experimental procedure, and reductions in chemical waste.2 At the same time, reactions using water as the solvent have attracted a great deal of attention, because water is an environmentally friendly, safe medium, which avoids the problems of pollution that are inherent with organic solvents.3 Thus, the development of small organic molecules that catalyze enantioselective reactions in water is currently a highly sought-after goal in chemistry.The catalytic asymmetric aldol reaction is one of the most powerful carbon–carbon bond forming reactions in modern organic synthesis.4 Since the pioneering findings by List, Barbas, and their co-workers that L-proline could work as an active organocatalyst in intermolecular direct aldol reactions, numerous organocatalysts have been developed for the direct asymmetric aldol reactions.5 In addition, there are several examples of the asymmetric aldol reaction using water as solvent, promoted by proline-related systems,3e,5k,l,6 tryptophan,7 serine derivatives,8 threonine derivatives,9 small peptides,10 pyrrolidine-based catalysts,11 or primary–tertiary diamine catalysts.12 However, it should be noted that a large amount of ketones are usually used in these aldol reactions, which hampers their practical application. In particular, the use of less volatile ketones in large excess, for example, cyclohexanone (bp 155.65 °C), complicated the reaction workup and product purification. Meanwhile, a number of catalysts suffered from the following drawbacks: some of them worked in mixed aqueous organic solvent6b,10 or required the use of surfactants,3e,10,11 and others were supported6a or dendritic systems6c whose preparation needed chemical manipulation. Therefore, it is interesting to investigate further organocatalytic aldol reactions in the presence of water.
Based on asymmetric organocatalysis of primary–tertiary diamine catalysts,10,13 it could be deduced that among the reported diamine catalysts, most were prepared with complex procedures and/or required expensive reagents,14 which imposed hurdles on the industrial application, and these organocatalysts were often only used in research laboratories. Meanwhile, Xiao and co-workers14 reported impressive results from a series of bifunctional prolinamide derivative organocatalysts for the direct asymmetric aldol reaction of various aromatic aldehydes and cyclohexanone and they also found that a subtle change in catalyst structure may affect the catalytic activation. Obviously, the development of new and effective primary–tertiary diamine catalysts, easily prepared in a few steps from inexpensive, commercially available, enantiopure materials, is urgently needed. Herein, we wish to report that a new primary–tertiary diamine could be easily obtained and applied as an efficient catalyst for direct aldol reaction, by using stoichiometric amounts of cyclic ketones and various aryl aldehydes in the presence of water from green chemistry and atom-economical perspective and this catalyst can be used in large-scale reactions with the enantioselectivity being maintained at the same level, which offers a great possibility for applications in industry.
2. Experiment
2.1 Materials and instruments
All chemicals were used as received unless otherwise noted. Reagent grade solvents were distilled prior to use. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates visualized with UV light and/or by staining with ethanolic phosphomolybdic acid (PMA) and/or ninhydrin both in ethanol stain. THF was freshly distilled from the sodium-benzophenone ketyl radical under an argon atmosphere immediately prior to use. Flash column chromatography was performed on silica gel (200–300 mesh). NMR spectra were recorded on a 300 MHz instrument. Chemical shifts (δ) are given in ppm relative to TMS as the internal reference, coupling constants (J) in Hz. IR spectra were recorded on a spectrometer. Melting points were measured on a digital melting-point apparatus. Mass spectra (MS) were measured with a spectrometer. Analytical high performance liquid chromatography (HPLC) was carried out on Agilent 1200 instrument using Chiralpak AD (4.6 mm × 250 mm), Chiralcel OD-H (4.6 mm × 250 mm) columns. Optical rotations were measured on a JASCO P-1010 Polarimeter at λ = 589 nm.2.2 The synthesis of the catalysts (Scheme 1)
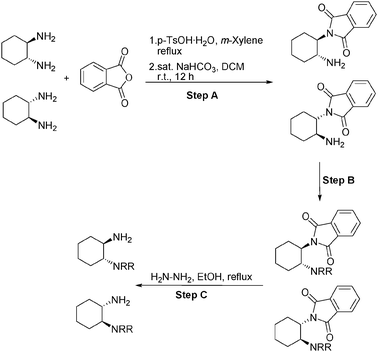 | ||
| Scheme 1 The synthetic route of the catalysts. | ||
A sample of (1R,2R)-N,N-dibenzyl-N′-phthaloyl-1,2-diaminocyclohexane (8 mmol) was refluxed with hydrazine hydrate (0.96 mL) in ethanol (16 mL) for 2 h. After cooling the solution was diluted with diethyl ether to precipitate phthaloyl hydrazide. The mixture was filtered and the filtrate was evaporated to dryness. The product was purified by extraction into the dilute HCl, followed by neutralization with saturated NaHCO3 solution and back extraction with dichloromethane. Product 1a was obtained as a yellow oil, yield 85%; [α]20D = −79.0 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 7.30–7.20 (m, 10H), 3.81 (d, J = 13.5 Hz, 2H), 3.37 (d, J = 13.5 Hz, 2H), 2.67 (dt, J = 3.9, 10.2 Hz; 1H), 2.12 (m, 1H), 1.98–1.94 (m, 2H), 1.79–1.58 (m, 4H), 1.27–0.84 (m, 4H); 13C NMR (75 MHz, CDCl3): δ = 140.2, 128.9, 128.3, 126.9, 64.6, 53.7, 51.1, 34.8, 25.7, 25.1, 22.5; FT-IR (KBr): [cm−1] = 3061, 3027, 2922, 2855, 1493, 1451, 748, 699. MS (ESI) m/z calcd. for (C20H26N2) 294.43, found 294.68.
A sample of (1R,2R)-N,N-dimethyl-N′-phthaloyl-1,2-diaminocyclohexane (7 mmol) was refluxed with hydrazine hydrate (0.84 mL) in ethanol (14 mL) for 2 h. After cooling the solution was diluted with diethyl ether to precipitate phthaloyl hydrazide. The mixture was filtered and the filtrate was evaporated to dryness. The products were purified by extraction into the dilute HCl, followed by neutralization with saturated NaHCO3 solution and back extraction with dichloromethane. Product 1d was obtained as a yellow oil, yield 85%; [α]20D = −38.7 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 2.59 (dt, J = 3, 9 Hz; 1H), 2.24 (s, 8H), 2.08–1.94 (m, 2H), 1.79–1.65 (m, 3H), 1.25–1.06 (m, 4H); 13C NMR (75 MHz, CDCl3): δ = 69.5, 51.3, 40.1, 34.8, 25.5, 25.0, 20.6; FT-IR (KBr): [cm−1] = 2929, 2858, 2825, 2778, 1668, 1593, 1512, 1452, 1358, 1333, 1270, 1203, 1153, 1116, 1089, 1046, 943, 901, 872, 848, 820, 774, 566, 543. MS (ESI) m/z calcd. for (C8H18N2) 142.24, found 143.63.
2.3 General procedure for aldol reaction of ketones with aldehydes
A mixture of organocatalyst 1a/TfOH (10 mol%) (prepared in diethyl ether with 1a and TfOH) and ketone (0.5 mmol) was stirred in water (0.5 mL) at room temperature. To this an aldehyde (0.5 mmol) was added after 30 min and the reaction mixture was stirred and the progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was partitioned between a saturated solution of ammonium chloride and dichloromethane. The organic layer was separated and dried over anhydrous Na2SO4. It was purified over silica gel by column chromatography to obtain the pure product. The chromatography purified aldol products were then examined by HPLC to determine the ee value.:1, ee = 98%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 80/20), 20 °C, 254 nm, 0.5 mL min−1; major enantiomer tR = 36.1 min and minor enantiomer tR = 47.2 min. 1H NMR (300 MHz, CDCl3): δ = 8.21 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.5 Hz, 2H), 4.91 (dd, J = 8.3 Hz and J = 3.0 Hz, 1H), 4.09 (d, 1H, J = 3.0 Hz), 2.40–2.64 (m, 2 H), 2.36 (td, J = 13.2 Hz and J = 5.7 Hz, 1H), 2.09–2.15 (m, 3 H), 1.78–1.85 (m, 1 H), 1.54–1.65 (m, 4H), 1.35–1.44 (m, 1H).
:6, ee = 98%. Enantiomeric excess was determined by HPLC with a Chiralpak OD-H column (hexane/2-propanol = 95/5), 20 °C, 254 nm, 0.5 mL min−1; major enantiomer tR = 47.2 min and minor enantiomer tR = 40.8 min. 1H NMR (300 MHz, CDCl3): δ = 7.84 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 7.8 Hz, 1H), 5.45 (d, J = 6.6 Hz, 1H), 3.90 (br, 1H), 2.82–2.70 (m, 1H), 2.50–2.40 (m, 1H), 2.34 (td, J = 12.3 Hz and J = 5.7 Hz, 1H), 2.15–2.06 (m, 1H), 1.90–1.55 (m, 4H).
:2, ee = 97%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 80/20), 20 °C, 254 nm, 0.5 mL min−1; major enantiomer tR = 30.6 min and minor enantiomer tR = 25.1 min. 1H NMR (300 MHz, CDCl3): δ = 8.21 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 4.90 (dd, J = 8.4 Hz and J = 3.0 Hz, 1H), 4.09 (d, J = 3.0 Hz, 1H), 2.65–2.45 (m, 2H), 2.36 (td, J = 13.2 Hz and J = 5.7 Hz, 1H), 2.17–2.06 (m, 1H), 1.87–1.78 (m, 1H), 1.67–1.51 (m, 3H), 1.45–1.31 (m, 1H).
:5, ee = 97%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 85/15), 25 °C, 254 nm, 1.0 mL min−1; major enantiomer tR = 26.3 min and minor enantiomer tR = 29.8 min. 1H NMR (300 MHz, CDCl3): δ = 8.75 (d, J = 2.4 Hz, 1H), 8.48 (dd, J = 8.4, 2.0 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.25 (d, J = 6.0 Hz, 2H), 5.06–4.90 (d, J = 8.4 Hz, 1H), 2.82–2.31 (m, 3H), 2.16–2.11 (m, 1H), 1.94–1.63 (m, 5H).
:2, ee = 97%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 21 °C, 254 nm, 0.5 mL min−1; major enantiomer tR = 27.3 min and minor enantiomer tR = 35.1 min. 1H NMR (300 MHz, CDCl3): δ = 7.74–7.55 (m, 3H), 7.40 (t, J = 7.2 Hz, 1H), 5.30 (d, J = 9.3 Hz, 1H), 4.03 (t, J = 3.0 Hz, 1H), 2.81–2.69 (m, 1H), 2.55–2.45 (m, 1H), 2.37 (td, J = 12.9 Hz and J = 4.8 Hz, 1H), 2.15–2.03 (m, 1H), 1.81–149 (m, 3H), 1.48–1.23 (m, 1H).
:2, ee = 90%. Enantiomeric excess was determined by HPLC with Chiralcel OD-H (hexane/i-PrOH = 95/5), 20 °C, 220 nm, flow rate 1.0 mL min−1, major anti enantiomer tR = 12.9 min and minor anti enantiomer tR = 10.9 min. 1H NMR (300 MHz, CDCl3): δ = 7.56 (d, J = 8.4 Hz, 1H), 7.20–7.34 (m, 3H), 5.35 (d, J = 8.0 Hz, 1H), 3.88 (s, 1H), 2.65–2.71 (m, 1H), 2.46–2.49 (m, 1H), 2.31–2.39 (m, 1H), 2.05–2.13 (m, 1H), 1.53–1.84 (m, 5H).
:6, ee = 93%. Enantiomeric excess was determined by HPLC with Chiralcel OD-H (hexane/i-PrOH = 96/4), 21 °C, 220 nm, flow rate 1.0 mL min−1, major anti enantiomer tR = 19.9 min and minor anti enantiomer tR = 15.8 min. 1H NMR (300 MHz, CDCl3): δ = 7.37 (s, 1H), 7.20–7.29 (m, 3H), 4.80 (d, J = 8.8 Hz, 1H), 3.88 (s, 1H), 2.30–2.45 (m, 3H), 1.31–2.08 (m, 6H).
:5, ee = 95%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 21 °C, 220 nm, 0.5 mL min−1; major enantiomer tR = 34.2 min and minor enantiomer tR = 40.7 min. 1H NMR (300 MHz, CDCl3): δ = 7.29 (dd, J = 20.4 Hz and J = 8.4 Hz, 4H), 4.76 (dd, J = 8.7 Hz and J = 2.7 Hz, 1H), 3.99 (d, J = 3.0 Hz, 1H), 2.61–2.44 (m, 2H), 2.35 (td, J = 12.9 Hz and J = 5.4 Hz, 1H), 2.15–2.05 (m, 1H), 1.85–1.75 (m, 1H), 1.70–1.50 (m, 3H), 1.37–1.20 (m, 1H).
:1, ee = 95%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 80/20), 20 °C, 254 nm, 0.5 mL min−1; major enantiomer tR = 36.7 min and minor enantiomer tR = 46.5 min. 1H NMR (300 MHz, CDCl3): δ = 7.65 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 4.85 (dd, J = 8.1 Hz and J = 3.0 Hz, 1H), 4.11 (d, J = 3.0 Hz, 1H), 2.65–2.44 (m, 2H), 2.37 (td, J = 12.9 Hz and J = 6.0 Hz, 1H), 2.17–2.06 (m, 1H), 1.88–1.77 (m, 1H), 1.72–1.47 (m, 3H), 1.44–1.31 (m, 1H).
:6, ee = 94%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 20 °C, 220 nm, 0.8 mL min−1; major anti enantiomer tR = 21.1 min and minor anti enantiomer tR = 25.4 min. 1H NMR (300 MHz, CDCl3): δ = 7.47 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.7 Hz, 2H), 4.75 (dd, J = 8.7 Hz and J = 2.7 Hz, 1H), 3.99 (d, J = 3.0 Hz, 1H), 2.61–2.44 (m, 2H), 2.35 (td, J = 12.9 Hz and J = 6.3 Hz, 1H), 2.15–2.04 (m, 1H), 1.85–1.75 (m, 1H), 1.70–1.50 (m, 3H), 1.37–1.20 (m, 1H).
:5, ee = 98%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 25 °C, 220 nm, 0.5 mL min−1; major enantiomer tR = 58.1 min and minor enantiomer tR = 52.5 min. 1H NMR (300 MHz, CDCl3): δ = 7.28–7.33 (m, 1H), 6.80–7.00 (m, 3H), 4.75–4.85 (d, J = 8.7 Hz, 1H), 3.85 (s, 3H), 2.30–2.75 (m, 3H), 2.00–2.15 (m, 1H), 1.55–1.90 (m, 4H), 1.20–1.40 (m, 1H).
:5, ee = 98%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 25 °C, 220 nm, 0.5 mL min−1; major enantiomer tR = 58.1 min and minor enantiomer tR = 52.5 min. 1H NMR (300 MHz, CDCl3): δ = 7.28–7.33 (m, 1H), 6.80–7.00 (m, 3H), 4.75–4.85 (d, J = 8.7 Hz, 1H), 3.85 (s, 3H), 2.30–2.75 (m, 3H), 2.00–2.15 (m, 1H), 1.55–1.90 (m, 4H), 1.20–1.40 (m, 1H).
:3, ee = 96%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 95/5), 20 °C, 254 nm, 1.0 mL min−1; major enantiomer tR = 45.0 min and minor enantiomer tR = 36.7 min. 1H NMR (300 MHz, CDCl3): δ = 7.70–7.90 (m, 4H), 7.40–7.55 (m, 2H), 4.90–5.05 (d, J = 8.7 Hz, 1H), 2.65–2.75 (m, 1H), 2.30–2.60 (m, 2H), 2.00–2.20 (m, 1H), 1.20–1.80 (m, 5H).
:8, ee = 97%. Enantiomeric excess was determined by HPLC with a Chiralpak OD-H column (hexane/2-propanol = 90/10), 20 °C, 220 nm, 0.5 mL min−1; major enantiomer tR = 29.3 min and minor enantiomer tR = 21.1 min. 1H NMR (300 MHz, CDCl3): δ = 7.50–7.24 (m, 5H), 4.80 (d, J = 9.0 Hz, 1H), 4.00 (m, 1H), 2.70–2.56 (m, 1H), 2.55–2.44 (m, 1H), 2.34 (td, J = 12.3, 5.4 Hz, 1H), 2.16–2.03 (m, 1H), 1.87–1.73 (m, 1H), 1.72–1.50 (m, 3H), 1.40–1.22 (m, 1H).
:12, ee = 88%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 95/5), 20 °C, 254 nm, 1.0 mL min−1; major enantiomer tR = 63.2 min, minor enantiomer tR = 67.2 min. 1H NMR (300 MHz, CDCl3): δ = 8.21 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 9.0 Hz, 2H), 4.85 (d, J = 9.0 Hz, 1H), 4.74 (s, 1H), 2.54–2.18 (m, 3H), 2.08–1.95 (m, 1H), 1.81–1.48 (m, 3H).
:1, ee (anti) = 94%. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (hexane/2-propanol = 90/10), 20 °C, 254 nm, 1.0 mL min−1; major enantiomer tR = 24.1 min, minor enantiomer tR = 23.2 min. 1H NMR (300 MHz, CDCl3): δ 8.18–8.23 (m, 2H), 7.47–7.52 (m, 2H), 4.92 (d, J = 8.6 Hz, 1H), 3.82 (br, 1H), 2.72–2.78 (m, 1H), 2.48–2.50 (m, 1H), 2.36–2.43 (m, 1H), 2.07–2.09 (m, 1H), 1.89–1.93 (m, 1H), 1.78–1.81 (m, 1H), 1.54–1.60 (m,1H), 1.33 (m, 1H), 1.05 (d, J = 6.9 Hz, 3H).
2.4 General procedure for large-scale aldol reactions
A mixture of organocatalyst 1a/TfOH (10 mol%) and ketone (20 mmol) was stirred in water (20 mL) at room temperature. To this an aldehyde (20 mmol) was added after 30 min and the reaction mixture was stirred and the progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was partitioned between a saturated solution of ammonium chloride and dichloromethane. The organic layer was separated and dried over anhydrous Na2SO4. It was purified over silica gel by column chromatography to obtain the pure product. The chromatography purified aldol products were then examined by HPLC to determine the ee value.3. Results and discussion
The catalytic activities for the asymmetric direct aldol reaction were investigated by performing a model reaction using 4-nitrobenzaldehyde with cyclohexanone in the presence of water at room temperature. As can been seen in Table 1, acidic additive such as TfOH was shown to be essential for catalysis. The reaction was very sluggish, affording only trace product if TfOH was absent (Table 1, entry 2 vs. 1). As depicted in Table 1 (entries 2, 3, 5 and 6) catalyst 1a exhibited high catalytic efficiency, whereas the other steric configurations (1b, 1d and 1e) showed lower diastereoselectivities and moderate enantioselectivities, which suggested that the steric property was important to the enantioselectivity. The catalyst 1c, with an electron-withdrawing group in the aromatic ring, afforded 5a with good enantioselectivity on the one hand, but moderate diastereoselectivity on the other hand (Table 1, entry 4). The poor catalytic performances observed for 2 highlighted the importance of the N,N-dialkylated diaminocyclohexane structure (Table 1, entry 7). Therefore, we chose 1a as a catalyst for the aldol reaction.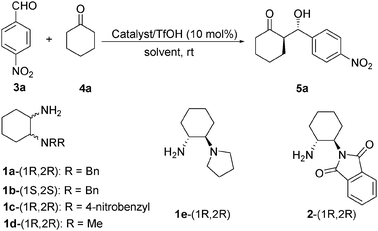
| Entry | Cat. | Solvent | Yieldb (%) | anti/sync | eed (%) |
|---|---|---|---|---|---|
| a Reaction conditions: cyclohexanone (1 mmol), aldehyde (0.5 mmol), catalyst/TfOH (0.05 mmol), solvent (0.5 mL) at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. e Without TfOH. f Cyclohexanone (0.5 mmol) was used. | |||||
| 1e | 1a | H2O | Trace | — | — |
| 2 | 1a | H2O | 83 | 99:1 |
98 |
| 3 | 1b | H2O | 79 | 95:5 |
−90 |
| 4 | 1c | H2O | 40 | 83:17 |
94 |
| 5 | 1d | H2O | 60 | 88:12 |
94 |
| 6 | 1e | H2O | 70 | 97:3 |
94 |
| 7 | 2 | H2O | 15 | 44:56 |
50 |
| 8 | 1a | DMF | 28 | 83:13 |
95 |
| 9 | 1a | CHCl3 | 60 | 93:7 |
95 |
| 10 | 1a | n-Hexane | 85 | 94:6 |
97 |
| 11f | 1a | H2O | 81 | 99:1 |
98 |
Solvent screening was then performed at room temperature to optimize the best reaction conditions (Table 1, entries 8–10). In polar organic solvents such as DMF and CHCl3, the reactions were generally sluggish affording low yields and diastereoselectivities (entries 8 and 9). While using nonpolar solvents such as n-hexane, the reactions indicated significant rate enhancement with high diastereoselectivity (94:6) and enantioselectivity (97%) (entry 10). The above results demonstrated that water was indispensable in the direct aldol reactions mediated by 1a/TfOH. The presence of water led to higher chemical yield, and improved diastereo- and enantioselectivity, compared to the reactions performed in organic solvents. To our surprise, using stoichiometric amounts of 4-nitrobenzaldehyde and cyclohexanone in the presence of water was sufficient to afford the reaction with no changes in diastereoselectivity and enantioselectivity (Table 1, entry 11). From the green chemistry perspective, 1:1 of donor (ketone) to acceptor (aldehyde) was adopted as the optimum reactant ratio.
It was well-known that a Brønsted acid additive could play an important role in the activation of the aldol acceptor by hydrogen bonding. Therefore, a series of Brønsted acids were examined as the cocatalysts in the direct asymmetric stoichiometric aldol reaction (Table 2). It became clear that optimal enantiocontrol and excellent yield could be achieved when trifluoromethane-sulfonic acid was used as the cocatalyst (entry 7). This experimental result indicated that strong acidic additive such as TfOH was essential for catalytic activity.
| Entry | Cocatalyst | Yieldb (%) | anti/sync | eed (%) |
|---|---|---|---|---|
| a Reaction conditions: cyclohexanone (0.5 mmol), aldehyde (0.5 mmol), catalyst 1a/acid (0.05 mmol), solvent (0.5 mL) at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. | ||||
| 1 | — | Trace | — | — |
| 2 | Formic acid | 57 | 71:29 |
88 |
| 3 | Acetic acid | 50 | 59:41 |
70 |
| 4 | 3-Nitrobenzoic acid | 80 | 86:14 |
97 |
| 5 | 4-Methylbenzenesulfonic | 65 | 98:2 |
94 |
| 6 | Trifluoroacetic acid | 70 | 97:3 |
97 |
| 7 | Trifluoromethanesulfonic acid | 81 | 99:1 |
98 |
Using catalyst 1a/TfOH as a mode, we investigated the effect of different amounts of water (Table 3). In the solvent-free stoichiometric aldol reaction, a comparable yield was achieved for the aldol product, accompanied with significantly decreased enantioselectivity and diastereoselectivity according to the same reaction in the presence of water (entries 1 and 2). Thus it indicated that water participated in the reaction transition states through hydrogen bonding and enhanced the stereoselectivity of the reaction. Further increasing the amount of water was detrimental to the reaction, resulting in much slower reactions, and slightly changing in diastereoselectivity and enantioselectivity (entries 2–7). When 0.5 mL water was employed, we obtained excellent yield, diastereo and enantioselectivity (entry 2). Considerations of yield and reaction time led to carry out our next study under 0.5 mL water.
| Entry | Water/mL | Yieldb (%) | anti/sync | eed (%) |
|---|---|---|---|---|
| a Reaction conditions: cyclohexanone (0.5 mmol), aldehyde (0.5 mmol), catalyst 1a/TfOH (0.05 mmol), H2O at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. | ||||
| 1 | — | 75 | 95:5 |
95 |
| 2 | 0.5 | 81 | 99:1 |
98 |
| 3 | 1.0 | 72 | 98:2 |
97 |
| 4 | 1.5 | 70 | 99:1 |
97 |
| 5 | 2.0 | 58 | 99:1 |
96 |
| 6 | 2.5 | 49 | 99:1 |
96 |
| 7 | 3.0 | 39 | 98:2 |
96 |
To examine the scope of the reaction, a series of aromatic aldehydes and cyclohexanone were studied by using of catalyst 1a/TfOH in the presence of water.18 As can be seen from the results summarized in Table 4, the reaction of cyclohexanone with aromatic aldehydes bearing various substituents afforded anti-aldol products with high diastereo and enantioselectivities.
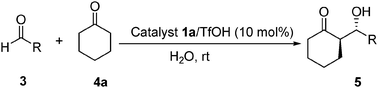
| Entry | R | Product | Time/h | Yieldb (%) | anti/sync | eed (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: cyclohexanone (0.5 mmol), aldehyde (0.5 mmol), catalyst 1a/TfOH (0.05 mmol), H2O (0.5 mL) at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. | ||||||
| 1 | 4-NO2-C6H4 | 5a | 50 | 81 | 99:1 |
98 |
| 2 | 2-NO2-C6H4 | 5b | 50 | 73 | 94:6 |
98 |
| 3 | 3-NO2-C6H4 | 5c | 55 | 85 | 98:2 |
97 |
| 4 | 2,4-(NO2)2-C6H4 | 5d | 55 | 75 | 95:5 |
97 |
| 5 | 4-CF3-C6H4 | 5e | 80 | 63 | 98:2 |
97 |
| 6 | 2-Cl-C6H4 | 5f | 90 | 70 | 98:2 |
90 |
| 7 | 3-Cl-C6H4 | 5g | 72 | 63 | 94:6 |
93 |
| 8 | 4-Cl-C6H4 | 5h | 96 | 55 | 95:5 |
95 |
| 9 | 4-CN-C6H4 | 5i | 72 | 75 | 99:1 |
95 |
| 10 | 4-Br-C6H4 | 5j | 96 | 62 | 94:6 |
94 |
| 11 | 4-F-C6H4 | 5k | 96 | 50 | 95:5 |
96 |
| 12 | 3-MeO-C6H4 | 5l | 96 | 55 | 95:5 |
98 |
| 13 | 1-Naphthyl | 5m | 96 | 42 | 97:3 |
96 |
| 14 | Ph | 5n | 90 | 65 | 92:8 |
97 |
To round out our initial study, cyclopentanone and 4-methylcyclohexanone were explored as aldol donors under the same conditions. As described in Table 5, the aldol products were obtained in high yields with moderate enantioselectivities.
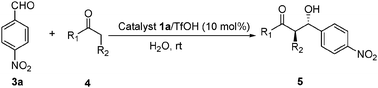
| Entry | Product | No. | Time/h | Yieldb (%) | drc | eed |
|---|---|---|---|---|---|---|
| a Reaction conditions: ketones (0.5 mmol), aldehyde (0.5 mmol), catalyst 1a/TfOH (0.05 mmol), H2O (0.5 mL) at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. | ||||||
| 1 |
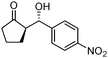
|
5o | 50 | 58 | 88:12 |
88 |
| 2 |
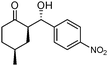
|
5p | 60 | 60 | 99:1 |
94 |
In order to show the practicality of the method, we further performed large-scale asymmetric aldol reactions with 20 mmol of aldehydes and 1 equivalent of ketones. The same catalyst loading of 10 mol% as in the experimental scale was used. The large-scale experiments can be facilely carried out according to the same procedure as for the experimental scale reactions. As can be seen from the results summarized in Table 6, delightfully, the enantioselectivity maintained at the same level for the large-scale reactions.
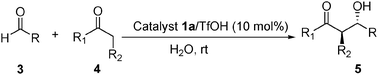
| Entry | Product | Time/h | Yieldb (%) | anti/sync | eed |
|---|---|---|---|---|---|
| a Reaction conditions: cyclohexanone (20 mmol), aldehyde (20 mmol), catalyst 1a/TfOH (2 mmol), H2O (20 mL) at room temperature. b The combined isolated yield of the diastereomers. c Determined by chiral HPLC analysis, major product is anti. d Determined by HPLC analysis of the anti product. | |||||
| 1 |
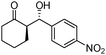
|
50 | 75 | 99:1 |
98 |
| 2 |
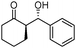
|
90 | 56 | 98:2 |
97 |
| 3 |
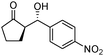
|
50 | 51 | 93:7 |
88 |
4. Conclusions
In summary, the results from the investigation demonstrate that the diamine salt 1a/TfOH is a robust and effective catalyst for direct aldol reactions, by using stoichiometric amounts of cyclic ketones and various aryl aldehydes in the presence of water. The advantages are clear from green chemistry and atom-economical perspectives. The catalyst can be easily prepared in a few steps from inexpensive, commercially available, enantiopure materials. Notably, this organocatalyzed direct asymmetric aldol reaction can be performed on a large-scale with the enantioselectivity being maintained at the same level, which offers a great possibility for applications in industry.Acknowledgements
This work was financially supported by National Ministry of Science and Technology Innovation Fund for High-tech Small and Medium Enterprise Technology (No. 09C26215112399) and National Ministry of Human Resources and Social Security Start-up Support Projects for Students Returned to Business, Office of Human Resources and Social Security Issued 2009 (143).References
- M. Movassaghi and E. N. Jacobsen, Nature, 2006, 298, 1904 Search PubMed.
- (a) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS; (b) P. L. Dalko and L. Moisan, Angew. Chem., 2004, 116, 5248 ( Angew. Chem., Int. Ed. , 2004 , 43 , 5138 ) CrossRef; (c) Y. Yang, Y.-H. He, Z. Guan and W.-D. Huang, Adv. Synth. Catal., 2010, 352, 2579 CrossRef CAS.
- (a) U. M. Lindstrom, Chem. Rev., 2002, 102, 2751 CrossRef; (b) C. J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (c) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 958 CrossRef CAS; (d) C. J. Li and L. Chen, Chem. Soc. Rev, 2006, 35, 68 RSC; (e) J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073 CrossRef CAS; (f) J. B. F. N. Engberts and M. J. Blandamer, Chem. Commun., 2001, 1701 RSC; (g) N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725 CrossRef CAS; (h) U. M. LindstrMm, Chem. Rev., 2002, 102, 2751 CrossRef; (i) S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209 CrossRef CAS; (j) D. Sinou, Adv. Synth. Catal., 2002, 344, 221 CrossRef CAS; (k) J. H. Lin, C. P. Zhang and J. C. Xiao, Green Chem., 2009, 11, 1750 RSC.
- B. List, R. A. Lerner and C. F. Barbas, III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- (a) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS; (b) B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573 CrossRef CAS; (c) A. Cordova, W. Notz and C. F. Barbas, III, Chem. Commun., 2002, 3024 CAS; (d) S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475 CrossRef CAS; (e) A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 CrossRef CAS; (f) Z. Tang, F. Jiang, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5755 CrossRef CAS; (g) U. Kazmaier, Angew. Chem., Int. Ed., 2005, 44, 2186 CrossRef CAS; (h) X. Y. Xu, Z. Tang, Y. Z. Wang, S. W. Luo, L. F. Cun and L. Z. Gong, J. Org. Chem., 2007, 72, 9905 CrossRef CAS; (i) A. Russo, G. Botta and A. Lattanzi, Tetrahedron, 2007, 63, 11886 CrossRef CAS; (j) F. Giacalone, M. Gruttadauria, P. L. Meo, S. Riela and R. Noto, Adv. Synth. Catal., 2008, 350, 2747 CrossRef CAS; (k) S. P. Zhang, X. K. Fu, S. D. Fu and J. F. Pan, Catal. Commun., 2009, 10, 401 CrossRef CAS; (l) S. P. Zhang, X. K. Fu and S. D. Fu, Tetrahedron Lett., 2009, 50, 1173 CrossRef CAS; (m) S. D. Fu, X. K. Fu, S. P. Zhang, X. C. Zou and X. J. Wu, Tetrahedron: Asymmetry, 2009, 20, 2390 CrossRef CAS; (n) C. S. Da, L. P. Che, Q. P. Guo, F. C. Wu, X. Ma and Y. N. Jia, J. Org. Chem., 2009, 74, 2541 CrossRef CAS; (o) Z. Jiang, H. Yang, X. Han, J. Luo, M. W. Wong and Y. Lu, Org. Biomol. Chem., 2010, 8, 1368 RSC.
- (a) Y. Wu, Y. Zhang, M. Yu, G. Zhao and S. Wang, Org. Lett., 2006, 8, 4417 CrossRef CAS; (b) G. Guillena, M. Hita and C. Najera, Tetrahedon: Asymmetry, 2006, 17, 1493 CrossRef CAS; (c) D. Font, C. Jimeno and M. A. Pericas, Org. Lett., 2006, 8, 4653 CrossRef CAS; (d) S. Guizzetti, M. Benag-lia, L. Raimondi and G. Celentano, Org. Lett., 2007, 9, 1247 CrossRef CAS.
- Z. Jiang, Z. Liang, X. Wu and Y. Lu, Chem. Commun., 2006, 2801 RSC.
- (a) X. Y. Wu, Z. Q. Jiang, H. M. Shen and Y. X. Lu, Adv. Synth. Catal., 2007, 349, 812 CrossRef CAS; (b) Y. C. Teo, Tetrahedron: Asymmetry, 2008, 18, 1155 CrossRef; (c) Y. C. Teo, G. L. Chua and C. Y. Poh, Tetrahedron Lett., 2009, 50, 4854 CrossRef CAS.
- C. L. Wu, X. K. Fu and S. Li, Eur. J. Org. Chem., 2011, 1291 CrossRef CAS.
- P. Dziedzic, W. Zou, J. Hafren and A. Cordova, Org. Biol. Chem., 2006, 4, 38 RSC.
- (a) S. Luo, X. Mi, S. Liu, H. Xu and J. P. Cheng, Chem. Commun., 2006, 3687 RSC; (b) N. Mase, N. Noshiro, A. Mokuya and K. Takabe, Adv. Synth. Catal., 2009, 351, 2791 CrossRef CAS.
- (a) J. H. Lin, C. P. Zhang and J. C. Xiao, Green Chem., 2009, 11, 1750 RSC; (b) F. Z. Peng, Z. H. Shao, X. W. Pu and H. B. Zhang, Adv. Synth. Catal., 2008, 350, 2199 CrossRef CAS; (c) M. Raj, G. S. Parashari and V. K. Singh, Adv. Synth. Catal., 2009, 351, 1284 CrossRef CAS.
- (a) S. Luo, H. Xu, L. Chen and J. P. Cheng, Org. Lett., 2008, 10, 1777 Search PubMed; (b) S. Luo, H. Xu, J. Li, L. Zhang and J. P. Cheng, J. Am. Chem. Soc., 2007, 129, 3074 CrossRef CAS; (c) B.-L. Zheng, Q.-Z. Liu, C.-S. Guo, X.-L. wang and L. He, Org. Biomol. Chem., 2007, 5, 2913–2915 RSC; (d) Q.-L. Liu, X.-L. Wang, S.-W. Luo, B.-L. Zheng and D.-B. Qin, Tetrahedron Lett., 2008, 49, 7434–7437 CrossRef CAS; (e) L.-W. Xu, Y.-D. Ju, L. Li, H.-Y. Qiu, J.-X. Jiang and G.-Q. Lai, Tetrahedron Lett., 2008, 49, 7037 CrossRef CAS; (f) S. Luo, H. Xu, L. Chen and J.-P. Cheng, Org. Lett., 2008, 10, 1775 CrossRef CAS; (g) J. Liu, Z. Yang, Z. Wang, F. Wang, X. Chen, X. Liu, X. Feng, Z. Su and C. Hu, J. Am. Chem. Soc., 2008, 130, 5654 CrossRef CAS; (h) J. Zhou, V. Wakchaure, P. Kraft and B. List, Angew. Chem., Int. Ed., 2008, 47, 7656 CrossRef CAS; (i) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC.
- (a) J. R. Chen, H. H. Lu, X. Y. Li, L. Cheng, J. Wan and W. J. Xiao, Org. Lett., 2005, 7, 4543 CrossRef CAS; (b) J. R. Chen, X. Y. Li, X. N. Xing and W. J. Xiao, J. Org. Chem., 2006, 71, 8198 CrossRef CAS.
- (a) M. Kaik and J. Gawroński, Tetrahedron: Asymmetry, 2003, 14, 1559 CrossRef CAS; (b) A. Berkessel, S. Mukherjee, T. N. Müllee, F. Cleemann, K. Roland, M. Brandenburg, J. M. Neudörfl and J. Lex, Org. Biomol. Chem., 2006, 4, 4319 RSC.
- (a) M. Kaik and J. Gawroński, Tetrahedron: Asymmetry, 2003, 14, 1559 CrossRef CAS; (b) A. Berkessel, S. Mukherjee, T. N. Müllee, F. Cleemann, K. Roland, M. Brandenburg, J. M. Neudörfl and J. Lex, Org. Biomol. Chem., 2006, 4, 4319 RSC.
- C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465 CrossRef CAS.
- Representative procedure for the enantioselective aldol reaction with catalyst 1a/TfOH: a mixture of organocatalyst 1a/TfOH (10 mol%) (prepared in ethylether with 1 eq. 1a and 1 eq. TfOH) and ketone (0.5 mmol) was stirred in water (0.5 mL) at room temperature. To this an aldehyde (0.5 mmol) was added after 30 min and the reaction mixture was stirred and the progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was partitioned between a saturated solution of ammonium chloride and dichloromethane. The organic layer was separated and dried over anhydrous Na2SO4. It was purified over silica gel by column chromatography to obtain the pure product. The chromatography purified aldol products were then examined by HPLC to determine the ee value.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra. See DOI: 10.1039/c1cy00318f |
| This journal is © The Royal Society of Chemistry 2012 |