Complete and partial oxidation of methane on ceria/platinum silicon carbide nanocomposites
Robert
Frind
a,
Lars
Borchardt
a,
Emanuel
Kockrick
a,
Lars
Mammitzsch
b,
Uwe
Petasch
b,
Mathias
Herrmann
b and
Stefan
Kaskel
*a
aDepartment of Inorganic Chemistry, Dresden University of Technology, Bergstrasse 66, D-01069 Dresden, Germany. E-mail: stefan.kaskel@chemie.tu-dresden.de; Fax: +49 351 46337287; Tel: +49 351 46333632
bFraunhofer Institute for Ceramic Technologies and Systems, Winterbergstrasse 28, D-01277 Dresden, Germany
First published on 5th October 2011
Abstract
We have studied the catalytic activity of CeO2/Pt–SiC composites in the total and partial oxidation as well as the dry reforming of methane. The composites were synthesized by an in situ functionalization strategy with variation in ceria and platinum contents and processing conditions. The impact of composition and pyrolysis temperature on the specific surface area and catalytic activity of the composite materials is studied. All catalysts have a high activity in the partial oxidation and dry reforming of methane close to the thermodynamic equilibrium composition. In the complete oxidation of methane, the T10% was lowered by 443 K compared to the non-catalyzed reaction.
Introduction
The scientific and technologic interest on syngas and hydrogen production has grown in the last few decades continuously. Syngas is used for the production of various industrially relevant chemicals such as methanol or liquid hydrocarbons in the Fischer–Tropsch process.1–5 Furthermore there are many other processes using hydrogen in large amounts e.g. hydrocracking and hydrotreating in refineries.6,7 The most promising route for the production of syngas is via methane from natural gas or biological fermentation, helping to reduce the emission of pollutants with origin in coal, e.g. sulfur compounds, heavy metals and fine airbrne particles.8 Generally the conversion of methane is performed via steam reforming.9,10 The reaction, shown in eqn (1), is highly endothermic and is typically carried out at temperatures higher than 900 °C.7,11| CH4 + H2O → CO + 3H2, ΔH = +206.2 kJ mol−1 | (1) |
| CH4 + 0.5O2 → CO + 2H2, ΔH = −35.7 kJ mol−1 | (2) |
In the following, we describe the synthesis of CeO2/Pt–silicon oxycarbide composites with variation in catalyst contents and their applications in the partial oxidation and dry reforming10 of methane. The synthesis developed by Kockrick et al.22,23,27 is based on an inverse microemulsion technique. The catalyst particles are formed in the water-phase and the polymeric polycarbosilane precursor is added to the oil-phase during the synthetic procedure. After the pyrolysis at different temperatures in order to form the ceramic silicon carbide, carbon residues were removed by oxidative treatment resulting in porous silicon oxycarbide composites. These composites have high specific surface areas (up to 482 m2 g−1) and show very good activity for the partial oxidation and dry reforming of methane.
Experimental
Synthesis of CeO2/Pt–silicon(oxy)carbide
All chemicals were used as received. The synthesis was already described in previous studies.22 Defined masses of Ce(NO3)3·6H2O (Aldrich, 99%) and H2PtCl6·6H2O (ABCR, 99%, 40 wt% (Pt)) listed in Table 1 were dissolved in 11.7 g of water. Then 136 g of n-heptane (Sigma-Aldrich, >99%), 18.16 g of the non-ionic surfactant Marlophen NP5 (RO(CH2CH2O)xH, x = 5, R = nonylphenyl, M = 440 g mol−1, SASOL) and the aqueous solution were mixed at 25 °C, giving a defined Rw value (molar water to surfactant ratio) of 16.4. The microemulsion systems containing cerium salt and hexachloro platinic acid were stirred for one hour in a closed 500 ml flask at 25 °C. Then, 1.2 g of diluted ammonia solution (Sigma-Aldrich, 2.5 wt%) were added drop-wise to the transparent microemulsion system and stirred for another hour at 25 °C. In the next step, 9.4 g of the liquid polycarbosilane precursor SMP-10 (Starfire Systems) was added.| Sample code | Target ωCeO2/wt% | Target ωPt/wt% | m Ce(NO3)3*6H2O/g | m H2PtCl6/g |
|---|---|---|---|---|
| A | 2.5 | 1 | 0.415 | 0.174 |
| B | 7.5 | 1 | 1.245 | 0.174 |
| C | 5.0 | 2 | 0.830 | 0.348 |
| D | 2.5 | 3 | 0.415 | 0.523 |
| E | 7.5 | 3 | 1.245 | 0.523 |
After one hour, the solid crosslinked polymer was isolated by distillation in order to remove the volatile compounds, water and n-heptane, using a rotary evaporator at temperatures up to 150 °C at 30 mbar. Pyrolysis of the CeO2/Pt–PCS composites were conducted in Ar 5.0 flow (3 l min−1) in a vertical furnace (FSW 315/400-2200-KS/SP, FCT Systeme GmbH) with maxima in temperatures of 840 °C, 1200 °C and 1500 °C according to previous studies.22,23 To remove residual carbon, pyrolysed CeO2/Pt–SiC composites were post-treated at 700 °C for 2 h with a heating ramp of 10 °C min−1 in a muffle furnace under a static air atmosphere.
Characterization
The composites were characterized by X-ray powder diffraction in transmission geometry using a Stoe Stadi-P diffractometer and Cu Kα1 radiation (λ = 0.15405 nm). Nitrogen physisorption isotherms were measured at 77 K using a Quantachrome Quadrasorb. Prior to the measurement the samples were activated under vacuum at 150 °C for 24 hours. Specific surface areas were calculated using the BET equation (p/p0 = 0.05–0.2). The total pore volume was determined at p/p0 = 0.95.The catalytic activity of the composites was investigated on a catalytic test reactor. 1 g of the post-treated sample was diluted with quartz wool to 5 cm height of the bed and transferred to a quartz reactor placed inside a furnace. The furnace was heated with a constant temperature ramp of 2 °C min−1 to 925 °C under a defined gas mixture of 100 sccm min−1 (GHSV = 1500 h−1). The flow rates of helium, oxygen and methane were regulated by a Bronkhorst El-Flow mass flow controller. Under total oxidation conditions (CH4/O2 ratio: 1/3) the gas mixture was set to 96 vol% helium, 3 vol% oxygen and 1 vol% methane. For investigations on the partial oxidation of methane (CH4/O2 ratio: 2) 94 vol% helium, 2 vol% oxygen and 4 vol% methane were used. Dry reforming (CH4/CO2 ratio: 1) was performed in 96 vol% helium, 2 vol% carbon dioxide and 2 vol% methane. The gas chromatograph was equipped with a 500 μl sample loop, two columns (A: 6′ × 1/8′′ stainless steel column molecular sieve 5A; B: 6′ × 1/8′′ stainless steel column Porapak Q), a thermal conductivity detector and a flame ionization detector (FID).
For comparison Pt@Al2O3 (Chempur, 0.5 wt% Pt) was investigated as a reference catalyst. For the estimation of thermodynamic equilibrium data the Cantera application MixMaster with the database GRI-Mech 3.0 was used.28
Investigations of the stability of the composites were performed by temperature programmed oxidation (TPO) of methane, described in previous studies.22
Results and discussion
Synthesis and characterization
The samples were investigated by nitrogen physisorption after pyrolysis and oxidative treatment. Specific surface areas and total pore volumes are summarized in Table 2. In general, composites pyrolysed at lower temperatures show higher specific surface areas. Comparing composites with different amounts of catalysts, composites with higher contents of ceria and platinum have higher specific surface areas with a maximum for the samples consisting of 2.5 wt% CeO2 and 3 wt% Pt. The differences in porosity are related to crosslinking degree of allyl-group-containing polycarbosilane intermediates by a Pt-induced catalytic hydrosilylation reaction. In agreement with previous studies the specific surface area increases with higher Pt contents.22 Nitrogen physisorption isotherms (Fig. 1) for lower catalyst contents show a characteristic type I shape. For higher loadings, higher mesopore volumes can be estimated by the appearance of a hysteresis in the isotherm.| Sample code | Specific surface areaa/m2 g−1 | Specific pore volumeb/cm3 g−1 | ||||
|---|---|---|---|---|---|---|
| 840 | 1200 | 1500 | 840 | 1200 | 1500 | |
| a Specific surface area estimated by the single point BET method at p/p0 = 0.3. b Specific pore volume calculated at a relative pressure of 0.975. | ||||||
| A | 212 | 29 | 33 | 0.112 | 0.023 | 0.086 |
| B | 298 | 112 | 33 | 0.157 | 0.061 | 0.063 |
| C | 373 | 345 | 51 | 0.230 | 0.248 | 0.257 |
| D | 482 | 405 | 87 | 0.384 | 0.334 | 0.353 |
| E | 428 | 370 | 68 | 0.340 | 0.350 | 0.313 |
 | ||
| Fig. 1 N2 physisorption isotherms of CeO2/Pt–SiC composites pyrolysed at (a) 840 °C, (b) 1200 °C and (c) 1500 °C. | ||
In composites pyrolysed at 1500 °C the crystalline β-SiC phase can be identified by X-ray powder diffraction patterns (Fig. 2). The catalyst content (especially the Pt-content) seems to affect the crystallinity of the resulting composites. With increasing platinum mass fraction the amorphous background 2θ = 20–30° rises suggesting a lower crystallinity. This could be explained with faster crosslinking of the polycarbosilane during the synthesis and the occurrence of defects in the structure preventing good ordering. Due to the low mass fraction of ceria and platinum, no reflections for the catalysts could be observed.
 | ||
| Fig. 2 X-Ray powder diffraction patterns: (a) samples B-1500 (violet), C-1500 (orange) and E-1500 (green); (b) sample A pyrolysed at 840 °C (blue), 1200 °C (black) and 1500 °C (red). | ||
The presence of platinum and cerium was proven by EDX spectroscopy. The results of this semiquantitative elemental analysis of samples A-840, A-1200 and A-1500 are presented in Table 3. The theoretical values were calculated assuming 2.5 wt% CeO2, 1 wt% platinum and 96.5 wt% silicon carbide. The observed contents of cerium and platinum are matching with the expected values. Although accuracy for light weight elements in EDX spectroscopy is limited, a decreasing content of oxygen can be detected with rising pyrolysis temperature. During the oxidative treatment of the pyrolysed composites partial oxidation of the ceramic takes place. This partial oxidation process is less pronounced if higher temperatures are used in the pyrolysis.22
| Element | Wt%a | |||
|---|---|---|---|---|
| Theoreticalb | A-840 | A-1200 | A-1500 | |
| a Elemental compositions were estimated as a mean value of five single EDX measurements at the same magnification. b Theoretical compositions were calculated assuming that the composite only consists of CeO2, Pt and SiC (ceramic yield SiC: 73% SMP-10). | ||||
| Si | 67.59 | 37.98 | 26.18 | 49.55 |
| C | 28.91 | 21.84 | 28.40 | 42.31 |
| O | 0.46 | 38.08 | 22.84 | 5.33 |
| Ce | 2.04 | 1.70 | 2.04 | 2.14 |
| Pt | 1.00 | 0.40 | 0.54 | 0.67 |
CO-chemisorption was performed on samples E-840, E-1200 and E-1500. The platinum surface area is negligible and only 0.0089 m2 g−1 for E-840. For the other samples no chemisorption of CO was observed, indicating the formation of Pt-compounds such as silicides instead of Pt metal.
Catalysis
The results of the combustion measurements (CH4/O2 ratio: 1/3) are given in Table 4. All composites show good catalytic activity for the oxidation of methane. The temperature of 10% conversion could be decreased by 443 K (E-840) in comparison to the noncatalysed reaction (T10% = 897 °C). Composites synthesized at 840 °C show the highest catalytic activity, with T10% of 536 °C and below. The analysis of the exhaust gas only shows the formation of carbon dioxide and water.| Sample code | T 10%/°C | ||
|---|---|---|---|
| 840 | 1200 | 1500 | |
| A | 536 | 664 | 653 |
| B | 515 | 632 | 589 |
| C | 520 | 610 | 669 |
| D | 486 | 523 | 533 |
| E | 454 | 555 | 532 |
The stability of the investigated composites was proved by temperature programmed oxidation (TPO) of methane. The procedure is described in previous studies.22 Even after 7 cycles of heating and cooling the onset temperature was stable.
Partial oxidation of methane was performed with a CH4/O2 ratio of 2/1 (eqn (2)). At a reactor temperature of 400 °C only composites pyrolysed at 840 °C are showing little conversion of methane to carbon dioxide and water. In contrast higher reactor temperatures promote the methane conversion. The yields of carbon dioxide and carbon monoxide (at reactor temperatures of 685 °C, 805 °C and 925 °C) are shown exemplarily for the composites A, C and E in Fig. 3–5. All investigations were also performed with a commercial Pt@Al2O3 catalyst (Chempur, 0.5 wt% Pt) as reference material. From Fig. 3(a) it can be seen that composites with the lowest content of catalyst are showing only little conversion of 50% at 685 °C for A-840 and less than 20% for A-1200 and A-1500, while the reference catalyst provides a conversion of 73%. Higher metal contents (B/C/D/E-840, D/E-1200) give better methane conversions above 75% of the equilibrium conversion. Samples pyrolysed at 1500 °C exhibit a conversion between 20–24%, independent of the catalyst content. Analyzing the composition of the exhaust gas, the yield of carbon dioxide is higher than the value determined for the thermodynamic equilibrium. At the same time the yield of the desired product carbon monoxide is significantly lower than the expected equilibrium. For all samples pyrolysed at 1500 °C the selectivity for carbon dioxide (not shown) is 1, whereas samples pyrolysed at 1200 °C or 840 °C show a selectivity for carbon dioxide, which decreases with increasing catalyst content, while the selectivity for carbon monoxide is rising. Obviously the carbon dioxide induced reforming of methane is lower than one can expect from the thermodynamic equilibrium. In the case of the samples A/C/E-1500 no carbon monoxide is detected. The increase of the reaction temperature to 805 °C (Fig. 4) results in higher conversions of methane for all composites. Especially the samples C/D/E-840 and D/E-1200 (85–93%) are nearly reaching the conversion given by the equilibrium (94%). The conversion of methane achieved with Pt@Al2O3 is 84%. The same tendency is obtained for the yield of carbon dioxide and carbon monoxide. There is only a little amount of carbon dioxide converted in the reforming processes and the yield of carbon monoxide is nearly 80% of the thermodynamic equilibrium. For all samples pyrolysed at 1500 °C and for A-1200 the selectivity for carbon dioxide is higher than 0.8. The other samples show selectivities for carbon monoxide over 0.75. At 925 °C (Fig. 5) the samples B/C/D/E-840 show conversions of methane of 97% and D/E-1200 96% (conversion in thermodynamic equilibrium: 98%, Pt@Al2O3: 97%). The materials do not promote the carbon dioxide production and the selectivity towards carbon monoxide is higher than 0.9. For samples B/C/D/E-1500 the selectivity for carbon dioxide is below 0.3 while the selectivity for carbon monoxide is over 0.6.
 | ||
| Fig. 3 Partial oxidation of methane (CH4/O2 = 2): (a) conversion of methane, (b) yield of CO2 and (c) yield of CO for composites A, C and E at a reaction temperature of 685 °C. | ||
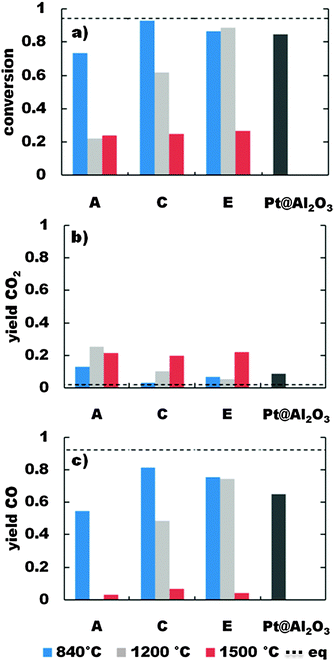 | ||
| Fig. 4 Partial oxidation of methane (CH4/O2 = 2): (a) conversion of methane, (b) yield of CO2 and (c) yield of CO for composites A, C and E at a reaction temperature of 805 °C. | ||
 | ||
| Fig. 5 Partial oxidation of methane (CH4/O2 = 2): (a) conversion of methane, (b) yield of CO2 and (c) yield of CO for composites A, C and E at a reaction temperature of 925 °C. | ||
From Fig. 3–5 it can be seen that composites pyrolysed at lower temperatures having a better performance in methane conversion, and higher yields and selectivity towards the desired product carbon monoxide, even higher than with the reference catalyst.
These composites also show the highest specific surface areas, so also the highest catalyst surface areas can be assumed in comparison to samples pyrolysed at 1200 °C or 1500 °C. Kockrick et al.23 suggested the formation of cerium silicate at higher pyrolysis temperatures, also reducing the active catalyst content. According to Tang et al.17 the formation of surface oxygen vacancies requires lower energy than forming bulk vacancies. The oxygen vacancies are needed for the oxidation of methane at the catalyst surface. Several studies showed a dependency between the energy of the formation of vacancies and the temperature of calcination: in high temperature calcined ceria oxygen is bound more strongly.17,29 On the other hand the doping of ceria with noble metals, such as Pt or Pd, leads to an increased mobility of oxygen vacancies in the lattice. This can also be observed in our studies concerning the partial methane oxidation. Samples D and E (3 wt% Pt) are showing nearly the same values for methane conversion and selectivity towards carbon monoxide, for pyrolysis temperature below 1500 °C approximately the equilibrium values. In contrast, a platinum content of 2 wt% leads to values near the equilibrium only for pyrolysis temperature of 840 °C. At 1 wt% platinum (samples A and B) higher CeO2 contents are required to reach the thermodynamic equilibrium composition for the most active composites. Craciun et al.30 showed a decreasing catalytic activity for steam reforming of methane when ceria-supported Pd was calcined at higher temperatures. Calcination at 1400 °C lowered the activity by a factor of 1000. This effect should be detectable by comparison of the H2/CO ratio in the product gas stream in the partial methane oxidation. According to the stoichiometry of the steam reforming or dry reforming of methane the H2/CO ratio should be 3 or 1, respectively. The overall stoichiometry of partial oxidation of methane consisting of total oxidation to carbon dioxide and water and following steam and dry reforming gives a H2/CO ratio of 2. If there is less activity for one of the reforming processes the H2/CO ratio for reduced activity of steam reforming or dry reforming should be lower or higher than 2, respectively.30
The H2/CO ratio of sample C pyrolysed at different temperatures is shown in Fig. 6. One can see that the ratio differs significantly at 685 °C whereas similar values result at 925 °C corresponding to the equilibrium value of 2 for this temperature. At 685 °C sample C-840 shows a H2/CO ratio of 3.1 which is not in agreement with the stoichiometry of steam reforming. Tang et al.17 explained higher H2/CO ratios with the occurrence of a water–gas shift reaction lowering the carbon monoxide concentration on the one hand and extending the hydrogen concentration on the other hand. For sample C-1200 the H2/CO ratio is approximately 2 for all reactor temperatures, so one can assume no preferentially dominating reforming processes. Deactivation of the catalysts by coking was not observed for all investigated composites.
 | ||
| Fig. 6 H2/CO ratio for the partial oxidation of methane by sample C at different pyrolysis conditions. | ||
The latter results for the partial oxidation of methane also offer the potential application of the introduced CeO2/Pt–SiC composites as advanced catalyst systems in the dry methane reforming. Hereby the oxygen in the feed of the reactor is substituted by carbon dioxide. The results for the three different reactor temperatures are shown in Fig. 7–9. In agreement with the previous results at the lowest reactor temperature of 685 °C composites C/D/E-840 show the best methane conversions and the highest CO yields. In contrast to the results of the partial oxidation, the samples A/B-840 and C/D/E-1200 show conversions less than 10% and only low CO yields. While the lower yield in the dry reforming implies that at this partial oxidation at 685 °C carbon monoxide is already formed, temperature steam reforming is the dominating reforming process for the production of syngas. At 805 °C and 925 °C the trends from the discussion of the partial oxidation above can be applied to the results of dry reforming. Samples C/D/E-840 and D/E-1200 are showing the best values for conversion of methane and yield of carbon monoxide near the thermodynamic equilibrium and the reference catalyst. At 925 °C also D/E-1500 shows a good conversion (over 70%) and yield, which was not expected from the previous results. In this case the total oxidation of methane obviously is the critical step. Since the dry reforming is an endothermic process the application of higher reactor temperatures results in higher conversions. On the other hand the total oxidation is an exothermic process, so higher reaction temperatures will lead to lower conversions of methane.
 | ||
| Fig. 7 (a) Conversion of methane and (b) yield of CO in dry reforming of methane for composites A, C and E at a reaction temperature of 685 °C. | ||
 | ||
| Fig. 8 (a) Conversion of methane and (b) yield of CO in dry reforming of methane for composites A, C and E at a reaction temperature of 805 °C. | ||
 | ||
| Fig. 9 (a) Conversion of methane and (b) yield of CO in dry reforming of methane for composites A, C and E at a reaction temperature of 925 °C. | ||
The H2/CO ratio for sample C pyrolysed at different temperatures is shown in Fig. 10. The values for C-840 and C-1200 are close to the thermodynamic equilibrium. Only sample C-1500 shows at 805 °C a H2/CO ratio of 0.5 which can be attributed to reverse water–gas shift reaction (CO2 + H2 → CO + H2O) producing additional carbon monoxide.17 Nakamura et al.31 proposed a mechanism based on reverse water–gas shift and steam reforming for the reaction of carbon dioxide and methane. The process of steam reforming seems badly catalyzed by samples pyrolysed at 1500 °C giving H2/CO ratios lower than 1.
 | ||
| Fig. 10 H2/CO ratio for the dry reforming of methane with composition C at different pyrolysis conditions. | ||
Conclusions
We have presented the synthesis of CeO2/Pt–SiC-composites via the microemulsion method developed by Kockrick et al.22,23 and their application in the total and partial oxidation, as well as dry reforming of methane. The composites were synthesized with contents of ceria varying between 2.5–7.5 wt% and platinum varying between 1–3 wt% and were pyrolysed at 840 °C, 1200 °C and 1500 °C. The porosity was tuned by the catalyst content and the temperature of pyrolysis. Samples with high metal contents and pyrolysed at 840 °C showed the highest specific surface areas and turned out to be the most active composites in total oxidation of methane, lowering the T10% by 443 K. The partial oxidation and dry reforming of methane were catalyzed nearly to the thermodynamic equilibrium composition already at 805 °C reactor temperature by these composites and the reference catalyst Pt@Al2O3. The activity in the partial oxidation of methane depends particularly on the platinum content; only at low Pt contents an influence of the ceria contents is observed. Due to the low pyrolysis temperature these samples were not stable against oxidation and exhibited high oxygen contents in EDX measurements. This disadvantage was minimized by increasing the pyrolysis temperature to 1500 °C which decreased the catalytic activity dramatically. The best compromise between stability against oxidation and catalytic activity was achieved using composites with high catalyst content pyrolysed at 1200 °C. These samples also reach nearly the equilibrium composition in the partial oxidation and dry reforming of methane at 805 °C.Acknowledgements
Financial support by the German Research Foundation in the NanoMat Program SPP 1181 is gratefully acknowledged.References
- Y. Liu, T. Hayakawa, T. Ishii, M. Kumagai, H. Yasuda, K. Suzuki, S. Hamakawa and K. Murata, Appl. Catal., A, 2001, 210, 301–314 CrossRef CAS.
- S. Murcia-Mascarós, R. M. Navarro, L. Gómez-Sainero, U. Costantino, M. Nocchetti and J. L. G. Fierro, J. Catal., 2001, 198, 338–347 CrossRef CAS.
- J. R. H. Ross, A. N. J. van Keulen, M. E. S. Hegarty and K. Seshan, Catal. Today, 1996, 30, 193–199 CrossRef CAS.
- P. J. A. Tijm, F. J. Waller and D. M. Brown, Appl. Catal., A, 2001, 221, 275–282 CrossRef CAS.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev. Sci. Eng., 1999, 41, 255–318 CrossRef.
- P. Grange, Catal. Rev. Sci. Eng., 1980, 21, 135–181 CAS.
- M. A. Pena, J. P. Gómez and J. L. G. Fierro, Appl. Catal., A, 1996, 144, 7–57 CrossRef CAS.
- B. C. Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef CAS.
- J. P. Van Hook, Catal. Rev. Sci. Eng., 1980, 21, 1–51 CrossRef CAS.
- K. Kochloefl, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, Germany, 1997, vol. 4, pp. 1819–1831 Search PubMed.
- K. Otsuka, Y. Wang, E. Sunada and I. Yamanaka, J. Catal., 1998, 175, 152–160 CrossRef CAS.
- A. P. E. York, T.-c. Xiao, M. L. H. Green and J. B. Claridge, Catal. Rev. Sci. Eng., 2007, 49, 511–560 CAS.
- A. P. E. York, T. Xiao and M. L. H. Green, Top. Catal., 2003, 22, 345–358 CrossRef CAS.
- R. Ramírez-López, I. Elizalde-Martinez and L. Balderas-Tapia, Catal. Today, 150, 358–362 Search PubMed.
- L. Pino, V. Recupero, S. Beninati, A. K. Shukla, M. S. Hegde and P. Bera, Appl. Catal., A, 2002, 225, 63–75 CrossRef CAS.
- L. Pino, A. Vita, M. Cordaro, V. Recupero and M. S. Hegde, Appl. Catal., A, 2003, 243, 135–146 CrossRef CAS.
- W. Tang, Z. Hu, M. Wang, G. D. Stucky, H. Metiu and E. W. McFarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- P. Colombo and M. Modesti, J. Am. Ceram. Soc., 1999, 82, 573–578 CAS.
- P. Krawiec and S. Kaskel, J. Solid State Chem., 2006, 179, 2281–2289 CrossRef CAS.
- Y. F. Shi, Y. Wan, Y. P. Zhai, R. L. Liu, Y. Meng, B. Tu and D. Y. Zhao, Chem. Mater., 2007, 19, 1761–1771 CrossRef CAS.
- S. Kaskel, G. Chaplais and K. Schlichte, Chem. Mater., 2005, 17, 181–185 CrossRef CAS.
- E. Kockrick, R. Frind, M. Rose, U. Petasch, W. Bohlmann, D. Geiger, M. Herrmann and S. Kaskel, J. Mater. Chem., 2009, 19, 1543–1553 RSC.
- E. Kockrick, P. Krawiec, U. Petasch, H. P. Martin, M. Herrmann and S. Kaskel, Chem. Mater., 2008, 20, 77–83 CrossRef CAS.
- P. Krawiec, D. Geiger and S. Kaskel, Chem. Commun., 2006, 2469 RSC.
- P. Krawiec, C. Schrage, E. Kockrick and S. Kaskel, Chem. Mater., 2008, 20, 5421–5433 CrossRef CAS.
- P. Krawiec, C. Weidenthaler and S. Kaskel, Chem. Mater., 2004, 16, 2869–2880 CrossRef CAS.
- E. Kockrick, C. Schrage, A. Grigas, D. Geiger and S. Kaskel, J. Solid State Chem., 2008, 181, 1614–1620 CrossRef CAS.
- J. M. Bergthorson, D. G. Goodwin and P. E. Dimotakis, Proc. Combust. Inst., 2005, 30, 1637–1644 CrossRef.
- G. Zhou, P. R. Shah, T. Montini, P. Fornasiero and R. J. Gorte, Surf. Sci., 2007, 601, 2512–2519 CrossRef CAS.
- R. Craciun, B. Shereck and R. J. Gorte, Catal. Lett., 1998, 51, 149–153 CrossRef CAS.
- J. Nakamura, K. Aikawa, K. Sato and T. Uchijima, Catal. Lett., 1994, 25, 265–270 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |