Exploring the versatility of a bis(phosphinimine) pincer ligand: effect of sterics on structure and lactide polymerization activity of cationic zinc complexes†
Craig A.
Wheaton
and
Paul G.
Hayes
*
Department of Chemistry and Biochemistry, University of Lethbridge, E866 University Hall, 4401 University Drive, Lethbridge, Alberta, Canada. E-mail: p.hayes@uleth.ca; Fax: (+1) 403-329-2057
First published on 10th October 2011
Abstract
Cationic zinc complexes of a neutral pincer framework 4,6-(ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh2)-dibenzofuran (L111: Ar = 2-iPrPh; L22: Ar = o-tolyl; L33: Ar = Ph), have been prepared and characterized. Crystallographic and NMR studies of the methylzinc complexes [LZnCH3+][BAr4−] (4a–6a: Ar = m-(CF3)2–C6H3; 4b–6b: Ar = Ph) demonstrated that the steric demands of the ligand dramatically affect the solid-state geometry. The cationic zinc–lactate complexes [LZnOR+][B(m-(CF3)2–C6H3)4−] (7: L = L22; 8: L = L33; R = CH(Me)CO2Me) were also studied, and their efficacy as lactide polymerization catalysts was examined. Polymerization using 7 requires heating to 60 °C, while complex 8 displays high activity at ambient temperature. The difference in activity can be attributed to κ2versus κ3 binding modes of the ligand, providing important insight into structure–activity relationships for this system. Complex 8 gives modestly heteroenriched PLA (Pr = 0.70), which represents the best stereocontrol yet achieved by a cationic metal catalyst.
PPh2)-dibenzofuran (L111: Ar = 2-iPrPh; L22: Ar = o-tolyl; L33: Ar = Ph), have been prepared and characterized. Crystallographic and NMR studies of the methylzinc complexes [LZnCH3+][BAr4−] (4a–6a: Ar = m-(CF3)2–C6H3; 4b–6b: Ar = Ph) demonstrated that the steric demands of the ligand dramatically affect the solid-state geometry. The cationic zinc–lactate complexes [LZnOR+][B(m-(CF3)2–C6H3)4−] (7: L = L22; 8: L = L33; R = CH(Me)CO2Me) were also studied, and their efficacy as lactide polymerization catalysts was examined. Polymerization using 7 requires heating to 60 °C, while complex 8 displays high activity at ambient temperature. The difference in activity can be attributed to κ2versus κ3 binding modes of the ligand, providing important insight into structure–activity relationships for this system. Complex 8 gives modestly heteroenriched PLA (Pr = 0.70), which represents the best stereocontrol yet achieved by a cationic metal catalyst.
1. Introduction
The preparation of highly modular pincer-type ligands for the stabilization of reactive metal species is a topical field, whereby the properties of such ligands can be readily modified to suit the needs of various catalytic applications.1,2 We have been exploring a neutral bis(phosphinimine) pincer architecture and coordinatively unsaturated cationic zinc complexes thereof, and have applied these compounds to the ring-opening polymerization of lactide (LA). It was expected that the electronically and coordinatively unsaturated metal centre would promote rapid monomer coordination, much like these properties promote rapid olefin polymerization in the well-known cationic metallocene catalysts of the early transition metals.3 Polylactide (PLA) has seen increasing attention in recent years due to its potential viability as an eco-friendly commodity plastic which is biodegradable and sourced from renewable resources.4 Many single-site catalysts for the controlled ring-opening polymerization of LA are known, and various reviews have been published.5 These systems are typically neutral complexes supported by formally anionic ancillary ligands, and most commonly incorporate zinc,6magnesium,7calcium,8 aluminium9 (and the heavier group 13 metals10), and the rare earth metals.11 Several cationic metal complexes have been employed for ring-opening polymerization of other lactones,12 but there have only been a handful of reports of the application of such complexes for the polymerization of LA.13 In general, these species have displayed poor activity or poorly defined structure of the active catalyst or the mechanism of polymerization. We have recently reported the first cationic metal complex capable of the controlled coordination-insertion polymerization of LA at ambient temperature.14 Shortly thereafter, a cationic calcium complex was reported by Mountford et al. which also exhibits high activity at ambient temperature and excellent molecular-weight control.13bOur initial foray into the field of LA polymerization catalysis utilized a mono(phosphinimine) architecture prepared by installation of a phosphinimine group on a dibenzofuran (dbf) backbone.15Zinc-alkyl complexes of this ligand exhibited bidentate coordination through the phosphinimine nitrogen and the dbf oxygen, but displayed relatively poor activity and control in polymerization experiments. However, substantially enhanced polymerization properties were achieved when a bis(phosphinimine) architecture, prepared by placement of phosphinimine moieties at the 4 and 6 positions of dibenzofuran, was employed.16 Interestingly, the ligand was shown to coordinate zinc in a bidentate manner at the phosphinimine sites, with no participation of the dbf oxygen. More recently, we began probing the steric properties of this pincer system by replacing the 2,4,6-trimethylphenyl (Mes) N-aryl substituents with the less sterically demanding para-isopropylphenyl (Pipp) group. Cationic zinc complexes of this less encumbered system exhibited tridentate chelation. When an appropriate initiating group (methyl-lactate) was installed on zinc, this less bulky system proved to be an extremely active and living catalyst for the polymerization of rac-LA at ambient temperature.14 Presently, it is not known whether this high activity is primarily due to decreased steric hindrance, or if the presence of the Zn–O bonding interaction also plays an important synergistic role.
In the current study, a wider investigation of the steric demands of the ancillary ligand has been undertaken, in hopes of obtaining a more complete understanding of structure–activity relationships for this novel class of cationic catalyst. Specifically, one ortho position of the N-aryl groups was left unsubstituted, while the substituent of the remaining ortho site was varied (H, CH3, iPr). We have prepared and studied the corresponding methylzinc complexes. Additionally, we have examined cationic zinc–lactate complexes in order to gauge the effect on lactide polymerization properties. All of the ligand variations employed have been found capable of stabilizing trigonal planar zinc cations, but significant variation in solid-state geometry, solution dynamics, and polymerization properties have been observed. The bulk of the ligand has been found to not only determine the binding mode (κ2 or κ3), but also the symmetry of the complex (Cs or C2). Only the less bulky zinc–lactate complex, in which the ligand adopts a tridentate coordination mode, is a highly active polymerization catalyst at ambient temperature. Additionally, the catalyst generates modestly heterotactic enriched PLA (Pr = 0.70), representing the best stereochemical control yet achieved by a cationic catalyst system.
2. Results and discussion
2.1 Ligand synthesis
The ligands L11–L33 were easily prepared from the diphosphine precursor and the appropriate aryl-azide, in isolated yields between 72.5–82.3%, giving analytically pure, thermally stable solids. All of the ligands exhibit similar features in the 31P{1H} NMR spectrum (C6D6 solvent), with each displaying one sharp singlet in the region between δ −6 and −4, suggesting C2v symmetry in solution. The isopropyl groups of L111 appear in the expected regions in the 1H NMR spectrum at δ 4.28 and 1.52, for the methine and methyl protons, respectively, while the characteristic o-tolyl methyl group of L22 resonates at δ 2.74. Single crystals suitable for X-ray diffraction were grown for each of these ligands, and their structures were verified by X-ray crystallography.†2.2 Ligand protonation
We have exploited the same synthetic methodology used in our previous studies of cationic zinc14–16 and magnesium species,17 and in the preparation of cationic zinc complexes of a neutral α-diimine ligand by Bochmann et al.12b,18 This route involves protonation of the ligand prior to complexation, thereby simultaneously installing a weakly coordinating anion while providing access to an efficient and irreversible alkane elimination pathway for complex preparation. As such, the complexes were readily prepared by reacting the protonated ligand with an appropriate alkyl-zinc precursor (Scheme 1). Brookhart's anion was installed using the oxonium acid reagent [H(OEt2)2][B(C6H3(CF3)2)4],19 giving compounds 1a–3a. Additionally, the ligands were protonated using HCl, followed by reaction with sodium tetraphenylborate to install the BPh4 counterion, giving compounds 1b–3b. This counterion is less stable and prone to Ph transfer, but facilitates growth of single crystals and thereby expedites the solid-state characterization of the complexes. All protonated ligand derivatives were isolated in high yield (ca. 90% for 1a–3a) as analytical pure powders, and are indefinitely stable at ambient temperature when stored under an inert atmosphere. As a general trend, introduction of the proton broadens the 1H and 31P{1H} NMR signatures associated with the ligand, presumably as a result of inter- and intramolecular proton exchange between phosphinimine sites. Each protonated ligand displays only a single peak in the 31P{1H} NMR spectrum, suggesting that the proton exchange process is rapid on the NMR timescale, generating an average C2v geometry in solution. As is usual for phosphinimine systems of this type, the protonation causes this resonance to shift downfield of the neutral ligand by approximately 20 ppm. The 31P resonance appears between δ 14.3 and 19.2 for 1a–3a (CD2Cl2), and between δ 12.6 and 14.5 for 1b–3b (acetone-d6). The acidic NH proton is difficult to observe in CD2Cl2 solvent, but is apparent as a broad singlet in the 1H NMR spectra of 1b–3b (acetone-d6) between δ 4 and 5.![Preparation of a series of cationic methylzinc and zinc–lactate complexes by first reacting the protonated ligand with (i) [H(OEt2)2][B(C6H3(CF3)2)4] (1a–3a) or (ii) HCl and Na[BPh4] (1b–3b) followed by reaction of the isolated protonated ligand with (iii) ZnMe2 (4–6) or (iv) EtZn(OCH(Me)CO2Me) (7, 8).](/image/article/2012/CY/c1cy00306b/c1cy00306b-s1.gif) | ||
| Scheme 1 Preparation of a series of cationic methylzinc and zinc–lactate complexes by first reacting the protonated ligand with (i) [H(OEt2)2][B(C6H3(CF3)2)4] (1a–3a) or (ii) HCl and Na[BPh4] (1b–3b) followed by reaction of the isolated protonated ligand with (iii) ZnMe2 (4–6) or (iv) EtZn(OCH(Me)CO2Me) (7, 8). | ||
2.3 Cationic methylzinc complexes
Generation of the cationic methylzinc complexes 4a and 4b was easily achieved by reaction of ZnMe2 with 1a and 1b (Scheme 1). Surprisingly, compound 4b was generated cleanly, with no evidence for back-transfer of a phenyl group from the anion. We attribute this observation to steric protection of the metal centre by the bulky isopropyl groups. The spectroscopic signatures of 4a and 4b are similar, with the organometallic methyl group appearing at δ −0.88. A single sharp peak appears in the 31P{1H} NMR spectrum at δ 22.7 (CD2Cl2), which is consistent with a tightly bound zinc cation in a left-right symmetric complex. The isopropyl CH3groups split into two resonances at δ 0.84 and 0.36 in the 1H NMR spectrum, which we suggest is due to conformational rigidity of the complex and reduction of symmetry from C2v to Cs (vide infra). This symmetry results in equivalent isopropyl groups, each with inequivalent methyls (Scheme 2). A variable-temperature 1H NMR study was performed for 4a (Fig. 1), and it was observed that the doublets morph into broad singlets at temperatures greater than 40 °C and coalesce to a single broad peak at 105 °C (C6D5Br). We propose that the coalescence of signals is due to a syn–syn fluxional process as displayed in Scheme 2, which has a calculated barrier ΔG‡ = 18.0(2) kcal mol−1.20 The coalesced peak remains very broad at temperatures up to 120 °C, at which point significant decomposition of the compound becomes evident. | ||
| Scheme 2 Proposed fluxional behaviour of 4a, viewing down the plane of the dbf backbone (represented by the solid black line). | ||
 | ||
| Fig. 1 Variable temperature 1H NMR spectra of the isopropyl methyl groups of 4a. | ||
An X-ray crystal structure of complex 4b has been obtained and is depicted in Fig. 2, with selected bond metrical data listed in Table 1. The zinc centre is κ2 bound via the phosphinimine N-atoms, with no Zn–Odbf interaction observed. The orientation of the ligand within complex 4b is unique compared with previously studied analogues, whereby the N-aryl groups bend away from the binding pocket in the same direction, resulting in a syn orientation and a Cs-symmetric complex. Conversely, complexes of the previously studied mesityl substituted ligand adopted an anti conformation, rendering the zinc complex C2-symmetric.16 Furthermore, the previously studied Pipp-substituted ligand was established to be κ3 bound due to the presence of a significant Zn–Odbf interaction.14 The syn orientation of the ligand in complex 4b creates a rather sharp bite angle [N(1)–Zn(1)–N(2) = 113.30(5) Å], though the coordination geometry is best described as trigonal planar. The Zn–N distances average 2.053(1) Å, which is noticeably longer than the same bonds in the analogous complexes of L22 and L33 (vide infra). We suggest that this apparent weaker binding of the ligand is a result of the isopropyl groups, which sterically crowd the metal centre.
 | ||
| Fig. 2 Molecular structure of the cation of 4b, with H-atoms omitted and ellipsoids drawn at the 30% probability level. | ||
| 4b | 5b/c | 6b/c | |
|---|---|---|---|
| a 4b: x = 79; 5b/c: x = 75; 6b/c: x = 49. | |||
| Zn(1)–N(1) | 2.050(1) | 2.013(2) | 2.004(2) |
| Zn(1)–N(2) | 2.056(1) | 2.020(2) | 2.002(2) |
| Zn(1)–O(1) | 3.125(1) | 3.326(2) | 3.276(2) |
| Zn(1)–C(x) | 1.961(2) | 1.967(4) | 1.977(2) |
| P(1)–N(1) | 1.601(1) | 1.599(2) | 1.601(2) |
| P(2)–N(2) | 1.607(1) | 1.602(2) | 1.600(2) |
| N(1)–Zn(1)–N(2) | 113.30(5) | 124.20(9) | 125.52(8) |
| N(1)–Zn(1)–C(x)a | 123.67(7) | 117.6(2) | 116.12(8) |
| N(2)–Zn(1)–C(x)a | 122.37(7) | 118.2(2) | 118.36(8) |
Reaction of dimethylzinc with the protonated ligands 2a and 2b rapidly afforded the desired methylzinc complexes 5a and 5b, respectively. Complex 5a was isolated as an analytically pure powder in 97% yield. Unlike complexes 4 and 6 (vide infra), however, 5a exhibits two relatively broad resonances in the ambient temperature 31P{1H} NMR spectrum (δ 24.0 and 22.0 (C6D5Br)) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This is not due to inequivalent phosphinimines in the complex, but rather, arises from the presence of two distinct isomers, as indicated by two peaks (δ −0.39 and −0.84) in the 1H NMR spectrum corresponding to the Zn–CH3 moiety. This conclusion has been further corroborated by variable temperature NMR studies (Fig. 3), whereby it was found that the two peaks in the 31P{1H} NMR spectrum (Δνmax = 327.6 Hz) coalesce to a single broad resonance at 56 °C. The energy barrier for this process was calculated to be ΔG‡ = 15.0(1) kcal mol−1. A concomitant coalescence and sharpening of the unique 1H NMR resonances was also noted, with the Zn–CH3 appearing at δ −0.40 (100 °C). Below ambient temperature, the NMR signatures of both isomers sharpen substantially while maintaining a 1:1 ratio. We propose that these peaks correspond to anti (C2) and syn (Cs) orientations of the ligand (Scheme 3). While the bulkier cations in 4a/4b appear locked in a syn geometry, it would seem that reducing the steric constraints on the system significantly enhances flexibility, resulting in little energy difference between the two conformations.
:1 ratio. This is not due to inequivalent phosphinimines in the complex, but rather, arises from the presence of two distinct isomers, as indicated by two peaks (δ −0.39 and −0.84) in the 1H NMR spectrum corresponding to the Zn–CH3 moiety. This conclusion has been further corroborated by variable temperature NMR studies (Fig. 3), whereby it was found that the two peaks in the 31P{1H} NMR spectrum (Δνmax = 327.6 Hz) coalesce to a single broad resonance at 56 °C. The energy barrier for this process was calculated to be ΔG‡ = 15.0(1) kcal mol−1. A concomitant coalescence and sharpening of the unique 1H NMR resonances was also noted, with the Zn–CH3 appearing at δ −0.40 (100 °C). Below ambient temperature, the NMR signatures of both isomers sharpen substantially while maintaining a 1:1 ratio. We propose that these peaks correspond to anti (C2) and syn (Cs) orientations of the ligand (Scheme 3). While the bulkier cations in 4a/4b appear locked in a syn geometry, it would seem that reducing the steric constraints on the system significantly enhances flexibility, resulting in little energy difference between the two conformations.
 | ||
| Fig. 3 Variable temperature 31P{1H} NMR spectra of compound 5a. | ||
 | ||
| Scheme 3 Isomers of complex 5a/5b with a view looking down the plane of the dbf backbone (represented by the solid black line). | ||
The synthesis of 5b from 2b and ZnMe2 does not proceed cleanly, giving rise to the phenylzinc species of the formula [L22ZnPh][MeBPh3] (5c) as a side product, in an approximate 60:40 ratio of 5b:5c. Single crystals were obtained from the mixture of 5b and 5c, and X-ray diffraction studies showed the crystal contained the cations of both compounds in a ratio of approximately 3:1. Only a single independent unit of the ligand and the zinc centre exist, and the presence of both components was modelled as a substitutional disorder of the methyl and phenyl groups. The structure of 5b is depicted in Fig. 4 with the disorder omitted for clarity; selected metrical parameters are presented in Table 1. The dbf ligand coordinates to the zinc centre via a κ2 bonding mode, with no notable interaction between zinc and the dbf oxygen. The coordination geometry at zinc is thus best described as trigonal planar. The ligand adopts the anti orientation rather than the syn orientation observed for 4b, and the complex is thereby pseudo-C2-symmetric. The unsubstituted ortho positions of the o-tolyl groups are oriented toward the dibenzofuran backbone, while the methyl groups closely approach the coordination sphere of the zinc centre, providing a reasonable degree of steric protection. Compared with the previously studied mesityl analogue, which was also C2-symmetric, the absence of a methyl group in the ortho site allows the ligand to adopt a sharper bite angle that is very close to ideal (120°).
 | ||
| Fig. 4 X-Ray crystal structure of the cation of 5b, with H-atoms omitted and ellipsoids drawn at the 30% probability level. | ||
The synthesis of the cationic methylzinc complexes of L33 (6a and 6b) was performed similarly to 4 and 5. The NMR spectra of 6a are rather unremarkable, with the ZnMe moiety resonating at δ −1.02 in the 1H NMR spectrum at ambient temperature. It is reasonable to suggest that interconversion between anti and syn isomers can occur in this complex even more readily than in complexes of L22 because of the less imposing N-aryl group. However, no evidence of this fluxional process was observed at ambient temperature, suggesting rapid interconversion between isomers on the NMR timescale. Likewise, only one peak was observed in the 31P{1H} NMR spectrum.
Similarly to 5b, the synthesis of compound 6b was plagued by the production of an appreciable quantity (∼20%) of the analogous phenylzinc compound [L33ZnPh][MeBPh3] (6c). Under the crystallization conditions employed, single crystals obtained from this mixture were enriched in the cation of 6c (93%). Determination of the solid-state structure revealed a single molecule in the asymmetric unit, with the two compounds again modelled as a substitutional disorder of the phenyl and methyl groups. The X-ray crystal structure of the cation of 6c is depicted in Fig. 5, while selected bond lengths and angles can be found in Table 2. Surprisingly, the ligand binds to the metal centre in a bidentate fashion, with no meaningful interaction between the dbf oxygen and zinc. The coordination geometry approaches trigonal planar. We attribute the lack of Zn–Odbf bonding in this relatively non-hindered complex to packing effects in the solid-state, which necessarily suggests that the Zn–Odbf interaction observed in the Pipp analogue14 is relatively weak. The ligand geometry exists primarily in a pseudo-C2-symmetric anti conformation. However, both phosphinimine groups exhibit significant disorder, with the minor component of each being in a syn conformation with respect to the major component of the other, lending support to the hypothesis that the complex undergoes rapid interconversion between anti and syn isomers in solution. This is modelled only as a positional disorder of the P and N atoms, in a ratio of 80:20 and 85:15 for P(1)–N(1) and P(2)–N(2), respectively.
 | ||
| Fig. 5 Molecular structure of the cationic component of the by-product 6c formed in the synthesis of 6b, with H-atoms and disordered atomic positions omitted for clarity and ellipsoids drawn at the 30% probability level. | ||
| Bond | 7 | 8 | Angle | 7 | 8 |
|---|---|---|---|---|---|
| Zn(1)–N(1) | 2.014(5) | 2.016(3) | N(1)–Zn(1)–N(2) | 124.8(2) | 116.2(1) |
| Zn(1)–N(2) | 1.996(4) | 1.988(3) | N(1)–Zn(1)–O(3) | 115.3(2) | 122.3(1) |
| Zn(1)–O(1) | 3.247(4) | 2.367(2) | N(2)–Zn(1)–O(3) | 118.6(2) | 117.6(1) |
| Zn(1)–O(2) | 2.105(4) | 2.131(3) | O(3)–Zn(1)–O(2) | 83.2(2) | 83.3(1) |
| Zn(1)–O(3) | 1.921(4) | 1.904(3) | N(1)–Zn(1)–O(2) | 99.6(2) | 103.2(1) |
| P(1)–N(1) | 1.599(4) | 1.600(2) | N(2)–Zn(1)–O(1) | N/A | 83.7(1) |
| P(2)–N(2) | 1.593(5) | 1.597(4) | O(1)–Zn(1)–O(2) | N/A | 168.3(1) |
2.4 Cationic zinc–lactate complexes
Complexes 4–6 are inactive for the polymerization of lactide, which is not surprising given our previous observation that cationic alkylzinc complexes are inefficient initiators.14–16 Therefore, in order to probe the effects of ligand structure on LA polymerization, it was necessary to install a suitable initiating group. This was accomplished by reaction of HL+ with EtZn(OCH(Me)CO2Me) to generate the corresponding methyl-lactate zinc complexes. The methyl-lactate moiety is an excellent initiating group, in addition to being a very close structural mimic of the growing polymer chain. Thus, studies of such complexes provide a great deal of insight into the nature of the active catalyst species. The synthesis of these complexes required rather harsh conditions relative to preparation of the related methylzinc compounds, and therefore the tetraphenylborate anion could not be employed. Additionally, the synthetic methodology proved ineffective for the most sterically hindered ligand L111. For this reason, only zinc–lactate complexes of L22 and L33 were prepared and studied.Reaction of 2a or 3a with racemic EtZn(methyl-DL-lactate) for a period of 1 h at 100 °C cleanly afforded complexes 7 and 8, respectively. Each was isolated as a thermally stable, analytically pure white crystalline solid in good yield (7: 76%, 8: 72%). Compound 7 displays fluxional solution behaviour similar to the methylzinc complex of L22, with broad resonances appearing in the 1H NMR spectrum. However, instead of two distinct isomers, the ambient temperature 31P{1H} NMR spectrum displayed three broad resonances of approximately equal intensity at δ 27.0, 26.6, and 26.0. These signals coalesce to a single broad resonance at 60 °C, corresponding to an activation barrier of approximately ΔG‡ = 16 kcal mol−1 (Fig. 6).21 This peak sharpens considerably at elevated temperatures, giving a singlet at δ 27.2 at 80 °C. The 1H NMR spectrum also exhibits sharp signals at this temperature with characteristic methyl-lactate peaks observed at δ 4.39 (methine), 2.89 (OMe), and 1.35 (CMe).22 At temperatures below 20 °C, the NMR spectra suffer from substantial broadening, but at −20 °C the 31P{1H} NMR spectrum simplifies into predominantly two relatively sharp peaks at δ 26.7 and 25.7. The presence of three isomers is readily explained by considering the symmetry of the ligand in the anti and syn orientations and how these interact with the asymmetric methyl-lactate moiety (Scheme 4). When the ligand exists in the C2-symmetric orientation, two unique diastereomers can be distinguished for a given methyl-lactate enantiomer (anti-A and anti-B), while for the Cs-symmetric orientation only a single diastereomer is possible (syn). For the opposite enantiomer of the methyl-lactate moiety the resulting isomers are enantiomers of the former.
 | ||
| Fig. 6 Variable temperature 31P{1H} NMR spectra of complex 7. | ||
 | ||
| Scheme 4 Possible isomers of complex 7 for a methyl-D-lactate moiety, with a view down the plane of the dbf backbone (represented by a solid black line). | ||
In contrast, the NMR spectra of complex 8 are relatively straightforward, including a single peak in the 31P{1H} spectrum at δ 29.3. However, examination of the 1H NMR spectrum suggests two unique isomers, as evidenced by two distinct sets of resonances. Specifically, resonances attributed to the methyl-lactate moiety appear at δ 3.42 (CH(CH3)), 3.44 (OCH3) and 0.87 (CH(CH3)), while a second set of equally intense signals resonate at δ 4.07, 3.93 and 1.36 (CD2Cl2). The identity of these isomers is presently unknown, but may be related to the relative positions of the two coordinating methyl-lactate oxygen atoms with respect to axial and equatorial trigonal bipyramidal sites (vide infra).
High quality single crystals of complexes 7 and 8 suitable for X-ray diffraction were obtained and their solid-state structures are depicted in Fig. 7 and 8, respectively. Selected metrical parameters are located in Table 2.
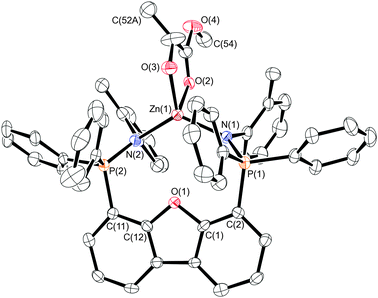 | ||
| Fig. 7 Molecular structure of the cation of 7, with H-atoms omitted and ellipsoids drawn at the 30% probability level. | ||
 | ||
| Fig. 8 Molecular structure of the cation of 8, with hydrogen atoms and an N-phenyl omitted for clarity and ellipsoids drawn at the 30% probability level. | ||
Interestingly, the structure of 7 finds L22 in the syn, as opposed to the anti conformation observed in the solid-state structure of 5b. The ancillary ligand is κ2 bound, as there is no evidence for a substantive Zn–Odbf interaction. Thus, the zinc centre is 4-coordinate with a geometry that is best described as trigonal pyramidal. The equatorial sites are occupied by the phosphinimine donors and the formally anionic oxygen atom (O(3)) of the lactate group, with bond lengths in the expected ranges, and a sum of angles about these equatorial sites of 358.7(3)°. The bite angle of the ligand in this pseudo-Cs-symmetric complex is 124.8(2)°, which is similar to that of the C2-symmetric ligand in the analogous methylzinc complex 5b. Although there is only one independent molecule in the unit cell, the compound is a racemic mixture, which has been modelled as a positional disorder of the methyl group C(52).
The X-ray crystal structure of complex 8 shows major differences compared with that of 7, despite the seemingly small change in the ligand. Specifically, the ligand is κ3 bound to the zinc centre, whereby a significant Zn–Odbf interaction occurs (Zn(1)–O(1) = 2.367(2) Å). This results in a zinc coordination geometry that is similar to the structure of the analogous complex of the Pipp ligand previously reported.14 Use of Addison and Reedijk's trigonality parameter23 gave τ = 0.72, suggesting a primarily trigonal bipyramidal coordination geometry. The equatorial sites are occupied by the phosphinimine N-donors and O(3), with a sum of angles about these sites of 356.1(2)°. The weaker, formally neutral oxygen donors occupy the axial sites at an angle of 168.3(1)°. As in complex 7, the compound is a racemic mixture, which is modelled as a position disorder of C(50).
2.5 Polymerization studies
Preliminary studies of the performance of complexes 7 and 8 as LA polymerization catalysts have been performed in order to establish the effect of ligand bulk and coordination mode. Compound 7 shows little activity for polymerization of rac-LA at ambient temperature, and must be heated to 60 °C to achieve polymerization at a reasonable rate. In CDCl3 solvent at 60 °C, with an initial monomer concentration of 1 M and a catalyst loading of 0.5 mol% ([LA]0/[7] = 200), polymerization occurs under well-behaved first-order kinetics with an observed rate constant of 3.65(2) × 10−4 s−1 (Table 3). This rate is only slightly better than that observed for the bulkier mesityl substituted complex previously reported, despite the significant reduction in steric bulk.16 Analysis of the stereochemistry of isolated polymer samples revealed slight stereocontrol and a modestly heteroenriched polymer chain (Pr = 0.61).24Gel permeation chromatography (GPC) of the same isolated polymer sample indicated reasonably good molecular weight control. The number-average molecular weight (Mn) was 18.0 × 103 g mol−1, and the polydispersity was 1.37. Furthermore, MALDI-ToF mass spectra of the polymer were obtained, which support a coordination-insertion mechanism, as the mass of all polymer peaks are consistent with a methyl-lactate end group. However, mass peaks are separated by m/z = 72, which suggests appreciable transesterification side-reactions.While complex 8 may appear to be only marginally less sterically encumbered than catalyst 7, it was found to be dramatically more active for the polymerization of rac-LA. Complex 8 displayed high activity at ambient temperature, and in CH2Cl2 solvent the observed first-order rate constant at 25 °C was determined to be 5.11(3) × 10−4 s−1. Interestingly, this rate is somewhat slower than that established for the previously studied Pipp analogue under identical conditions (8.65(4) × 10−4 s−1), despite their similar steric bulk (Table 3). As such, the moderate reduction in activity cannot simply be attributed to steric differences, and we therefore propose that electronic effects are responsible. The lack of a para-isopropyl group on the N-aryl rings somewhat reduces the electron donating capacity of the ligand, thereby rendering the zinc centre in 8 slightly more electropositive. Thus, the activity of these catalysts is highly dependent on the electrophilicity of the metal centre, and suggests that better moderation of electron density may lead to improved activity. Hence, we conclude that the reason for the large difference in activity between 7 and 8, which is not commensurate with the difference in steric bulk, is due largely to the difference in the coordination geometry of the ligand. Specifically, the interaction between the cationic zinc centre and the dbf oxygen, that is only present in sterically open complexes (8 and the Pipp analogue) is strong enough to help moderate the electrophilicity of the metal. We thus conclude that reduced steric demand of the ligand with concomitant reduction in metal electrophilicity work synergistically to promote enhanced LA polymerization activity in cationic zinc complexes stabilized by dbf-based bis(phosphinimine) pincer ligands.
Analysis of polymer samples produced using 8 as catalyst showed very similar properties compared with those generated using 7. At ambient temperature under otherwise identical conditions, the molecular weight was found to be 18.9 × 103 g mol−1, and the molecular weight distribution low (PDI = 1.35). However, analysis of the tetrad sequences revealed significantly enhanced stereocontrol, with isolated polymer samples exhibiting substantial heterotactic enrichment (Pr = 0.70), presumably due to a chain-end control mechanism.25 This level of stereoregularity is uncommon for zinc catalysts active at ambient-temperature, and represents the best stereocontrol yet reported for polymerization of rac-lactide by a cationic metal complex.
3. Conclusions
In summary, a series of closely related bis(phosphinimine) ligands, L11–L33, which differ only in the nature of the substituents in the ortho positions of N-aryl groups, have been constructed from a dibenzofuran backbone. Cationic methylzinc complexes of each ligand were prepared and structurally characterized. All species were found to have low-coordinate trigonal planar geometry, and exist as well separated ion pairs. The structural investigations established that the bulkiness of the N-aryl groups has a substantial impact on ligand orientation in the complexes.The synthesis of zinc–lactate complexes was only possible using the two less bulky ligands (L22 and L33). These complexes were fully characterized and their efficacy for polymerization of rac-LA was explored. Notably, polymerization activity is heavily dependent upon the steric bulk of the ancillary ligand; removal of only a single methyl substituent from each N-aryl group resulted in a dramatic improvement in activity. This increased activity is attributed not only to the reduced steric constraints, but also to the additional stabilizing interaction between zinc and the dibenzofuran oxygen, which occurs only within the less bulky complex. The study has thus significantly advanced our knowledge of fundamental structure–activity relationships, which will allow further rational development of this relatively new pincer ligand, whereby catalytic activity and control may be simultaneously optimized. Remarkably, complex 8 also displays improved stereoselectivity, giving substantially heteroenriched PLA (Pr = 0.70), which is unprecedented for well-defined cationic metal complexes.
4. Experimental section
4.1 General procedures
All manipulations of air-sensitive materials and reagents were conducted using high-vacuum techniques under a purified argon atmosphere or in a glove box (MBraun Labmaster 130). Protio solvents were purified using an MBraun solvent purification system (MB-SPS), stored in Teflon-sealed glass vessels over appropriate drying agents, and vacuum transferred directly to reaction vessels. Deuterated solvents (Cambridge Isotopes) were dried with appropriate drying agents, vacuum transferred, and stored under an inert atmosphere prior to use. All other materials were obtained in high purity (Sigma-Aldrich, Acros Organics) and used without additional purification. NMR spectra (1H (300.13 MHz), 13C{1H} (75.47 MHz), 31P{1H} (121.48 MHz), 19F (282.42 MHz), and 11B (96.29 MHz)) were collected using a Bruker Avance II NMR spectrometer equipped with a variable-temperature unit. Spectra were collected at ambient temperature and referenced to residual solvent resonances (1H and 13C{1H}), or an external standard (triphenylphosphine (31P{1H}), trifluorotoluene (19F), or boron trifluoride diethyl etherate (11B)). 1H and 13C NMR peak assignments were facilitated by DEPT, COSY, and HSQC experiments. X-ray crystal structures were collected using a Bruker AXS SMART APEX II single crystal X-ray diffractometer (Mo-Kα (λ = 0.71073 Å)). Elemental analyses were performed using an Elementar Vario Microcube. MALDI mass spectra were performed on an ultraflexXtreme™ MALDI-TOF/TOF (Bruker Daltonics, Billerica, MA, USA) mass spectrometer in positive MS mode using 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) as the matrix compound. GPC data were obtained on a Varian PL-GPC 50 Plus instrument and measured against polystyrene standards. The molecular weight values were scaled by the accepted Mark-Houwink parameter of 0.58.26Phenyl-azide27 and 2-isopropylphenyl-azide28 were prepared according to published literature procedures,29 and the spectral data match literature values. The ligand diphosphine precursor was prepared by the previously reported procedure.304.2 Details of polymerization studies
4.3 Synthesis and characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) PPh2)2-dbf (L111).
In a 100 mL round bottom flask attached to a swivel-frit apparatus, 4,6-bis(diphenylphosphino)dibenzofuran (1.00, 1.86 mmol) was dissolved in toluene (40 mL). 2-isopropylphenyl-azide (0.63 g, 3.9 mmol) was added, and the resulting solution was stirred for 18 h at a temperature of 70 °C. After cooling to ambient temperature, the solution was filtered through the frit to remove any trace undissolved impurities, and was then concentrated to a volume of 5 mL. To this 30 mL of pentane was added, causing precipitation of the product as a yellow powder. Vigorous stirring for 1 h produced a fine yellow powder, which was collected by filtration, washed with pentane (3 × 5 mL), and dried under vacuum. Yield: 1.08 g (1.35 mmol), 72.5%. 1H NMR (C6D6): δ 8.26 (dd, 2H, 3JPH = 13.6 Hz, 3JHH = 6.4 Hz, 3,7-H dbf), 7.64 (dd, 8H, 3JPH = 12.8 Hz, 3JHH = 7.0 Hz, o-Ph), 7.49 (d, 2H, 3JHH = 7.7 Hz, 1,9-H dbf), 7.37 (m, 2H, 4-NAr), 7.00–6.80 (ov m, 10H, p-Ph + 2,8-H dbf + 2-NAr + 3-NAr), 6.73 (td, 8H, 3JHH = 7.6 Hz, 4JPH = 3.0 Hz, m-Ph), 6.63 (m, 2H, 1-NAr), 4.28 (sp, 2H, 3JHH = 6.9 Hz, CH(CH3)2), 1.52 (d, 12H, 3JHH = 6.9 Hz, CH(CH3)2); 31P{1H} NMR (C6D6): δ −5.2 (s); 13C{1H} NMR (C6D6): δ 156.76 (d, J = 2.6 Hz), 149.07 (d, J = 1.1 Hz), 143.28 (d, 2JCP = 21.7 Hz), 135.06 (d, 2JCP = 6.6 Hz, 3,7-dbf), 132.66 (d, 3JCP = 10.2 Hz, o-Ph), 131.94 (d, 1JCP = 107.6 Hz, ipso-Ph), 131.83 (d, 4JCP = 2.9 Hz, p-Ph), 129.02 (d, 3JCP = 12.6 Hz, m-Ph), 126.44 (s, 3-NAr), 126.18 (d, 4JCP = 2.2 Hz, 4-NAr), 125.14 (dd, JCP = 6.3 Hz, JCP = 0.9 Hz), 125.00 (d, 4JCP = 2.5 Hz, 1,9-dbf), 124.35 (d, 3JCP = 9.9 Hz, 2,8-dbf), 121.78 (d, 3JCP = 10.1 Hz, 1-NAr), 118.90 (d, 4JCP = 0.5 Hz, 2-NAr), 117.60 (d, 1JCP = 85.6 Hz, 4,6-dbf), 29.20 (s, CH(CH3)2), 23.70 (s, CH(CH3)2); elemental analysis calcd (%) for C54H48N2OP2: C 80.78, H 6.03, N 3.49; found: C 80.48, H 6.03, N 3.29.
PPh2)2-dbf (L22).
L22 was prepared similarly to L111 from 4,6-bis(diphenylphosphino)dibenzofuran (1.00 g, 1.86 mmol) and o-tol-N3 (0.52 g, 3.90 mmol), affording L22 as a light yellow powder in 80.6% yield (1.12 g, 1.50 mmol). 1H NMR (C6D6): δ 8.27 (dd, 2H, 3JPH = 13.4 Hz, 3JHH = 7.6 Hz, 3,7-H dbf), 7.64 (dd, 8H, 3JPH = 12.7 Hz, 3JHH = 7.4 Hz, o-Ph), 7.49(d, 2H, 3JHH = 7.6 Hz, 1,9-dbf), 7.32(d, 2H, 3JHH = 7.0 Hz, 4-o-Tol), 6.95 (t, 2H, 3JHH = 7.6 Hz, 2,8-dbf), 6.92–6.85 (ov m, 6H, p-Ph + 2-o-Tol), 6.81 (2H, 3JHH = 7.1 Hz, 3-o-Tol), 6.74(td, 8H, 3JHH = 7.6 Hz, 4JPH = 2.8 Hz, m-Ph), 6.58(d, 2H, 3JHH = 7.7 Hz, 1-o-Tol), 2.74 (s, 6H, CH3o-Tol); 31P{1H} NMR (C6D6): δ −5.9; 13C{1H} NMR (C6D6): δ 156.80 (d, JCP = 2.6 Hz), 150.30 (d, JCP = 1.0 Hz), 135.02 (d, 2JCP = 6.5 Hz, 3,7-dbf), 133.47 (d, JCP = 22.4 Hz), 132.67 (d, 2JCP = 10.2 Hz, o-Ph), 131.96 (d, 1JCP = 106.8 Hz, ipso-Ph), 131.85 (d, 4JCP = 3.0 Hz, p-Ph), 131.03 (d, 4JCP = 2.0 Hz, 4-o-Tol), 129.02 (d, 3JCP = 12.5 Hz, m-Ph), 126.83 (s, 2-o-Tol), 125.10 (dd, JCP = 6.5 Hz, JCP = 0.9 Hz), 125.00 (d, 4JCP = 2.3 Hz, 1,9-dbf), 124.30 (d, 3JCP = 10.2 Hz, 2,8-dbf), 121.28 (d, 3JCP = 9.7 Hz, 1-o-Tol), 118.52 (s, 3-o-Tol), 117.52 (d, 1JCP = 87.4 Hz, 4,6-dbf), 20.62 (s); elemental analysis calcd (%) for C50H40N2OP2: C 80.41, H 5.40, N 3.75; found: C 80.34, H 5.31, N 3.78.
PPh2)2-dbf (L33).
L33 was prepared similarly to L111 from 4,6-bis(diphenylphosphino)dibenzofuran (1.00 g, 1.86 mmol) and phenyl-azide (0.47 g, 3.95 mmol). This reaction was much more facile; reaction at ambient temperature for a 2 h period proved sufficient to give complete conversion of the phosphine starting material, affording L33 as a pale yellow powder in 82.3% yield (1.10 g, 1.53 mmol). 1H NMR (C6D6): δ 8.24 (dd, 2H, 3JPH = 13.9 Hz, 3JHH = 7.6 Hz, 3,7-dbf), 7.68 (dd, 8H, 3JPH = 12.6 Hz, 3JHH = 7.6 Hz, o-Ph), 7.48 (d, 2H, 3JHH = 7.6 Hz, 1,9-dbf), 7.18–7.04 (ov m, 8H, o+m-NPh), 6.98–6.88 (ov m, 6H, p-Ph + 2,8-dbf), 6.87–6.75 (ov m, 10H, m-Ph+p-NPh); 31P{1H} NMR (C6D6): δ −4.2 (s); 13C{1H} NMR (C6D6): δ 156.86 (d, JCP = 2.2 Hz), 152.08 (d, JCP = 1.9 Hz), 134.92 (d, 2JCP = 7.1 Hz, 3,7-dbf), 132.92 (d, 2JCP = 10.2 Hz, o-Ph), 131.87 (d, 4JCP = 3.0 Hz, p-Ph), 131.67 (d, 1JCP = 103.7 Hz, ipso-Ph), 129.54 (d, 4JCP = 1.2 Hz, m-NPh), 129.06 (d, 3JCP = 12.4 Hz, m-Ph), 125.14 (dd, JCP = 6.4 Hz, JCP = 0.9 Hz), 124.96 (d, 4JCP = 2.5 Hz, 1,9-dbf), 124.42 (d, 3JPC = 17.8 Hz, o-NPh), 124.14 (d, 3JPC = 10.4 Hz, 2,8-dbf), 118.42 (d, 5JPC = 0.8 Hz, p-NPh), 116.96 (d, 1JCP = 93.0 Hz, 4,6-dbf); Elemental analysis calcd (%) for C48H36N2OP2: C 80.21, H 5.05, N 3.90; found: C 80.23, H 5.30, N 3.48.
:1 ratio of the methylzinc and phenylzinc products, respectively. Only the resonances of the primary product (5b) are listed below. 1H NMR (CD2Cl2): δ 8.49 (d, 2H, 3JHH = 7.9 Hz, 1,9-dbf), 7.70–7.40 (br ov m, 14H, o-Ph + p-Ph + 2,8-dbf), 7.38–7.22 (br ov m, 16H, m-Ph + o-BPh4), 7.12 (br s, 2H, 3,7-dbf), 7.00 (t, 8H, 3JHH = 7.5 Hz, m-BPh4), 6.85 (t, 4H, 3JHH = 7.5 Hz, p-BPh4), 6.77 (br ov m, 4H, 3-o-Tol+4-o-Tol), 6.39 (br s, 2H, 2-o-Tol), 6.14 (br s, 1H, 1-o-Tol), 5.95 (br s, 1H, 1-o-Tol), 1.78 (br s, 3H, o-Tol CH3), 1.61 (br s, 3H, o-Tol CH3), −0.60 (br s, 1.5H, ZnCH3), −0.93 (br s, 1.5H, ZnCH3); 31P{1H} NMR (CD2Cl2): δ 22.2 (br s), 23.9 (br s); 11B{1H} NMR (CD2Cl2): δ −6.6 (s).
PPh2)2-dbf (L111).
In a 100 mL round bottom flask attached to a swivel-frit apparatus, 4,6-bis(diphenylphosphino)dibenzofuran (1.00, 1.86 mmol) was dissolved in toluene (40 mL). 2-isopropylphenyl-azide (0.63 g, 3.9 mmol) was added, and the resulting solution was stirred for 18 h at a temperature of 70 °C. After cooling to ambient temperature, the solution was filtered through the frit to remove any trace undissolved impurities, and was then concentrated to a volume of 5 mL. To this 30 mL of pentane was added, causing precipitation of the product as a yellow powder. Vigorous stirring for 1 h produced a fine yellow powder, which was collected by filtration, washed with pentane (3 × 5 mL), and dried under vacuum. Yield: 1.08 g (1.35 mmol), 72.5%. 1H NMR (C6D6): δ 8.26 (dd, 2H, 3JPH = 13.6 Hz, 3JHH = 6.4 Hz, 3,7-H dbf), 7.64 (dd, 8H, 3JPH = 12.8 Hz, 3JHH = 7.0 Hz, o-Ph), 7.49 (d, 2H, 3JHH = 7.7 Hz, 1,9-H dbf), 7.37 (m, 2H, 4-NAr), 7.00–6.80 (ov m, 10H, p-Ph + 2,8-H dbf + 2-NAr + 3-NAr), 6.73 (td, 8H, 3JHH = 7.6 Hz, 4JPH = 3.0 Hz, m-Ph), 6.63 (m, 2H, 1-NAr), 4.28 (sp, 2H, 3JHH = 6.9 Hz, CH(CH3)2), 1.52 (d, 12H, 3JHH = 6.9 Hz, CH(CH3)2); 31P{1H} NMR (C6D6): δ −5.2 (s); 13C{1H} NMR (C6D6): δ 156.76 (d, J = 2.6 Hz), 149.07 (d, J = 1.1 Hz), 143.28 (d, 2JCP = 21.7 Hz), 135.06 (d, 2JCP = 6.6 Hz, 3,7-dbf), 132.66 (d, 3JCP = 10.2 Hz, o-Ph), 131.94 (d, 1JCP = 107.6 Hz, ipso-Ph), 131.83 (d, 4JCP = 2.9 Hz, p-Ph), 129.02 (d, 3JCP = 12.6 Hz, m-Ph), 126.44 (s, 3-NAr), 126.18 (d, 4JCP = 2.2 Hz, 4-NAr), 125.14 (dd, JCP = 6.3 Hz, JCP = 0.9 Hz), 125.00 (d, 4JCP = 2.5 Hz, 1,9-dbf), 124.35 (d, 3JCP = 9.9 Hz, 2,8-dbf), 121.78 (d, 3JCP = 10.1 Hz, 1-NAr), 118.90 (d, 4JCP = 0.5 Hz, 2-NAr), 117.60 (d, 1JCP = 85.6 Hz, 4,6-dbf), 29.20 (s, CH(CH3)2), 23.70 (s, CH(CH3)2); elemental analysis calcd (%) for C54H48N2OP2: C 80.78, H 6.03, N 3.49; found: C 80.48, H 6.03, N 3.29.
PPh2)2-dbf (L22).
L22 was prepared similarly to L111 from 4,6-bis(diphenylphosphino)dibenzofuran (1.00 g, 1.86 mmol) and o-tol-N3 (0.52 g, 3.90 mmol), affording L22 as a light yellow powder in 80.6% yield (1.12 g, 1.50 mmol). 1H NMR (C6D6): δ 8.27 (dd, 2H, 3JPH = 13.4 Hz, 3JHH = 7.6 Hz, 3,7-H dbf), 7.64 (dd, 8H, 3JPH = 12.7 Hz, 3JHH = 7.4 Hz, o-Ph), 7.49(d, 2H, 3JHH = 7.6 Hz, 1,9-dbf), 7.32(d, 2H, 3JHH = 7.0 Hz, 4-o-Tol), 6.95 (t, 2H, 3JHH = 7.6 Hz, 2,8-dbf), 6.92–6.85 (ov m, 6H, p-Ph + 2-o-Tol), 6.81 (2H, 3JHH = 7.1 Hz, 3-o-Tol), 6.74(td, 8H, 3JHH = 7.6 Hz, 4JPH = 2.8 Hz, m-Ph), 6.58(d, 2H, 3JHH = 7.7 Hz, 1-o-Tol), 2.74 (s, 6H, CH3o-Tol); 31P{1H} NMR (C6D6): δ −5.9; 13C{1H} NMR (C6D6): δ 156.80 (d, JCP = 2.6 Hz), 150.30 (d, JCP = 1.0 Hz), 135.02 (d, 2JCP = 6.5 Hz, 3,7-dbf), 133.47 (d, JCP = 22.4 Hz), 132.67 (d, 2JCP = 10.2 Hz, o-Ph), 131.96 (d, 1JCP = 106.8 Hz, ipso-Ph), 131.85 (d, 4JCP = 3.0 Hz, p-Ph), 131.03 (d, 4JCP = 2.0 Hz, 4-o-Tol), 129.02 (d, 3JCP = 12.5 Hz, m-Ph), 126.83 (s, 2-o-Tol), 125.10 (dd, JCP = 6.5 Hz, JCP = 0.9 Hz), 125.00 (d, 4JCP = 2.3 Hz, 1,9-dbf), 124.30 (d, 3JCP = 10.2 Hz, 2,8-dbf), 121.28 (d, 3JCP = 9.7 Hz, 1-o-Tol), 118.52 (s, 3-o-Tol), 117.52 (d, 1JCP = 87.4 Hz, 4,6-dbf), 20.62 (s); elemental analysis calcd (%) for C50H40N2OP2: C 80.41, H 5.40, N 3.75; found: C 80.34, H 5.31, N 3.78.
PPh2)2-dbf (L33).
L33 was prepared similarly to L111 from 4,6-bis(diphenylphosphino)dibenzofuran (1.00 g, 1.86 mmol) and phenyl-azide (0.47 g, 3.95 mmol). This reaction was much more facile; reaction at ambient temperature for a 2 h period proved sufficient to give complete conversion of the phosphine starting material, affording L33 as a pale yellow powder in 82.3% yield (1.10 g, 1.53 mmol). 1H NMR (C6D6): δ 8.24 (dd, 2H, 3JPH = 13.9 Hz, 3JHH = 7.6 Hz, 3,7-dbf), 7.68 (dd, 8H, 3JPH = 12.6 Hz, 3JHH = 7.6 Hz, o-Ph), 7.48 (d, 2H, 3JHH = 7.6 Hz, 1,9-dbf), 7.18–7.04 (ov m, 8H, o+m-NPh), 6.98–6.88 (ov m, 6H, p-Ph + 2,8-dbf), 6.87–6.75 (ov m, 10H, m-Ph+p-NPh); 31P{1H} NMR (C6D6): δ −4.2 (s); 13C{1H} NMR (C6D6): δ 156.86 (d, JCP = 2.2 Hz), 152.08 (d, JCP = 1.9 Hz), 134.92 (d, 2JCP = 7.1 Hz, 3,7-dbf), 132.92 (d, 2JCP = 10.2 Hz, o-Ph), 131.87 (d, 4JCP = 3.0 Hz, p-Ph), 131.67 (d, 1JCP = 103.7 Hz, ipso-Ph), 129.54 (d, 4JCP = 1.2 Hz, m-NPh), 129.06 (d, 3JCP = 12.4 Hz, m-Ph), 125.14 (dd, JCP = 6.4 Hz, JCP = 0.9 Hz), 124.96 (d, 4JCP = 2.5 Hz, 1,9-dbf), 124.42 (d, 3JPC = 17.8 Hz, o-NPh), 124.14 (d, 3JPC = 10.4 Hz, 2,8-dbf), 118.42 (d, 5JPC = 0.8 Hz, p-NPh), 116.96 (d, 1JCP = 93.0 Hz, 4,6-dbf); Elemental analysis calcd (%) for C48H36N2OP2: C 80.21, H 5.05, N 3.90; found: C 80.23, H 5.30, N 3.48.
:1 ratio of the methylzinc and phenylzinc products, respectively. Only the resonances of the primary product (5b) are listed below. 1H NMR (CD2Cl2): δ 8.49 (d, 2H, 3JHH = 7.9 Hz, 1,9-dbf), 7.70–7.40 (br ov m, 14H, o-Ph + p-Ph + 2,8-dbf), 7.38–7.22 (br ov m, 16H, m-Ph + o-BPh4), 7.12 (br s, 2H, 3,7-dbf), 7.00 (t, 8H, 3JHH = 7.5 Hz, m-BPh4), 6.85 (t, 4H, 3JHH = 7.5 Hz, p-BPh4), 6.77 (br ov m, 4H, 3-o-Tol+4-o-Tol), 6.39 (br s, 2H, 2-o-Tol), 6.14 (br s, 1H, 1-o-Tol), 5.95 (br s, 1H, 1-o-Tol), 1.78 (br s, 3H, o-Tol CH3), 1.61 (br s, 3H, o-Tol CH3), −0.60 (br s, 1.5H, ZnCH3), −0.93 (br s, 1.5H, ZnCH3); 31P{1H} NMR (CD2Cl2): δ 22.2 (br s), 23.9 (br s); 11B{1H} NMR (CD2Cl2): δ −6.6 (s).
4.4 X-Ray structure determination
Crystals were grown from a concentrated CH2Cl2 solution of the compound at −35 °C for L111, by slow diffusion of pentane into a benzene solution at 25 °C for L22 and L33, by slow diffusion of benzene into a benzene/bromobenzene solution at 25 °C for 4b, 5b/c and 6b/c, from a concentrated bromobenzene solution spiked with a small amount of benzene at −35 °C for 8, and by slow diffusion of pentane into a CH2Cl2 solution of the compound at 25 °C for 7. X-ray intensities were measured on a Bruker SMART APEX II (Mo-Kα radiation; λ = 0.71073 Å) instrument at a temperature of 173(2) K. Unit cell parameters were determined and refined on all reflections and data were integrated with APEX2 software.31 Data reduction and correction for Lorentz polarization were performed using Saint-plus,32 and scaling and absorption correction were performed using the SADABS software package.33 Structure solution by direct methods and least-squares refinement on F2 were performed using the SHELXTL software suite.34Non-hydrogen atoms were refined with anisotropic displacement parameters, while hydrogen atoms were placed in calculated positions and refined with a riding model. The SQUEEZE subroutine of the PLATON software package35 was used to model disordered solvent molecules in L22 and 6b, and these solvent molecules are included in the respective formulae. Multi-scan absorption correction was employed in all cases. Structural figures were generated with ORTEP-3.36![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 14.3702(7), b = 15.2542(7), c = 17.2588(8) Å, α = 81.451(1)°, β = 66.469(1)°, γ = 76.867(1)°, V = 3370.8(3) Å3, ρc = 1.262 Mg m−3, θmax = 25.03°, 32672 reflections measured, 11879 independent [Rint = 0.0203], 10320 with I > 2σ(I), μ = 0.462 mm−1, Tmax = 0.9157, Tmin = 0.7684, 834 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0302/0.0785, R1/wR2 (all refl.): 0.0370/0.0825, residual electron density between −0.287 and 0.337 eÅ−3.
(no. 2), a = 9.9214(8), b = 17.118(1), c = 18.475(1) Å, α = 86.754(1)°, β = 78.725(1)°, γ = 80.859(1)°, V = 3036.9(4) Å3, ρc = 1.270 Mg m−3, θmax = 25.03°, 36687 reflections measured, 10686 independent [Rint = 0.0643], 6972 with I > 2σ(I), μ = 0.505 mm−1, Tmax = 0.7456, Tmin = 0.6780, 784 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0445/0.0849, R1/wR2 (all refl.): 0.0878/0.0987, residual electron density between −0.327 and 0.338 eÅ−3.
(no. 2), a = 9.9023(5), b = 16.8670(9), c = 20.711(1) Å, α = 101.974(1)°, β = 91.114(1)°, γ = 95.296(1)°, V = 3366.9(3) Å3, ρc = 1.199 Mg m−3, θmax = 25.03°, 40833 reflections measured, 11870 independent [Rint = 0.0212], 10042 with I > 2σ(I), μ = 0.459 mm−1, Tmax = 0.7456, Tmin = 0.6873, 829 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0387/0.1026, R1/wR2 (all refl.): 0.0465/0.1073, residual electron density between −0.421 and 0.684 eÅ−3.
(no. 2), a = 11.466(1), b = 19.564(3), c = 19.662(3) Å, α = 99.640(2)°, β = 103.416(2)°, γ = 92.916(2)°, V = 4210(1) Å3, ρc = 1.458 Mg m−3, θmax = 25.12°, 41317 reflections measured, 14955 independent [Rint = 0.1034], 7374 with I > 2σ(I), μ = 0.487 mm−1, Tmax = 0.9770, Tmin = 0.8476, 1170 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0785/0.1742, R1/wR2 (all refl.): 0.1704/0.2148, residual electron density between −0.665 and 0.724 eÅ−3.
702 reflections measured, 14960 independent [Rint = 0.0443], 11787 with I > 2σ(I), μ = 0.612 mm−1, Tmax = 0.8489, Tmin = 0.7608, 1339 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0613/0.1516, R1/wR2 (all refl.): 0.0797/0.1617, residual electron density between −0.636 and 0.852 eÅ−3.
(no. 2), a = 14.3702(7), b = 15.2542(7), c = 17.2588(8) Å, α = 81.451(1)°, β = 66.469(1)°, γ = 76.867(1)°, V = 3370.8(3) Å3, ρc = 1.262 Mg m−3, θmax = 25.03°, 32672 reflections measured, 11879 independent [Rint = 0.0203], 10320 with I > 2σ(I), μ = 0.462 mm−1, Tmax = 0.9157, Tmin = 0.7684, 834 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0302/0.0785, R1/wR2 (all refl.): 0.0370/0.0825, residual electron density between −0.287 and 0.337 eÅ−3.
(no. 2), a = 9.9214(8), b = 17.118(1), c = 18.475(1) Å, α = 86.754(1)°, β = 78.725(1)°, γ = 80.859(1)°, V = 3036.9(4) Å3, ρc = 1.270 Mg m−3, θmax = 25.03°, 36687 reflections measured, 10686 independent [Rint = 0.0643], 6972 with I > 2σ(I), μ = 0.505 mm−1, Tmax = 0.7456, Tmin = 0.6780, 784 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0445/0.0849, R1/wR2 (all refl.): 0.0878/0.0987, residual electron density between −0.327 and 0.338 eÅ−3.
(no. 2), a = 9.9023(5), b = 16.8670(9), c = 20.711(1) Å, α = 101.974(1)°, β = 91.114(1)°, γ = 95.296(1)°, V = 3366.9(3) Å3, ρc = 1.199 Mg m−3, θmax = 25.03°, 40833 reflections measured, 11870 independent [Rint = 0.0212], 10042 with I > 2σ(I), μ = 0.459 mm−1, Tmax = 0.7456, Tmin = 0.6873, 829 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0387/0.1026, R1/wR2 (all refl.): 0.0465/0.1073, residual electron density between −0.421 and 0.684 eÅ−3.
(no. 2), a = 11.466(1), b = 19.564(3), c = 19.662(3) Å, α = 99.640(2)°, β = 103.416(2)°, γ = 92.916(2)°, V = 4210(1) Å3, ρc = 1.458 Mg m−3, θmax = 25.12°, 41317 reflections measured, 14955 independent [Rint = 0.1034], 7374 with I > 2σ(I), μ = 0.487 mm−1, Tmax = 0.9770, Tmin = 0.8476, 1170 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0785/0.1742, R1/wR2 (all refl.): 0.1704/0.2148, residual electron density between −0.665 and 0.724 eÅ−3.
702 reflections measured, 14960 independent [Rint = 0.0443], 11787 with I > 2σ(I), μ = 0.612 mm−1, Tmax = 0.8489, Tmin = 0.7608, 1339 parameters were refined, R1/wR2 [I > 2σ(I)]: 0.0613/0.1516, R1/wR2 (all refl.): 0.0797/0.1617, residual electron density between −0.636 and 0.852 eÅ−3.
Acknowledgements
P.G.H. thanks NSERC, Canada Foundation for Innovation (CFI), Canada School of Energy and Environment and GreenCentre Canada for financial support. C.A.W. acknowledges NSERC and the Alberta Ingenuity Fund (Alberta Innovates) for student awards. Thanks also to Yun Yang of GreenCentre Canada for GPC measurements and Tony Montina (University of Lethbridge) for expert technical assistance.Notes and references
- See for example: (a) W. Piers and D. Emslie, Coord. Chem. Rev., 2002, 233, 131–155 CrossRef; (b) T. Hanusa, Coord. Chem. Rev., 2000, 210, 329–367 CrossRef CAS; (c) V. Gibson and S. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS.
- See for example: (a) D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS; (b) J. L. Bolliger, O. Blacque and C. M. Frech, Angew. Chem. Int. Ed., 2007, 46, 6514–6517 CrossRef CAS; (c) A. Moncada, S. Manne, J. Tanski and L. Slaughter, Organometallics, 2006, 25, 491–505 CrossRef CAS; (d) E. Peris and R. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CAS; (e) S. Gründemann, M. Albrecht, J. Loch, J. Faller and R. Crabtree, Organometallics, 2001, 20, 5485–5488 CrossRef.
- For selected reviews see: (a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS; (b) G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS; (c) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS; (d) H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed., 1995, 34, 1143–1170 CrossRef CAS.
- For selected reviews see: (a) K. Sudesh and T. Iwata, Clean: Soil Air Water, 2008, 36, 433–442 Search PubMed; (b) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS; (c) J. Bogaert and P. Coszach, Macromol. Symp., 2000, 153, 287–303 CrossRef CAS; (d) R. Drumright, P. Gruber and D. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- (a) J. Dijkstra, H. Du and J. Feijen, J. Polym. Chem., 2011, 2, 520–527 Search PubMed; (b) N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC; (c) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC; (d) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 Search PubMed; (e) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS; (f) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- Zinc complexes: See for example: (a) D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360–2371 CrossRef CAS; (b) F. Drouin, P. O. Oguadinma, T. J. J. Whitehorn, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139–2147 CrossRef CAS; (c) H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS; (d) C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132–2133 RSC; (e) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS; (f) M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 2, 333–224 Search PubMed.
- Magnesium complexes: See for example: (a) F. Drouin, T. J. J. Whitehorn and F. Schaper, Dalton Trans., 2011, 40, 1396–1400 RSC; (b) L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775–2789 CrossRef CAS; (c) J. Ejfler, K. Krauzy-Dziedzic, S. Szafert, L. B. Jerzykiewicz and P. Sobota, Eur. J. Inorg. Chem., 2010, 3602–3609 CrossRef CAS; (d) M.-Y. Shen, Y.-L. Peng, W.-C. Hung and C.-C. Lin, Dalton Trans., 2009, 9906–9913 RSC; (e) J. Ejfler, M. Kobylka, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2005, 2047–2050 RSC; (f) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- Calcium complexes: (a) See for example: H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Polymer, 2007, 48, 2257–2262 Search PubMed; (b) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS; (c) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (d) Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS.
- Aluminium complexes: See for example: (a) A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, C. Alonso-Moreno, J. A. Castro-Osma, I. Márquez-Segovia, L. F. Sánchez-Barba, A. M. Rodríguez and J. C. Garcia-Martinez, Organometallics, 2011, 30, 1507–1522 CrossRef CAS; (b) A. D. Schwarz, Z. Chu and P. Mountford, Organometallics, 2010, 29, 1246–1260 CrossRef CAS; (c) D. J. Darensbourg and O. Karroonnirun, Organometallics, 2010, 29, 5627–5634 CrossRef CAS; (d) H. Du, A. H. Velders, P. J. Dijkstra, Z. Zhong, X. Chen and J. Feijen, Macromolecules, 2009, 42, 1058–1066 CrossRef CAS; (e) X. Pang, H. Du, X. Chen, X. Wang and X. Jing, Chem. Eur. J., 2008, 14, 3126–3136 CrossRef CAS; (f) H. Ma, G. Melillo, L. Oliva, T. P. Spaniol, U. Englert and J. Okuda, Dalton Trans., 2005, 721–727 RSC; (g) M. H. Chisholm, N. J. Patmore and Z. Zhou, Chem. Commun., 2005, 127–129 RSC.
- Ga and In Complexes: See for example: (a) M. P. Blake, A. D. Schwarz and P. Mountford, Organometallics, 2011, 30, 1202–1214 CrossRef CAS; (b) P. Horeglad, P. Kruk and J. Pécaut, Organometallics, 2010, 29, 3729–3734 CrossRef CAS; (c) A. Pietrangelo, S. C. Knight, A. K. Gupta, L. J. Yao, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 11649–11657 CrossRef CAS; (d) J.-C. Buffet, J. Okuda and P. L. Arnold, Inorg. Chem., 2010, 49, 419–426 CrossRef CAS; (e) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem. Int. Ed., 2008, 47, 2290–2293 CrossRef CAS.
- Rare earth Complexes: See for example: (a) A. Buchard, R. H. Platel, A. Auffrant, X. F. Le Goff, P. Le Floch and C. K. Williams, Organometallics, 2010, 29, 2892–2900 CrossRef CAS; (b) H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602–3621 CrossRef CAS; (c) H. Ma, T. P. Spaniol and J. Okuda, Inorg. Chem., 2008, 47, 3328–3339 CrossRef CAS; (d) N. Barros, P. Mountford, S. M. Guillaume and L. Maron, Chem. Eur. J., 2008, 14, 5507–5518 CrossRef CAS; (e) X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS; (f) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013–2030 CrossRef CAS; (g) A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem. Eur. J., 2006, 12, 169–179 CrossRef CAS; (h) H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem. Int. Ed., 2006, 45, 7818–7821 CrossRef.
- (a) M. Reinmuth, U. Wild, D. Rudolf, E. Kaifer, M. Enders, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2009, 4795–4808 CrossRef CAS; (b) M. D. Hannant, M. Schormann and M. Bochmann, J. Chem. Soc., Dalton Trans., 2002, 4071–4073 RSC; (c) Y. Sarazin, M. Schormann and M. Bochmann, Organometallics, 2004, 23, 3296–3302 CrossRef CAS; (d) S. Dagorne, M. Bouyahyi, J. Vergnaud and J.-F. Carpentier, Organometallics, 2010, 29, 1865–1868 CrossRef CAS; (e) S. Dagorne, S. Bellemin-Laponnaz and R. Welter, Organometallics, 2004, 23, 3053–3061 CrossRef.
- (a) Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS; (b) M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276–2278 RSC; (c) Y. Sarazin, V. Poirier, T. Roisnel and J.-F. Carpentier, Eur. J. Inorg. Chem., 2010, 3423–3428 CrossRef CAS; (d) J. Boerner, S. Herres-Pawlis, U. Floeke and K. Huber, Eur. J. Inorg. Chem., 2007, 5645–5651 CrossRef; (e) S. Dagorne, F. Le Bideau, R. Welter, S. Bellemin-Laponnaz and A. Maisse-Francois, Chem. Eur. J., 2007, 13, 3202–3217 CrossRef CAS.
- C. A. Wheaton and P. G. Hayes, Chem. Commun., 2010, 46, 8404–8406 RSC.
- C. A. Wheaton, B. J. Ireland and P. G. Hayes, Organometallics, 2009, 28, 1282–1285 CrossRef CAS.
- C. A. Wheaton and P. G. Hayes, Dalton Trans., 2010, 39, 3861–3869 RSC.
- B. J. Ireland, C. A. Wheaton and P. G. Hayes, Organometallics, 2010, 29, 1079–1084 CrossRef CAS.
- M. D. Hannant, M. Schormann, D. L. Hughes and M. Bochmann, Inorg. Chim. Acta, 2005, 358, 1683–1691 CrossRef.
- M. Brookhart, B. Grant and A. F. Volpe, Jr., Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- J. Sandstrøm, Dynamic NMR Spectroscopy, Academic Press, New York, 1982 Search PubMed.
- The calculated barrier is taken as an approximate value due to an inability to achieve full peak separation.
- A second set of much weaker resonances for the methyl-lactate moiety also consistently appears regardless of temperature. See the experimental details for more information.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- P r is the probability of racemic linkages, where Pr = 0.5 for atactic PLA, Pr = 1 for completely heterotactic PLA, and was calculated from the formula Pr2/2 = [rmr]. Tetrad sequences were observed in the methine region of homonuclear decoupled 1H NMR spectra. For more details see: (a) M. Zell, B. Padden, A. Paterick, K. Thakur, R. Kean, M. Hillmyer and E. Munson, Macromolecules, 2002, 35, 7700–7707 CrossRef CAS; (b) M. Chisholm, S. Iyer, D. McCollum, M. Pagel and U. Werner-Zwanziger, Macromolecules, 1999, 32, 963–973 CrossRef CAS.
- M. Chisholm, N. Patmore and Z. Zhou, Chem. Commun., 2005, 127–129 RSC.
- T. Biela, A. Duda and S. Penczek, Macromol. Symp., 2002, 183, 1–10 CrossRef CAS; A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- K. Barral, A. D. Moorehouse and J. E. Moses, Org. Lett., 2007, 9, 1809–1911 CrossRef CAS.
- A. Ouchi, B. Z. S. Awen, L. Hongxia, Y. Araki and I. Osamu, J. Photochem. Photobiol. A: Chem., 2006, 182, 238–244 CrossRef CAS.
- T. Tsuritani, H. Mizuno, N. Nonoyami, S. Kii, S. Akao, K. Sato, N. Yasuda and T. Mase, Org. Process Res. Dev., 2009, 13, 1407–1412 Search PubMed.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- APEX2, Version 2.1-4; Data Collection and Refinement Program. Bruker AXS Inc., Madison, WI, 2006.
- SAINT-Plus, Version 7.23a; Data Reduction and Correction Program. Bruker AXS Inc., Madison, WI, 2004.
- G. M. Sheldrick, SADABS, Area-Detector Absorption Correction, v2.10, Universität Göttingen, Germany, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148–155 CAS.
- M. N. Burnett, C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996.
Footnote |
| † CCDC reference numbers 833323–833330. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cy00306b |
| This journal is © The Royal Society of Chemistry 2012 |