Modified zeolite ZSM-5 for the methanol to aromatics reaction†
Marco
Conte
a,
Jose A.
Lopez-Sanchez
a,
Qian
He
b,
David J.
Morgan
a,
Yulia
Ryabenkova
a,
Jonathan K.
Bartley
a,
Albert F.
Carley
a,
Stuart H.
Taylor
a,
Christopher J.
Kiely
b,
Karim
Khalid
c and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: Hutch@cardiff.ac.uk
bCenter for Advanced Materials and Nanotechnology, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015-3195, USA
cSABIC Technology & Innovation, Riyadh 11551, Saudi Arabia
First published on 27th October 2011
Abstract
Catalysts comprising zeolite ZSM-5 impregnated with precious metals including Ag, Cu, Ni, Pd, Ir and Ru, have been tested for the methanol to hydrocarbons reaction in a continuous flow fixed bed reactor. Comparison with the activity of unmodified ZSM-5 showed that Ag, Cu and Ni enhanced the selectivity to C6–C11 aromatic products by a factor of two or higher. Moreover, Ag/ZSM-5 showed improved selectivity for the C6–C7 fraction of aromatic products. Ni/ZSM-5 was found to be selective to naphthalene, while Cu/ZSM-5 was selective for C9–C11 aromatic products. It was ascertained that all the impregnated metals were present as metal oxides in the starting materials. It is therefore proposed that the enhanced selectivity to aromatic products is due to the interaction of the acid sites of the zeolite with the basic sites of the metal oxide at the edge of the zeolite crystals, as well as the possible coordination of propene molecules formed during the reaction, that are likely to be the building blocks for the formation of aromatics.
1. Introduction
The conversion of methanol to hydrocarbons is one of the most investigated reactions in the field of industrial chemistry since its discovery in 1977.1 However, despite significant improvements in the activity of zeolite-based materials for aromatics formation, particularly by doping the zeolites with Ga based precursors,2 few results have been obtained that demonstrate an increase in the selectivity to a specific aromatic product, such as toluene, ethyl benzene or naphthalene. This target is complicated by the existence of dynamic equilibria between these aromatic molecules,3 as in the case of the xylenes,4 as well as the possibility of cracking and isomerisation reactions in which branched alkyl groups can easily migrate on the aromatic ring.5This prompted us to focus in the preparation of new materials capable of demonstrating higher selectivity and to investigate the effect of the impregnation of metal nitrates or chlorides of Pd, Ir, Ni, Cu, Ru and Ag on commercial ZSM-5. These materials at present find application in other chemical processes rather than the methanol to aromatics reaction. Pd and Ir modified ZSM-5 are active for the isomerisation of alkanes6 and propane aromatisation;7 Ni and Cu in the methylation of toluene with methanol8 or in the aromatics formation from propene respectively;9 Ru is used in the deoxygenation of methanol10 or in ethane aromatisation,11 while Ag has received limited attention for the methanol to aromatics reaction,12 but these studies were mainly focused in the field of materials science13 rather than catalysis.
Here we report a comparative study of Ag, Ni, Cu, Pd, Ir and Ru doped ZSM-5 for the methanol to aromatics reaction by carefully examining selectivity to a specific aromatic product versus time, and using XRPD to gain structural information on the nature of the active sites of these materials. Particularly, we observed that Ag, Ni and Cu doped zeolites have a higher selectivity to aromatics than commercial ZSM-5, by a factor of at least two under our experimental conditions, as well as enhanced selectivity to specific aromatics such as naphthalene. In contrast, Pd, Ir and Ru showed no significant enhancement. This highlighted the importance of olefins as building block to aromatic products, and the relevance of these metals to coordinate propene and similar olefins, by investigating the propene consumption in the products selectivity.
2. Experimental
2.1 Catalyst preparation
2.2 Catalytic tests and characterization of the products
All the catalysts were prepared in pellet form, obtained by pressing the solids twice at 2 tons cm−2 for 1 min. The pellets were then ground and sieved, collecting the fraction between 850 and 600 μm. Catalytic tests for the methanol to aromatics reaction were carried out using a glass reactor at 450 °C under N2 flow (88 mL min−1, inlet pressure 4.5 bar) with a methanol feeding of 320 μL h−1 for up to 12 h time on stream.Prior to the start of the reaction, all catalysts were pre-treated in air at 500 °C for 1 h. The reaction mixture was analysed via gas chromatography using a Varian CP-3800 Gas Chromatograph and a CP-Porabond Q column (25 m length, 0.53 mm diameter, 10 μm film thickness). Identification of the reaction products was carried out by comparison of the retention time with standard materials. Quantification of the hydrocarbons was carried out using a flame ionization detector (FID), while H2 was quantified using a thermal conductivity detector (TCD).
2.3 Characterization of the catalysts
3. Results and discussion
3.1 Methanol to aromatics selectivity over precious metal doped ZSM-5 catalysts
All the catalysts prepared showed conversion values close to 100% during a time on-stream of 12 h under our test conditions (Fig. 1), showing virtually no difference with the undoped zeolite ZSM5 used for comparison. In contrast, the addition of different metals had clear effects on the selectivity to aromatics for the total fraction of C6–C11 carbon atoms, making it possible to divide these materials into two groups: (i) a group including Pd, Ir and Ru which do not display any improvement to total aromatic selectivity (C6–C11 selectivities between 25 and 30%) when compared to zeolite ZSM-5 (Fig. 2 and Table S1) and (ii) a group including Ag, Ni and Cu, which in contrast, are capable of enhancing the selectivity to aromatics up to ca. 45, 50 and 60% respectively for the C6–C11 fraction (Fig. 3 and Table S1). It should also be noted, that in all cases a closed carbon mass balance, compatible with 100% within the experimental error, was observed (Table S2). | ||
| Fig. 1 Selectivity to aromatics for the fraction C6–C11 of: (■) ZSM-5, (◆) Pd/ZSM-5, (◀) Ir/ZSM-5, (▶) Ru/ZSM-5, (●) Ag/ZSM-5, (▲) Ni/ZSM-5 and (▼) Cu/ZSM-5. Open symbols correspond to methanol conversion (the metal loading for all catalysts was 1% wt). | ||
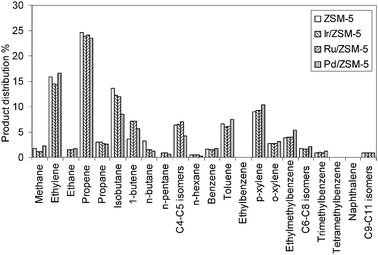 | ||
| Fig. 2 Product distribution for Ir/ZSM-5, Ru/ZSM-5 and Pd/ZSM-5. ZSM-5 is reported for comparison. No significant difference is detected when the zeolite is impregnated with Ir, Ru or Pd (C9–C11 isomers: aromatic products; for a detailed description of the identified products see ESI,† Table S1). | ||
 | ||
| Fig. 3 Product distribution for Ag/ZSM-5, Cu/ZSM-5 and Ni/ZSM-5. ZSM-5 is reported for comparison. Ag impregnated zeolite promotes toluene formation, while Cu promotes the faction C9–C11 carbon atoms, and Ni shows tuned selectivity to naphatalene (C9–C11 isomers: aromatic products; for a detailed description of the identified products see ESI,† Table S1). | ||
The use of transition metals as dopants for zeolites is not unprecedented in the area of aromatics formation for Ru or Cu,20,21 using alkanes as substrates and this has been so far confirmed for metals such as Mo, Fe, V, W and Cr.22 However, in our tests, using methanol as substrate, Pd, Ir and Ru were completely ineffective. A possible explanation for this behaviour could be that while in reactions such as methane conversion to aromatics the first step of the reaction is a hydrogen abstraction process from CH4 to CH3˙ or H−–CH3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20,23 in the methanol to aromatics reaction, the first step is invariably a dehydration step from CH3–OH to dimethylether CH3–O–CH3 and from here to aromatics, independently from which overall reaction mechanism is considered.24 In contrast, Ag, Ni and Cu were capable of enhancing the selectivity to aromatic products. However, it should be noted that due to the preparation method used, all these metals are expected to be present in oxide form on the ZSM-5 framework, and this is confirmed by XPS analysis (Table 1). We have previously demonstrated that, in the case of Ga/ZSM-5 enhanced selectivity to aromatics was the result of an interaction between the metal oxide species Ga2O3 and the Brønsted acid sites on the zeolite25 with an effect denoted as ‘contact synergy’,26 which has been observed in mixed oxides comprising MnMoO4 and MoO3 for the oxidation of C4 hydrocarbons. We consider that it is likely this effect is taking place also in the present case.
20,23 in the methanol to aromatics reaction, the first step is invariably a dehydration step from CH3–OH to dimethylether CH3–O–CH3 and from here to aromatics, independently from which overall reaction mechanism is considered.24 In contrast, Ag, Ni and Cu were capable of enhancing the selectivity to aromatic products. However, it should be noted that due to the preparation method used, all these metals are expected to be present in oxide form on the ZSM-5 framework, and this is confirmed by XPS analysis (Table 1). We have previously demonstrated that, in the case of Ga/ZSM-5 enhanced selectivity to aromatics was the result of an interaction between the metal oxide species Ga2O3 and the Brønsted acid sites on the zeolite25 with an effect denoted as ‘contact synergy’,26 which has been observed in mixed oxides comprising MnMoO4 and MoO3 for the oxidation of C4 hydrocarbons. We consider that it is likely this effect is taking place also in the present case.
In order to probe the catalytic behaviour of these metals, the selectivity to all the reaction products versus time on stream was carefully examined. For the set of metals including Pd, Ir and Ru there are no significant differences when compared with zeolite ZSM-5, even when comparing the fine details of the product distribution (Fig. 2). The only distinction concerns the absence of methane formation in the initial stages of the reaction, with methane only being detected above a 10% level after 10 h of time on stream. Methane formation can either be related to metal sintering27 or coke deposition with a decrease in the number of the acid sites of the zeolite.28
In contrast, Ag, Ni and Cu enhance the overall selectivity to aromatics, but also display different tuning properties within the total aromatics distribution (Fig. 3). In fact, Ag appears to be particularly selective for the fraction C6–C8 (ca. 40%, while Ni leads to high amounts of naphthalene (ca. 7%). Finally, Cu displays an intermediate behaviour between Ag and Ni enhancing the C9–C11 fraction up to 20% when compared to ZSM-5, which is selective to these aromatic fractions, in the range of 25% for the C6–C8 group, ca. 1% for C9–C11, and no naphthalene was observed. Control tests comparing the catalytic performance of these materials with Ga/ZSM-5 and Zn/ZSM-5, which are well known catalysts for the conversion of methanol to aromatics29,30 were also carried out (Fig. S1–S4). These data showed that Ag, Ni, and Cu can be useful alternatives to Ga and Zn, as in fact these two catalysts are capable of producing ca. 50 and 55% of aromatics respectively under our experimental conditions.
3.2 Structure of the catalysts and comments on the nature of the active sites
It should be noted that despite its apparent simplicity, the impregnation of metal precursors over zeolite is a complex process that can lead to the formation of metal nanoparticle (or metal oxides) either inside or outside the zeolite pores.31 Moreover, it must be emphasised that impregnation involves a calcination step above 500 °C; it is therefore unlikely that the metal precursor remains in metallic form on the zeolite material, but rather is present as metal oxide.In view of this, and to obtain structural information on the nature of the active sites and in turn the selectivity, all the catalysts were characterized using X-ray powder diffraction, before and after reaction. No significant differences are observed in terms of changes in the zeolite framework (Fig. 4), unit cell parameters or dealumination (Fig. S5 and Table S3), with the only exception of Ni/ZSM-5 where a slight increase in the unit cell volume was detected (ca. 0.6%). This could also contribute to the observed product distribution for this metal. However, these data are consistent overall with the preparation procedure used, which is not expected to lead to any incorporation of the metal cations in the zeolite framework,32 but rather inside the channels or at the edges of the zeolite. In contrast, the most important feature from the XRPD patterns is the presence of the doping metal in the form of metal oxide before the reaction and in reduced, metallic, form after the reaction for both: the set comprising Pd, Ir and Ru impregnated zeolite (Fig. 5) and the set comprising Ag, Cu and Ni impregnated materials (Fig. 6). This was further confirmed by means of XPS (Table 1) which also allowed unambiguous assignment of the oxidation states of the doping metals before reaction, and therefore ruling out the existence of mixed metal oxides formation.13
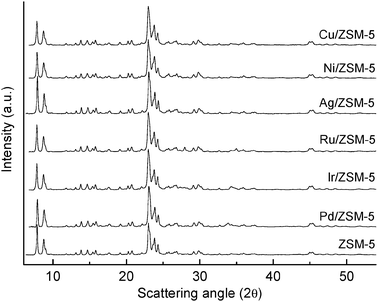 | ||
| Fig. 4 XRPD patterns of ZSM-5, Pd/ZSM-5, Ir/ZSM-5, Ru/ZSM-5, Ag/ZSM-5, Ni/ZSM-5 and Cu/ZSM-5, from 6 to 55° 2θ. | ||
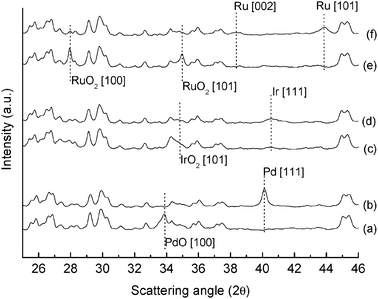 | ||
| Fig. 5 Enlarged 2θ region from 25 to 46° for: (a) Pd/ZSM-5 before reaction, and (b) Pd/ZSM-5 after reaction; (c) Ir/ZSM-5 before reaction and (d) Ir/ZSM-5 after reaction; (e) Ru/ZSM-5 before reaction and (f) Ru/ZSM-5 after reaction; PdO,33 Pd,34 IrO2,35 Ir36 RuO2,37 and Ru38 before and after reaction for each doping metal are detected respectively. | ||
 | ||
| Fig. 6 Enlarged 2θ region from 25 to 46° for: (a) Ag/ZSM-5 before reaction and (b) Ag/ZSM-5 after reaction; (c) Ni/ZSM-5 before reaction, and (d) Ni/ZSM-5 after reaction; (e) Cu/ZSM-5 before reaction, and (f) Cu/ZSM-5 after reaction; AgO,39 Ag,40 NiO,41 and Ni,42 before and after reaction for each doping metal are detected respectively; In the case of CuO,43 and Cu, for the Cu/ZSM-5 sample after reaction, Cu,44 is not detected due to fluorescence under our experimental conditions. | ||
Previous studies on the fine tuning of selectivity by doped ZSM-5 using metals such as Ga, Zn and Co,45–47 showed that the alkenes, which are also formed during the methanol to aromatic process, are formed via alkane dehydrogenation, while aromatics are mainly formed by dehydrocyclization reactions of alkenes. For these metals the enhanced selectivity was due to removal of hydrogen as dihydrogen by the metal doping atoms during the reaction.45 It is considered that this phenomenon occurs also for the metals investigated in the present study, and it is responsible for the reduction of the initial metal oxide species to the metal, as a small amount of hydrogen (ca. 2%) in the effluent from the reactor was detected. It should also be noted that no CO or CO2 were detected at any stage of the reaction. Control tests for Ag/ZSM-5 on the oxidation state of the metal using XRPD (Fig. S6) also showed that the reduction of the metal occurs simultaneously to the start of methanol feeding.
This can also explain the behaviour and the differences between the metals chosen to impregnate the zeolite. In fact, as the metals are present in metal oxide form in the fresh catalysts, and therefore as metal cations, these can display different coordination capability to olefins, which are known to be intermediates in the formation of the aromatic products48 and consequently a modified selectivity. Indeed, Ag2+, Ni2+ and Cu2+ are all effective metals for coordinating olefins,49 and this could promote the aromatic formation. Furthermore, among these metals, Ni2+ in also an excellent aromatic/benzene coordinating species,50 and this could explain the build up of naphthalene units from a benzene precursor over the metal surface.
In addition, control tests on a fully reduced Ag/ZSM-5 catalyst (Fig. S7) showed this material was not active for the formation of C6–C9 aromatics, with the formation of high amounts of dimethyl ether, C9+ products and coke, thus showing the importance of the presence of cationic species in the starting material in order to ensure reactivity to the required product range.
However, the situation appears more complex. Zeolites are shape selective catalysts, with the geometry of the zeolite pores as well as the opening of the zeolite channels, able to modify the final product distribution.24 Therefore, if the pore diameters are modified, i.e. reduced by the presence of metal oxide clusters, a different selectivity is expected. Conversely, if the intermediates cannot be formed inside the pores, because completely occupied by the metal oxide nanoparticles, acid or metal sites located over the zeolite crystals will be substantially inactive.51 In view of this, particle sizes of both the starting metal oxides and final reduced metal nanoparticle were calculated from the XRPD patterns (Table 2).
| Zeolite | Dopant metal phase and particle size (nm) before reaction | Dopant metal phase and particles size (nm) after reactiona |
|---|---|---|
| a For Cu the particle size determination was not possible due to fluorescence under our experimental conditions. | ||
| Pd/ZSM-5 | PdO (58) | Pd (29) |
| Ir/ZSM-5 | IrO2 (68) | Ir (11) |
| Ru/ZSM-5 | RuO2 (25) | Ru (19) |
| Ag/ZSM-5 | AgO (82) | Ag (52) |
| Ni/ZSM-5 | NiO (51) | Ni (17) |
| Cu/ZSM-5 | CuO (51) | Cu (−) |
Zeolite ZSM-5 consists of two perpendicular intersecting channel systems having cross sections of 5.4 × 5.6 Å and 5.1 × 5.4 Å,24 and a channel intersection diameter of ca. 9 Å. However, from the data reported in Table 1, the average metal and metal-oxides particle sizes are far larger then these values, ranging from 11 nm for Ir to 82 nm for AgO, thus suggesting that these species are outside the pores and possibly at the edge of the zeolite crystals.31 Although the total error associated to the particle size determinations was estimated to be ca. 30%, this still allows the determination of the location of the metal and metal oxide clusters on the final doped zeolite material, because the final size is from 1 to 2 order of magnitude bigger that the zeolite pore size. On the other hand, this does not preclude the existence of smaller clusters, which could not be detected using XRPD. This was confirmed for Ag/ZSM-5 where TEM images were acquired (Fig. 6). Electron microscopy of Ag/ZSM-5 showed a large dispersion of the silver oxide nanoparticles spanning from 20 to 100 nm, and matching the particle size obtained using XRPD and therefore constricting the particles to the outside of the zeolite crystals (Fig. 7A and B). However, a subset of particles below 10 nm and centred in the range from 1 to 2 nm is also detected (Fig. 7C and D). For this set of particles, a smaller diameter also means that the fraction of the total surface area represented by non basal plane of silver oxide surfaces in the crystallites is much higher, and these may be either catalytically more reactive or more easily converted to Ag metal.52 It is reasonable to consider that this occurs also for all the other metals used to impregnate the zeolite.45 All these particles and clusters are at the edge of the zeolite crystals, and this can explain the trends observed for the impregnated metals tested in the present study via the concept of contact synergy postulated above.23,24 In fact for Pd, Ir and Ru with the exception of a higher methane formation, no other difference is present when compared to undoped ZSM-5 under our experimental conditions. This suggests that no interaction is present at the junction between the clusters of the metal oxides PdO, IrO2, RuO2 and the crystallites of ZSM-5. In contrast the enhanced aromatics formation observed for Ag, Ni and Cu would be the result of the interaction between the basic centres of AgO, NiO and CuO with the acid sites of the zeolites.
 | ||
| Fig. 7 TEM of Ag/ZSM-5. Two sets of particles population are detected. (A and B) a set containing large particles in the range 20 to 100 nm and (C and D) a set containing smaller particles in the range 1 to 2 nm. | ||
In view of this, the observed changes in selectivity would not be a consequence of narrower channel sizes,53 and the data could also explain the lack of formation of large highly substituted aromatics like in the case of Ag/ZSM-5, as well as preventing the blockage of the pores by subsequent coke formation. In order to verify this assumption, surface area and micropore volume were systematically determined for all the catalysts prepared before and after the methanol to aromatics reaction (Table 3).
| Catalyst | Micropore volume (mL g−1) | Surface area (m2 g−1) | ||
|---|---|---|---|---|
| Before reaction | After reactiona | Before reaction | After reactiona | |
| a The values in bracket after reaction are obtained subtracting the carbon content, as determined from TGA, as correction factor. | ||||
| ZSM-5 | 0.293 | 0.249 (0.250) | 360 | 272 (273) |
| Pd/ZSM-5 | 0.293 | 0.279 (0.284) | 310 | 278 (283) |
| Ir/ZSM-5 | 0.302 | 0.295 (0.297) | 317 | 312 (314) |
| Ru/ZSM-5 | 0.301 | 0.280 (0.281) | 332 | 302 (304) |
| Ag/ZSM-5 | 0.315 | 0.273 (0.273) | 327 | 286 (286) |
| Ni/ZSM-5 | 0.320 | 0.237 (0.250) | 345 | 207 (213) |
| Cu/ZSM-5 | 0.321 | 0.292 (0.293) | 329 | 311 (312) |
We observe that no significant changes are present among the catalysts before reaction in terms of micropore volume, but there is a systematic reduction of the micropore volume after reaction. This is important, because it suggests that the metal oxide nanoparticles are not formed inside the pore channels, thus corroborating the results obtained by XRPD and TEM. No significant difference in surface area is observed for all fresh catalysts, but there is a reduction of the surface area after reaction, with the most marked decrease being for Ni/ZSM-5, and this is consistent with the formation of naphthalene based products during the reaction for this catalyst. Moreover, for all the catalysts coke deposition was detected (Table 4), and Ni/ZSM-5 is most affected by coking (ca. 4.7%), then Pd/ZMS-5 (ca. 1.6%) while Ag is the additive less affected by carbonaceous products formation (ca. 0.14%). It is likely that coke formation starts at the metal oxide surface first and then propagates to the zeolite channels.
| Catalyst | Coke content after reaction (%) | Maximum selectivity to aromatics (%) | Selectivity to aromatics after 11 h time on stream (%) |
|---|---|---|---|
| a Estimated value due to concomitant CuO formation during TGA conditions. | |||
| ZSM-5 | 0.18 | 30.1 | 27.6 |
| Pd/ZSM-5 | 1.60 | 33.6 | 32.1 |
| Ir/ZSM-5 | 0.49 | 31.1 | 28.2 |
| Ru/ZSM-5 | 0.40 | 33.1 | 30.1 |
| Ag/ZSM-5 | 0.14 | 55.1 | 39.7 |
| Ni/ZSM-5 | 4.69 | 59.6 | 52.3 |
| Cu/ZSM-5 | 0.28a | 55.1 | 53.8 |
This further sustains the model of contact synergy. Among the set of metal oxides present in our materials, AgO, CuO and NiO are markedly basic compared with PdO, IrO2 and RuO2, and this may then explain why they are active. In contrast, for PdO, IrO2 and RuO2 as contact synergy is not operating, it is irrelevant if coke formation occurs or not because this will take place on sites which are not active anyway, and this explain why these catalysts, unlike in the case of AgO, CuO and NiO, do not deactivate with time on stream. On the other hand, the variability in coke formation for the active metals does not result in a corresponding loss of selectivity to aromatics (Table 4), except for Ni/ZSM-5, therefore it is also possible that the reduction of the metal, particularly in the case of Ag/ZSM-5 can be responsible of the decreased selectivity at the end of the reaction.
3.3 Comments on the selectivity to aromatics and propene consumption
Several models have been so far proposed in order to explain the selectivity, and in particular the shape selectivity, of zeolites, regardless of whether they are metal doped or not. These models include: configurational diffusion controlled selectivity,54 reverse molecular selectivity,55 molecular traffic control56 and a hydrocarbon pool mechanism.57 The latter is one of the most endorsed, particularly in the methanol to hydrocarbons reaction. The essential feature of the hydrocarbon pool route postulates that methanol reacts with hydrocarbon species in the catalyst to begin a sequence of steps leading to olefins and regeneration of the original hydrocarbon in a catalytic cycle.58,59 Therefore, in this model the role and presence of olefins such as propene is essential. In view of this, we wanted to establish if any correlation between propene consumption and aromatics formation was present, i.e. if propene can be a building block to aromatics.If the mean selectivity to total aromatics, for the fraction C6–C11, is plotted against the whole series of catalysts reported in the present study (Fig. 8), it is possible to observe that as long as more aromatics are obtained, then less propene is detected, thus suggesting that the aromatics are obtained at the expense of propene, this is also supported by experiments in which feeding Cu/ZSM-5 with propene led to aromatics.9 We consider this to be a key correlation in the methanol to aromatics process, suggesting that although the catalytic process must involve methanol dehydration to start the reaction, aromatics formation can also occur with propene as building block. This would also support co-feeding experiments that make use of methanol/propene mixtures to increase the formation of aromatic products.60 In order to support this experimental observation further, propene, propane and n-butane were used as substrates over ZSM-5 (Fig. S8). Formation of aromatics products up to 40% was observed when propene was used. In contrast, no clear trend from the amount of ethylene detected can be drawn (Fig. 8) and our data cannot be used for mechanistic considerations for this the origin of this molecule. At present, the most accredited mechanism for ethylene formation is the one proposed by Dessau61 in which ethylene is an alkene cracking product. On the other hand, more recent reports would suggest that ethylene is formed independently from cracking of higher alkenes.59 It is considered likely both mechanisms are operating.
 | ||
| Fig. 8 Average propene and ethylene formation for ZSM-5, Ir/ZSM-5, Ru/ZSM-5, Pd/ZSM-5, Ag/ZSM-5, Cu/ZSM-5 and Ni/ZSM-5 ordered per total aromatics C6–C11 formation. The fraction, C1–C6 carbon atoms, is also reported for comparison. | ||
4. Conclusions
Metal doped ZSM-5 zeolites comprising Ag, Ni and Cu showed improved selectivity to aromatics with methanol as a feedstock, compared with ZSM-5. All these metals are present in the form of metal oxide in the starting doped materials, and the enhanced selectivity of Ag, Ni and Cu, could be explained by a more efficient coordination of olefinic intermediates, which are building blocks to aromatic products.62,63 It is likely that this process takes place on metal oxide clusters at the edge of the zeolite crystals. In contrast, metals like Pd, Ir and Ru, despite being efficient doping agents to aromatisation processes including alkanes, did not display any enhanced selectivity to aromatics. This is possibly due to the lack of any contact interaction between the metal oxide clusters and the zeolite crystals. Moreover, fine tuning of the selectivity to aromatics within the C6–C11 fraction was observed using different metals. In particular, Ni/ZSM-5 displayed a selectivity centred on naphthalene derivatives, while Cu was centred on the fraction of C9–C11 species. Finally for Ag selectivity was to the C6–C8 fraction. We consider these findings important for the design of zeolites with the desired characteristics towards the enhancement of aromatics formation and the fine tuning of the selectivity to a specific product.Acknowledgements
The authors thank SABIC for financial support.References
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS.
- D. Freeman, R. P. K. Wells and G. J. Hutchings, Catal. Lett., 2002, 82, 217–225 CrossRef CAS.
- J. Das, B. S. Bhat and A. B. Halgeri, Catal. Lett., 1994, 23, 161–168 CrossRef CAS.
- L. M. Polinskit and M. J. Ballard, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 540–544 CrossRef.
- T. F. Degnan Jr, C. M. Smith and C. R. Venkat, Appl. Catal., A, 2001, 221, 283–294 CrossRef.
- A. de Lucas, J. L. Valverde, P. Sánchez, F. Dorado and M. J. Ramos, Ind. Eng. Chem. Res., 2004, 43, 8217–8225 CrossRef CAS.
- R. Maggiore, S. Scirè, S. Galvagno, C. Crisafulli and G. Toscano, Appl. Catal., A, 1991, 79, 29–40 CrossRef CAS.
- S. Faramawy, Pet. Sci. Technol., 1999, 17, 249–271 CrossRef CAS.
- Z. Popova, K. Aristirova and C. Dimitrov, React. Kinet. Catal. Lett., 1990, 41, 369–374 CrossRef CAS.
- A. Mahay, F. Simard, G. Lemay, A. Adnot, S. Kaliaguine and J. Monnier, Appl. Catal., 1987, 33, 55–71 CrossRef CAS.
- U. Mroczek, F. Roessner and K. H. Steinberg, React. Kinet. Catal. Lett., 1991, 43, 559–563 CrossRef CAS.
- Y. Inoue, K. Nakashiro and Y. Ono, Micropor. Mesopor. Mater., 1995, 4, 379–383 CAS.
- D. Zeng, J. Yang, J. Wang, J. Xu, Y. Yang, C. Ye and F. Deng, Microporous Mesoporous Mater., 2007, 98, 214–219 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-Ray Diffraction, Prentice-Hall Inc, Upper Saddle River, 3rd edn, 2001, p. 236 Search PubMed.
- J. I. Langford, J. Appl. Crystallogr., 1978, 11, 10–14 CrossRef CAS.
- K. Sing, Colloids Surf., A, 2001, 187–188, 3–9 CrossRef CAS.
- A. Sayari, E. Crusson, S. Kaliaguine and J. R. Brown, Langmuir, 1991, 7, 314–317 CrossRef CAS.
- H. E. Robson, Verified syntheses of zeolitic materials, Elsevier Science B. V., Amsterdam, 2nd edn, 2001, p. 64 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- O. V. Bragin, T. V. Vasina, A. V. Preobrazhenskii and Kh. M. Minachev, Izv. Akad. Nauk. SSSR. Ser. Khim., 1989, 3, 750–751 Search PubMed.
- T. V. Vasina, A. V. Preobrazhenskii, S. A. Isaev, O. V. Chetina, O. V. Masloboishchikova and O. V. Bragin, Kinet. Katal., 1994, 35, 106–109 CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338–346 CrossRef CAS.
- Y. Xu, W. Liu, S. Wong, L. Wang and X. Guo, Catal. Lett., 1996, 40, 207–214 CrossRef CAS.
- N. Y. Chen, T. F. Degnan Jr. and C. Morris Smith, Molecular transport and reactions in zeolites, VCH, New York, 1994, p. 135 Search PubMed.
- D. Freeman, R. P. K. Wells and G. J. Hutchings, J. Catal., 2002, 205, 358–365 CrossRef CAS.
- S. Ozkan, M. R. Smith and S. A. Driscoll, Stud. Surf. Sci. Catal., 1992, 72, 363–377 CrossRef.
- J. A. Z. Pieterse, R. W. van den Brink, S. Booneveld and F. A. de Bruijn, Appl. Catal., B, 2002, 39, 167–179 CrossRef CAS.
- G. Cheng, Q. Liang, G. Cai, S. Zhao and M. Ying, in New Developments in Zeolite Science and Technology, ed. Y. Murakami, A. Lijima and J. W. Ward, Elsevier, Tokyo, 1986, p. 907 Search PubMed.
- E. Lalik, X. Liu and Jacek Klinowski, J. Phys. Chem., 1992, 96, 805–809 CrossRef CAS.
- Y. Ono, H. Adachi and Y. Senoda, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1091–1099 RSC.
- S. Hashimoto, T. U. wada, H. Masuhara and T. Asahi, J. Phys. Chem. C, 2008, 112, 15089–15093 CAS.
- R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2406 CrossRef CAS.
- D. Grier and G. McCarthy, ICDD Grant-in-Aid, 1991, in International Centre for Diffraction Data, Powder Diffraction File, Entry 43-1024, 1996.
- A. Kern and W. Eysel, ICDD Grant-in-Aid, 1993, in International Centre for Diffraction Data, Powder Diffraction File, Entry 46-1043, 1996.
- US Natl. Bur. Stand. Monogr., 1965, in International Centre for Diffraction Data, Powder Diffraction File, Entry 15-870, 1996.
- A. Kern and W. Eysel, ICDD Grant-in-Aid, 1993, in International Centre for Diffraction Data, Powder Diffraction File, Entry 46-1044, 1996.
- J. Welton and G. McCarthy, ICDD Grant-in-Aid, 1989, in International Centre for Diffraction Data, Powder Diffraction File, Entry 40-1290, 1996.
- US Natl. Bur. Stand. Circ., 1955, in International Centre for Diffraction Data, Powder Diffraction File, Entry 6-663, 1996.
- D. Grier and G. McCarthy, ICDD Grant-in-Aid, 1991, in International Centre for Diffraction Data, Powder Diffraction File, Entry 43-1038, 1996.
- US Natl. Bur. Stand. Circ., 1953, in International Centre for Diffraction Data, Powder Diffraction File, Entry 4-783, 1996.
- US Natl. Bur. Stand. Circ., 1953, in International Centre for Diffraction Data, Powder Diffraction File, Entry 4-835, 1996.
- G. Carturan, G. Cocco, S. Enzo, R. Ganzerla and M. Lenarda, Mater. Lett., 1988, 7, 47-50, in International Centre for Diffraction Data, Powder Diffraction File, Entry 45-1027, 1996.
- US Natl. Bur. Stand. Circ., 1953, in International Centre for Diffraction Data, Powder Diffraction File, Entry 5-661, 1996.
- US Natl. Bur. Stand. Circ., 1953, in International Centre for Diffraction Data, Powder Diffraction File, Entry 4-836, 1996.
- E. Iglesia, J. E. Baumgartner and G. L. Price, J. Catal., 1992, 134, 549–571 CrossRef CAS.
- J. A. Biscardi and E. Iglesia, J. Catal., 1999, 182, 117–128 CrossRef CAS.
- S. Y. Yu, G. J. Yu, W. Li and E. Iglesia, J. Phys. Chem. B, 2002, 106, 4714–4720 CrossRef CAS.
- A. J. Marchi and G. F. Froment, Appl. Catal., 1991, 71, 139–151 CrossRef CAS.
- F. A. Cotton, G. Wilkinson and P. L. Gaus, Basic Inorganic Chemistry, John Wiley & Sons, Inc., New York, 3rd edn, 1995, p. 609 Search PubMed.
- F. A. Cotton, G. Wilkinson, G. A. Morillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, Inc., New York, 6th edn, 1990, pp. 852–853 Search PubMed.
- P. Cañizares, A. de Lucas, F. Dorado and D. Pérez, Appl. Catal., A, 2000, 190, 233–240 CrossRef.
- M. Conte, T. Davies, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 252, 23–29 CrossRef CAS.
- L. Quian and Z. F. Yan, Colloids Surf., A, 2001, 180, 311–316 CrossRef.
- P. B. Weisz, Chemtech., 1973, 3, 498–505 CAS.
- E. G. Derouane and Z. Gabelica, J. Catal., 1980, 65, 486–489 CrossRef CAS.
- L. D. Rollman, J. Catal., 1997, 47, 113–121 CrossRef.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Petter Lillerud, S. Kolboe and B. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS.
- K. P. Möller, W. Böhringer, A. E. Schnitzler and C. T. O'Connor, Stud. Surf. Sci. Catal., 2000, 130, 2711–2716 CrossRef.
- R. M. Dessau, J. Catal., 1986, 99, 111–116 CrossRef CAS.
- W. O. Haaga, R. M. Lagoa and P. G. Rodewalda, J. Mol. Catal., 1982, 17, 161–169 CrossRef.
- O. A. Anunziata, L. B. Pierella and R. G. Marino, React. Kinet. Catal. Lett., 1995, 54, 229–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chromatographic data, products identification, control tests with Ga and Zn/ZSM-5, details of the crystal structure of the catalysts, reduced catalysts and hydrocarbons feeding over ZSM-5. See DOI: 10.1039/c1cy00299f |
| This journal is © The Royal Society of Chemistry 2012 |