Oxidative esterification of 1,2-propanediol using gold and gold-palladium supported nanoparticles
Received
5th July 2011
, Accepted 12th August 2011
First published on 31st August 2011
Introduction
The utilisation of 1,2-propanediol for the synthesis of fine chemicals has recently attracted significant attention from the scientific community.1–3 A possible route to 1,2-propanediol from hydrogenolysis of glycerol is attractive and opens the possibility for increased usage from a renewable source.4Glycerol production is expected to increase significantly in the near future as it is a by-product (10 wt/%) from biodiesel production.5–8 The most studied heterogeneous catalysts for catalytic applications of aerobic oxidation of polyols, such as 1,2-propanediol and glycerol are based on supported, noble metal nanoparticles, particularly gold.9–21
The high catalytic efficiency of gold-based supported nanoparticles has been demonstrated for a variety of oxidation and hydrogenation reactions such as CO,22–28alcohol29–37 and alkene/alkane oxidation,38–43hydrogen peroxide synthesis44–49 and hydrogenation reactions.50 In the case of polyol oxidation, it has been demonstrated that the utilisation of supported gold nanoparticles is successful for achieving high catalytic activity and selectivity to specific products. Systematic studies based on the influence of the metal particle size and the metal support have shown that the oxidation of polyols is sensitive to the reactant/structure.13–17,19,20,51–53 Moreover, the influence of a variety of preparation methods has been studied for the synthesis of active supported metal catalysts, such as impregnation, deposition-precipitation and colloidal methods. Colloidal methods typically produce the most active catalytic systems, due the formation of small sized, narrow-dispersed metal nanoparticles and the presence of the metallic state.15,17,19 In addition, the combination of two metals, such as gold-palladium or gold-platinum has shown promising results in terms of activity, selectivity and resistance to poisoning.
The oxidative esterification of polyols was first reported by Christensen et al.54 They demonstrated that it was possible to oxidise glycerol or 1,2-propanediol in the presence of methanol forming dimethyl mesoxalate or methyl lactate respectively in high yields using commercial gold catalysts. Methanol was chosen as a solvent/reactant as it is a solvent in the formation of glycerol production from biodiesel production using alkaline conditions. Therefore, oxidation of the basic glycerol/methanol solution offered a facile route to value added products. However, in these studies the substrate/metal molar ratio was low, ca. 56, and therefore we consider that a higher substrate to metal molar ratio would be highly desirable. In the present work we report the utilisation of gold-based supported catalysts synthesised by colloidal methods and we discuss the effect of particle size, oxidation state, support and preparation method on the oxidative esterification of 1,2-propanediol. We extend this study and demonstrate that using low temperatures and catalysts with low metal loadings are highly beneficial compared to literature examples.54 In addition, we indicate that the utilisation of a commercial nano-ceria support is beneficial for depositing gold-based nanoparticles and significantly enhances catalytic activity and selectivity to methyl lactate.
Experimental
Materials
1,2-Propanediol (99%), methanol, 30 wt% sodium methoxide in methanol and all the intermediates were purchased from Sigma Aldrich and used as received. NaBH4 (96%) PVA (M = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolysed, tetrakis (hydroxypropyl)phosphonium chloride (THPC) (98% solution) were similarly obtained. TiO2 (P25) was purchased from Degussa, G60 Darco, SiO2 and sodium hydroxide from Sigma Aldrich. CeO2 was purchased from Evonic Degussa- (Adnano Ceria). PdCl2 and HAuCl4·3H2O were purchased from Johnson Matthey. Pure oxygen (99.9%) was from BOC. In addition, we have contrasted the reactivity with that of a standard gold catalyst prepared by the World Gold Council comprising 4.5% Au/Fe2O3 (Lot No. 02-04) and 1.5% Au/TiO2 (Lot No. 02-9).55
000, 80% hydrolysed, tetrakis (hydroxypropyl)phosphonium chloride (THPC) (98% solution) were similarly obtained. TiO2 (P25) was purchased from Degussa, G60 Darco, SiO2 and sodium hydroxide from Sigma Aldrich. CeO2 was purchased from Evonic Degussa- (Adnano Ceria). PdCl2 and HAuCl4·3H2O were purchased from Johnson Matthey. Pure oxygen (99.9%) was from BOC. In addition, we have contrasted the reactivity with that of a standard gold catalyst prepared by the World Gold Council comprising 4.5% Au/Fe2O3 (Lot No. 02-04) and 1.5% Au/TiO2 (Lot No. 02-9).55
Sol-immobilisation method.
NaBH4/PVA colloidal method.
Au, Pd and Au–Pd catalysts supported on carbon or titania were prepared using a sol-immobilisation method based on NaBH4/PVA methodology. For the monometallic Au and Pd supported catalysts the detailed experimental procedure has been described previously.17 Briefly, for the bimetallic gold-palladium carbon-supported catalyst the following procedure was followed. An aqueous solution of PdCl2 (Johnson Matthey) and HAuCl4·3H2O of the desired concentration was prepared. Polyvinyl alcohol (PVA) (1 wt% solution, Aldrich, weight average molecular weight MW = 9000–10000 g mol−1, 80% hydrolysed) was added (PVA/Au (wt/wt) = 1.2); a 0.1 M freshly prepared solution of NaBH4 (>96%, Aldrich, NaBH4/Au (mol/mol) = 5) was then added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilised by adding activated carbon (acidified to pH 1 by sulfuric acid) under vigorous stirring. The amount of support material required was calculated so as to have a total final metal loading of 1% wt. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and dried at 120 °C overnight under static air. In the case where we used SiO2 as the chosen support, the immobilisation time was extended from 2 h to 8 h. Further catalysts were prepared using molar ratio or weight ratio of Au:Pd and denoted with the symbol SIm and SIw. Monometallic catalysts containing gold or palladium are denoted with the symbol SI.
THPC/NaOH colloidal method.
Fresh aqueous solutions of THPC (0.0675 M) and NaOH (0.2 M) were prepared. The desired amounts of NaOH and THPC were added in an aqueous solution. After stirring for 3 min, the desired amount of an aqueous solution of HAuCl4 was then added under vigorous stirring. The formation of orange-brown hydrosols of gold was observed. The final concentration of gold solution was [Au] = 1 × 10−3 M and the molar ratio of NaOH/THPC/metal was 5.91:1.33:1. For the synthesis of bimetallic Au–Pd colloids, the desired amounts of aqueous solutions of HAuCl4 and PdCl2 were added to the NaOH/THPC aqueous solution. The formation of grey-brown hydrosols of gold-palladium was observed. The final concentration of gold-palladium solution [M] = 1 × 10−3 M and the molar ratio of NaOH/THPC/metal was 5.91:1.33:1.
After the reduction step the hydrosols were stirred for 30 min and the desired amount of support was added (acidified to pH 2 by sulfuric acid) for obtaining 1 wt% of metal loading. The amount of support material required was calculated so as to have a total final metal loading of 1% wt. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and dried at 120 °C overnight under static air.
UV-vis spectra (200–900 nm) of the Au–Pd sols were recorded in H2O using a UV spectrometer (V-570, JASCO).
Samples were characterized using a Kratos Axos Ultra-DLD photoelectron spectrometer, using monochromatic Al kα radiation, at 120 W power. High resolution and survey scans were performed at pass energies of 40 and 160 eV respectively. Spectra were calibrated to the C(1s) signal for adventitious carbon at 284.7 eV and quantified using CasaXPS v2.3.15, utilising sensitivity factors supplied by the manufacturer.
TEM analysis
Samples for examination by transmission electron microscopy (TEM) were prepared by dispersing the dry catalyst powder onto a holey carbon film supported by a 300 mesh copper TEM grid. Samples were then subjected to bright field and phase contrast imaging experiments using a 200 kV JEOL 2200FS transmission electron microscope.
The reactor was charged with methanol (0.354 mol), 1,2-propanediol (6 mmol), NaOMe (0.6 mmol). Catalyst (alcohol/metal ratio = 500–1000, mol/mol) was added to the solution and the autoclave was sealed, pressurised with oxygen (3 bar pressure), heated at 100 °C, and stirred at 1200 rpm for 24 h. The reactor vessel was cooled to room temperature and the reaction mixture (after centrifugation of the sample) was analysed by GC without prior removal of NaOMe, (Varian 3800 fitted with a CPwax52 column). Products were identified by comparison with authentic samples. For the quantification of the amounts of reactants consumed and products generated, an external calibration method was used.
Results and discussion
Extensive and systematic characterisation of the sol-immobilized catalysts was carried out in our previous studies using a combination of TEM, STEM-XEDS, HAADF imaging and XPS techniques.17,43,49 We aimed to contrast the reactivity of supported Au nanoparticles on different supports (activated carbon and titania) and Pd and Au–Pd supported nanoparticles synthesised by the sol immobilisation method for the oxidative esterification of 1,2-propanediol in the presence of methanol. The mean and median AuPd particle in catalysts prepared by sol-immobilisation on various supports are shown in Table 1. Representative particle size distribution data and TEM micrographs for the Au–Pd/C and Au–Pd/TiO2 materials are shown in Fig. 1. The AuPd particle size distribution was found to be quite narrow i.e. between 2–7 nm. Detailed STEM-HAADF studies17,43 showed that the supported Au–Pd catalysts prepared using sol-immobilisation were homogeneous random alloys. The majority of the colloidal particles before the immobilisation step were icosahedral or decahedral (multiply twinned), whereas a small percentage of the particles were cuboctahedral. After the immobilisation step onto carbon, it was observed that the particles retained their spherical shape and showed a low propensity to wet the amorphous carbon support. On the contrary, when immobilising the colloids onto titania, it was observed that the particles were highly faceted with the majority possessing a cub-octahedral structure or singly twinned morphology. In addition, it was observed that the metal nanoparticles were forming an extended flat interface structure with the crystalline titania support particles.56 In terms of oxidation state, XPS analysis confirmed the sol-immobilised monometallic and bimetallic catalysts dried at 120 °C all contain Au in the metallic state and Pd predominantly in the metallic state.17,49 In the case when ceria was used as the support, initial experiments for depositing the same gold or gold-palladium sol using the NaBH4/PVA methodology were not successful due to the fact that immobilisation onto the support was not taking place. Therefore, we utilised the THPC/NaOH methodology for preparing and immobilising sols onto the CeO2 support. In Fig. 2 the UV-visible spectrum of the gold-palladium sol is shown. The spectrum resulting from the mixture of gold and palladium indicates the absence of the gold surface plasmon band (490–520 nm). This indicates the possible formation of an alloyed phase between gold and palladium as previous studies have shown.15XPS analysis of Au/CeO2 and AuPd/CeO2 (Fig. 3) prepared using the THPC/NaOH method revealed that in the monometallic catalyst, where the gold loading is 0.5 at%, the metal is present in both metallic and cationic states (binding energies of 83.9 and 85.5 eV, respectively in ca. 80:20 ratio). The bimetallic preparation produced a catalyst with approximately the expected nominal Au:Pd ratio of 1:1 (ca. 0.2 at% each) and the gold is considered to be metallic. Whilst the Pd signal is extremely weak (see Fig. 3), the signal is seen to undergo a reduction of intensity during X-ray analysis (not shown), suggesting that the Pd is present as Pd(II) (binding energy ca. 338 eV). Previous studies using the THPC/NaOH methodology have shown that single particles contained both metals were evident together with the presence of PdO.56 We considered that these series of well-characterised materials provide a useful basis for this study of the selective oxidative esterification of 1,2-propanediol.
Table 1 Mean and median particle sizes of the various immobilized Au, Pd and Au–Pd supported catalysts used for catalytic studies
|
Catalysts
|
Au/C |
Pd/C |
Au–Pd/C |
Au/TiO2 |
Pd/TiO2 |
Au–Pd/TiO2 |
| Mean size (nm) |
4.7 |
6.3 |
3.7 |
4.6 |
4.8 |
3.9 |
| Median size (nm) |
4.0 |
5.0 |
3.3 |
3.9 |
3.8 |
3.5 |
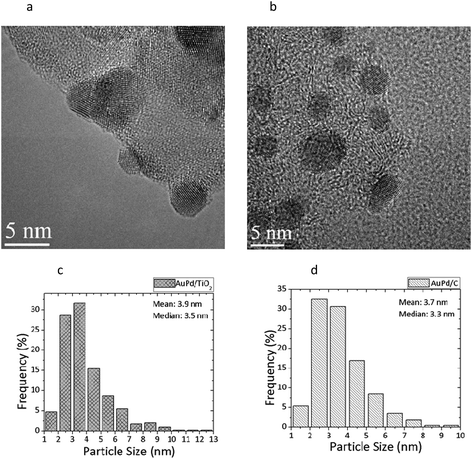 |
| | Fig. 1
TEM micrographs and particle size distributions for sol–immobilized Au–Pd/TiO2 (a,c) dried at 120 °C and sol–immobilized Au–Pd/C dried at 120 °C (b,d). | |
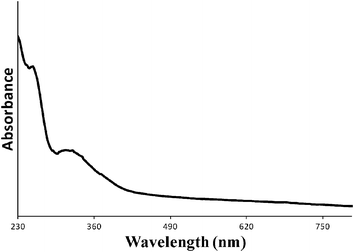 |
| | Fig. 2
UV-Vis spectrum obtained for the AuPdSIw (1:1.85 molar ratio) colloidal solution. | |
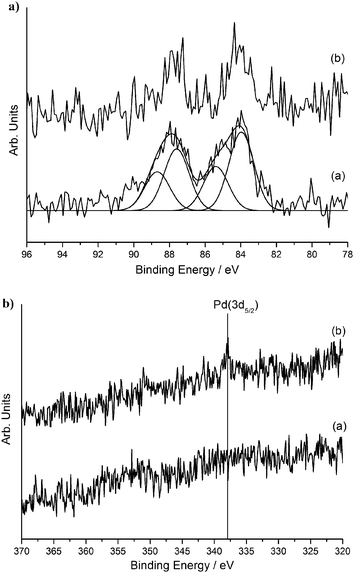 |
| | Fig. 3
Au(4f) and Pd(3d) XP spectra for (a) 1% Au/CeO2 and (b) 1% AuPd/CeO2SIw catalysts. | |
Oxidation of 1,2-propanediol was carried out using a pressurised glass reactor at 100 °C in the presence of sodium methoxide in methanol. The molar ratio of substrate to sodium methoxide was in the range 1–10 and substrate/metal molar ratio was between 500–1000. Initially, the catalytic performance of a standard reference 1.5 wt% Au/TiO2 catalyst supplied by the World Gold Council was investigated.55 The reaction profile is shown in Fig. 4. The main products of this reaction were methyl lactate, hydroxyacetone and methyl pyruvate, in agreement with recent published reports.53 The major product was hydroxyacetone (47%) followed by methyl lactate (38%) and methyl pyruvate (15%). We did not observe any formation of lactaldehyde which is expected to be an intermediate that is rapidly oxidised to methyl lactate or due to tautomerisation equilibrium, transformed to hydroxyacetone. The formation of 1-hydroxyacetone at high levels at the initial stages of the reaction indicates that it can be a possible intermediate for the formation of methyl lactate. For verifying the reaction network we have performed the oxidation of intermediates (Table 2) and we observed that 1-hydroxyacetone was transformed to methyl lactate (71%) and methyl pyruvate (29%), and therefore we consider that lactate was formed via1-hydroxyacetone. The stability of methyl lactate was investigated and we observed the formation of methyl pyruvate. In addition, we performed the reaction of lactic acid in the presence of methanol in order to determine if the reaction pathway involves an esterification process of lactic acid and methanol (Table 3). It is evident that the esterification pathway from lactic acid is also possible for the formation of methyl lactate. Another possibility is the reaction of 1-methoxy-1,2-propanediol and pyruvaldehydevia a Canizzaro reaction, which could form methyl lactate and hydroxyacetone as is indicated in Scheme 1. Therefore, in the case of oxidative esterification of 1,2-propanediol we expect that the formation of methyl lactate and methyl pyruvate can proceed via five distinct pathways. One pathway is vialactaldehyde, which is an intermediate that is rapidly oxidised under alkaline conditions; the second one is viahydroxyacetone, formed by direct oxidation of 1,2-propanediol or by base-induced tautomeric equilibrium with the intermediate enediol derived from lactaldehyde. Hydroxyacetone could further react to form methyl lactateviapyruvaldehyde. The third pathway is via the formation of 1-methoxy-1,2-propanediol from lactaldehyde and methanol and then via a Cannizaro reaction to from methyl lactate and hydroxyacetone. The fourth pathway is vialactic acid and methanol esterification. Finally, a fifth reaction pathway for the formation of methyl pyruvateviamethyl lactate is also feasible, (Scheme 1).42,53 Moreover, the effect of 1,2-propanediol/molar ratio was investigated in the range of 500–1000 and the conversion increased linearly with an increase of the amount of catalyst used. In terms of selectivity, by decreasing the substrate/metal molar ratio, the selectivity to methyl lactate was not affected significantly (Table 4).
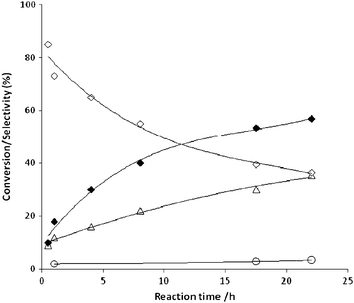 |
| | Fig. 4 Oxidative esterification of 1,2-propanediol using 1.5 wt%Au/TiO2 (WGC). Conversion of 1,2-propanediol (◆), selectivity to hydroxyacetone (⋄) Selectivity to methyl lactate (△), Selectivity to methyl pyruvate (○). Reaction conditions: methanol (0.354 mol), 1,2-propanediol (6 mmol), NaOMe (0.6 mmol) 1,2-propanediol/total metal mol ratio = 500, T = 100 °C, pO2 = 3 bar, stirring rate 1200 rpm, reaction time = 24 h. | |
|
Catalyst
|
Time (h) |
Yield (%) |
|
Methyl lactate
|
|
Reaction conditions: methanol (0.354 mol), lactic acid (6 mmol), NaOMe (0.6 mmol) 1,2-propanediol/total metal mol ratio = 500, T = 100 °C, pO2 = 3 bar, stirring rate 1200 rpm, reaction time = 24 h.
|
|
Au/TiO2SI |
1 |
3.3 |
| 4 |
6.7 |
| 24 |
35.2 |
|
AuPd/TiO2SIw |
1 |
2.2 |
| 4 |
11.2 |
| 24 |
29.6 |
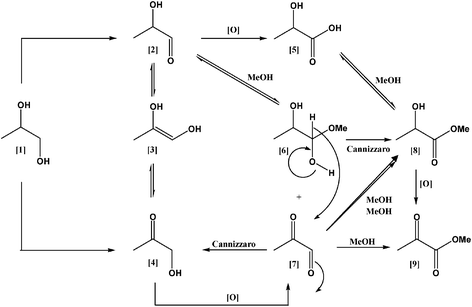 |
| | Scheme 1 Reaction pathway in the oxidative esterification of 1,2-propanediol. (1) 1,2-propanediol, (2) lactaldehyde, (3) enediol, (4) hydroxyacetone, (5) lactic acid, (6) 1-methoxy-1,2-propanediol, (7) pyruvaldehyde, (8) methyl lactate, (9) methyl pyruvate. | |
Table 4 Effect of substrate metal molar ratio in the liquid phase oxidative esterification of 1,2-propanediol in methanol using 1.5 wt% Au/TiO2 (WGC)a
A series of gold supported catalysts were investigated for the oxidative esterification of 1,2-propanediol and a comparison with known commercial samples was carried out (Table 5). For the synthesis of gold supported catalysts we used mainly colloidal methods. Using the colloidal method based on the NaBH4/PVA methodology, we successfully immobilised gold colloids onto carbon, titania and silica. In the case of nanoceria as a support, we used the THPC/NaOH methodology for successfully immobilising gold colloids. Catalytic data are shown in Table 5. The most active gold catalysts were the 1.5 wt% Au/TiO2 (WGC) and the 1 wt% Au/TiO2 synthesised by the NaBH4/PVA methodology. The order of catalytic activity was the following: 1.5 wt% Au/TiO2 (WGC), 1 wt% Au/TiO2SI, 5 wt% Au/Fe2O3 (WGC), 1 wt% Au /CeO2SI, 1 wt% Au/CSI, 1.5 wt% Au/SiO2SI. In terms of selectivity and at a similar range of conversion (31–38%), the 1 wt% Au/TiO2SI material showed the highest selectivity to methyl lactate (41–50%). Gold supported nanoparticles on carbon, ceria and silica showed a similar range of conversion (10–18%). However, the distribution of products was affected considerably by the choice of support. 1 wt% Au/CSI showed high selectivity to hydroxyacetone and methyl lactate, 1 wt% Au/SiO2SI showed high selectivity to methyl pyruvate (64%), whereas 1 wt% Au/CeO2SI showed excellent selectivity to methyl lactate (84%). From the data obtained it seems that using metal oxides as supports for the deposition of gold nanoparticles results in an improvement in the catalytic performance of gold nanoparticles for the oxidative esterification of 1,2-propanediol in the presence of methanol. Methyl lactate or methyl pyruvate can be produced in high selectivities by choosing the appropriate supports. Taking into account the facts that (i) the particle size distribution has a similar range for all the gold supported catalysts (3–7 nm) and (ii) the oxidation state of gold is zero on titania, carbon and silica, the significant enhancement in terms of reactivity for the gold supported nanoparticles might be attributable to a number of factors including metal-support interactions, active sites at the metal-support interface and the morphology of the nanoparticles. In the case of 1 wt% Au/CeO2 we observed lower catalytic activity but an enhanced selectivity to methyl lactate. The lower activity and enhanced selectivity to methyl lactate could be attributed to the presence of cationic gold (20%).57 It seems that presence of metallic and cationic gold is beneficial for the improvement in the selectivity of methyl lactate. Moreover, the catalytic behaviour of monometallic Au, Pd and bimetallic Au–Pd-supported nanoparticles was investigated to reveal the effect of Pd. As is shown in Table 6, addition of Pd improved the conversion of 1,2-propanediol with respect to its monometallic gold analogue by a factor of 1.3–3.7, which is in agreement with our previous reported studies of glycerol and 1,2-propanediol oxidation.17,18 The effect of Au/Pd molar ratio was systematically studied with Au–Pd/CeO2 catalysts. As is shown in Table 7, decreasing the Au/Pd molar ratio led to an increase in conversion of 1,2-propanediol and a shift in the distribution of products, with the selectivity to methyl lactate increasing with increasing Pd content.
|
Catalyst
|
Conversion (%) |
Selectivity (%) |
|
Hydroxyacetone
|
Methyl lactate
|
Methyl pyruvate
|
Others |
|
Reaction conditions: methanol (0.354 mol), 1,2-propanediol (6 mmol), NaOMe (0.6 mmol) 1,2-propanediol/total metal mol ratio = 500, T = 100 °C, pO2 = 3 bar, stirring rate 1200 rpm, reaction time = 24 h.
|
| Blank |
7.7 |
48.6 |
25.1 |
0 |
26.3 |
| 5%Au/Fe2O3 (WGC) |
30.6 |
30.4 |
40.3 |
29.3 |
0 |
| 1.5%Au/TiO2 (WGC) |
35.1 |
47.2 |
37.8 |
15.0 |
0 |
| 1%Au/TiO2SI |
38.5 |
27.5 |
47.1 |
8.7 |
16.6 |
| 1%Pd/TiO2SI |
27.4 |
34.6 |
46.7 |
2.5 |
16.2 |
| 1%Au/CSI |
13.6 |
45.7 |
51.8 |
2.5 |
0 |
| 1%Au/CeO2SI |
18.1 |
2.8 |
83.5 |
6.0 |
7.8 |
| 1.5%Au/SiO2SI |
10.1 |
17.9 |
18.0 |
64.0 |
0 |
|
Catalyst
|
Conversion (%) |
Selectivity /% |
|
Hydroxyacetone
|
Methyl lactate
|
Methyl pyruvate
|
Others |
|
Reaction conditions: methanol (0.354 mol), 1,2-propanediol (6 mmol), NaOMe (0.6 mmol) 1,2-propanediol/total metal mol ratio = 500, T = 100 °C, pO2 = 3 bar, stirring rate 1200 rpm, reaction time = 24 h.
|
| 1%Au/TiO2SI |
38.5 |
27.5 |
47.1 |
8.7 |
16.6 |
| 0.5%Au0.5%Pd/TiO2SIw |
50.2 |
34.2 |
61.4 |
2.1 |
2.3 |
| 1%Pd/TiO2SI |
27.4 |
34.6 |
46.7 |
2.5 |
16.2 |
| 1%Au/CSI |
13.6 |
45.7 |
51.8 |
2.5 |
0.0 |
| 0.5%Au0.5%Pd/CSIw |
50.2 |
45.1 |
47.0 |
4.5 |
3.4 |
| 1%Au/CeO2SI |
18.1 |
2.8 |
83.5 |
6.0 |
7.8 |
| 0.5%Au0.5%Pd/CeO2SIw |
39.6 |
13.1 |
71.1 |
13.2 |
2.6 |
|
Catalyst
|
Au–Pd molar ratio |
Conversion (%) |
Selectivity (%) |
|
Hydroxyacetone
|
Methyl lactate
|
Methyl pyruvate
|
Others |
|
Reaction conditions: methanol (0.354 mol), 1,2-propanediol (6 mmol), NaOMe (0.6 mmol) 1,2-propanediol/total metal mol ratio = 500, T = 100 °C, pO2 = 3 bar, stirring rate 1200 rpm, reaction time = 24 h.
|
| 1%Au/CeO2SI |
1:0 |
12.6 |
4.9 |
74.6 |
12.4 |
8.1 |
| 1%AuPd/CeO2SIm |
2:1 |
22.6 |
15.0 |
67.7 |
16.0 |
1.3 |
| 1%AuPd/CeO2SIm |
1:1 |
30.9 |
7.1 |
75.7 |
14.4 |
2.8 |
| 1%AuPd/CeO2SIm |
1:85 |
39.6 |
13.1 |
71.1 |
13.2 |
2.6 |
| 1%Pd/CeO2SI |
0:1 |
56.0 |
44.1 |
54.0 |
0 |
0 |
Conclusions
The oxidative esterification of 1,2-propanediol was studied using gold based supported nanoparticles. We found that the distribution of products is significantly affected by the choice of support and metal. Using gold-palladium catalysts led to a significant enhancement in terms of catalytic activity as well as selectivity to methyl lactate. We used nanoceria as a support and we found that pronounced selectivity to methyl lactate could be obtained (70–75%) at 30–39% conversion. Moreover, we successfully utilised higher substrate/metal molar ratios than in previous studies and have demonstrated that Au–Pd supported nanoparticles possess higher catalytic efficiency than gold supported nanoparticles. Mechanistic studies indicated that one of the main pathways for methyl lactate formation is via oxidative esterification of hydroxyacetone/lactaldehyde without excluding the possibility of forming methyl lactatevialactic acid and methanol. Furthermore, we have demonstrated that by careful tuning of the catalyst support and choice of metals it is possible to further oxidise methyl lactate to form methyl pyruvate, a previously unreported oxidation. Future studies based on the encouraging results obtained with the Au, Au–Pd/CeO2 catalysts will focus on the detailed characterisation of these materials in order to reveal the morphology and structure of the gold and gold-palladium nanoparticles.
Acknowledgements
This work formed part of the Glycerol Challenge. The Sasol Company and the Technology Strategy Board are thanked for their financial support. This project is co-funded by the Technology Strategy Board's Collaborative Research and Development programme, following an open competition. The Technology Strategy Board is an executive body established by the Government to drive innovation.
References
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- T. Suijino, T. Ohigashi, S. Sugiyama, K. Kawashiro and H. Hayashi, J. Mol. Catal., 1992, 71, 25 CrossRef.
- H. H. C. M. Pinxt, B. F. M. Kuster and G. B. Marin, Appl. Catal., A, 2000, 191, 45 CrossRef CAS.
- T. Miyazawa, Y. Kasunoki, K. Kunimori and K. Tomisihige, J. Catal., 2006, 240, 213 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434 CrossRef CAS.
- C. H. C. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Top. Catal., 2009, 52, 258 CrossRef CAS.
- H. Kimura, A. Kimura, I. Kubo, T. Wakisaka and Y. Mitsuda, Appl. Catal., A, 1993, 95, 143 CrossRef CAS.
- H. Kimura, K. Tsoto, T. Wakisaka, Y. Kazumi and Y. Inaya, Appl. Catal., A, 1993, 96, 217 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696 RSC.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329 RSC.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397 CrossRef CAS.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, J. M. Anthonykutty, G. Brett, A. F. Carley, R. C. Tiruvalam, A. A. Herzing, C. J. Kiely, D. W. Knight and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 4952 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, J. M. Anthonykutty, G. Brett, A. F. Carley, S. H. Taylor, D. W. Knight and G. J. Hutchings, Green Chem., 2009, 11, 1209 RSC.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499 CrossRef CAS.
- Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Chem.–Eur. J., 2010, 16, 7368 CrossRef CAS.
- C.-H. Zhou, J. N. Beltramini, C.-X. Lin, Z.-P. Xu, G. Q. Lu and A. Tanksale, Catal. Sci. Technol., 2011, 1, 111 CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405 CrossRef.
- P. Landon, J. Ferguson, B. E. Solsona, T. Garcia, A. F. Carley, A. A. Herzing, C. J. Kiely, S. E. Golunski and G. J. Hutchings, Chem. Commun., 2005, 3385 RSC.
- P. Landon, J. Ferguson, B. E. Solsona, T. Garcia, S. Al-Sayari, A. F. Carley, A. Herzing, C. J. Kiely, M. Makkee, J. A. Moulijn, A. Overweg, S. E. Golunski and G. J. Hutchings, J. Mater. Chem., 2006, 16, 199 RSC.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105 CrossRef CAS.
- W. Yan, B. Chen, S. M. Mahurin, S. Dai and S. H. Overbury, Chem. Commun., 2004, 1918 RSC.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538 CrossRef CAS.
- J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS.
- D. I. Enache, J. Edwards, P. Landon, B. Solsana-Espriu, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. Garcia, Tetrahedron, 2006, 62, 6666 CrossRef CAS.
- A. Biffis, S. Cunial, P. Spontoni and L. Prati, J. Catal., 2007, 251, 1 CrossRef CAS.
- P. Haider and A. Baiker, J. Catal., 2007, 248, 175 CrossRef CAS.
- C. D. Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384 CrossRef.
- W. Hou, N. A. Dehm and R. W. Scott, J. Catal., 2008, 253, 22 CrossRef CAS.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- P. Lignier, F. Morfin, S. Mangematin, L. Massin, J.-L. Rousset and V. Gaps, Chem. Commun., 2007, 186 RSC.
- D. Gajan, K. Guillois, P. Delichère, J.-M. Basset, J.-P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667 CrossRef CAS.
- S. Bawaked, N. F. Dummer, N. Dimitratos, D. Bethell, Q. He, C. J. Kiely and G. J. Hutchings, Green Chem., 2009, 11, 1037 RSC.
- L. Kesavan, R. Tiruvalam, M. I. Bin Saiman, D. I. Enache, R. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195 CrossRef CAS.
- T. Ishikara, Y. Ohura, S. Yoshida, Y. Hata, H. Nishiguchi and Y. Takita, Appl. Catal., A, 2005, 291, 215 CrossRef.
- Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619 CrossRef CAS.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225 RSC.
- J. K. Edwards, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS.
- C. Ketchie, Y. L. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94 CrossRef.
- W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 264 CrossRef CAS.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538 RSC.
- E. Taarning, A. T. Madsen, J. M. Marchetti, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 408 RSC.
- For details of the reference catalyst: http://www.utilisegold.com/uses_applications/catalysis/reference_catalysts/.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191 CAS.
- A. Abad, P. Conception, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.