Catalytic conversion of compounds representative of lignin-derived bio-oils: a reaction network for guaiacol, anisole, 4-methylanisole, and cyclohexanone conversion catalysed by Pt/γ-Al2O3†
Ron C.
Runnebaum
a,
Tarit
Nimmanwudipong
a,
David E.
Block
ab and
Bruce C.
Gates
*a
aDepartment of Chemical Engineering and Materials Science, University of California, Davis, Davis CA, USA. E-mail: bcgates@ucdavis.edu; Fax: (+ 1) 530-752-1031
bDepartment of Viticulture and Enology, University of California, Davis, Davis CA, USA
First published on 7th September 2011
Abstract
The conversion of compounds representative of lignin and lignin-derived bio-oils (guaiacol, anisole, 4-methylanisole, and cyclohexanone), catalysed by Pt/Al2O3 in the presence of H2 at 573 K is described by a reaction network indicating a high selectivity for platinum-catalysed aromatic carbon–oxygen bond cleavage accompanied by acid-catalysed methyl group transfer reactions.
1. Introduction
Among the potential routes for production of fuels and chemicals from lignocellulosic biomass, fast pyrolysis accompanied by or followed by catalytic upgrading offers excellent potential because the number of conversion steps is small and the processing may be cost effective.1Catalytic upgrading is required because the pyrolysis products are characterised by thermal and chemical instability, low heating values, corrosiveness, and immiscibility with petroleum-derived fuels.2 Research on catalytic upgrading of bio-oils has focused on cellulose-derived fractions,3 with less attention paid to lignin-derived fractions—which offer attractive prospects for production of aromatic chemicals such as phenolics.4Lignin-derived bio-oils can also be converted into fuels, with a key processing challenge being the removal of oxygen—because bio-oils derived from lignocellulose typically contain 35–40 wt% oxygen.2,5 The literature of bio-oils conversion is largely lacking in fundamental chemistry.6 The complexity of the feedstocks limits the usefulness of the available data for predicting catalyst performance. Even data characterising the reactions of individual biomass-derived compounds7 are scarce, and the catalytic reaction networks are largely limited to primary products or products lumped into classes of similar compounds.8,9Our goal was to determine a reaction network to account for the reactions of a group of compounds prototypical of lignin and compounds derived from it, incorporating the representative aromatic rings, hydroxyl groups, keto groups, and ether linkages—the compounds are guaiacol, anisole, 4-methylanisole, and cyclohexanone. These reactants were converted in the presence of a solid catalyst, platinum on γ-Al2O3 (Pt/γ-Al2O3).
2. Experimental
2.1 Catalytic reaction experiments
The Pt/γ-Al2O3 catalyst (1 wt% Pt, Sigma-Aldrich) had a BET surface area of 206 ± 1 m2 g−1 and a platinum dispersion determined by H2 chemisorption of 0.25.10 The catalyst was used in powder form.In the reaction experiments, carried out with a once-through flow reactor with reactant vapors flowing at steady state, the catalyst (0.001–0.100 g, mixed with particles of inert nonporous α-Al2O3) was pre-treated in flowing gases at 573 K. The pretreatment gas, flowing at 100 mL min−1, was 30% H2/70% N2. In a typical experiment, liquid reactant (guaiacol, flowing at 0.015 mL min−1, or anisole, 4-methylanisole, or cyclohexanone, each flowing at 0.030 mL min−1) was vaporised into a gas stream (30% H2/70% N2) flowing at 100 mL min−1 into the packed-bed reactor. The reactor temperature was 573 K, and the pressure was 140 kPa. The effluent was condensed at 283–288 K.
Analysis of the products in the liquid samples was performed with a gas chromatograph (GC, Agilent 7890) equipped with a mass selective detector (MSD, Agilent 5795) and a flame ionisation detector. Analysis of the gas-phase products was performed with a GC refinery gas analyser (Agilent 7980). Details of the analysis are given in ESI.
Mass balance closures were typically greater than 95%.
3. Results
3.1 Oxygen removal in reactions catalysed by Pt/γ-Al2O3
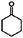
The products formed in the reactions catalysed by Pt/γ-Al2O3 in the presence of H2 are listed in Tables 1 and 2. In the conversion of each of the reactants individually with H2, we observed dozens of products. The data determine conversions and selectivities of the products that were formed in relatively high yields at conversions < 10%. Conversion of the four individual reactants as a function of time on stream, shown in Fig. 1, demonstrates similar rates of catalyst deactivation. Plots of selectivity as a function of conversion (e.g., Fig. 2) demonstrate which of the major products are primary and which are not; the methodology for these determinations is summarised in the papers by Bhore et al.11| Reactant | Products formed in conversion of prototypical reactant | Product structure |
|---|---|---|
| Guaiacol (2-methoxyphenol) | Anisole (methoxybenzene) |

|
| Cyclohexanone |
|
|
| Phenol |

|
|
| o-Cresol (2-methylphenol) |
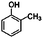
|
|

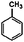
| a A list of the trace products is given in the ESI. | ||
|---|---|---|
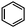
| Cyclohexanone | Benzene |
|
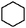
| Cyclohexane |
|
|
| Cyclohexene |
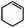
|
| Reactant | Products formed in conversion of prototypical reactant | Product structure |
|---|---|---|
| Guaiacol | Catechol (benzene-1,2-diol) |
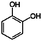
|
| 3-Methylcatechol (3-methyl-1,2-benzenediol) |

|
|
| 3-Methylguaiacol (2-methoxy-3-methylphenol) |

|
|
| 6-Methylguaiacol (2-methoxy-6-methylphenol) |
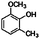
|
|
| Veratrole (1,2-dimethoxybenzene) |
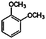
|
|
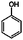
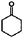

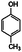

| 4-Methylanisole | 4-Methylphenol |
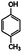
|
| Methylcyclohexanone |
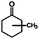
|
|
| 2,4-Dimethyphenol |
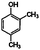
|
|
| 2,4,6-Trimethylphenol |

|
|
| Tetramethylphenol |
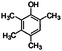
|
| a A list of the trace products is given in the ESI. | ||
|---|---|---|
| Cyclohexanone | Cyclohexanol |

|
| Cyclohexenone |

|
|
| Phenol |

|
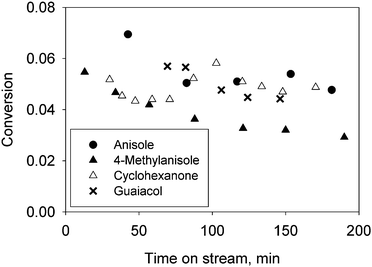 | ||
| Fig. 1 Change in conversion of anisole (●), 4-methylanisole (▲), cyclohexanone (△), and guaiacol (✗) during operation of a flow reactor. Reactants were fed individually and all conversions were catalysed by Pt/γ-Al2O3 in the presence of H2 at 573 K. The WHSV, in units of (g of reactant)/(g of catalyst × h), of the feed streams was chosen to achieve similar initial conversions: anisole, 18; 4-methylanisole, 17; cyclohexanone, 160; and guaiacol, 20. | ||
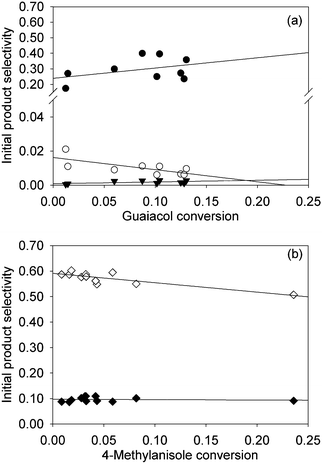 | ||
| Fig. 2 (a) Selectivity for the formation of phenol (●), anisole (○), and benzene (▼) in the conversion of guaiacol. (b) Selectivity for the formation of 4-methylphenol (◇) and toluene (◆) in the conversion of 4-methylanisole. Each conversion was catalysed by Pt/γ-Al2O3 in the presence of H2 at 573 K. Data for each product were fitted with a straight line and extrapolated to zero conversion; intercepts of regression lines significantly different from zero selectivity at zero conversion (determined with 95% confidence limits) indicate primary products, in this case phenol, anisole, 4-methylphenol, and toluene. | ||
The data show that one of the dominant classes of reactions observed with guaiacol, anisole, and 4-methylanisole was transalkylation. Reported results indicate that with HY zeolite catalyst, transalkylation was the only kinetically significant reaction class.10,12 This observation implies that the alumina support in Pt/γ-Al2O3 catalysed the transalkylation reactions.
The other important reaction classes observed with Pt/γ-Al2O3 (but only when H2 was a coreactant) were hydrogenation (e.g., the conversion of cyclohexanone to cyclohexanol and of benzene to cyclohexene), dehydrogenation (e.g., the conversion of cyclohexanone to phenol and hydrogen), and hydrogenolysis (e.g., the reaction of guaiacol to give catechol and methane).
There was no evidence of hydroxylation reactions under our experimental conditions; for example, anisole was not observed to produce guaiacol.
C–O bond cleavage reactions that removed oxygen from the organic reactant were observed, as were C–O bond cleavage reactions that did not remove oxygen from the organic reactant. The former are categorised as hydrodeoxygenation (HDO) and the latter as hydrogenolysis. In the HDO reactions, oxygen was removed as methanol or water (exemplified by the reaction of 4-methylanisole to give toluene and methanol). In contrast, in hydrogenolysis, one of the products was an alkane (as in the reaction of anisole to give phenol and methane). In the conversion of cyclohexanone, HDO was observed, but, because of the lack of substitutents such as methyl, hydrogenolysis was not.
The observation of these three reaction classes when the catalyst was the supported metal and not when it was the zeolite demonstrates that they were catalysed by the platinum.
3.2 Identification of primary reactions and determination of kinetics: first step towards elucidation of reaction networks
The data determine quantitative conversions and selectivities of the products that were formed in relatively high yields at conversions < 10%.In the conversion of guaiacol with H2, phenol and anisole are primary products, and benzene was a non-primary product, as shown by the plot of Fig. 2a. The plot of Fig. 2b shows that 4-methylphenol and toluene are primary products in the reactions of 4-methylanisole and H2. (We emphasise that the designations of primary and non-primary products are empirical and do not provide details about intermediates that were too reactive to be detected.)
The data also determine approximate kinetics of the primary reactions. As illustrated by the data of Fig. 3, which are typical, the reactions are satisfactorily represented as first-order in the organic reactant. However, because the conversions were kept low in almost all of our experiments to simplify the analyses, we lack the data to determine the reaction orders precisely and use pseudo-first-order kinetics as a matter of convenience. The rate constants are summarised in Table 3.
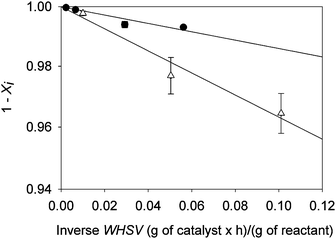 | ||
| Fig. 3 Conversion of guaiacol to give phenol (△) and conversion of anisole to give benzene (●); both conversions were catalysed by Pt/γ-Al2O3 in the presence of H2 at 573 K. The term Xi represents the conversion to product i. WHSV is weight hourly space velocity. The vertical scale is logarithmic. | ||
| Reaction number (keyed to Fig. 4) | Reactant | Rate constanta | Reaction class |
|---|---|---|---|
| a Rate constants unit in L (g catalyst)−1 h−1. | |||
| 1 | Guaiacol | 0.11 | Hydrodeoxygenation |
| 2 | Guaiacol | 4.4 | Hydrodeoxygenation |
| 3 | Guaiacol | 6.5 | Hydrogenolysis |
| 4 | Guaiacol | 1.8 | Transalkylation |
| 5 | Guaiacol | 0.50 | Bimolecular transalkylation |
| 6 | Guaiacol | 0.21 | Bimolecular transalkylation |
| 7 | Guaiacol | 0.26 | Bimolecular transalkylation |
| 8 | Anisole | 12 | Hydrogenolysis |
| 9 | Anisole | 0.86 | Hydrodeoxygenation |
| 10 | Anisole | 2.8 | Transalkylation |
| 11 | Anisole | 0.039 | Transalkylation |
| 12 | Anisole | 0.14 | Bimolecular transalkylation |
| 13 | 4-Methylanisole | 0.76 | Hydrodeoxygenation |
| 14 | 4-Methylanisole | 4.2 | Hydrogenolysis |
| 15 | 4-Methylanisole | 2.2 | Transalkylation |
| 16 | Cyclohexanone | 77 | Dehydrogenation |
| 17 | Cyclohexanone | 5.5 | Hydrogenation |
| 18 | Cyclohexanone | 0.05 | Hydrodeoxygenation |
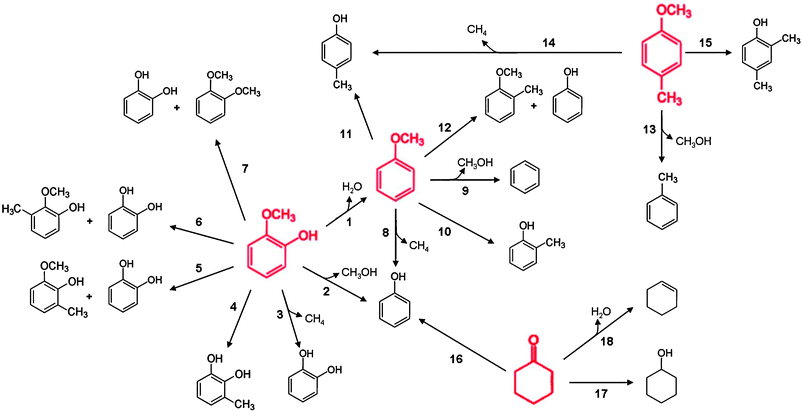 | ||
| Fig. 4 Reaction network accounting for formation of primary products determined from analysis of selectivity-conversion plots for the conversion of individual reactants (shown in red), guaiacol, anisole, 4-methylanisole, and cyclohexanone, catalysed by Pt/γ-Al2O3 in the presence of H2 at 573 K. H2, as a reactant, is omitted for simplicity. Pseudo-first-order rate constants for the individual primary products formed in the conversion of the individual reactants catalysed by Pt/γ-Al2O3 are shown in Table 3; the numbers next to the arrows in this figure are keyed to the list in Table 3. | ||
4. Discussion
4.1 Combined reaction network
The data form the basis for determining a reaction network to summarise the quantitative results, as shown in Fig. 4. This network shows all the primary products formed from each reactant.The data provide a comparison of the reactivities of the various reactants. For example, the rate constants representing the reactions of guaiacol and anisole to remove the methoxy group are 4.4 and 0.86 L (g of catalyst)−1 h−1, respectively. This comparison indicates that the methoxy group becomes easier to remove when a hydroxy group is present in the ortho position. The rate constant for removal of the methyl group from the methoxy substituent is greater for anisole, 12 L (g of catalyst)−1 h−1, than for guaiacol, 6.5 L (g of catalyst)−1 h−1.
4.2 Evaluation of the reaction network and comparison with reported data
A statement of the reaction network including the minor and trace products is presented in Fig. 5; we inferred this network by recognising which compounds were primary products and by using our chemical judgment of the most likely pathways for formation of the minor and trace compounds by presuming that the important reaction classes are methyl group transfer, hydrodeoxygenation, hydrogenolysis, and hydrogenation.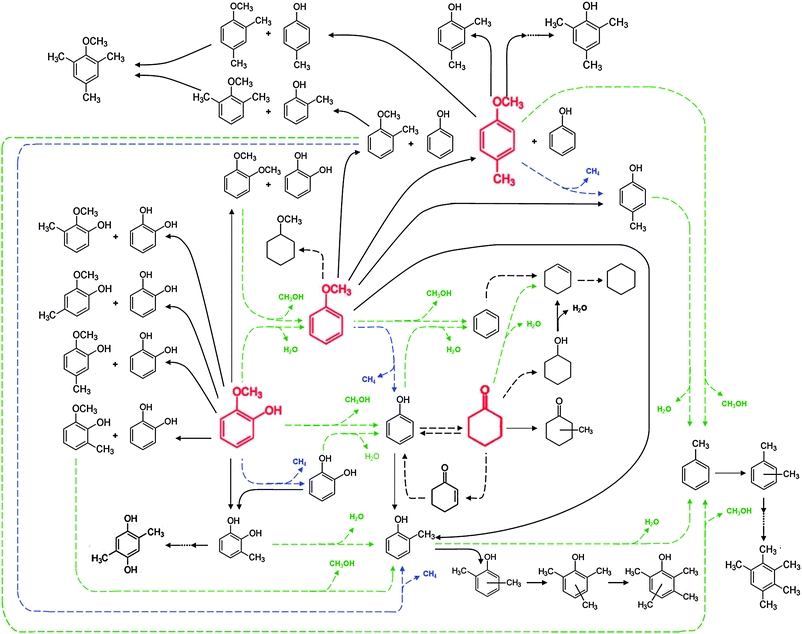 | ||
| Fig. 5 Reaction network for the conversion of lignin-derived compounds (each compound shown in red was used as a reactant) with H2 catalysed by Pt/γ-Al2O3 at 573 K and 140 kPa. HDO, hydrogenolysis, and hydrogenation (or dehydrogenation) reactions are represented by dashed green, blue, and black arrows, respectively. Transalkylation reactions are represented by solid black arrows. H2 as a reactant is omitted for simplicity. The representation in this network is simplified: for example, in transalkylation reactions in which two guaiacol molecules are involved (e.g., 2-guaiacol → catechol + veratrole), the stoichiometry is not represented here. | ||
The network of Fig. 5 provides a more complete description than that given in Fig. 4, but it is not quantitative. The inference that the most important reaction classes are hydrodeoxygenation, transalkylation, hydrogenolysis, and hydrogenation is in good agreement with earlier reports,8 but the results provide detailed new evidence of the reactions. This is a more detailed representation of the catalytic reactions of lignin-derived compounds than any reported.
As represented in Fig. 5, for example, the data show that some of the reactions are reversible (e.g., cyclohexanone produces phenol), but others appeared to be irreversible at the relatively low conversions observed in our experiments (e.g., 4-methylanisole gave negligible yields of anisole). The data are too few to identify reversible reactions involving trace products.
The results indicate that methyl group transfer reactions were negligible when the methyl group was bonded to the aromatic ring; for example, only a trace of 2-methylanisole was observed to form from 4-methylanisole. We contrast this observation with the relatively rapid methyl group transfer reactions occurring when the methyl group migrated from the methoxy group to the aromatic ring, as, for example, in the formation of 2,4-dimethylphenol from 4-methylanisole. The data also imply the simultaneous occurrence of intermolecular and intramolecular methyl group transfers; for instance, both 2-methylanisole and o-cresol were identified as primary products in the reactions of anisole. Similarly, methylguaiacols (formed by intermolecular transalkylation) and 3-methylcatechol (formed by intramolecular transalkylation) were identified as primary products in the conversion of guaiacol.
The data characterising the conversion of anisole and of 4-methylanisole show that the conversions of reactants in homologous series give homologous products (e.g., benzene and toluene, respectively). These results suggest that the data may be extrapolated with some confidence to increasingly higher homologues including those found in pyrolysis oils.13
The results highlight the requirement of higher partial pressures of H2 than we used when selective deoxygenation is a goal; for example, deoxygenated compounds such as toluene and xylenes are valuable high-octane-number fuel components. The data also point to potentially useful routes for production of chemicals. For example (Fig. 5), the products phenol and cyclohexanone could be useful as chemical intermediates.
The results also clearly demonstrate the value of metals as catalysts for HDO reactions. We suggest further that the results presented here may be useful in guiding the conversion of lignin fractions produced in new processes for biomass conversion, such as those using molten salt catalysts.4,14
Conclusions
In summary, the reaction network of Fig. 5 represents our data well, accounting quantitatively for the kinetically important reaction classes. This reaction network is much more extensive than any reported for the catalytic conversion of lignin-derived compounds. The data show that the hydrogenolysis reactions (including HDO) are kinetically significant under our operating conditions and require a metal function and H2. Transalkylation reactions occur in the presence of an acid such as our catalyst support (γ-Al2O3), which is not active for oxygen removal reactions. Thus, the identification of the roles of the acid and metal functions in the bifunctional catalyst implies an opportunity to optimize the catalyst for a particular application by balancing these functions and optimizing processes for the conversion of lignin-derived bio-oils to fuels and chemicals.Acknowledgements
We thank Jennifer Heelan of the University of California, Davis, for help with the analytical instrumentation. Support of this work was provided by Chevron Technology Ventures, a division of Chevron USA, Inc. An Agilent Technologies Foundation Research Project Gift provided a GC7890 Refinery Gas Analyzer. Tarit Nimmanwudipong thanks the Fulbright Foundation for an Open Competition scholarship.Notes and references
- T. P. Vispute, H. Zhang, A. Sanna and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS; M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS; S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef.
- For examples see: G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS; G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., 2004, 116, 1575–1577 ( Angew. Chem., Int. Ed. , 2004 , 43 , 1549 ) CrossRef; P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef; Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2009, 23, 631–637 CrossRef CAS.
- For examples see: R. K. Sharma and N. N. Bakhshi, Fuel Process. Technol., 1991, 27, 113–130 CrossRef CAS; R. K. Sharma and N. N. Bakhshi, Bioresour. Technol., 1991, 35, 57–66 CrossRef; R. K. Sharma and N. N. Bakhshi, Energy Fuels, 1993, 7, 306–314 CrossRef; R. K. Sharma and N. N. Bakhshi, Biomass Bioenergy, 1993, 5, 445–455 CrossRef; J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 161–183 CrossRef; D. C. Elliot, T. R. Hart, G. G. Neuenschwander, L. J. Rotness and A. H. Zacher, Environ. Prog. Sustainable Energy, 2009, 28, 441–449 CrossRef.
- For examples see: C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS; D. Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC; A. Gutierrez, R. K. Kaila, M. L. Honkela, R. Siloor and A. O. I. Krause, Catal. Today, 2009, 147, 239–246 CrossRef.
- P. D. Chantal, S. Kaliaguine and J. L. Grandmaison, Appl. Catal., 1985, 18, 133–145 CrossRef CAS; X. Zhu, R. G. Mallinson and D. E. Resasco, Appl. Catal., A, 2010, 379, 172–181 CrossRef.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610–2618 CrossRef CAS; A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2619–2626 CrossRef.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Catal. Lett., 2011, 41, 779–783 CrossRef.
- N. A. Bhore, M. T. Klein and K. B. Bischoff, Ind. Eng. Chem. Res., 1990, 29, 313–316 CrossRef CAS; N. A. Bhore, M. T. Klein and K. B. Bischoff, Chem. Eng. Sci., 1990, 45, 2109–2116 CrossRef.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catal. Lett., 2011, 41, 817–820 CrossRef.
- G. M. Loudon, Organic Chemistry, Benjamin/Cummings, Menlo Park, 1988, p. 57 Search PubMed.
- B. A. Simmons, D. Loque and J. Ralph, Curr. Opin. Plant Biol., 2010, 13, 313–320 CrossRef CAS; S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The details of materials and methods, table showing product list from the catalytic reactions of each of the prototypical compounds. See DOI: 10.1039/c1cy00169h |
| This journal is © The Royal Society of Chemistry 2012 |