Enzyme structure and catalytic properties affected by the surface functional groups of mesoporous silica†
Kazuki
Murai
ab,
Takayuki
Nonoyama
bc,
Takao
Saito
ab and
Katsuya
Kato
*b
aDepartment of Applied Chemistry, Chubu University, 1200, Matsumoto-cho, Kasugai-si Aichi, Japan
bNational Institute of Advanced Industrial Science and Technology, 2266-98 Anagahora, Shimo-Shidami moriyama-ku, Nagoya, Aichi, Japan. E-mail: katsuya-kato@aist.go.jp; Fax: +81-52-736-7405; Tel: +81-52-736-7551
cDepartment of Frontier Materials, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya, Aichi, Japan
First published on 14th November 2011
Abstract
The enzyme subtilisin from Bacillus licheniformis (4.1 nm × 7.8 nm × 3.7 nm) was easily immobilized onto a mesoporous silica (MPS) surface by a direct one-step method and the amount of subtilisin immobilized on each functionalized MPS surface was similar (approximately 0.30 mg of enzyme/mg of MPS support). The catalytic performance (hydrolytic activity and enantioselectivity) of the immobilized subtilisin was found to depend on the properties of the organofunctional group on the MPS surface. In particular, the hydrolytic activity of enzyme immobilized on ethyl-group-modified MPS increased relative to the behavior of free subtilisin (relative activity 143%). The activity of subtilisin immobilized on the modified MPS was improved by facilitation of contact between enzyme and hydrophobic substrate by increase in hydrophobicity with an immobilized carrier. On the other hand, the enantioselectivity of subtilisin immobilized on 3-mercaptopropyl-group-modified MPS significantly decreased (enantioselectivity of 2.6 compared to 4.3 for free subtilisin). This decrease in enantioselectivity indicated that the mercapto group on the MPS surface was changed in the secondary structure of enzyme by interacting between enzyme and immobilized support. The denaturation temperature of subtilisin immobilized on no-substituted MPS increased (65 °C compared with 57 °C for free subtilisin). The denaturation temperature of immobilized subtilisin was dependent on the absorbed fraction of thermal energy by functional groups on the MPS surface.
1. Introduction
Mesoporous silica (MPS) is an inorganic material with uniformly nanosized mesopores (pore size 2–50 nm, as classified by IUPAC) upon which a self-assembly of surfactants or polymers can be applied. MPS has been used as a host for enzyme immobilization1–4 and a carrier for drug delivery5,6 because of its advantages of extremely large surface area, controllable pore diameter, and uniform pore-size distribution. MPS pore structures range from two-dimensional,7,8 to three-dimensional hexagonal9,10 to cubic.8 MPS nanoparticles, since their first synthesis in 1990 by Kuroda et al.,11,12 have been formed into rods, mesocellular mesoporous silica and thin films.13–16 By using bridged organic silanes, they have also been formed into organic–inorganic hybrid materials that exhibit the properties of organofunctional groups, acid catalysts17 or metal immobilizers.18 In addition, crystallization of wall of MPS has been achieved by introduction of an organic group.19 MPS syntheses have been performed recently by sol–gel17 and biomimetic20 methods.Catalysis of organic reaction by enzymes is favored because the reactions proceed at ordinary temperatures and pressure, and is typically accomplished by uncomplicated procedures. Enzymes are excellent catalysts for achieving chemo-, regio-, stereo-, and enantioselectivity.21,22 For example, the biocatalysts (cytochrome c, laccase, lipase, and subtilisin) have been used for the asymmetric synthesis and polymerization of organic and inorganic molecules.20,23Lipase and subtilisin in particular exhibit high enantioselectivity. Subtilisin is a serine protease having serine, histidine and aspartic acid in active sites. This enzyme is composed of a single strand of polypeptide containing around 275 amino acids, and has a wide range of substrate selectivity. However, biocatalysts can be inactivated by changes in external factors such as temperature, pH and organic solvents. Moreover, subtilisin as serine protease triggers an irreversible denaturation by autodigestion. To address this problem, biocatalysts are often immobilized, and immobilization to solid supports may be the most commonly used strategy to improve the operational stability of biocatalysts. Several immobilization methods exist, including cross linking,24,25 covalent attachment,26–28 physical entrapment,23 and physical adsorption.29 Solid supports used for immobilization are usually polymer resins, natural polymeric derivatives, organic gels, fibers, zeolites, and MPS. Enzyme immobilization has two main advantages: (1) easy separation of enzymes by centrifugation, filtration, or magnetic separation, facilitating better handling and reuse,15,30,31 and (2) good operational and storage stability of enzymes, reducing process costs. However, it also has some disadvantages. For example, the catalytic activity of enzyme protein depends on its higher-order (typically secondary- and tertiary-) structures. In a previous study, activity of enzyme immobilized on an inorganic support was decreased by a conformational change of the secondary structure of enzyme.26 In another case, activity of immobilized enzyme was retained by maintenance of enzyme structure.23 Therefore, maintenance of higher-order structures of biocatalyst immobilized on supports is a possible means of maintaining the excellent catalytic performance of the enzyme. Tuning of enzyme properties via immobilization has been often reported.32–34 Guisan et al. immobilized lipase and penicillin acylase on a carrier such as an agarose gel, and demonstrated their activity, immobilization yield and enantiomeric excess.
In this paper, we describe the catalytic performance (hydrolytic activity and enantioselectivity) of the hydrolytic enzyme subtilisin immobilized on an organo-functionalized MPS surface and the influence of the organic chain structure of MPS surface on conformational changes of the enzyme inside the MPS pore. To the best of our knowledge, the influence on catalytic performance of changes in the higher-order structure of an enzyme immobilized on mesoporous inorganic materials has not yet been reported.
2. Experimental
Detailed experimental procedures are presented in the ESI.†3. Results and discussion
3.1. Characterization of synthesized mesoporous silica
Various MPS materials with different pore-size distributions were synthesized from a cationic surfactant (cetyltrimethylammonium bromide, CTAB) or triblock co-polymer (poly(ethylene glycol)-block poly(propylene glycol)-block poly(ethylene glycol), Pluronic P123, EO20PO70EO20) template. Fig. S1 (ESI†) shows field-emission scanning electron microscope (FE-SEM) images of each MPS material after calcination at 500 °C. The material denoted MPS-1 is composed of uniform rods approximately 500 nm in size, as measured from the FE-SEM images. The materials denoted MPS-2 and MPS-3 are composed of uniform nanoparticles approximately 50 nm in size. MPS-2 nanoparticles form aggregates >1 μm in size whereas MPS-3 nanoparticles form secondary block-like particles approximately 500 nm in size. The MPS particle size is reduced if the pore diameter is controlled by a swelling reagent during synthesis.A typical method for separating an immobilized enzyme from its substrate is by centrifugation. For a simpler method, we created a composite material composed of coated magnetic nanoparticles (MN) on MPS (ESI†, Fig. S2). The particle size of the MN–MPS composite is the same as that of MPS before MN modification. Fig. 1 shows transmission electron microscope (TEM) images of the MPS materials and composite; the figure clearly shows that in the composite, MN is absorbed onto the MPS surface. MPS-1 and MPS-2 contain small, uniform cylindrical domains containing very small (∼3 nm) and small (∼6 nm) pores. MPS-3 contains large homogenous pores (∼10 nm). Fig. S3 (ESI†) shows pore-size distribution curves and nitrogen adsorption–desorption isotherms for MPS-1, MPS-2, and MPS-3 obtained by the Barrett–Joyner–Halenda (BJH) and Brunauer–Emmett–Teller (BET) methods. Table S1 (ESI†) lists the associated values. The MPS materials have pore sizes ranging from 2.7–12.3 nm, an extremely large surface area ranging from 501–963 m2 g−1, and a pore volume of 1.4–1.9 cm3 g−1. Nitrogen adsorption–desorption isotherms show that these materials are a type IV. The maximum pore size of MPS-2 increased from 2.7 to 5.3 nm when synthesis was performed in the presence of swelling reagent 1,3,5-trimethylbenzene (TMB). This value is sufficiently large to trap the subtilisin molecule (molecular weight 27 kDa: dimensions 4.1 nm × 7.8 nm × 3.7 nm).
 | ||
| Fig. 1 Transmission electron microscopy images: (a) MPS-1; (b) MPS-2; (c) MPS-3; (d) MN–MPS-2 composite. Scale bar = 100 nm. | ||
The MPS pore structure was investigated by small-angle X-ray diffraction (XRD). Fig. S4a (ESI†) shows the patterns for MPS-1, MPS-2, and MPS-3. Three well-resolved peaks on MPS-1 and MPS-3 are evident, corresponding to the (100), (110), and (200) reflections characteristic of mesoporous materials with ordered two-dimensional hexagonal mesostructures (p6mm).35 The pattern for MPS-2 shows a weak, broad peak in the low-angle diffraction region, suggesting that the pore structures of materials synthesized with the swelling reagent (TMB) are not well ordered.
The surface phase structure was also investigated by wide-angle XRD. Fig. S4b (ESI†) shows the patterns for the MN–MPS-2 composite, with several peaks that are typical of Fe3O4.36 The amount of coated MN (particle size of MN is 10 nm) is about 20% of the total mass of the composite.
The saturation magnetization of the samples was measured by a vibrating sample magnetometer. Fig. S5 (ESI†) shows the curves for pure MN, MN–MPS-2 composite, and washed MN–MPS-2 composite are 49.58, 10.07, and 10.85 emu g−1, respectively, suggesting that the initial magnetization properties of MN are retained during composite formation. Also, magnetization of all samples (pure MN, MN–MPS-2 and washed MN–MPS-2) shows no hysteresis, which is typical of superparamagnetic nanomaterials.37
3.2. Catalytic activity and structural evaluation of subtilisin immobilized on MPS
Subtilisin was immobilized on three types of MPS material (pore size 2.7, 5.3, and 12.3 nm) by physical adsorption and the presence of subtilisin on the MPS surface was verified by using Fourier transform infrared (FT-IR) spectrometry and thermogravimetric/differential thermal analysis (TG-DTA). Fig. S6 (ESI†) shows the various FT-IR spectra. The spectrum of the MPS-2 material (curve a) shows dominant peaks attributable to MPS (1063 and 796 cm−1) and water molecules (1621 cm−1).38 The spectrum of subtilisin immobilized on no-substituted MPS-2 (No-MPS-2, curve b) shows bands in the regions of amide I and II (1644 and 1530 cm−1), respectively,39,40 as do the spectra of all other samples of subtilisin immobilized on MPS materials (curves b, c and d). The spectra of subtilisin immobilized on ethyl-group-modified MPS-2 (Et-MPS-2, curve c) and 3-mercaptopropyl-group-modified MPS-2 (SH-MPS-2, curve d) show peaks attributable to CH3 (1370 cm−1), CH2 (2885 and 2964 cm−1) and SH (2550 cm−1). Fig. S7 (ESI†) shows the TG-DTA analysis results. Exothermic DTA peaks attributable to the ethyl-group, mercaptopropyl-group, and subtilisin are evident at 278, 288, and 400–500 °C, respectively. The zeta-potential of MPS at pH 7 was −21.06 (data not shown) but subtilisin, with an isoelectric point of approximately 10, has a positive charge. Therefore, the immobilization of subtilisin on MPS was multipoint immobilization by electrostatic interaction between the hydroxyl group of MPS surface and amino group of enzyme surface.41,42The hydrolytic activity of immobilized subtilisin was measured by hydrolyzing racemic 1-phenylethyl acetate at room temperature for two days. Relative activities of free and immobilized subtilisin were calculated from eqn (1):
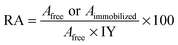 | (1) |
Fig. 2 shows the relative activities of subtilisin immobilized on the various MPS materials. The amounts of subtilisin immobilized on MPS-1, MPS-2, and MPS-3 were 0.10, 0.30, and 0.26 mg of enzyme/mg of MPS support, respectively. Maximum relative activity (126%) was observed for subtilisin immobilized on MPS-2 (pore size 5.3 nm). Interestingly, the result of BET measurements using MPS-2 immobilized subtilisin demonstrated that the pore diameter was decreased from 5.3 nm diameter to approximately 4.0 nm, indicating that subtilisin was preferentially immobilized into the mesopore (ESI†, Fig. S8). However, subtilisin on MPS-1 may be immobilized on the outer surface of mesopores since the enzyme size is larger than the pore size. Also, the enzyme on MPS-3 may be immobilized on the inner surface of mesopores for larger mesopore size. When the subtilisin was immobilized into mesopores fitted to the subtilisin molecule size (4.1 nm × 7.8 nm × 3.7 nm), the enzyme stability may increase to prevent enzyme conformational changes resulting from contact with an organic solvent. Thus, an important factor for enzyme immobilization seems to be the relationship between the MPS pore size and enzyme molecular size. The optimal immobilized carrier for subtilisin, MPS-2, was therefore used in subsequent experiments.
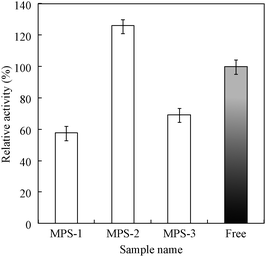 | ||
| Fig. 2 Catalytic activity of subtilisin immobilized on various MPS materials with different pore diameters. Relative activity was calculated by eqn (1). “Free” denotes subtilisin solution before immobilization. | ||
The MPS-2 surface was organo-functionalized using the silane coupling reagents methyltrimethoxysilane, (3-mercaptopropyl)-trimethoxysilane, and 3-isocyanatepropyltriethoxysilane. Subtilisin was immobilized on each functionalized MPS-2 with similar amounts (approximately 0.3 mg of enzyme/mg of MPS). Fig. 3 shows the hydrolytic activities and enantioselectivity E values21 of subtilisin immobilized on organo-functionalized MPS-2. E values of free and immobilized subtilisin were calculated according to eqn (2):
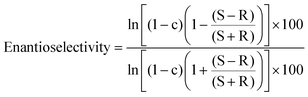 | (2) |
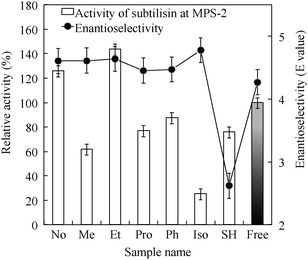 | ||
| Fig. 3 Catalytic activity and enantioselectivity of subtilisin immobilized on several organofunctionalized MPS-2 materials. Relative activity and E value were calculated by eqn (1) and (2). “Free” denotes subtilisin solution before immobilization. | ||
The secondary and tertiary structures of native and immobilized subtilisin were studied by circular dichroism (CD) spectroscopy and fluorescence emission spectra of Trp residues. Fig. 4a shows the CD spectra of each enzyme measured at wavelengths of 190–260 nm for native subtilisin and for immobilized subtilisin suspended in 10 mM phosphate-buffer (pH7.0, PBS); the results are summarized in Table 1. The spectrum of free subtilisin (dotted line) shows broad negative peaks from 208 to 222 nm, suggesting a combination of two structures: α-helical with negative maxima at 222 and 208 nm,43 and β-sheet with negative maximum at 217 nm.44 The spectrum of the secondary structure of subtilisin immobilized on Et-MPS-2 (dashed line) shows the highest hydrolytic activity because the structure does not undergo major structural changes. However, the conformation of subtilisin immobilized on SH-MPS-2 (dot-dashed line) has a lower E value, suggesting significant conformation changes such as decrease in the proportion of the α-helical structure and increase in the proportion of the β-sheet structure compared to native subtilisin. Fig. 4b shows the fluorescence emission spectra of Trp residues from each immobilized subtilisin. The highest emission intensities are as follows: for free subtilisin at 303 nm; for subtilisin immobilized on No-MPS-2, Et-MPS-2, and SH-MPS-2, at 307, 307 and 308 nm, respectively. The peaks are slightly red-shifted, suggesting partial unfolding of the tertiary structure or greater exposure of Trp residues to the solvent.45,46
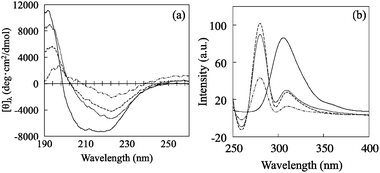 | ||
| Fig. 4 (a) Circular dichroism spectra of free subtilisin and subtilisin immobilized on various MPS-2 materials. For comparison, the corrected spectrum of free subtilisin in aqueous buffer is also shown (solid line). (b) Fluorescence emission curve for the Trp residue of free and immobilized subtilisin. Free subtilisin is denoted by solid line, Et-MPS-2 by dotted line, No-MPS-2 by dash line, and SH-MPS-2 by dot-dashed line. | ||
| Sample | α-Helix (%) | β-Sheet (%) | β-Turn (%) | Random coil (%) |
|---|---|---|---|---|
| Free | 9.6 | 29.4 | 22.7 | 38.3 |
| No-MPS-2 | 6.8 | 36.6 | 20.6 | 36.0 |
| Et-MPS-2 | 8.9 | 37.1 | 22.6 | 31.4 |
| SH-MPS-2 | 2.6 | 40.6 | 19.0 | 37.8 |
3.3. Assays of subtilisin stability under various conditions
The stability of immobilized subtilisin was investigated using three types of assays under various conditions. Protein biocatalysts are known to undergo thermal denaturation. We therefore measured the thermal denaturation temperature of subtilisin immobilized on MPS-2 by differential scanning calorimetry (DSC) analysis. Fig. 5a shows endothermic curves for free subtilisin (A1) and for subtilisin immobilized on No-MPS-2 (A2), Et-MPS-2 (A3) and SH-MPS-2 (A4). The denaturation temperatures, estimated from the curve minima, are 57, 65, 53, and 55 °C, respectively. The differences in these values are due to different thermal-energy absorptions of functional groups on the MPS-2 surface. For example, subtilisin immobilized on Et-MPS-2 becomes denatured at a relatively low-temperature because the ethyl group on the MPS-2 surface has a lower rate of absorption of thermal-energy than hydroxyl and mercapto groups. On the other hand, hydroxyl group on No-MPS-2 has a high rate of absorption of thermal-energy.30 Therefore, subtilisin immobilized on No-MPS-2 was denatured at a relatively high temperature for decrease a thermal load to enzyme. Fig. 5b shows the remaining activity of free subtilisin and each immobilized subtilisin after thermal treatment at 60 °C for different times. Subtilisin immobilized on No-MPS-2 exhibits the best catalytic activity, retaining ∼50% of its initial activity even after 60 min. This result exhibited that subtilisin immobilized on No-MPS-2 becomes stabilized with respect to temperature by having a higher thermal denaturation temperature compared with free and other immobilized subtilisin.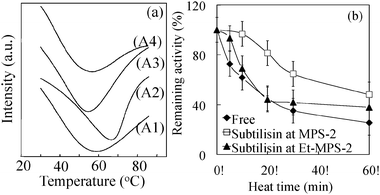 | ||
| Fig. 5 (a) Differential scanning calorimetry curves for free subtilisin (A1) and subtilisin immobilized on No-MPS-2 (A2), Et-MPS-2 (A3), and SH-MPS-2 (A4). (b) Thermal stability as a function of time for free subtilisin and subtilisin immobilized on No-MPS-2 and Et-MPS-2 at 60 °C in 10 mM phosphate buffer (pH 7.0). Initial activities are considered as 100%. | ||
Fig. 6 shows the catalytic activity of subtilisin immobilized on a MN–MPS-2 composite after various reaction cycles. Each immobilized subtilisin retains ∼40% of its initial activity after five recycle reactions. These results suggest that subtilisin gets stripped from the pore during the multiple soakings, separations and washing steps involved in recycling because it is attached to the silica wall by only relatively weak interaction forces. Therefore, the supernatant from the recycled samples was tested for hydrolytic activity and protein content. The total protein content, after five recycles, was about 40% of its value before recycling (0.30 → 0.12 mg of subtilisin/mg of MN–MPS-2 or MN–Et-MPS-2 support). To prevent a release of enzyme from MPS, subtilisin would be held by encapsulation on MPS with peptide, poly-ionic polymer or inorganic materials.41,47–49 The catalytic activity of subtilisin is the same when it is immobilized on No-MPS-2 and MN–MPS-2, suggesting that MN does not affect its catalytic activity. Fig. 7 shows the catalytic activity of subtilisin free and immobilized on No-MPS-2 in the acetone solvent of various concentrations. The activity of free subtilisin decreases to <50% of its initial value as concentration of acetone increases to 30%. The activity of subtilisin immobilized on Et-MPS-2 decreases to ∼70% and 40% of its initial value as acetone concentration increases to 30% and 50%, respectively.
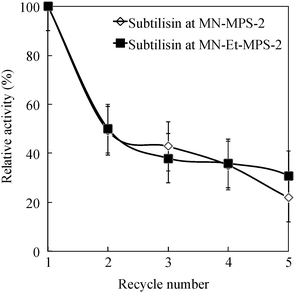 | ||
| Fig. 6 Relative activity of the subtilisin, immobilized on MN–MPS-2 and MN–Et-MPS-2, as a function of catalyst recycle number. | ||
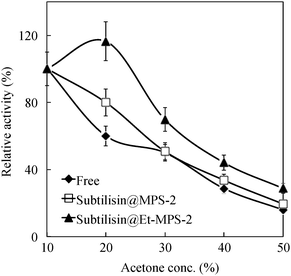 | ||
| Fig. 7 Relative activity of subtilisin, free, and immobilized on No-MPS-2 and Et-MPS-2 as a function of acetone solvent concentration. Initial activities are considered as 100%. | ||
4. Conclusions
In summary, MPS materials with different pore sizes were synthesized using a cationic surfactant (CTAB) as the organic template or triblock co-polymer (Pluronic P123) where particle morphology and pore diameter were controlled by a swelling reagent. The MPS materials were surface-functionalized using a silane coupling reagent and MN. Subtilisin was immobilized on the organofunctionalized MPS materials by physical adsorption. The hydrolytic activity, measured by hydrolysis with racemic 1-phenylethylacetate, is highest when subtilisin was immobilized on Et-MPS-2 (pore size: 5.3 nm). The E value decreases significantly when subtilisin is immobilized on SH-MPS-2. Conformational changes in subtilisin caused by the organofunctional group on the MPS material, estimated by CD spectra and fluorescence emission spectra of Trp residues, suggest that the secondary structure of subtilisin is retained when it is immobilized on Et-MPS-2 but not when it is immobilized on SH-MPS-2. The thermal stability of immobilized subtilisin is affected by the thermal energy absorption of the MPS material.In conclusion, we have presented herein significant information regarding the conformation changes and stabilities of biocatalysts such as enzyme and antibody biocatalyst immobilized on MPS materials.
Notes and references
- Y. Li, G. Zhou, C. Li, D. Qin, W. Qiao and B. Chu, Colloids Surf., A, 2009, 341, 79 CrossRef CAS.
- M. Hartmann, Chem. Mater., 2005, 17, 4577 CrossRef CAS.
- T. Itoh, R. Ishii, T. Ebina, T. Hanaoka, Y. Fukushima and F. Mizukami, Bioconjugate Chem., 2006, 17, 236 CrossRef CAS.
- S. Seelan, A. Sinha, K. Kato and Y. Yokogawa, Adv. Mater., 2006, 18, 3001 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S.-Y. Lin, Adv. Funct. Mater., 2007, 17, 1225 CrossRef CAS.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398 CrossRef CAS.
- S. Shen, P. S. Chow, S. Kim, K. Zhu and R. B. H. Tan, J. Colloid Interface Sci., 2008, 321, 365 CrossRef CAS.
- K. Kato and S. Seelan, J. Biosci. Bioeng., 2010, 109, 615 CrossRef CAS.
- K. Kato, S. Seelan and T. Saito, J. Biosci. Bioeng., 2009, 108, 310 CrossRef CAS.
- R. Atluri, Y. Sakamoto and A. E. Garcia-Bennett, Langmuir, 2009, 25, 3189 CrossRef CAS.
- C. Ispas, I. Sokolov and S. Andreescu, Anal. Bioanal. Chem., 2009, 393, 543 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 393, 543 Search PubMed.
- K. Kosuge, T. Sato, N. Kikukawa and M. Takemori, Chem. Mater., 2004, 16, 899 CrossRef CAS.
- J. Lee, H. B. Na, B. C. Kim, J. H. Lee, B. Lee, J. H. Kwak, Y. Hwang, J. Park, M. B. Gu, J. Kim, J. Joo, C. Shin, J. W. Grate, T. Hyeon and J. Kim, J. Mater. Chem., 2009, 19, 7864 RSC.
- J. Lee, J. Kim, J. Kim, H. Jia, M. I. Kim, J. H. Kwak, S. Jin, A. Dohnalkova, H. G. Park, H. N. Chang, P. Wang, J. W. Grate and T. Hyeon, Small, 2005, 1, 744 CrossRef CAS.
- T. Clark Jr., J. D. Ruiz, H. Fan, C. J. Brinker, B. I. Swanson and A. N. Parikh, Chem. Mater., 2000, 12, 3879 CrossRef.
- M. P. Kapoor, Q. Yang and S. Inagaki, J. Am. Chem. Soc., 2002, 124, 15176 CrossRef CAS.
- C. Yoshina, T. Asefa, N. Coombs, M. MacLachlan and G. A. Ozin, Chem. Commun., 1999, 2539 RSC.
- S. Inagaki, S. Guan, T. Ohsuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS.
- K. Kato, S. Nakagaki, M. Nishida and K. Hirao, J. Ceram. Soc. Jpn., 2011, 119, 140 CrossRef CAS.
- D. Yu, Z. Wang, L. Zhao, Y. Cheng and S. Cao, J. Mol. Catal. B: Enzym., 2007, 48, 64 CrossRef CAS.
- Y. Han, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 643 CrossRef CAS.
- K. Kato, M. Suzuki, M. Tanemura and T. Saito, J. Ceram. Soc. Jpn., 2010, 118, 410 CrossRef CAS.
- M. Thust, M. J. Schoning, J. Vetter, P. Kordos and H. Luth, Anal. Chim. Acta, 1996, 323, 115 CrossRef CAS.
- L. Cao, F. V. Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361 CrossRef CAS.
- B. Kranz, J. Burck, M. Franzreb, R. Koster and A. S. Ulrich, J. Colloid Interface Sci., 2007, 316, 413 CrossRef CAS.
- L. Ferreira, M. A. Ramos, J. S. Doedick and M. H. Gil, J. Mol. Catal. B: Enzym., 2003, 21, 189 CrossRef CAS.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019 CrossRef CAS.
- A. Salis, D. Meloni, S. Ligas, M. F. Casula, M. Monduzzi, V. Solinas and E. Dumitriu, Langmuir, 2005, 21, 5511 CrossRef CAS.
- K. Murai and K. Kato, Appl. Surf. Sci., 2011, 258, 1725 Search PubMed.
- J. Huang, Y. Liu and X. Wang, J. Mol. Catal. B: Enzym., 2009, 57, 10 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS.
- R. C. Rodrigues, B. C. C. Pessela, G. Volpato, R. Fernandez-Lafuente, J. M. Guisan and M. A. Z. Ayub, Process Biochem., 2010, 45, 1268 CrossRef CAS.
- M. Terreni, G. Pagani, D. Ubiali, R. Fernandez-Lafuente, C. Mateo and J. M. Guisan, Bioorg. Med. Chem. Lett., 2001, 11, 2429 CrossRef CAS.
- G. Zhou, Y. Chen, J. Yang and S. Yang, J. Mater. Chem., 2007, 17, 2839 RSC.
- Q. Gan, X. Lu, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and C. Liu, Biomaterials, 2011, 32, 1932 CrossRef CAS.
- K. Liu, L. Zhao, P. Klavins, F. E. Osterloh and H. Hiramatsu, J. Appl. Phys., 2003, 93, 7951 CrossRef CAS.
- L. Chen, J. Di, C. Cao, Y. Zhao, Y. Ma, J. Luo, Y. Wen, W. Song, Y. Song and L. Jiang, Chem. Commun., 2011, 47, 2850 RSC.
- Z. An, S. Lu, J. He and Y. Wang, Langmuir, 2009, 25, 10704 CrossRef CAS.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405 CrossRef CAS.
- K. Hernanez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107 CrossRef.
- M. Higuchi, K. Nagata, S. Abiko, M. Tanaka and T. Kinoshita, Langmuir, 2008, 24, 13359 CrossRef CAS.
- Y. Zhao, M. Tanaka, T. Kinoshita, M. Higuchi and T. Tan, J. Controlled Release, 2010, 147, 392 CrossRef CAS.
- F. Farivar, A. A. Moosavi-Movahedi, Y. Sefidbakht, K. Nazari, J. Hong and N. Sheibani, Biochem. Eng. J., 2010, 49, 89 CrossRef CAS.
- V. Bansal, Y. Delgado, E. Fasoli, A. Ferrer, K. Griebenow, F. Secundo and G. L. Barletta, J. Mol. Catal. B: Enzym., 2010, 64, 38 CrossRef CAS.
- Z. Cabrera, G. Fernandez-Lorente, R. Fernandez-Lafuente, J. M. Palomo and J. M. Guisan, Process Biochem., 2009, 44, 226 CrossRef CAS.
- V. Vamvakaki, M. Hatzimarinaki and N. Chaniotakis, Anal. Chem., 2008, 80, 5970 CrossRef CAS.
- L. Betancor and H. R. Luckarift, Trends Biotechnol., 2008, 26, 566 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods, and additional data. See DOI: 10.1039/c1cy00258a |
| This journal is © The Royal Society of Chemistry 2012 |