Speciation of Pd(OAc)2 in ligandless Suzuki–Miyaura reactions†
Luis A.
Adrio
a,
Bao N.
Nguyen
a,
Gemma
Guilera‡
b,
Andrew G.
Livingston
c and
King Kuok (Mimi)
Hii
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: mimi.hii@imperial.ac.uk; Fax: +44-20-7594 5804; Tel: +44-20-7594 1142
bEuropean Synchrotron Radiation Facility (ESRF), P. O. Box 220, 38043 Grenoble Cedex, France
cDepartment of Chemical Engineering and Chemical Technology, Imperial College London, UK
First published on 8th September 2011
Abstract
Generation of catalytically active Pd(0) species from Pd(OAc)2 has been examined, in the context of Suzuki–Miyaura reactions involving substitution of aryl bromides under aerobic and ambient conditions. Using a combination of spectroscopic, microscopic and kinetic measurements, the role of each reaction component is delineated in the speciation of the palladium species. Among the key findings are the effects of O2, H2O and inorganic base, and implications for catalytic activity.
1. Introduction
The discovery of the Suzuki–Miyaura (SM) cross-coupling reaction is undoubtedly one of the important landmarks of organic chemistry for the 20th century. Providing a reliable protocol for the formation of aryl–aryl bonds, it has stimulated great progress in modern material1–4 and medicinal chemistry.5–11 Accordingly, there is sustained interest in developing Pd catalysts that can offer the requisite reactivity with high TON and TOF. To this end, a number of ligands were developed, which can generate highly active catalysts with wider scope,12–14 allowing these processes to be viable on an industrial scale.15Conversely, there is significant interest in the development of ‘ligandless’ protocols.16–23 Although the scope of the catalytic system is presently limited to aryl iodides, bromides and certain activated chlorides, these are very often adequate for most transformations, and offer a practical alternative to ligand-promoted reactions, as they can be performed aerobically and bypasses issues related with cost and removal of the ligand.
Very often, Pd(OAc)2 is employed as a catalyst precursor in these reactions, which is reduced in situ to form catalytically active Pd(0) species, in molecular, colloidal and/or nanoparticle forms, that can catalyse cross-coupling reactions at ambient temperature.17,24 Further aggregation of these species to form larger particles deactivates the catalyst, eventually leading to the deposition of Pd black (Fig. 1). The process can be dependent upon the reaction medium, oxygen, moisture and additives/stabilizers.
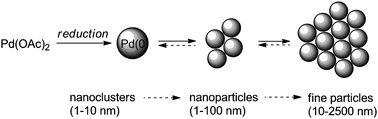 | ||
| Fig. 1 Process of catalyst activation and deactivation by aggregation. | ||
A plethora of additives/stabilizers has been reported for ‘ligandless’ SM reactions,21,23,25–28 but this is not always necessary; we29 and others17,24 have shown that Pd(OAc)2 alone is sufficient for high turnovers. Notably, these reactions occur in protic solvents such as alcohols, ethylene glycol derivatives, or mixtures of water with organic solvents. Previously, the formation of Pd nanoparticles from Pd(OAc)2 under Suzuki coupling reaction conditions was reported to be promoted by O2 in PEG-400.27,28 More recently, PdCl2 can also be activated towards catalysis in an aqueous media under an aerobic atmosphere.30 In these papers, the beneficial effects of O2 and H2O were illustrated by the comparison of isolated yields, rather than kinetic profiles, and it was not clear how these promoters exert a beneficial effect for catalysis. In the present work, we will delineate the role of each of the reacting components in catalyst activation, stability and eventual deactivation.
2. Experimental
2.1 Materials
All chemical reagents were procured commercially and used as received unless otherwise indicated. Anhydrous solvents were dried in a purification system by passing through columns of activated molecular sieves under N2. Pd(OAc)2 (Pd-111) was obtained from Johnson Matthey plc through an academic loan scheme.2.2 Experimental methods
Catalytic reactions were monitored by recording 1H (400 MHz) and 19F (376 MHz) NMR spectra of reaction mixtures in CDCl3 on Bruker Avance-400 spectrometers. High-resolution TEM images were recorded using a JEOL 2010 instrument, fitted with an analytical pole-piece (resolution 0.23 nm, +/− 30° tilt), in bright field mode. Stopped-flow experiments were performed using a Bio-Logic SFM-300 multi-mixer fitted with a MPS-52 syringe controller and Bio-Kine32 software, and equipped with a TC-100 quartz cuvette (10 × 1.5 mm, transparency between 185–2500 nm), with a total volume of 45 μL. UV-Vis spectra were measured using a deuterium tungsten light source and a multiwavelength J&M TIDAS diode array for spectra collection with an integration time of 15 ms. EXAFS experiments were conducted at the dispersive undulator beamline ID24 of the ESRF, Grenoble, France, operating at an average current of ca. 90 mA. The absorption measurements were performed using a Si (311) polychromator in the Bragg configuration. EXAFS spectra at the Pd K-edge (24.35 keV) were collected in transmission mode with 1 s (1 ms × 1000 accumulations) resolution. An FReLoN2k (Fast Readout Low Noise) CCD camera working in kinetic mode was used to collect the data.31 A Pd reference foil was measured regularly to provide the energy calibration for the spectra and was used to determine the multiple electron reduction factor to be employed for the EXAFS analysis of the samples (S02 = 0.78). The EXAFS data were background subtracted and normalised following standard procedures using the software package Viper v. 10.1 for Windows.32 Radial distribution functions were obtained by Fourier transformation of the k2-weighted EXAFS functions. Amplitudes and phases for single and multiple scattering contributions to the EXAFS signal have been obtained by applying the FEFF7 code to different crystallographic model compounds. The fitting procedure has been performed in back Fourier k space and crossed-checked on R space. High-resolution TEM images were recorded using a JEOL 2010 instrument. ICP-OES (Pd, P) and combustion analyses (C, H) were carried out by a commercial provider.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of THF–H2O (degassed for reactions under N2) was then added, and the resultant mixture was stirred at room temperature for up to 2 h. During this period, aliquots of the reaction mixture (0.2 mL) were extracted and diluted with CH2Cl2 (1.5 mL), dried over MgSO4, filtered through cotton wool, and evaporated. The residue was dissolved in CDCl3 and subjected to 1H NMR and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
:1 v/v, 5.9 mL) (Solution B). A magnetic stir bar was added, and the resultant mixture was stirred at room temperature. Periodically, aliquots of the reaction mixture (0.2 mL) were extracted and diluted with CH2Cl2 (1 mL) and H2O (1 mL), dried over MgSO4, filtered through cotton wool, and evaporated. The residue was dissolved in CDCl3 and subjected to 1H NMR and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
:1 v/v, 4 mL) or THF–water (2:1 v/v, 4 mL) was added. The reaction vial was then fitted with a rubber septum to which a balloon of O2 was attached, and the mixture was stirred at room temperature for 16 h. The solvent was then evaporated, and the residue was subjected to 1H and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
:1 v/v mixture of THF–H2O (degassed for reactions under N2) was then added, and the resultant mixture was stirred at room temperature for up to 2 h. During this period, aliquots of the reaction mixture (0.2 mL) were extracted and diluted with CH2Cl2 (1.5 mL), dried over MgSO4, filtered through cotton wool, and evaporated. The residue was dissolved in CDCl3 and subjected to 1H NMR and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
:1 v/v, 5.9 mL) (Solution B). A magnetic stir bar was added, and the resultant mixture was stirred at room temperature. Periodically, aliquots of the reaction mixture (0.2 mL) were extracted and diluted with CH2Cl2 (1 mL) and H2O (1 mL), dried over MgSO4, filtered through cotton wool, and evaporated. The residue was dissolved in CDCl3 and subjected to 1H NMR and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
:1 v/v, 4 mL) or THF–water (2:1 v/v, 4 mL) was added. The reaction vial was then fitted with a rubber septum to which a balloon of O2 was attached, and the mixture was stirred at room temperature for 16 h. The solvent was then evaporated, and the residue was subjected to 1H and 19F NMR analyses. Product conversion was determined by 1H NMR integration.
2.3 Stopped-flow experiments
Prior to the experiment, the stopped-flow system was purged with the solvent three times. Solutions of Pd3(OAc)6 (8.9 mM) and arylboronic acid (17.8 mM) were prepared under N2 and anhydrous conditions using Schlenk techniques unless otherwise specified. UV-vis spectra were collected every 0.5 s between 200–700 nm, observing absorbance peaks due to Pd3(OAc)6 (300 nm), biaryl (λmax = 300 nm) and Pd–Pd formation (450 nm). The kinetic model was fitted using Berkeley MadonnaTM programme (Modelling and Analysis of Dynamic Systems).3. Results and discussion
3.1 Effect of O2
The reaction between 4-chlorobromobenzene (1) and 4-fluorophenylboronic acid (2) in a mixture of THF–H2O was chosen as the model system for our initial study (Scheme 1), where the C–C bond formation occurs specifically at the C–Br position.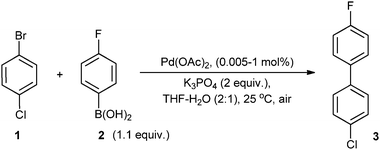 | ||
| Scheme 1 Model SM reaction used in this work. | ||
Using 1 mol% of Pd(OAc)2 as the catalyst precursor, reactions were conducted under an atmosphere of O2 or N2 (Fig. 2). No induction period was observed in either of these reactions, signifying that the generation of an active catalyst is extremely rapid. Significantly, the results clearly indicated that the role of O2 is to prevent catalyst deactivation: under an inert N2 atmosphere, very fast catalytic turnovers were observed initially, reaching >80% conversion within 10 min, but the reaction was curtailed at ca. 86% conversion. In contrast, the reaction performed under O2 has a slower initial rate, but turnover was maintained until it reached completion within 60 min. Thus, the presence of O2 suppresses both catalyst activation and deactivation processes, affording a higher overall conversion.
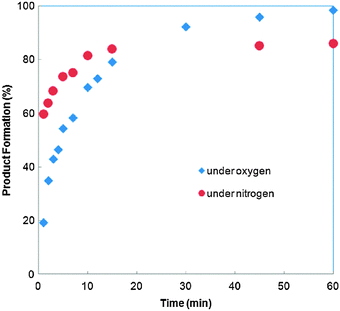 | ||
| Fig. 2 Effect of O2 on the reaction (at 1 mol% catalyst loading). | ||
3.2 Homo- vs. cross-coupling
Homocoupling of the arylboronic acid is also reported to be catalysed by ligandless Pd(OAc)2, facilitated by moisture and air,33 but it was not a significant process in any of the experiments conducted in the present study.34 This was investigated by exposing arylboronic acid 2 to O2 and 3 mol% of Pd(OAc)2 under reaction conditions prescribed for homo-33 and cross-29 coupling conditions, I and II, respectively (Scheme 2). The reaction conducted using K2CO3 proceeded with quantitative conversion of 2 to biaryl 4, whereas only 32% of the homocoupled product was obtained when K3PO4 was employed. Thus, the choice of an inorganic base and a solvent system has a profound effect on the partition of the reaction pathway––i.e. when K3PO4 is employed as a base, the homocoupling process can be considered as non-competitive.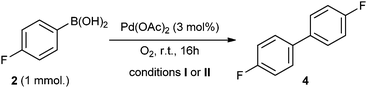 | ||
| Scheme 2 Homocoupling of arylboronic acid 2 by Pd(OAc)2: Conditions I: K2CO3, acetone–water (1:1), >99% conversion. Conditions II: K3PO4, THF–water (2:1), 32% conversion. | ||
3.3 Catalyst activation/stability
To interrogate the effect of other reaction components on catalyst stability/activation, catalytic loading was lowered to 0.05 mol% to ensure observable and comparable rates.35 A stock solution of Pd(OAc)2 was prepared in THF under aerobic conditions, to which individual components of the reaction (water, base or aryl boronic acid) was added (Solution A). This was injected into a mixture of reactants (aryl bromide, aryl boronic acid and base) in a 2:1 mixture of THF–H2O36 (Solution B). The reaction progress was monitored by 1H NMR analysis of extracted reaction aliquots (Fig. 3). The catalyst activity of solution A was evaluated when freshly prepared, and reassessed after it has been ‘aged’ for 48 h.
 | ||
| Fig. 3 Assessment of catalytic performance of Pd(OAc)2. | ||
Freshly prepared solutions A (used within 5 minutes of preparation) showed similar temporal profiles within the first 60 min of the reaction, signifying that the amount of the active catalyst generated is little affected by the presence of water, inorganic base and arylboronic acid (Fig. 4a). After 48 h, the Pd(OAc)2 precursor in solutions A evolved into separate species, reflected in different observed reaction profiles (Fig. 4b); while the solution containing arylboronic acid initiated catalytic activity instantaneously, notable induction periods were observed when the precursor was left exposed to water or K3PO4.
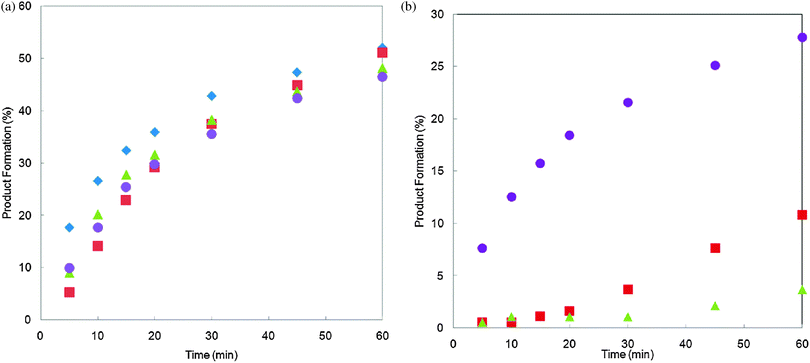 | ||
Fig. 4
Catalytic activities of solutions A prepared in: THF ( ), THF–H2O ( ), THF–H2O ( ), K3PO4, in H2O only ( ), K3PO4, in H2O only ( ) and arylboronic acid (2), THF ( ) and arylboronic acid (2), THF ( ). Effective catalyst loading = 0.05 mol%. Reaction progress with: (a) freshly prepared solution (left), and (b) after standing at room temperature for 48 h (right). ). Effective catalyst loading = 0.05 mol%. Reaction progress with: (a) freshly prepared solution (left), and (b) after standing at room temperature for 48 h (right). | ||
3.4 Effect of a base (K3PO4)
Tripotassium phosphate (K3PO4) is a general inorganic base used in many ligandless protocols for SM29 and also Heck37 reactions. It is important to note that although THF and water are miscible to a certain extent, the dissolution of the reactants causes them to separate, such that the reaction occurs in a biphasic solution. By adding K3PO4 to a solution of Pd(OAc)2 in aqueous THF,38 a light-orange solid slowly precipitates as more K3PO4 is added, signifying a formation of a new compound (Fig. 5), which is water-soluble.39 Elemental analyses of the precipitate revealed a Pd:P:C ratio of 4:1:1,40 suggesting the formation of a complex salt through an anion-exchange process, likely to be a network of bridged oxo-/hydroxopalladium units, interspersed by phosphate and acetate anions.
 | ||
| Fig. 5 Solutions containing Pd(OAc)2 (0.5 mM) with varying amounts of K3PO4 in THF–H2O (1:1). | ||
3.5 Solution structure of Pd(OAc)2
Due to the ambidentate nature of the acetate anion, Pd(OAc)2 can exist in several aggregated forms (Fig. 6). While trimeric41 (5) and polymeric42 structures have both been found in the solid state, its solution structure remains controversial. Early independent studies by Wilkinson and Pandey suggested, by molecular weight measurements, that it exists as a trimer (5) in benzene43 and acetic acid44 at ambient temperatures. A much later study by Stoyanov using infrared spectroscopy, however, suggested the existence of an equilibrium process between 5 and a linear dimer (6) in chloroform–acetic acid.45 A few years later, the monomeric form of Pd(OAc)2 (7) was detected in N-methyl-2-pyrrolidinone (NMP) by Evans et al., by examining the EXAFS at the Pd K-edge.46 This was closely followed by a 1H NMR study by Cotton and co-workers, which showed that the trimeric structure (1) is largely retained in methanol, chloroform and benzene.47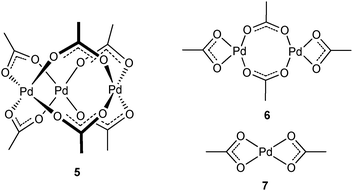 | ||
| Fig. 6 Suggested structures of Pd(OAc)2. | ||
The dipole moment or dielectric constant of a solvent is frequently invoked to describe its polarity, although the two properties are intrinsically different. This can be demonstrated by comparing methanol with NMP, which have markedly different dipole moments, yet very similar dielectric constants (Table 1). Conversely, water has a similar dipole moment to THF, but dramatically different dielectric constants.
| Solvent | Dipole moment (D) | Dielectric constant |
|---|---|---|
| Benzene | 0 | 2.28 |
| Toluene | 0.36 | 2.38 |
| Chloroform | 1.15 | 4.81 |
| Acetic acid | 1.68 | 6.15 |
| THF | 1.75 | 7.5 |
| Methanol | 2.87 | 32.7 |
| N,N-Dimethylformamide | 3.86 | 36.7 |
| NMP | 4.09 | 32.2 |
| Water | 1.85 | 80 |
An initial assessment of results of previous studies with the values presented in Table 1 appears to suggest a greater correlation between the nuclearity of the Pd(OAc)2 and the dipole moment of the solvent in which it is dissolved. With this in mind, the structure of Pd(OAc)2 was examined using Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy at the Pd K-edge (24.35 keV). EXAFS spectra were recorded of the sample as a solid, and also as solutions in toluene and DMF, chosen for their very different polarities.
The data of the solid sample fit well with the crystallographic data reported for the trimeric structure. Due to the limited solubility of the palladium salt in toluene, solutions of a lower concentration (40 mM) were used compared to that used previously in NMP (50 mM).46 Structural parameters extracted from the refinement fits of the EXAFS signals are shown in Table 2. The fitted curves to the experimentally obtained EXAFS signals can be found in the ESI.†
| Sample | Neighbour | CNb | R/Åc | σ 2/Å−2d | ΔE0/eVe |
|---|---|---|---|---|---|
| a Parameters in italics were restricted in the fit (see Experimental). b Coordination number. c Interatomic distance. d Debye–Waller factor. e Energy variation in the Fermi level from theory. f 3.84 < k < 16.69; ΔR = 2.38 Å. g 3.84 < k < 11.58; ΔR = 2.07 Å. h 3.84 < k < 11.58; ΔR = 1.91 Å. | |||||
| Solidf | O | 4.8(4) | 2.016(6) | 0.003(1) | −1.85(3) |
| R = 10.6% | C | 4 | 2.954(6) | 0.005 | −4.85 |
| Pd | 2.2(3) | 3.106(4) | 0.006(1) | −4.85 | |
| In tolueneg | O | 4.38(30) | 2.018(6) | 0.004(2) | 0.18(2) |
| R = 15.4% | C | 4 | 2.898(7) | 0.005 | −2.81 |
| Pd | 0.65(40) | 3.085(12) | 0.006(2) | −2.81 | |
| In DMFh | O | 4.03(15) | 2.046(5) | 0.005(1) | 2.05(1) |
| R = 17.1% | C | 4 | 2.895(7) | 0.005 | −0.95 |
| Pd | 0.45(20) | 3.134(29) | 0.012 | −0.95 |
Dissolution in toluene caused a significant reduction of the FT peak corresponding to the Pd–Pd distance (indicated by the dashed line, Fig. 7), concomitant with a decrease in the Pd–Pd coordination number, and the shortening of Pd–C contact; both these parameters reduced by switching the solvent from toluene to DMF (indicated by the blue line, Fig. 7). Interestingly, there is an observable lengthening of the Pd–O bond length in the order: solid < toluene solution < DMF solution. Additionally, the quality of the fits to 5, although reasonable, worsens the same order. These observations suggest the existence of an equilibrium process between the trimer and other species. The data suggest that the presence of the dimeric structure (6) is unlikely to be significant. As well as being a poor fit with the experimental data, its Pd–Pd distance (2.38–2.58 Å) was also not observable in the FT spectrum.
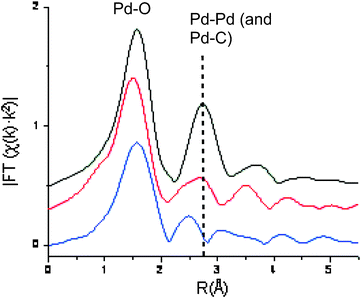 | ||
| Fig. 7 Pd K-edge k2-weighted FT modulus (uncorrected) of Pd(OAc)2 in the solid state (black), and as 40 mM solutions in toluene (red) and DMF (blue). Pd–Pd distance is indicated by the vertical dashed line. | ||
Principal Component Analysis (PCA) was subsequently performed using linear combinations of monomeric and trimeric Pd species, with or without the presence of a third component, which may be Pd particles or 6. The result showed that the inclusion of a third component did not influence the global outcome of the analysis (Table 3), i.e. if a third species is present, it is beyond the detection limit of the technique (<8%). Some considerations upon this analysis can be found in the ESI.†
It is important to acknowledge that these experiments were conducted in an X-ray beam, where local heating cannot be ruled out. Nevertheless, it supports the ability of the Pd(OAc)2 trimer 5 to dissociate to (primarily) monomers in solution, the degree of dissociation increases along with the dipole moment of the solvent, in the order: toluene (21%, 0.36 D) < DMF (42%, 3.86 D) < NMP (100%, 4.09 D); while earlier studies conducted in relatively less polar solvents (benzene, acetic acid and methanol, all <2.9 D) supported (predominantly) the trimeric structure.
3.6 Stability of Pd(OAc)2 in the presence of water and arylboronic acid
The stability of the Pd(OAc)2 precursor was found to be affected by water and arylboronic acid in the earlier part of this study (Fig. 4a and b). To assess these processes in detail, stopped-flow equipment coupled with a UV-spectrometer was used to examine the reaction between palladium(II) acetate with water and arylboronic acid. Using anhydrous THF as a solvent,49 the trimeric structure 5 gives rise to a unique UV absorption peak at 300 nm, which is stable over a period of time. The addition of water triggers two consecutive processes: the first step gives rise to an intermediate species, which decays via a unimolecular process. The first of these processes is dependent upon [H2O], related by a first order relationship (Fig. 8). Subsequent curve fitting yielded values of 4.5 × 10−3 M−1 s−1 and 1.7 × 10−3 s−1 for k1 and k2, respectively, for the reaction conducted in 4:1 THF–H2O (see ESI†). The result of our stopped-flow experiment concurs with a fast nucleophilic attack of water on the Pd trimer 5, followed by the slow dissociation to the less stable monomer 7 (Scheme 3), through a series of equilibrating processes. A similar phenomenon was previously observed, where the formation of heterobimetallic complexes from Pd3(OAc)6 was found to proceed via an initial hydrolytic cleavage of the trimeric structure.50
![Reaction rate vs. [H2O] for the dissociation of Pd trimer (5).](/image/article/2012/CY/c1cy00241d/c1cy00241d-f8.gif) | ||
| Fig. 8 Reaction rate vs. [H2O] for the dissociation of Pd trimer (5). | ||
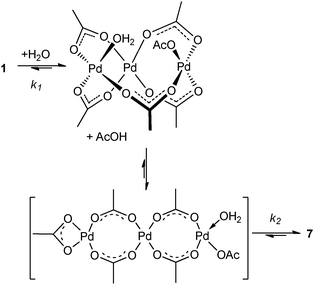 | ||
| Scheme 3 Reaction between Pd3(OAc)6 and water. | ||
3.7 Reduction of Pd(II) to Pd(0)
In previous reports, water was found to accelerate the reduction of tertiary phosphine (PR3) complexes of palladium(II) acetate to LnPd(0), but at elevated temperatures.51,52 Recently, two independent studies of the transmetalation of arylboronic acids to phosphine-ligated palladium complexes were reported,53,54 which implicated the involvement of a hydroxylpalladium intermediate for the transfer of an aryl group from ArB(OH)2.The formation of Pd(0) species was not observed by the introduction of water, under the reaction conditions adopted by the stopped-flow experiments described above. The addition of two equivalents of arylboronic acid 2, which contains small traces of water, also did not affect the appearance of the UV spectrum of Pd3(OAc)6 in anhydrous THF over the timescale of the experiment (75 min, see Fig. S1, ESI†). However, by adding 2 equivalents of arylboronic acid to a solution in 25% H2O–THF, the formation of biaryl 4 was spontaneously observed, signifying a fast reduction of Pd(II) to Pd(0)via double transmetalation and reductive elimination. This was followed by a slow evolution of an absorption peak at ca. 450 nm (Fig. 9), which is attributed to the nucleation of Pd(0).55,56
![Evolution of Pd nanoparticles. Conditions: [Pd(OAc)2] = 3.4 mM, 25% H2O–THF, arylboronic acid 2 (2 equivalents).](/image/article/2012/CY/c1cy00241d/c1cy00241d-f9.gif) | ||
| Fig. 9 Evolution of Pd nanoparticles. Conditions: [Pd(OAc)2] = 3.4 mM, 25% H2O–THF, arylboronic acid 2 (2 equivalents). | ||
Based on these observations, we can deduce that the reduction of palladium(II) acetate is accelerated by water, which first induces a rapid dissociation to monomer 7 (Scheme 4). Water then exchanges with acetate anions to form palladium(II) hydroxide species,57 which undergoes transmetalation with the arylboronic acid to produce palladium(0). The overall process is limited by the highly unfavourable acid–base equilibrium, which is dependent upon the amount of water present and the presence of a base. The reduction of the Pd–OH species by arylboronic acid is extremely rapid (no induction period). At high dilution/low catalytic loading, subsequent formation of Pd⋯Pd clusters is a slow process.56,58
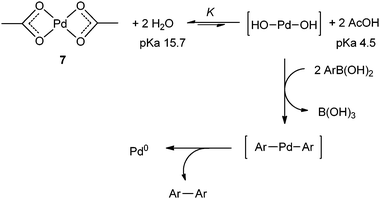 | ||
| Scheme 4 Reduction of palladium acetate to Pd(0). | ||
The effect of solvents on the reduction of Pd(OAc)2 can be clearly illustrated by recording TEM images of solutions generated in THF–H2O or dry toluene in the presence of arylboronic acid (Fig. 10). While the formation of small clusters of colloidal Pd between 2–5 nm is clearly observed in the former, large crystalline Pd(OAc)2 remained largely intact in the anhydrous toluene solution.
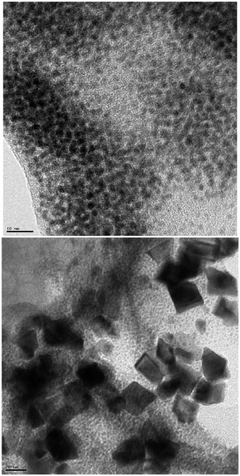 | ||
| Fig. 10 TEM images of solutions generated from solution of Pd(OAc)2 in arylboronic acid, THF–H2O (top, scale bar = 10 nm) and arylboronic acid, toluene (bottom, scale bar = 20 nm). | ||
4. Conclusions
Our study has unravelled the effect of organic solvent and water on the speciation of Pd(OAc)2 in ligandless SM reactions. Most significantly, water was found to have several roles in the catalyst activation process. The presence of this protic solvent causes a fast dissociation of the Pd3(OAc)6 trimer, whereupon acid–base equilibrium occurs to generate hydroxopalladium(II) species, which is subsequently reduced by the arylboronic acid to catalytically active Pd(0) species, leading to fast generation of an active catalyst. Conversely, the presence of water maintains the inorganic base in a separate phase, and could create a reservoir for Pd(II)via the formation of palladium-phosphate salts (Scheme 5), which may be slowly released into the organic layer over the course of the reaction. The presence of O2 appears to suppress the catalyst activation process, but also prevents catalyst deactivation, probably by inhibiting the formation of Pd–Pd bonds through the adsorption of O2.59,60 Under an N2 atmosphere, the very fast generation of the catalytically active Pd(0) species is counteracted by an equally rapid Pd aggregation, leading to premature catalyst deactivation (Fig. 2).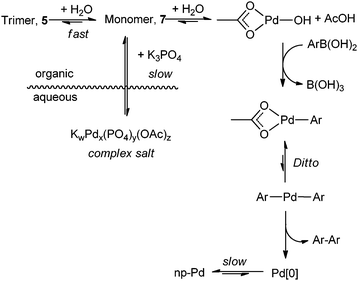 | ||
| Scheme 5 Summary of interactions of Pd(OAc)2 with H2O, K3PO4 and arylboronic acid. | ||
Thus, under working catalytic conditions, the presence of water controls the amount of catalytically active Pd(0) released into the reaction mixture, by a series of reversible equilibria. Under SM reaction conditions, the generation of an active catalyst from Pd(OAc)2 is extremely rapid, compared to its transformation by water, arylboronic acid or base (Fig. 3a and b). The presence of O2 also appears to exert an important effect on catalyst activation (Fig. 2). In both cases, the major catalytic deactivation pathway appears to be the aggregation of Pd to form clusters and nanoparticles.
At this juncture, it is not possible to comment on the nature of the active catalyst in solution, although it is highly likely a mixture of Pd(0) species, ranging from molecular to colloidal species, each with their different reactivities. This inherent complexity makes this a very challenging topic for further research. Nevertheless, work is currently underway to understand this process, as well as to develop effective post-reaction treatment for the removal of palladium residues from the product.61
Acknowledgements
LAA is a recipient of a postdoctoral fellowship from Xunta Galicia (Angeles Alvariño program). BNN's postdoctoral fellowship was supported by an EPSRC grant: ‘Elucidate and Separate’ (ELSEP) (grant number: EP/G070172/1), with further contributions from the Pharmacat Consortium (AstraZeneca, GlaxoSmithKline and Pfizer). We thank Johnson Matthey plc for the provision of Pd salts, and the ESRF for the provision of beam time (project number: CH-2555) and facilities at ID24. We are also grateful to Dr M. Ardakani and Ms E. Ware for help with TEM experiments.Notes and references
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schluter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS.
- M. R. Huang, P. Gao and X. G. Li, Prog. Chem., 2010, 22, 113–118 CAS.
- J. Soloducho, J. Cabaj, K. Idzik, A. Nowakowska-Oleksy, A. Swist and M. Lapkowski, Curr. Org. Chem., 2010, 14, 1234–1244 CrossRef CAS.
- W. W. H. Wong, C. Q. Ma, W. Pisula, C. Yan, X. L. Feng, D. J. Jones, K. Mullen, R. A. J. Janssen, P. Bauerle and A. B. Holmes, Chem. Mater., 2010, 22, 457–466 CrossRef CAS.
- O. Baudoin, M. Cesario, D. Guenard and F. Gueritte, J. Org. Chem., 2002, 67, 1199–1207 CrossRef CAS.
- U. V. Mentzel, D. Tanner and J. E. Tonder, J. Org. Chem., 2006, 71, 5807–5810 CrossRef CAS.
- S. S. Kulkarni and A. H. Newman, Bioorg. Med. Chem. Lett., 2007, 17, 2074–2079 CrossRef CAS.
- E. Perissutti, F. Frecentese, A. Lavecchia, F. Fiorino, B. Severino, F. De Angelis, V. Santagada and G. Caliendo, Tetrahedron, 2007, 63, 12779–12785 CrossRef CAS.
- R. Severinsen, G. T. Bourne, T. T. Tran, M. Ankersen, M. Begtrup and M. L. Smythe, J. Comb. Chem., 2008, 10, 557–566 CrossRef CAS.
- H. Fuwa, A. Saito, S. Naito, K. Konoki, M. Yotsu-Yamashita and M. Sasaki, Chem.–Eur. J., 2009, 15, 12807–12818 CrossRef CAS.
- Y. Nakama, O. Yoshida, M. Yoda, K. Araki, Y. Sawada, J. Nakamura, S. Xu, K. Miura, H. Maki and H. Arimoto, J. Med. Chem., 2010, 53, 2528–2533 CrossRef CAS.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366–374 CrossRef CAS.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS.
- V. F. Slagt, A. H. M. de Vries, J. G. de Vries and R. M. Kellogg, Org. Process Res. Dev., 2010, 14, 30–47 CrossRef CAS.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- A. Arcadi, G. Cerichelli, M. Chiarini, M. Correa and D. Zorzan, Eur. J. Org. Chem., 2003, 4080–4086 CrossRef CAS.
- Y. J. Deng, L. Z. Gong, A. Q. Mi, H. Liu and Y. Z. Jiang, Synthesis, 2003, 337–339 CAS.
- B. W. Xin, Y. H. Zhang, L. F. Liu and Y. G. Wang, Synlett, 2005, 3083–3086 CAS.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS.
- B. W. Xin, J. Chem. Res., 2008, 412–415 CrossRef CAS.
- A. Del Zotto, F. Amoroso, W. Baratta and P. Rigo, Eur. J. Org. Chem., 2009, 110–116 CAS.
- F. Durap, M. Rakap, M. Aydemir and S. Ozkar, Appl. Catal., A, 2010, 382, 339–344 CrossRef CAS.
- Y. Y. Peng, J. B. Liu, X. L. Lei and Z. L. Yin, Green Chem., 2010, 12, 1072–1075 RSC.
- C. Li, X. Q. Li and C. Zhang, J. Chem. Res, 2008, 525–527 CrossRef CAS.
- L. F. Liu, Y. H. Zhang and Y. G. Wang, J. Org. Chem., 2005, 70, 6122–6125 CrossRef CAS.
- R. Tatumi, T. Akita and H. Fujihara, Chem. Commun., 2006, 3349–3351 RSC.
- W. Han, C. Liu and Z. L. Jin, Org. Lett., 2007, 9, 4005–4007 CrossRef CAS.
- W. Han, C. Liu and Z. L. Jin, Adv. Synth. Catal., 2008, 350, 501–508 CrossRef CAS.
- J. M. A. Miguez, L. A. Adrio, A. Sousa-Pedrares, J. M. Vila and K. K. Hii, J. Org. Chem., 2007, 72, 7771–7774 CrossRef CAS.
- C. Liu, Q. Ni, P. Hu and J. Qiu, Org. Biomol. Chem., 2011, 9, 1054–1060 CAS.
- J. C. Labiche, O. Mathon, S. Pascarelli, M. A. Newton, G. Guilera-Ferré, C. Curfs, G. Vaughan, A. Homs and D. F. Carreiras, Rev. Sci. Instrum., 2007, 78, 091301 CrossRef.
- (a) K. V. Klementiev, VIPER for Windows, freeware; (b) K. V. Klementiev, J. Phys. D: Appl. Phys., 2001, 34, 209 CrossRef.
- Z. Xu, J. C. Mao and Y. W. Zhang, Catal. Commun., 2008, 9, 97–100 CrossRef CAS.
- <4% at 1 mol% catalyst loading, and not detected (<0.1%) at 0.05 mol%.
- Homocoupled product 4 was not observed under these reaction conditions.
- Water is necessary to ensure solubility of the inorganic base. Also, arylboronic acid dehydrates to arylboroxine under anhydrous conditions.
- Q. Yao, E. P. Kinney and Z. Yang, J. Org. Chem., 2003, 68, 7528–7531 CrossRef CAS.
- Palladium(II) acetate is soluble in THF but practically insoluble in water.
- A. El Maadi, J. Bennazha, J. M. Réau, A. Boukhari and E. M. Holt, Mater. Res. Bull., 2003, 38, 865–874 CrossRef CAS.
- Elemental analysis by ICP and combustion analysis: Pd, 57.69; P, 4.24; C, 1.71 and H, 3.99%.
- A. C. Skapski and M. L. Smart, J. Chem. Soc. D, 1970, 658 RSC.
- S. D. Kirik, R. F. Mulagaleev and A. I. Blokhin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, M449–M450 CrossRef.
- T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632–3640 RSC.
- R. N. Pandey and P. M. Henry, Can. J. Chem., 1974, 52, 1241–1247 CrossRef CAS.
- E. S. Stoyanov, J. Struct. Chem., 2000, 41, 440–445 CrossRef CAS.
- J. Evans, L. O'Neill, V. L. Kambhampati, G. Rayner, S. Turin, A. Genge, A. J. Dent and T. Neisius, J. Chem. Soc., Dalton Trans., 2002, 2207–2212 RSC.
- V. I. Bakhmutov, J. F. Berry, F. A. Cotton, S. Ibragimov and C. A. Murillo, Dalton Trans., 2005, 1989–1992 RSC.
- J. A. Ruddick and W. B. Bunger, in “Techniques of Chemistry”, ed. A. Weissberger, Wiley-Interscience, New York, 1986 Search PubMed.
- Chosen for its low polarity, miscibility with water and the ability to dissolve arylboronic acid.
- N. S. Akhmadullina, N. V. Cherkashina, N. Y. Kozitsyna, I. P. Stolarov, E. V. Perova, A. E. Gekhman, S. E. Nefedov, M. N. Vargaftik and Moiseev, II, Inorg. Chim. Acta, 2009, 362, 1943–1951 CrossRef CAS.
- F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 2177–2180 CrossRef CAS.
- B. Fors and B. P. Fors, Org. Lett., 2008, 10, 3505 CrossRef CAS.
- C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS.
- J. Wang, H. F. M. Boelens, M. B. Thathagar and G. Rothenberg, ChemPhysChem, 2004, 5, 93–98 CrossRef CAS.
- A. V. Gaikwad and G. Rothenberg, Phys. Chem. Chem. Phys., 2006, 8, 3669–3675 RSC.
- A. Kitamura and M. Yui, J. Nucl. Sci. Technol., 2010, 47, 760–770 CrossRef CAS.
- E. E. Finney and R. G. Finke, J. Colloid Interface Sci., 2008, 317, 351–374 CrossRef CAS.
- A. N. Salanov, A. I. Titkov and V. N. Bibin, Kinet. Catal., 2006, 47, 430–436 CrossRef CAS.
- A. N. Salanov and E. A. Suprun, Kinet. Catal., 2009, 50, 31–39 CrossRef CAS.
- S. Malladi, B. N. Nguyen, K. K. Hii and A. G. Livingston, unpublished results.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of UV-vis stopped-flow and EXAFS experiments, and associated data fitting. See DOI: 10.1039/c1cy00241d |
| ‡ Dr Gemma Guilera, current affiliation: ALBA Synchrotron Light Source, Experiments Division, Crta. BP 1413 Km 3.3, 08290 Cerdanyola del Vallès, Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2012 |