Polymeric monolith supported Pt-nanoparticles as ligand-free catalysts for olefin hydrosilylation under batch and continuous conditions†
Rajendar
Bandari
a and
Michael R.
Buchmeiser
*ab
aLehrstuhl für Makromolekulare Stoffe und Faserchemie, Institut für Polymerchemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: michael.buchmeiser@ipoc.uni-stuttgart.de; Fax: +49 (0)-711-685-64050
bInstitut für Textilchemie und Chemiefaser Denkendorf (ITCF), Körschtalstr. 26, D-73770 Denkendorf, Germany
First published on 3rd November 2011
Abstract
Pt-nanoparticles were generated within the mesopores of polymeric monolithic supports. These monoliths were prepared viaring-opening metathesis polymerization (ROMP) from (Z)-9-oxabicyclo[6.1.0]non-4-ene (OBN) and tris(cyclooct-4-en-1-yloxy)methylsilane (CL) using the 3rd-generation Grubbs-initiator RuCl2(pyridine)2(IMesH2)(CHPh) (1) in the presence of a macro- and microporogen. To generate the Pt nanoparticles inside the porous system, the epoxy-groups of the monolithic supports were hydrolyzed into the corresponding vic-diol with the aid of 0.1 N sulfuric acid. Loading with Pt4+ and reduction with NaBH4 resulted in the formation of Pt nanoparticles <7 nm in diameter that were exclusively located within the mesopores of the support as demonstrated by transmission electron microscopy/EDX spectrometry. The nanoparticles were stabilized by both the vic-diols and the polymeric double bonds as evidenced by control experiments with poly(glycidylmethacrylate)-based monoliths that lack the double bonds. Typical metal loadings of around 1.7 mg g−1 (8.7 μmol g−1) were obtained. Pt(0)-loaded monoliths were used in the hydrosilylation of terminal alkenes, norborn-2-ene and norbornadiene applying both batch and continuous flow conditions. Hydrosilylation of olefins under continuous flow conditions resulted in constant product formation in 98% yield at T = 45 °C applying a linear flow of 12.0 mm min−1. Metal leaching was very low, resulting in Pt-contamination in the addition products of less than 4 ppm throughout.
Introduction
The immobilization of metal nanoparticles on micro- and mesoporous supports such as Al(OH)3 and silica,1zeolites,2–7 polymeric micelles,8–14carbon nanotubes (CNTs)15–18 or within ionic liquids19–24 is a well-established technique. Compared to these “standard” supports, polymeric monoliths, originally developed for applications in separation science,25–30 have entered the field of heterogeneous catalysis at a comparable late stage.28,31–33Ring-opening metathesis polymerization (ROMP) turned out to be an attractive alternative to the free radical polymerization-based synthesis of such supports. So far, ROMP-derived supports34–40 have been employed in catalysis as continuous-flow devices and disk-shaped supports for the immobilization of well-defined transition metal complexes such as Pd-based Heck catalysts as well as Schrock and Grubbs catalysts.Pt(0)-nanoparticles have been immobilized onto polysilane-supports by micro-encapsulation,14 on mesoporous polymer colloids,41 on conducting polymers,42 and in ionic liquid-modified magnetic nanoparticles.43 Moreover, they have been immobilized on hollow porous carbon shells44 and on poly(vinyl alcohol)-based polymers.45,46 More recently, our group has developed a method for selectively immobilizing Pt(0) and Pd(0) nanoparticles inside the open pores of electron-beam (EB) and ROMP-derived monoliths.45–48 In contrast to any other polymerization technique, ROMP provides unsaturated sites along the polymer chain with one double bond per repeat unit.49 Here, we report on the immobilization of Pt-nanoparticles within the pores of ROMP-derived monoliths in which particularly the unsaturated backbone provides stabilization, thereby completely avoiding the use of any other organic ligand. The thus-prepared supported Pt-catalysts were successfully used in olefin hydrosilylation reactions both under batch and continuous conditions.
Results and discussion
Synthesis and characterization of monoliths
Epoxy-based monolithic columns were prepared according to published procedures.48 In brief, monoliths were prepared by ROMP using (Z)-9-oxabicyclo[6.1.0]non-4-ene (OBN) as the monomer and tris(cyclooct-4-enyl-1-oxy)methylsilane (CL)34,50 as the cross-linker. 2-Propanol (2-PrOH) was used as a macroporogen, toluene was used as a microporogen, RuCl2(pyridine)2(IMesH2)(CHPh) (1) was used as an initiator. For monolith synthesis, OBN and the CL were dissolved in 2-PrOH. A second solution was prepared by dissolving the initiator in toluene. Both solutions were chilled to 0 °C, mixed for 30 s and the final mixture was transferred into 4.6 × 100 mm or 4.6 × 150 mm i.d. steel columns, respectively (Scheme 1). Polymerization was allowed to proceed for 30 min at 0 °C, followed by 16 h at room temperature. The columns were cleaned and closed with end fittings; then the monolith was cleaned by flushing with a solution of ethyl vinyl ether (20 wt%) in dimethylsulfoxide. This end-capping procedure ensured an effective removal of all Ru-compounds as evidenced by ICP-OES measurements. Thus, the final Ru content in the monolithic rod was <17 ppm. | ||
| Scheme 1 Monolith synthesis. | ||
After this procedure, the monoliths were characterized by using both scanning electron microscopy (SEM) and inverse size-exclusion chromatography (ISEC).51 A SEM image of the cross-section of such a monolithic structure (Scheme 1) revealed microglobules with an average diameter of 1.3 μm. ISEC provided detailed structural data in terms of the inter-microglobule porosity (εz), pore porosity (εp) and total porosity (εt). These measurements revealed that the monolith had a bimodal pore-size distribution typical for all monolithic materials with 40% of macro- and mesopores as well as 66% of micropores (<7 nm). Moreover, the pore volume (Vp, mL g−1) and the mean pore size (Φm, Å) was determined. The mean pore size Φm of the monoliths was calculated from the graph of log Φav (Å) vs. ΔR (%). A summary of the relevant structural data is provided in Table 1 and Fig. 1 (M0), respectively.
| ε p (vol%) | ε z (vol%) | ε t (vol%) | V p/μL g−1 | Φ m/Å |
|---|---|---|---|---|
| Structural data (before hydrolysis of the epoxide groups) | ||||
| 20 | 50 | 70 | 530 | 620 |
| Structural data (after hydrolysis of the epoxide groups) | ||||
| 20 | 47 | 67 | 510 | 570 |
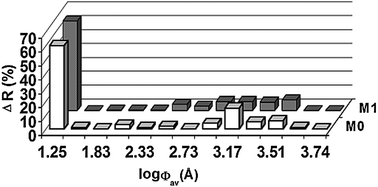 | ||
| Fig. 1 log Φaverage (Å) vs. ΔR (%) for the unmodified monolith (M0) and after hydrolysis with 0.1 N H2SO4 (M1). | ||
Hydrolysis of the epoxide groups of the monolith
The complete conversion of the monolith's epoxide groups into vic-diols was accomplished via acidic hydrolysis according to a published procedure.52,53 Briefly, H2SO4 acidic solution (5 mL, 0.1 N H2SO4, pH 1) was injected into the monolithic column, which was then closed with end fittings. The column was kept at 65 °C for 24 h. After the reaction had been completed, the monolith was washed with deionized water until reaching a neutral eluent; then it was washed with methanol and n-hexane (Scheme 2a). | ||
| Scheme 2 (a) Hydrolysis of the epoxy groups, (b) metal loading in the pores and reduction of Pt4+ by NaBH4 leads to Pt-nanoparticles immobilized within the pores. | ||
After hydrolysis, the pore size distribution was again determined viaSEC. The same bimodal character of the distribution was retained, however, all pore diameters were shifted toward smaller pore sizes, concomitantly, the amount of micropores increased from 60 to 65% (Table 1, Fig. 1 (M1)). This reduction in meso- and macroporosity can be explained by the higher hydrophilicity of the vic-diol based monolith, which is much higher than that of an epoxide based monolith. Consequently, larger amounts of polar solvent molecules will be present around these diols and the support will experience a higher degree of swelling. As a result, also the mean pore diameter was reduced from 620 Å to 570 Å.
Immobilization of PtIV within the pores
A solution of PtCl4 (1 wt% in THF) was pumped through the hydrolyzed monolith. Pt-nanoparticles were generated viareduction with an aqueous 10 wt% solution of NaBH4 (Scheme 2b). The nanoparticles that formed within the pores were approximately 7 nm in diameter as evidenced by transmission electron spectroscopy (TEM) (Fig. 2a; Fig. S1(a), ESI†). After extensive washing, the final Pt-loading was determined by ICP-OES and found to be 1.7 mg of Pt/g of monolith. The existence of Pt-nanoparticles was confirmed by energy dispersive X-ray spectrometry (EDXS) (Fig. S2, ESI†). | ||
| Fig. 2 TEM micro graphs of Pt(0) immobilized on monolithic materials. (a) Before, (b) after use in olefin hydrosilylation reactions. | ||
Application of monolith-supported Pt nanoparticles in hydrosilylation reactions
The polymeric monolith-supported Pt-nanoparticles were used in the catalytic hydrosilylation of 1-alkenes, styrene, norborn-2-ene and norbornadiene using bis(trimethylsilyloxy)methylsilane and triethoxysilane at 45 °C in hexane (Scheme 3). | ||
| Scheme 3 Pt-nanoparticle mediated olefin hydrosilylation reactions. | ||
In a batch setup, olefins were treated with an equimolar amount of bis(trimethylsilyloxy)methylsilane or triethoxysilane in the presence of monoliths with a Pt-loading of 1.3 × 10−4 mmol (15 mg of monolith) in n-hexane at 45 °C for 4 h. The desired products were obtained in excellent yields up to 98%. The results of the hydrosilylation of representative alkenes, catalyzed by monolith-supported Pt-nanoparticles, are summarized in Table 2. Assuming a density of face-centered cubic (fcc) platinum (21.45 g cm−3) for the Pt-nanoparticles, the approximate number of surface Pt-atoms per Pt-nanoparticle can be calculated as 630 (Table S1, ESI†). This allows for calculating turn-over numbers (TONs) per active Pt atom.
| # | Olefin | Product | Conversion (%)a | TONb | TONc |
|---|---|---|---|---|---|
| a Only product and no byproducts were observed. b Assuming every Pt atom participates in the reaction. c Assuming that only the surface Pt-atoms participate in the reaction. d The reaction was run for 24 h. | |||||
| 1 | 1-Octene |
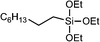
|
94 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
404000 |
| 2 | 1-Octene |

|
98 | 33700 |
609000 |
| 3 | Allylbenzene |

|
92 | 21800 |
395000 |
| 4 | Allylbenzene |

|
98 | 33700 |
609000 |
| 5 | Styrene |

|
72 | 22300 |
404000 |
| 6 |

|
22 | |||
| 7 | Styrene |
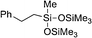
|
83 | 30600 |
553000 |
| 8 |

|
6 | |||
| 9d | Norborn-2-ene |

|
26 | 6200 | 112000 |
| 10d | Norborn-2-ene |

|
52 | 17900 |
323000 |
| 11 | trans-3-octene | — | — | — | — |
| 12 | trans-3-octene | — | — | — | — |
| 13d | 2,5-Norbornadiene |

|
87/12 (exo/endo) | 23400 |
425000 |
| 14d | 2,5-Norbornadiene |

|
2 | 690 | 12400 |
In batch experiments, the hydrosilylation of all terminal olefins resulted in excellent yields of the desired addition products (Table 2). Except for styrene, high regiocontrol and selective formation of the Markovnikov product were observed. Kinetic investigations revealed that all hydrosilylation reactions were completed within 4 h. A representative graph of conversion vs. time for the hydrosilylation of 1-octene with 1,1,1,3,5,5-heptamethyltrisiloxane shows a constant increase in product formation (Figure S3, ESI†). Turnover numbers (TONs) with respect to the number of Pt surface atoms present in all Pt-nanoparticles up to 609000 were obtained. Internal alkenes such as trans-3-octene, however, were inert under these conditions (Table 2, entries 11 and 12). It is worth noting that norborn-2-ene could be hydrosilylated with TONs up to 323000 by extending the reaction time from 4 h to 24 h. All hydrosilylations catalyzed by Pt(0)-nanoparticles afforded crude products that were typically ≤99% pure as evidenced by NMR (ESI†). Since no byproducts were observed neither by GC-MS nor by NMR, conversions are equivalent to yields. Equally important, the supported Pt (0)-nanoparticles were still active at the end of the reaction. Indeed, Pt-loaded monoliths could be removed from a reaction mixture, re-fed with reactants and the reaction proceeds with virtually equal efficiency (ESI†). Inductively coupled plasma spectrometry (ICP-OES) analysis indicated that Pt-leaching into the products was very low (<4 ppm). To shed light onto the question, whether Pt clusters in solution or the surface-immobilized Pt-nanoparticles were the actually active species, a leaching experiment was conducted.41,54 Thus, a Pt-loaded monolith was removed from the hot mother liquor obtained from the reaction mixture of 1-octene and 1,1,1,3,5,5,5-heptamethyltrisiloxane after 4 h of reaction, reactants were added to this solution and the mixture was again kept at 45 °C for 16 h. No further reaction was observed, suggesting that the reaction in fact takes place at the surface or close to the surface of the Pt-nanoparticles. However, a significant increase in the average Pt-nanoparticle diameter from 7 nm to 12 nm during catalysis was observed by TEM (Fig. 2b; Fig. S1(b), ESI†), indicating Ostwald ripening during the catalytic process.55–57
In principle, PtIV can be expected to Π-coordinate to the double bonds of the ROMP-derived polymer backbone. Additional stabilization is provided by the vic-diols.58–60 To shed some light onto the role of the double bonds present in ROMP-derived monoliths, poly(glycidylmethacrylate)-based monoliths were prepared via thermally triggered free radical polymerization. Depending on conversion, such monoliths contain no or a comparably low number of double bonds. A brief synthetic procedure for the synthesis of the corresponding monolithic supports and the immobilization of Pt(0) on these carriers as well as selected results are shown in the ESI.† Though the thus prepared heterogeneous Pt(0) catalysts were also active in the hydrosilylation of various olefins, the TONs obtained were throughout significantly lower than those obtained with the ROMP-derived monoliths (Table S4, ESI†). Since Pt-nanoparticles approx. 7 nm in diameter are formed with both monolithic supports (Fig. S5–S6, Table S3, ESI†) the unsaturated polymeric backbone of the ROMP-derived monolithic support apparently provides an additional Π-stabilization for the nanoparticles or intermediary Pt(II)/Pt(IV) species in course of the reaction.
In order to check for any catalytically active Pt in the reaction mixture, the hydrosilylation reaction of 1-octene with bis(trimethylsilyloxy)methylsilane was carried out in the presence of HgCl2. Complementarily, the above-mentioned hydrosilylation reaction was carried out as described in the Experimental section, then the reaction mixture was isolated by filtration, fresh reactants were added and the reaction was restarted. In none of these control experiments any significant hydrosilylation occurred, indicating the absence of any catalytically active Pt-species. These results also correlate well with the low Pt-leaching and suggest that the hydrosilylation reactions in fact either occur at the surface of the Pt-nanoparticles or that any active, solubilized Pt-species is rapidly “recaptured” by the Pt-nanoparticles.
Hydrosilylation of olefins under continuous flow conditions
Pt-nanoparticles containing ROMP-derived monolithic supports were also used as flow through reactors. Model reactions between 1-octene and bis(trimethylsilyloxy)methylsilane were carried out in n-hexane as a solvent and tert-butylbenzene as an internal standard using a continuous flow setup at 45 °C. There, mixtures of 1-octene and bis(trimethylsilyloxy)methylsilane were constantly converted into 1,1,1,3,5,5,5-heptamethyl-3-octyltrisiloxane over a period of 8 h in 98% yield (Fig. 3). Again, the Pt leaching as determined by ICP-OES was very low (<4 ppm), further illustrating the high immobilization efficiency. | ||
| Fig. 3 Hydrosilylation of 1-octene under continuous flow conditions. Column dimensions: 150 × 4.6 mm i.d. steel column; flow rate: 0.2 mL min−1; T = 45 °C. | ||
Conclusions
In summary, Pt-nanoparticles were immobilized within the pores of both a ROMP- and a free radical polymerization-derived polymeric monolithic support. In the case of the ROMP-derived support, the Pt(0) particles were additionally stabilized by the double bonds present in the polymeric backbone. The thus supported Pt-nanoparticles were successfully used in the hydrosilylation of olefins both under batch and continuous conditions allowing for both high yields and TONs. In all cases, Pt-leaching was low (∼4 ppm).Experimental section
General
1,5-Cyclooctadiene, m-chloroperbenzoic acid, LiAlH4, cis-cyclooctene, ethyl vinyl ether (EVE) [RuCl2(PCy3)(IMesH2)(CHPh)] (IMesH2 = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene), (PCy3 = tricyclohexylphosphine) were obtained from Aldrich Chemical Co. (Karlsruhe, Germany) and used as received. [RuCl2(Py)2(IMesH2)(CHPh)] (1; py = pyridine), (Z)-9-oxabicyclo[6.1.0]non-4-ene (OBN),48tris(cyclooct-4-en-1-yloxy)methylsilane (CL)34 were prepared according to the literature. Triethylamine, CHCl3 (99.5%) and 2-propanol were purchased from Aldrich Chemical Co. (Germany) and dried over CaH2 prior to use. Toluene and CH2Cl2 were dried by an MBraun SPS solvent purification system (MBraun, Garching, Germany). Narrow polystyrene (PS) standards (972 < Mw < 4110000 g mol−1) used for inverse size exclusion chromatography (ISEC) were purchased from Polymer Standard Service, PSS (Mainz, Germany). ICP-OES measurements were carried out on a VISTA-MPX, CCD-Simultaneous ICP-OES (Varian Inc.). An ETHOS 1800 (MLs GmbH, Germany) was used for the microwave-assisted dissolution of the polymeric samples. Programming: 20–180 °C within 15 min, 15 min at 180 °C, then to 20 °C within 1 h. NMR data were obtained at 250.13 MHz for proton and at 62.90 MHz for carbon in the indicated solvent at 25 °C on a Bruker Spectrospin 250 and are listed in parts per million downfield from tetramethylsilane for proton and carbon. GC-MS investigations were carried out on an Agilent GC-MS-7890A instrument equipped with a 7693 Autosampler using an SPB-5 fused silica column (30 m × 0.25 mm × 0.25 μm film thickness). The injection temperature was set to 150 °C; the initial column temperature was set to 45 °C and then increased to 250 °C within 8 min, then held for another 5 min. The column flow was set to 1.10 mL min−1. For the investigation of the microstructures of the monolithic material, powdered samples placed on a conductive carbon sticker of a QUANTA FEG 250 (FEI, Eindhoven, Netherlands) combined with an Oxford Inca x-act (Oxford instruments, Oxfordshire, UK) were used. EDX measurements were done on a scanning electron microscope (SEM) with energy dispersive X-ray analysis (EDX) and low-voltage/high-contrast detection (vCD). The software for the EDX-analysis was INCA 4.13 and microanalysis suite issue 18b+SP1.
Synthesis of monoliths
All monoliths were prepared as follows: stainless steel columns (100 × 4.6 mm or 150 × 4.6 mm i.d.) were cleaned, rinsed and sonicated in a 1:1 mixture of ethanol, acetone then dried for 2 h in vacuo. The columns were closed at one end with frits and end fittings and cooled to 0 °C. Separately, two different solutions (A, B) were prepared and cooled to 0 °C. Solution A consisted of 20 wt% of OBN, 20 wt% of CL and 50 wt% of 2-propanol, while solution B consisted of a 0.2 wt% of initiator 1 in 10 wt% of toluene. Both solutions were merged at 0 °C and mixed for ∼30 s. The column was filled with the polymerization mixture, sealed with Teflon caps and kept at 0 °C for 30 minutes. After rod formation was completed, the column was removed from the ice bath and stored at room temperature for 16 h. In order to remove initiator and excess of OBN and CL, the column was provided with new frits and flushed with a mixture of EVE (20 vol.%) in DMSO.
Hydrolysis of the epoxy groups
For the hydrolysis of the epoxy groups, a 5 mL of acidic solution of 0.1 N H2SO4 (pH 1) was injected into the monolithic column, which was then closed with end fittings and kept at 65 °C for 24 h. The monolith-containing column was connected to an HPLC pump and washed with deionized water at a flow rate of 0.3 mL min−1 until reaching a neutral eluent. Then, the monolithic column was washed with 10 mL of methanol and 10 mL of THF. These columns were characterized by ISEC and used for the immobilization of platinum.Inverse size exclusion chromatography (ISEC)
After synthesis, monolithic columns were characterized by ISEC in terms of intermicroglobule porosity (εz), microporosity (εP), total porosity (εt), pore volume (Vp, mL g−1) and mean pore diameter (Φm, Å). 10 μL samples of individual PS standards (0.25 mg mL−1) dissolved in the mobile phase (CHCl3) were injected into each column applying a flow rate of 0.6 mL min−1. For the determination of the total accessible porosity of the column, a 10 μL of sample benzene was injected. All chromatograms were recorded at a wavelength of 254 nm. Retention times and volumes corresponding to each injection were determined from the peak maximum. All retention volumes were corrected for the extra-column volume of the equipment. Calculations were carried out by assuming that the hydrodynamic radii of the PS standards in CHCl3 do not differ significantly from those reported in CH2Cl2.51,61 Similarly, measurements were carried out after hydrolysis of the epoxy groups.Preparation of Pt-loaded monoliths
A solution of [PtCl4] (25 mg, 0.074 mmol) in THF (2.5 mL) was introduced into the hydrolyzed monolith. After 1 h, washing was carried out using 50 mL of THF. Finally, the support was dried in vacuum for 4 h. The Pt-content was measured by ICP-OES.Determination of the Pt content by ICP-OES
15–30 mg samples of the monoliths were dissolved in a minimum amount of aqua regia (typically 5–7 mL) applying microwave irradiation. The digest was transferred into a volumetric flask and the volume of the solution was adjusted to 25.000 mL. Pt was measured at λ = 214.424 nm, (average of at least two consecutive measurements). The background was measured and averaged at λ1 = 214.35 and λ2 = 214.55 nm. The limit of detection (LOD) was 0.0001 mg L−1. For calibration, Pt-containing aqueous standards (pH = 1, nitric acid) with Pt concentrations of 0, 0.004, 0.14, 0.5, and 40 mg L−1 were used. Ru was measured at λ = 240.272 nm the background was measured at λ1 = 240.273 and λ2 = 245.657 nm. The limit of detection (LOD) was 0.0001 mg L−1. For calibration, Ru-containing aqueous standards (pH = 1, nitric acid) with Ru concentrations of 0, 0.004, 0.14, 0.5, and 40 mg L−1 were used.Hydrosilylation of olefins
In an 8 mL vial, the terminal alkene, norborn-2-ene or norbornadiene (4.4 mmol) were mixed with bis(trimethylsilyloxy)methylsilane (4.4 mmol) or triethoxysilane (4.4 mmol), tert-butylbenzene (1.03 mmol, internal standard), 15 mg of the Pt-loaded monolith and with 5 mL of hexane. The vial was closed, and the reaction mixture was stirred at 45 °C for 4 h for terminal alkenes and 24 h for norborn-2-ene and norbornadiene, respectively. After completion of the reaction, the crude product was filtered through a Whatman filter paper, then n-hexane was removed in vacuo. The products were characterized by NMR-spectroscopy; yields were determined by GC-MS using tert-butylbenzene as internal standard.Hydrosilylation under continuous flow conditions
A mixture of 1-octene (5.04 g, 44.9 mmol), bis(trimethylsilyloxy)methylsilane (10.0 g, 44.9 mmol) and tert-butylbenzene (0.50 g, 3.7 mmol) in 30 mL of n-hexane was introduced into the column at a flow rate of 0.2 mL min−1 (linear flow 12.0 mmmin−1). The temperature of the column was maintained at 45 °C. Samples were collected in 10 min intervals; conversion was determined by GC-MS using tert-butylbenzene as internal standard.Poisoning experiments (hydrosilylation in the presence of HgCl2)
Hydrosilylation experiments were carried out both in the presence and absence of HgCl2 to check for any active Pt-ions in the reaction mixture. Experiments were carried out in 8 mL vials using a solution of n-hexane, THF (5 mL, 1:1), tert-butyl benzene (50 mg, 0.373 mmol), 1-octene (0.504 g, 4.4 mmol), bis(trimethylsilyloxy)methylsilane (1.0 g, 4.4 mmol) and 15 mg of monolithic material (1.3 × 10−4 mmol of Pt). The reaction mixture was stirred at 45 °C for 4 h. Finally, it was filtered through celite and the filtrate was divided into two equal parts A and B. Fresh substrates were added to both filtrates A and B followed by the addition of HgCl2 (60 mg, 0.22 mmol) to filtrate B, then both A and B were stirred for another 4 h at 45 °C. Aliquots were taken from both reaction mixtures and conversions were determined by GC-MS. No conversion was observed. In case the first hydrosilylation experiment was also conducted in the presence of HgCl2 (60 mg, 0.22 mmol), no hydrosilylation product was observed by GC-MS, too.
Acknowledgements
We are grateful to Apl. Prof. Dr Michael Hunger (Institute of Chemical Technology, University of Stuttgart, Germany) and Dr Ralf Thomann (FMF- Freiburg, Germany) for their kind help with ICP-OES and TEM measurements, respectively. This work was supported by a grant provided by the DFG (Deutsche Forschungsgemeinschaft, grant no. BU 2174/1-2).References
- B. M. L. Dioos, I. F. J. Vankelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413–1446 CrossRef CAS.
- R. B. Bedford, U. G. Singh, R. I. Walton, R. T. Williams and S. A. Davis, Chem. Mater., 2005, 17, 701–707 CrossRef CAS.
- L. Djakovitch, H. Heise and K. Köhler, J. Organomet. Chem., 1999, 584, 16–26 CrossRef CAS.
- M. S. Kwon, N. Kim, C. M. Park, J. S. Lee, K. Y. Kang and J. Park, Org. Lett., 2005, 7, 1077–1079 CrossRef CAS.
- A.-H. Lu, W.-C. Li, Z. Hou and F. Schuth, Chem. Commun., 2007, 1038–1040 RSC.
- C. P. Mehnert and J. Y. Ying, Chem. Commun., 1997, 2215–2216 RSC.
- J. P. M. Niederer, A. B. J. Arnold, W. F. Hölderich, B. Spliethof, B. Tesche, M. Reetz and H. Bönnemann, Top. Catal., 2002, 18, 265–269 CrossRef CAS.
- L. De Zan, D. Gasparovicova, M. Kralik, P. Centomo, M. Carraro, S. Campestrini, K. Jerabek and B. Corain, J. Mol. Catal. A: Chem., 2007, 265, 1–8 CrossRef CAS.
- Z. Guo, L. L. Henry, V. Palshin and E. J. Podlaha, J. Mater. Chem., 2006, 16, 1772–1777 RSC.
- Y. Hong and A. Sen, Chem. Mater., 2007, 19, 961–963 CrossRef CAS.
- J. l. Le Bars, U. Specht, J. S. Bradley and D. G. Blackmond, Langmuir, 1999, 15, 7621–7625 CrossRef.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., 2006, 118, 827–830 CrossRef.
- K. Okamoto, R. Akiyama, H. Yoshida, T. Yoshida and S. Kobayashi, J. Am. Chem. Soc., 2005, 127, 2125–2135 CrossRef CAS.
- H. Oyamada, R. Akiyama, H. Hagio, T. Naito and S. Kobayashi, Chem. Commun., 2006, 4297–4299 RSC.
- H. Jiang, L. Zhu, K.-s. Moon and C. P. Wong, Carbon, 2007, 45, 655–661 CrossRef CAS.
- B. M. Quinn, C. Dekker and S. G. Lemay, J. Am. Chem. Soc., 2005, 127, 6146–6147 CrossRef CAS.
- J. Zhang and L. Gao, J. Alloys Compd., 2010, 505, 604–608 CrossRef CAS.
- S. Zhang, Y. Shao, G. Yin and Y. Lin, J. Mater. Chem., 2010, 20, 2826–2830 RSC.
- X. d. Mu, J. q. Meng, Z. C. Li and Y. Kou, J. Am. Chem. Soc., 2005, 127, 9694–9695 CrossRef CAS.
- C. C. Cassol, A. P. Umpierre, G. Machado, S. I. Wolke and J. Dupont, J. Am. Chem. Soc., 2005, 127, 3298–3299 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and F. Montingelli, J. Org. Chem., 2005, 70, 6040–6044 CrossRef CAS.
- E. T. Silveira, A.-P. Umpierre, L. M. Rossi, G. Machado, J. Morais, G. V. Soares, I. J. R. Baumvol, S. R. Teixeira, P. F. P. Fichtner and J. Dupont, Chem.–Eur. J., 2004, 10, 3734–3740 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli, A. Detomaso and P. Iliade, Organometallics, 2003, 33, 4193–4197 CrossRef.
- M. S. Carvalho, R. A. Lacerda, J. P. B. Leão, J. D. Scholten, B. A. D. Neto and P. A. Z. Suarez, Catal. Sci. Technol., 2011, 1, 480–488 RSC.
- M. R. Buchmeiser, Polymer, 2007, 48, 2187–2198 CrossRef CAS.
- A. Jungbauer, J. Chromatogr., A, 2005, 1065, 3–12 CrossRef CAS.
- A. Jungbauer and R. Hahn, J. Chromatogr., A, 2008, 1184, 62–79 CrossRef CAS.
- F. Švec, T. B. Tenikova and Z. Deyl, Monolithic Materials: Preparation, Properties and Application, Elsevier, Amsterdam, 2003 Search PubMed.
- S. H. Lubbad and M. R. Buchmeiser, J. Sep. Sci., 2009, 32, 2521–2529 CrossRef CAS.
- S. H. Lubbad and M. R. Buchmeiser, J. Chromatogr., A, 2010, 1217, 3223–3230 CrossRef CAS.
- S. Xie, F. Švec and J. M. J. Fréchet, Biotechnol. Bioeng., 1999, 62, 30–35 CrossRef CAS.
- P. Krajnc, J. F. Brown and N. R. Cameron, Org. Lett., 2002, 4, 2497–2500 CrossRef CAS.
- J. A. Tripp, F. Svec and J. M. J. Fréchet, J. Comb. Chem., 2001, 3, 604–611 CrossRef CAS.
- R. Bandari, A. Prager-Duschke, C. Kuhnel, U. Decker, B. Schlemmer and M. R. Buchmeiser, Macromolecules, 2006, 39, 5222–5229 CrossRef CAS.
- M. R. Buchmeiser, S. Lubbad, M. Mayr and K. Wurst, Inorg. Chim. Acta, 2003, 345, 145–153 CrossRef CAS.
- J. O. Krause, S. Lubbad, O. Nuyken and M. R. Buchmeiser, Adv. Synth. Catal., 2003, 345, 996–1004 CrossRef CAS.
- J. O. Krause, S. H. Lubbad, O. Nuyken and M. R. Buchmeiser, Macromol. Rapid Commun., 2003, 24, 875–878 CrossRef CAS.
- M. Mayr, B. Mayr and M. R. Buchmeiser, Angew. Chem., 2001, 113, 3957–3960 CrossRef.
- M. Mayr, D. Wang, R. Kröll, N. Schuler, S. Prühs, A. Fürstner and Michael R. Buchmeiser, Adv. Synth. Catal., 2005, 347, 484–492 CrossRef CAS.
- L. Yang, M. Mayr, K. Wurst and M. R. Buchmeiser, Chem.–Eur. J., 2004, 10, 5761–5770 CrossRef CAS.
- Y. Zhang, L. Zhao, P. K. Patra and J. Y. Ying, Adv. Synth. Catal., 2008, 350, 662–666 CrossRef CAS.
- H. J. Salavagione, C. Sanchís and E. Morallón, J. Phys. Chem. C, 2007, 111, 12454–12460 CrossRef CAS.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Adv. Synth. Catal., 2007, 349, 2145–2150 CrossRef CAS.
- S. Ikeda, S. Ishino, T. Harada, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., 2006, 118, 7221–7224 CrossRef.
- C. Luo, Y. Zhang and Y. Wang, J. Mol. Catal. A: Chem., 2005, 229, 7–12 CrossRef CAS.
- Y. Luo and X. Sun, Mater. Lett., 2007, 61, 2015–2017 CrossRef CAS.
- R. Bandari, T. Höche, A. Prager, K. Dirnberger and M. R. Buchmeiser, Chem.–Eur. J., 2010, 16, 4650–4658 CrossRef CAS.
- R. Bandari, A. Prager, T. Höche and M. R. Buchmeiser, ARKIVOC, 2011, part (iv), 54–70.
- M. R. Buchmeiser, Ring-Opening Metathesis Polymerization, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2009 Search PubMed.
- R. Bandari, W. Knolle, A. Prager-Duschke, H.-J. Gläsel and M. R. Buchmeiser, Macromol. Chem. Phys., 2007, 208, 1428–1436 CrossRef CAS.
- I. Halász and K. Martin, Angew. Chem., 1978, 90, 901 CrossRef.
- H. Hrudková, F. Švec and J. Kálal, Br. Polym. J., 1977, 9, 238–240 CrossRef CAS.
- V. Smigol, F. Svec and J. M. J. Frechet, Macromolecules, 1993, 26, 5615–5620 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS.
- M. T. Reetz and J. G. d. Vries, Chem. Commun., 2004, 1559–1563 RSC.
- J. G. d. Vries, Dalton Trans., 2006, 421–429 RSC.
- J. Hubert, A. L. Beauchamp and T. Theophanides, Can. J. Chem., 1973, 51, 604–608 CrossRef CAS.
- H. Rüegger, Magn. Reson. Chem., 1991, 29, 113–119 CrossRef.
- P. E. Slade and H. B. Jonassen, J. Am. Chem. Soc., 1957, 79, 1277–1279 CrossRef CAS.
- I. Halász and K. Martin, Ber. Bunsen-Ges. Phys. Chem, 1975, 79, 731–734 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00351h |
| This journal is © The Royal Society of Chemistry 2012 |