Iron(III) metal–organic frameworks as solid Lewis acids for the isomerization of α-pinene oxide
Amarajothi
Dhakshinamoorthy
a,
Mercedes
Alvaro
a,
Hubert
Chevreau
b,
Patricia
Horcajada
b,
Thomas
Devic
b,
Christian
Serre
b and
Hermenegildo
Garcia
*a
aInstituto Universitario de Tecnología Química CSIC-UPV and Departamento de Química, Universidad Politécnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es; Fax: (+)34 9638 7807
bInstitut Lavoisier, UMR 8180 CNRS-Université de Versailles Saint Quentin en Yvelines, 78035 Versailles, France
First published on 23rd December 2011
Abstract
A series of Fe3+-containing porous metal–organic frameworks (MOFs), including the commercial iron trimesate Basolite F-300 or Fe(BTC) (BTC: 1,3,5-benzenetricarboxylate) and the synthetic iron terephthalate MIL-88B (Fe3O(BDC)3X, X = Cl, OH, BDC = 1,4-benzenedicarboxylate), iron naphthalenedicarboxylate MIL-88C (Fe3O(NDC)3X, X = Cl, OH, NDC = 1,6-naphthalenedicarboxylate), iron trimesate MIL-100 (Fe3O(BTC)2X, X = Cl, OH) and iron azobenzenetetracarboxylate soc-MOF(Fe) or MIL-127 (Fe6O2(Tazb)3X2, X = Cl, OH, Tazb = 3,3′,5,5′-azobenzenetetracarboxylate; MIL stands for Materials from Institut Lavoisier), have been tested for the rearrangement of α-pinene oxide to camphonelal and isopinocamphone in the absence of solvent. Conversions of about 10% with 50% selectivity towards camphonelal were obtained. This catalytic performance has been compared with that of the copper trimesate Basolite C-300 or HKUST-1 of formula Cu3(BTC)2 and the aluminium terephthalate Basolite A-100 or MIL-53(Al) of formula Al(OH)(BDC) as well as with some homogeneous (ZnCl2, Cu(NO3)2, Al(NO3)3) and heterogeneous (Fe3+-exchanged Y-zeolite) Lewis acids. Fe(BTC) also exhibits catalytic activity for the rearrangement of other epoxides (styrene, cyclohexene and norbornene oxides) under solventless conditions. Some partial deactivation of the Fe(BTC) with a slight degradation of the structure has nevertheless been observed.
Introduction
There is a continued interest in developing solid acids that can act as heterogeneous catalysts for organic reactions in the liquid phase requiring Lewis acidity.1 In particular, zeolites have been widely applied as solid acids for a large number of organic reactions with application in the production of fine chemicals.2,3 In this regard transformation of pinene oxide into camphonelal and camphones is one of the most important reactions in the fragrance industry, which is typically promoted by aluminium based catalysts.4,5 Metal–organic frameworks or MOFs exhibit several key advantages with respect to zeolites: their easy tuneable composition (metal ions and organic linkers), their versatility in the synthetic preparation conditions and their easy design.6,7 Besides, porous MOFs8 are currently evaluated as heterogeneous catalysts for a wide range of reactions in the liquid phase.6,9–18 The presence of coordinatively unsaturated metal sites (CUS) in some of these MOFs makes possible their interaction with substrates, the metal ion or cluster acting as Lewis acid or redox center.19–21 Recently, De Vos and co-workers have reported that Cu3(BTC)2 is a suitable heterogeneous catalyst to promote this transformation in various organic solvents.19 Thus, it is of interest to study the activity of other MOFs, particularly those containing iron metal sites, which have been previously proved their ability to act as Lewis acids.22–25 The commercial iron(III) trimesate Fe(BTC) of molecular formula C9H3FeO6 is widely available from BASF (Basolite F-300), and exhibits high activity for a large variety of organic transformations.23,24,26 Nevertheless, the unknown crystal structure of this commercial sample and poor local characterization of this solid render difficult the understanding of the reaction mechanisms. Moreover, although the availability of the solid is obviously a key point for the use of these hybrid materials in industrial applications (catalysis, separation, etc.), many potentially interesting MOFs are now produced at a multi grams-laboratory-scale. For these reasons, we examine herein the rearrangement of α-pinene oxide to camphonelal and isopinocamphone in the absence of solvent using different iron-based MOFs either commercially available (Fe(BTC)) or produced in the laboratory MIL-100(Fe),13 MIL-88B, MIL-88C and Soc-MOF(Fe) or MIL-127.27,28 Their catalytic activities have been compared with the ones of two other commercially available MOFs (Al(OH)(BDC) and Cu3(BTC)219 and homogeneous (ZnCl2, Cu(NO3)2) or heterogeneous (Fe3+-exchanged Y-zeolite) as reference catalysts.Results and discussion
Table 1 contains the list of MOFs and zeolite employed in the present study as well as relevant data concerning surface area and pore size. Finally, in order to show the generality of the epoxide rearrangement observed for pinene oxide, we have expanded our study to other substrates that, although of lower industrial importance, are also useful to assess the scope of MOFs as heterogeneous catalysts for Lewis acid promoted epoxide rearrangement.| MOFs or zeolite | Ligand | Formula | S BET/m2 g−1 | Pore dimensions/Å |
|---|---|---|---|---|
| a According to Aldrich. b Determined here by N2 adsorption isotherms. c Accessible through microporous windows (4.5 × 5.5 and 8.5 Å). d Simulated surface areas since these highly flexible solids exhibit closed pores when dried, and thus no significant accessible surface area to N2 gas. e Pore diameter varies due to breathing. | ||||
| Fe(BTC) |
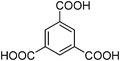
|
Fe(BTC) a | 840b | 21b |
| MIL-100 |
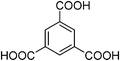
|
Fe3O(BTC)2X (X = OH, Cl) | 1950 | Cages: 25 and 29c |
| Windows 5.5 and 8.6 | ||||
| MIL-88B |
|
Fe3O(BDC)3X (X = OH, Cl) | 3040d | 9 |
| MIL-88C |
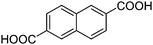
|
Fe3(NDC)3X (X = OH, Cl) | 4030d | 13 |
| MIL-127 |
|
Fe3O(Tazb)4/3X (X = OH, Cl) | 1400 | 10 |
| Cu3(BTC)2 |
|
Cu3(BTC)2 | 1019 | Cages; 25, Windows 6 |
| Al(OH) (BDC) |
|
Al(OH) (BDC) | 950 | —e |
| Fe-Y | Crystalline aluminosilicates | Fe12.3Na17-(Si178Al55O395)·xH2O | 345 | 7.4 |
In the present study, the isomerization of α-pinene oxide (1) to industrially relevant fragrance products, namely campholenic aldehyde (2) and isopinocamphone (4), has been performed using iron containing MOFs as solid Lewis acids under neat conditions and mild temperatures. A precedent in the literature has shown that exchangeable coordination positions around the nodal metal ion or metal clusters can act as catalytic sites with moderate Lewis acid strength.13 Besides isomerization to aldehyde 2, Lewis acid sites can also induce the ring opening of epoxide 1 to verbenol (3), which further undergoes rearrangement to give isopinocamphone (4). In addition, an unknown isomer of m/z 154 has also been observed in a small percentage. Scheme 1 indicates the products observed upon treatment of epoxide 1 under solventless conditions. The results obtained are summarized in Table 2. The obtained activity and selectivity showed that commercial Fe(BTC) is a suitable and convenient solid catalyst to promote this type of reaction.
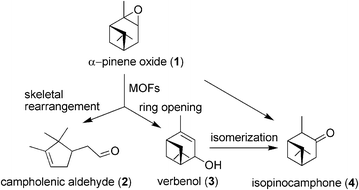 | ||
| Scheme 1 Products derived from α-pinene oxide isomerization promoted by MOFs as Lewis acids. | ||
| Run | Catalyst | Time/h | Conv. of 1b (%) | Selectivity (%)b,c | TONd | ||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | |||||
| a Reaction conditions: α-pinene oxide (0.5 mL), 50 mg catalyst activated at 150 °C for 2 h under vacuum before use, 6 h, 70 °C. b Determined by GC. c An unknown isomer of m/z 154 is also formed with a selectivity ranging from 1% to 11%. d Estimated by dividing the moles of 1 converted by the number of theoretical centers in 50 mg of the catalyst. TON could not be calculated for Fe(BTC) due to its unknown structure. e In the presence of 0.1 mL of pyridine. f Reaction conditions: α-pinene oxide (3 mL), 400 mg of Fe(BTC) activated at 150 °C for 2 h under vacuum, 70 °C. g Reaction conditions: α-pinene oxide (0.5 mL), 100 mg of Fe-Y zeolite activated at 250 °C for 7 h under vacuum; t-carveol and p-cymene were also observed in significant amount. h 50 mg was used; 4% of isomer with m/z 154 was observed and other isomerized products were observed. i 50 mg was used and 22% of isomer with m/z 154 was observed. j 90 mg was used and 37% of other isomers were observed. | |||||||
| 1 | Fe(BTC) | 6 | 10 | 50 | 5 | 35 | 1.9 |
| 2 | Fe(BTC) e | 6 | 4 | 52 | 2 | 45 | |
| 3 | Cu3(BTC)2 | 6 | 8 | 48 | 6 | 41 | 1.2 |
| 4 | Al(OH)(BDC) | 6 | 9 | 52 | 7 | 33 | 0.9 |
| 5 | Al(OH)(BDC) e | 6 | 5 | 56 | 7 | 33 | — |
| 6 | MIL-100 (Fe) | 6 | 22 | 45 | 4 | 40 | 14.3 |
| 7 | MIL-88B | 6 | 9.5 | 47 | 9 | 38 | 5.8 |
| 8 | MIL-88C | 6 | 11 | 48 | 11 | 37 | 7.0 |
| 9 | MIL-127 | 6 | 10 | 45 | 7 | 43 | 6.4 |
| 10 | Fe(BTC) f | 1 | 13 | 45 | 10 | 42 | — |
| 11 | Fe(BTC) | 3 | 14 | 47 | 7 | 44 | — |
| 12 | Fe(BTC) | 5 | 14 | 45 | 6 | 43 | 2.1 |
| 13 | Fe(BTC) | 1 | 11 | 46 | 7 | 41 | — |
| 14 | Fe(BTC) | 3 | 12 | 46 | 7 | 41 | — |
| 15 | Fe(BTC) | 5 | 13 | 45 | 7 | 44 | 2 |
| 16 | Fe-Y zeolite g | 6 | 80 | 62 | 4 | 12 | 18.3 |
| 17 | ZnCl2h | 4 | 57 | 50 | 3 | 7 | 3.5 |
| 18 | Cu(NO3)2·3H2Oi | 4 | 82 | 50 | 2 | 5 | 9.1 |
| 19 | Al(NO3)3·9H2Oj | 4 | 96 | 52 | 5 | 6 | 15.0 |
In the precedent study describing the use of Cu3(BTC)2 as catalyst for the isomerization of 1, the role of the solvent in the selectivity to 2 was pointed out.19 Conversion of oxide 1 was much higher in ethyl acetate and dichloromethane than in toluene. It was also established that, except for zinc bromide, the selectivity to camphonelal 2 was higher with Cu3(BTC)2 than for the other tested homogeneous catalysts, while the reaction rate was higher for homogeneous catalysts.
Continuing with our ongoing interest in exploiting the catalytic activity of commercially available MOFs,11,22,29 we initially checked the isomerization of 1 under neat conditions in the presence of Fe(BTC) and Al(OH)(BDC) and compared the results with the activity of Cu3(BTC)2. After 6 h of reaction using Fe(BTC) as catalyst, there were mainly two products, namely 2 and 4, with a higher selectivity for the former. However, a certain amount of compound 3 was always observed under the present experimental conditions. Interestingly, the percentage conversion of 1 was increased with time, observing 10% of conversion at 6 h.
Controlling the selectivity in the absence of solvent seems to be quite challenging as the skeletal isomerization of 1 to 2 and the ring opening of epoxide 1 to 3 are parallel reactions, whose relative reaction rate strongly depends on the solvent. Thus, it has been reported that this rearrangement of 1 to 2 can be highly selective depending on the nature of the solvent.19 It has been proposed that solvent molecules such as acetonitrile can interact with the Lewis acid sites and, therefore, the observed high selectivity towards 2 in the presence of solvent has been attributed to the controlled replacement in certain sites of the adsorbed solvent molecule by α-pinene oxide. In the present report we are using solventless conditions, which represents a considerable advantage for the greenness of the process, and the selectivity towards the target products 2 and 4 should be a consequence of the catalytic activity.
To confirm the Lewis acid nature of the active sites promoting the reaction of α-pinene oxide, we performed a test in the presence of pyridine. Addition of pyridine to the reaction mixture should strongly coordinate to the iron metal sites,30 reducing the conversion of 1. Observation of the effect of the addition of pyridine can be attributed to the poisoning of active sites and supports that the active sites are the Lewis acid centres. It is interesting to comment that in contrast to the catalytic activity of Fe(BTC), the use of iron nitrate nonahydrate as catalyst with an equal amount of iron content as per entry 1 in Table 2 promotes the isomerization reaction of 1 with the appearance of many unidentified reaction products but where compounds 2 and 4 were absent. Thus, apparently, site isolation of Fe3+ centers and their immobilization in a matrix change completely the catalytic activity of this metal ion.
Next, the well studied Cu3(BTC)2 was used as catalyst under solvent free conditions and observed 8% conversion with 48% and 41% selectivities for 2 and 4, respectively. Although it may seem that the percentage conversion of 1 is lower compared to that reported in the earlier study (up to 90%),19 it should be noted that here the reactions were carried out under solventless conditions. In addition, the higher selectivity towards 2 is in accordance with the expected product distribution.19 A similar trend was observed with Al(OH)(BDC) yielding 9% conversion with 52% selectivity for 2 after 6 h. In this case, it was assumed that each of the Al3+ can act as a potential Lewis acid site. The activity data of Al(OH)(BDC) are very similar in terms of percentage conversion and selectivity to that achieved for Fe(BTC). Addition of 0.1 mL of pyridine to the rearrangement of 1 promoted by Al(OH)(BDC) also reduces the percentage conversion to 5% suggesting again that pyridine is acting as poison to the active Lewis acid sites present in the catalyst.
Although lab-scale MIL-100(Fe) and the commercial Fe(BTC) are polymorphs, the use of MIL-100(Fe) with its known mesoporous cubic structure13 makes easier the rationalization of the catalytic activity. As shown in Table 2, MIL-100 (Fe) exhibited about twice higher conversion than Fe(BTC) with somewhat different selectivity for 2 and 4, respectively. The higher conversion could be related with the higher surface area of MIL-100 (Fe) and/or may be to a larger number of accessible Lewis metal sites. However, in this case, the presence of a large percentage of the unknown isomer was observed (11%).
We further extended this study to other three iron(III) based solids: the microporous flexible iron terephthalate MIL-88B, the microporous flexible iron naphthalene dicarboxylate MIL-88C and the microporous rigid iron 3,3′,5,5′-azobenzene tetracarboxylate MIL-127 or Soc-MOF(Fe). Although the expected surface area of these catalysts is different, they showed a similar catalytic activity converting around 10% of 1 in 6 h. Considering that the flexible character of the framework of MIL-88B and MIL-88C allows a modulation of their pore aperture to fit the size of the adsorbed molecule, adsorption of α-pinene oxide might lead here to a partial-opening of both MIL-88 structures to reach textural properties close to those of MIL-127. In all the three catalysts, the selectivity toward product 2 was higher than 4 accompanied with a small amount of 3 and its unknown isomer. These results show the general applicability of MOFs containing iron Lewis acid sites to promote the rearrangement of α-pinene oxide.
The advantage of using MOFs as catalysts for isomerization of 1 in terms of combined selectivity to 2 and 4 was visualized when the reaction was performed with an iron-exchanged Y zeolite (Fe-Y) and homogeneous Lewis acids zinc chloride and copper nitrate. It should be noted that due to the differences in chemical compositions, catalytic tests using Fe-Y were performed with twice as much catalyst weight as for MOFs and that this difference in mass can be responsible for a larger conversion value compared to MOFs.
In fact, when comparing the relative catalytic performance of the various catalysts, the turnover number (TON) defined as the moles of substrate converted per active site should be the value to be considered. The use of TON values provides a valid comparison of different materials since the activity per active site and not per mass unit is considered. Thus, even though the amount of zeolite as well as homogeneous catalysts used is different from those of MOFs, the number of molecules reacted per active site can be compared directly for all the catalysts in Table 2. Fe-Y zeolite has the highest TON value, which is comparable with that of MIL-100 (Fe) and notably higher than those of the other solids, including the homogeneous ZnCl2 and Cu(NO3)2·3H2O reference catalysts.
However, besides conversion, product selectivity to 2 and 4 is a key parameter to evaluate the catalytic performance. Although Fe-Y zeolite resulted in higher selectivity for 2 and 4 than Fe(NO3)3·9H2O and the other homogeneous catalysts, the value of 74% was still unsatisfactory and significantly lower than that exhibited for MOFs that is systematically above 85%. In addition, it was also observed the formation of t-carveol and p-cymene, the typical products expected from zeolites as catalysts and that are detrimental for the quality of the reaction mixture.31 In contrast, ZnCl2 exhibited much higher selectivity for 2 than for 4, but other isomers were also formed in significant percentage. Finally, copper nitrate trihydrate and aluminium nitrate nonahydrate showed the formation of 2 with about 50% selectivity, but formation of the significant amounts of an unknown isomer with m/z 154 amu was observed. Interestingly, the selectivity of copper nitrate trihydrate for 2 coincides with the selectivity reported for Cu3(BTC)2 in the presence of solvents.
Heterogeneity of the catalysis was ascertained by performing an experiment under the typical reaction conditions described in Table 2, but the solid catalyst was removed by filtration whilst hot after 30 min. The filtered solution was allowed to react further at 70 °C for additional 6 h without observing further pinene oxide conversion.
One of the main advantages of using heterogeneous catalysts is their possible reuse for consecutive runs if the catalytic activity is not completely lost. Reusability of Fe(BTC) to promote the isomerization of 1 was investigated using 3 mL of this compound in the presence of 400 mg of Fe(BTC) as catalyst. The temporal profile of the time-conversion plot is shown in Fig. 1. Although the catalyst showed high reactivity in initial times, the conversion of 1 remained almost constant after the first hour. After 5 h, the reaction was stopped and the catalyst was washed, filtered and dried. When the Fe(BTC) catalyst was reused for the second time, a slight decrease in the initial reaction rate and final conversion of 1 was observed without any change in the selectivity towards products 2 and 4. It should be noted, however, that deactivation is usually more severe when the reaction is carried out in the absence of solvent as is the case considered here. The recovered catalyst after its second run was characterised by powder XRD (Fig. 2) and BET surface area measurement. The comparison of XRD patterns of the fresh and the reused catalyst indicates changes in the relative Bragg peak intensities for the reused catalyst. This is either due to a partial degradation or to a change in the pore content. Thus, it is likely that the remaining products might block the accessibility to some metal sites within the pores in further catalytic runs. This possibility is supported by the notable decrease in the BET surface area after use of Fe(BTC) as catalyst that gave a value of 170 m2 g−1. Thus, although the catalytic activity of the used Fe(BTC) is similar to that of the fresh material, characterization data, particularly the notable decrease in surface area, suggest that the catalytic activity may arise in a large extent from the external surface area that will be less sensitive to pore blocking or restructuring of the material.
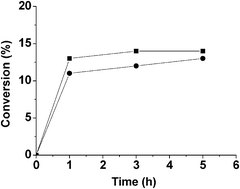 | ||
| Fig. 1 Time conversion plot showing deactivation of Fe(BTC) in its use to promote the isomerization of 1 under solvent free conditions. Square and circle indicates first and second run, respectively. | ||
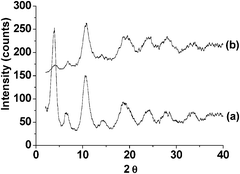 | ||
| Fig. 2 Powder XRD pattern of (a) fresh and (b) used Fe(BTC) catalyst. | ||
To address this possibility, IR spectra of the fresh and reused Fe(BTC) samples were recorded and compared (Fig. 3). The fact that no new peaks were observed indicates that co-adsorbed substrates and reaction products should act as poisons in very minor amounts below the sensitivity of this spectroscopic technique. However, a slightly higher relative intensity of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at around 1700 cm−1 suggests the presence of a higher amount of free carboxylic groups, pointing out the possibility of a slight structural degradation of Fe(BTC) in agreement with the changes observed in powder XRD.
O) at around 1700 cm−1 suggests the presence of a higher amount of free carboxylic groups, pointing out the possibility of a slight structural degradation of Fe(BTC) in agreement with the changes observed in powder XRD.
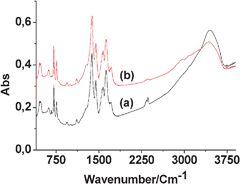 | ||
| Fig. 3 FT-IR spectra of Fe(BTC) (a) fresh and (b) reused. | ||
The scope of Fe(BTC) in isomerization of epoxides was expanded by studying also the rearrangement of three other epoxides, namely styrene oxide, cyclohexene oxide and norbornene oxide (Table 3). Under solventless conditions Fe(BTC) exhibits 23% conversion of styrene oxide resulting in a remarkable selectivity of 84% for phenylacetaldehyde and only 14% of benzaldehyde. On the other hand, cyclohexene oxide showed no conversion in the absence of solvent. Using acetonitrile as solvent a 7% conversion of cyclohexene oxide was observed with Fe(BTC) at 6 h resulting in 35% of 2-methoxycyclohexanol, 55% of 2-hydroxycyclohexanol and 10% of 2-cyclohexenol. Similarly, norbornene oxide in acetonitrile showed 6% conversion using Fe(BTC) as catalyst with 13% of norborneol and 85% of norborneone.
| Substrate | Conversion (%)a | Products (% selectivity)a |
|---|---|---|
| a Determined by GC using external standard. b Styrene oxide (1 mL), 100 mg of Fe(BTC) preactivated at 150 °C for 3 h under vacuum, 100 °C, 6 h. c Substrate (0.1 mL), 100 mg of Fe(BTC) preactivated at 150 °C for 3 h under vacuum, acetonitrile (4 mL), 70 °C, 6 h. | ||
| Styrene oxide b | 23 | Phenylacetaldehyde (84) |
| Benzaldehyde (14) | ||
| Cyclohexene oxide c | 7 | 2-Methoxycyclohexanol (35) |
| 2-Hydroxycyclohexanol (55) | ||
| Cyclohexanol (10) | ||
| Norbornene oxide c | 6 | Norborneol (13) |
| Norborneone (85) |
Conclusion
Fe(BTC) can be conveniently used for the isomerization of pinene oxide to campholenic aldehyde (2) and isopinocamphone (4) in the absence of solvent. MOFs showed a high selectivity towards these products 2 and 4. Other homogeneous or heterogeneous Lewis acid catalysts such as Fe3+-exchanged Y zeolite exhibit higher conversion under the same conditions, but lower selectivity. Fe(BTC) could be reused with somewhat lower activity due to a partial degradation of the catalyst. The present protocol could also be applied in the rearrangement of other epoxides leading to products which can be of interest in organic synthesis.Experimental procedure
All epoxides, reagents, starting materials and Fe(BTC) were obtained commercially from Sigma-Aldrich and used as received. Fe-Y was prepared starting from Na-Y (Si/Al 2.4) by two consecutive room temperature ion exchanges using aqueous solutions of Fe(NO3)3 of 0.2 and 0.4 M. The initial pH value of the solutions was set at 3 using HNO3. The solid to liquid weight ratio of the ion exchanges was 1 to 10. The Fe content of the resulting Fe-Y was 5.3% as determined by ICP-EAS chemical analysis after dissolving a known weight of the zeolite using HF-HNO3. Synthetic MOFs (MIL-100(Fe), MIL-88B and MIL-88C) were prepared following the reported procedures13,27 and their corresponding XRDs were in total agreement with their reported pattern. All the XRDs in this work were recorded as powders in a Philips X'Pert diffractometer using the CuKα radiation at a scan rate of 0.2° min−1.Soc-MOF(Fe) or MIL-127 is isostructural to soc-MOF(In) reported earlier by Eddaoudi et al.28Fig. 4–6 show the XRD, thermogravimetry and N2 adsorption isotherms of this MOF. It was synthesized in a round bottom flask from a solution of 28 g of Fe(ClO4)3·6H2O (Aldrich) and 14 g of 3,3′,5,5′-azobenzene tetracarboxylic acid (laboratory synthesized, see below) in 100 mL of dimethylformamide (DMF) under reflux for 20 h. The orange solid was recovered by filtration. The product was then purified by washing under reflux in 100 mL of absolute ethanol and drying at 150 °C for 16 h. As it can be seen in Fig. 4, the experimental XRD agrees well with the simulated XRD based on the reported MIL-127 structure.
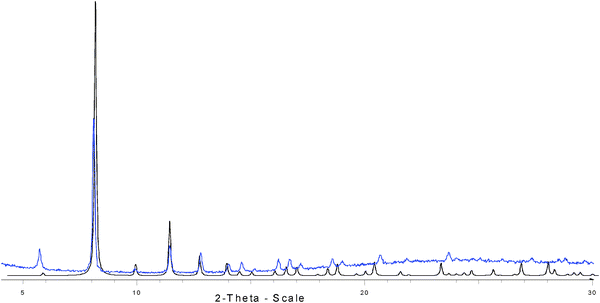 | ||
| Fig. 4 X-Ray powder diffraction of soc-MOF(Fe) or MIL-127 (simulated: blue; experimental: black; conventional high resolution (θ–2θ) D5000 Siemens X'Pert MDP diffractometer (λCu Kα1–Kα2)),from 5 to 30° (2θ) using a step size of 0.02° and 4 s per step in continuous mode). | ||
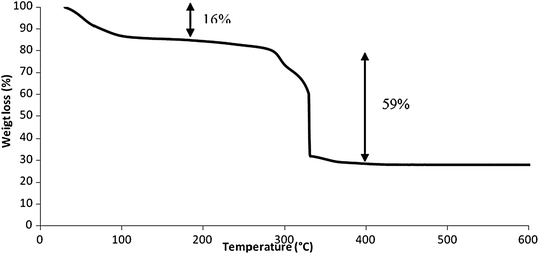 | ||
| Fig. 5 Thermogravimetric analysis of soc-MOF(Fe) or MIL-127 under oxygen flow (20 mL min−1), using a Perkin Elmer Diamond TGA/DTA. The heating rate was 2 °C min−1. | ||
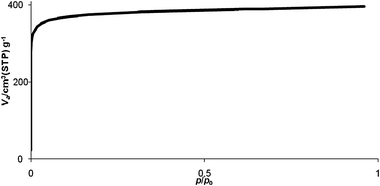 | ||
| Fig. 6 Nitrogen sorption isotherm at 77 K of soc-MOF(Fe) or MIL-127 activated overnight at 423 K (P0 = 1 atm; apparatus: BEL Japan, BELSORP Mini). | ||
Synthesis of 3,3′,5,5′-azobenzene tetracarboxylic acid (TazbH4): TazbH4 was prepared according to the procedure developed by Ameerunisha and Zacharias for other azobenzene derivatives.32 19 g of 5-nitroisophthalic acid, 50 g of sodium hydroxide, NaOH, and 250 mL of distilled water were placed into a 1 L 3-neck round bottom flask and stirred at 60 °C continually. This formed first a pink slurry that turns yellow after 1 hour. Meanwhile 100 g of glucose were dissolved into 150 mL of distilled water at 60 °C. The glucose solution was added slowly to the slurry, which becomes immediately dark brown. This was left to cool down for 10 minutes, and air was bubbled into it for 16 hours under stirring at room temperature. The reaction mixture was cooled in an ice-bath and the sodium salt of Tazb recovered by filtration. The solid was then dissolved in 200 mL of distilled water and then this solution was acidified to pH = 1, using 37% HCl. This yielded a bright orange precipitate. The product was recovered by filtration, washed with distilled water dried at 120 °C.
Typical procedure
In a 25 mL round bottomed flask, 0.5 mL of pinene oxide was added to the required amount of Fe(BTC) preactivated at 150 °C for 2 h under vacuum. This slurry was magnetically stirred at 70 °C for the required time. An aliquot (10 μL) of the sample was taken from the reaction mixture at 1, 3, and 5 h reaction time, diluted with cold acetonitrile and filtered through nylon filters to remove catalyst particles. This sample was analyzed by GC (Hewlett Packard 5890 series II gas chromatograph with an FID detector and high purity helium as carrier gas) as soon as possible and the products were confirmed by GC-MS (Hewlett Packard 6890 series spectrometer). At the final time, the reaction mixture was extracted with acetonitrile and filtered. The recovered catalyst was subjected to spectroscopic analyses. A similar procedure was followed for homogeneous catalysts and for zeolites. It is relevant to mention that most of the homogeneous catalysts were only partly soluble in pinene oxide. It was estimated that about 2 g of the inorganic salts dissolve in 1 L of pinene oxide.Acknowledgements
Financial support by the Spanish DGI (CTQ2009-11587, CTQ2010-18671 and CONSOLIDER MULTICAT) is gratefully acknowledged. Funding of European Commission through an integrated FP7 project MACADEMIA (FP7/2007-2013 Nr. 228862) is also acknowledged. Authors thank G. Maurin for kindly simulating accessible surface areas of the open form of flexible MIL-88 solids.References
- A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077–1104 CrossRef CAS.
- K. Maruoka, S. Nagahara, T. Ooi and H. Yamamoto, Tetrahedron Lett., 1989, 30, 5607–5610 CrossRef CAS.
- Y. Kita, A. Furukawa, J. Futamura, K. Ueda, Y. Sawama, H. Hamamoto and H. Fujioka, J. Org. Chem., 2001, 66, 8779–8786 CrossRef CAS.
- A. Corma, H. Garcia and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- A. Dhakshinamoorthy, M. Alvaro, A Corma and H. Garcia, Dalton Trans., 2011, 40, 6344–6360 RSC.
- Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, J. Catal., 2009, 267, 1–4 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2009, 351, 2271 CrossRef CAS.
- P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Greneche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820 RSC.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS.
- E. Pérez-Mayoral and J. Čejka, ChemCatChem, 2011, 3, 157–159 CrossRef.
- M. Savonnet, S. Aguado, U. Ravon, D. Bazer-Bachi, V. Lecocq, N. Bats, C. Pinel and D. Farrusseng, Green Chem., 2009, 11, 1729–1732 RSC.
- F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. De Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- L. Alaerts, E. Seguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353 CrossRef CAS.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J.-M. Grenèche and J.-M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259 CrossRef.
- J. W. Yoon, Y. K. Seo, J.-S. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 3022–3030 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem.–Eur. J., 2010, 16, 8530 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 711 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem.–Eur. J., 2011, 17, 6256–6262 CrossRef CAS.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS.
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, Ch. V. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS.
- A. Dhakshinamoorhty, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856–867 Search PubMed.
- H. Leclerc, A. Vimont, J.-C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llewellyn, P. Horcajada, G. Férey and C. Serre, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC.
- N. Ravasio, F. Zaccheria, A. Gervasini and C. Messi, Catal. Commun., 2008, 9, 1125–1127 CrossRef CAS.
- S. Ameerunisha and P. S. Zacharias, J. Chem. Soc., Perkin Trans. 2, 1995, 1679 RSC.
| This journal is © The Royal Society of Chemistry 2012 |