Aurophilic interactions as a subject of current research: an up-date
Hubert
Schmidbaur
ab and
Annette
Schier
a
aDepartment Chemie, Technische Universität München, Lichtenbergstreet 4, 85747 Garching b. M., Germany. E-mail: H.Schmidbaur@lrz.tum.de
bFaculty of Science, Department of Chemistry, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
First published on 24th August 2011
Abstract
Recently accumulated experimental evidence for aurophilic interactions in and between molecular gold(I) compounds and the results of pertinent theoretical calculations are reviewed for the period from 2007 to mid-2011. The influence of the intra- and intermolecular bonding contacts between the closed-shell metal centres, Au–Au, on the molecular and crystal structures, and the consequences of these effects for the chemical and physical properties of gold compounds are summarized for the various classes of mono- and polynuclear systems. The literature survey builds on the contents of previous reviews and relates new experimental and theoretical findings to earlier observations (353 references).
Hubert Schmidbaur is presently Professor Emeritus at Department Chemie of Technische Universität München, Germany, and Adjunct Professor at King Abdulaziz University, Jeddah, Saudi Arabia. In his career he held positions at the universities of Munich, Marburg and Würzburg in Germany, and was a visiting professor at several universities in Australia, Finland, India, Japan, New Zealand, Singapore, South Africa, the UK, and the USA. In research he has contributed to many areas of inorganic, organometallic and coordination chemistry as reflected by more than 850 widely cited publications and acknowledged by many awards, including the Centennial and Ludwig Mond medals of the RSC, and memberships in several academies and learned societies. He is the founder of the series of conferences on the Chemistry and Technology of Gold now continued under the auspices of the World Gold Council. |
Annette Schier has been engaged in the crystallography unit and in advanced teaching assignments at the Inorganic Chemistry Institute of Technische Universität München. She was in charge of the organization of several major international chemistry conferences, and is a co-author of more than 200 publications and associated with the editorial boards of scientific journals. |
1. Introduction
In the 1970s evidence had accumulated in the structural chemistry of molecular gold compounds for significant attractive interactions between linearly two-coordinate gold atoms in the +1 oxidation state. In the absence of steric hindrance, the gold(I) centres contained within a given molecule, or of different molecules, were found to be drawn together to intra- or intermolecular equilibrium distances in the range from ca. 2.50–3.50 Å, well below the sum of two van der Waals radii (3.80 Å), and often below the distance between gold atoms in cubic close-packed gold metal (2.89 Å). Such attractive interactions were unexpected since gold(I) centres have a [5d10] closed-shell electronic configuration and should thus experience only weak van der Waals forces. Moreover, the interactions between the (+1) electrical charges of the gold cations or between gold atoms with the same polarisation in their covalent bonds should result in a significant Coulomb repulsion at any shorter distances. Already in early publications, reviews and monographs these observations were occasionally addressed and briefly discussed, but the full scope of this phenomenon had not yet been recognized.1–9Preparative and structural work in the following decade has then shown that these “closed-shell interactions” not only can be found throughout the structural chemistry of gold, but can even be deliberately employed for the synthesis of novel multinuclear gold compounds with unusual stoichiometries and structures, and the results became a major incentive for research in gold chemistry. In 1987 the terms “aurophilic bonding” and “aurophilicity” were introduced for these interactions which clearly play a role in almost all areas of gold chemistry.11–15 In the late 1980s gold chemistry generally underwent a rapid growth, owing mainly to emerging new applications of gold compounds.16,17 As a consequence, a large set of structural data became available. The new type of intra- and intermolecular interactions evident from these compilations, and their consequences for the physical properties of the materials,18–21 became the subject of dedicated investigations which afforded a rapidly growing number of examples for “aurophilicity at work”.
The progress has been summarized periodically in review articles14,15,21–27 which have reflected the broad acceptance of this interesting facet of chemical bonding in all kinds of solids. In a still small number of cases, the effect was finally also observed in the solution state where it was possible to study the association equilibria allowing first estimates and then measurements of the bond energies involved. These data have shown that aurophilic bonding can be associated with about the same energy as standard hydrogen bonding, leaving little doubts that its effect is highly significant for the description of gold-based systems.23
As might be expected, the interactions concerned have their counterparts in the chemistry of other metals with a similar electronic configuration, some examples in fact dating back to much earlier periods of coordination chemistry. Many of these related phenomena are now consequently summarized under the term “metallophilicity”, for which examples can be found not only for the homologues of gold, viz.silver and copper, but also for iridium, platinum, mercury, and thallium.18,19 Extensive theoretical studies have confirmed the validity of the concept and the estimated energies involved. Since the most obvious examples were found with the very heavy elements (Ir–Pt–Au–Hg–Tl), the phenomenon appeared to be related to relativistic effects which reach a maximum at gold.28–34 Although theoretical calculations have confirmed the importance of these effects, observations with the lighter congeners Cu and Ag suggest that significant metallophilic bonding is also relevant to compounds of these metals.35,36 The decrease or increase of the bond energies involved along the triad Cu–Ag–Au is still a matter of debate.32,35
The fact that metallophilic bonding is most readily discernible in compounds of gold may therefore not only have its reason in a maximum of the Au–Au bond energy among the M–M contacts and in the maximum relativistic contraction of gold orbitals, but also in the smallest possible coordination number of gold(I)—linear two-coordination—which leaves the coordination sphere of the metal centre wide open for an approach of its congeners.23 Maybe in the future this aspect should be given more consideration in order to avoid misinterpretations when comparing metallophilicity phenomena. The metal–metal contacts thus established may be homo-metallic M–M or heterometallic M–M′, with M = Au and M′ = Ag being a particularly common combination.18
The present review summarizes the progress made in the time span since the publication of a similar briefing in this Journal by the same authors.23 The literature has been screened for the period from 2007 to mid-2011 for developments in the molecular and supramolecular chemistry of low-valent gold, i.e. systems with homometallic Au–Au contacts. Older references are only included where it appeared to be necessary for putting new results into proper perspectives. Molecules with heterometallic Au–M combinations have not been included in order to keep the size of the review within the editorial limits, and because some recent coverage is available.17,18,27
A summary of the current theoretical interpretations of aurophilic bonding is given here in Chapter 3.1 on unsupported aurophilic interactions, because it is there that the phenomenon can be observed in its purest form.
2. Intramolecular aurophilic interactions in gold(I) compounds
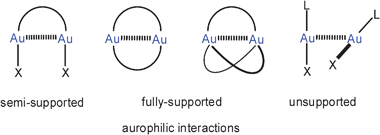
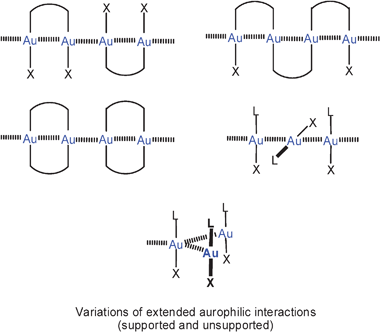
Intramolecular AuI–AuI interactions may be observed in molecules and molecular ions containing more than one gold atom and are recognized by short Au–Au distances in the solid state or by restricted structural flexibility in solution. All examples in this category are classified “supported” aurophilic interactions because the gold atoms are also connected by the molecular framework which allows for the mutual approach of the gold atoms and can support it through entropic contributions or structural rigidity. The gold atoms may be separated by one or more bridging atoms.For the most common cases, where two gold atoms are linearly two-coordinate, two categories of intramolecular aurophilic bonding may be distinguished, in which one or two difunctional ligands connect the gold atoms, and these are described as semi-supported and fully supported Au–Au contacts, respectively. In the rare cases where the coordination number of the gold(I) centres rises to three, even three ligands may be supportive. By contrast, any intermolecular aurophilic bonding is described as unsupported (Scheme 1). It is important to note that a given gold(I) centre may not only become attached to one other gold(I) centre, but rather also become a member in a shorter or longer chain of gold atoms, or enter in multicentric systems (Scheme 2).
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
2.1. Geminal auration and the clustering of gold atoms at a common ligating atom
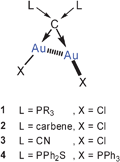
Two or more gold atoms separated by only one other atom E are held in close proximity with Au–Au distances depending on the Au–E bond lengths and on the Au–E–Au bond angles. All structure determinations carried out in recent years have shown that the clustering of gold atoms at a given atom E and the structure of the product E(Au)n are strongly influenced by aurophilic bonding.Recent examples include the analogues of the known di-aurated carbodiphosphoranes which have been prepared following the concept of “coordination at carbon atoms”.39–45 Substitution of one or both tertiary phosphines R3P in a compound of type 1 by a carbene ligand R2C leads to dinuclear complexes of type 2 of which several representative examples have been structurally characterized.44,45 These structures provide evidence for the twofold donor capacity of the carbon atom which also functions as a twofold acceptor for the phosphine and/or the carbene, respectively. It should be noted that complexes of diaurated malodinitrile (3)46 and diaurated bis(diorganophosphino)methane disulfides (4)47 and related compounds have been known from earlier work and should be considered in the same context.48 In 3, two cyanide donors and two gold cations can be viewed as the donors and acceptors to a carbon atom, respectively. In all cases the short intramolecular Au–Au distances (<3.1 Å) associated with small Au–C–Au angles (≪109.5°) indicate the influence of aurophilic bonding.
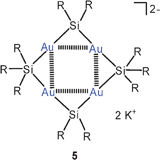
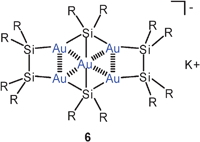
Geminal diauration of silicon in the coordination numbers four and five has recently been accomplished by the reaction of potassium bis(supersilyl)aurate(I) with supersilyl chloride.49 In the structure of the dianion of 5 two gold atoms are attached to a pentacoordinated silicon atom with very short Au–Au contacts of 2.752 Å (R = SiMe3). These units are linked to geminally diaurated units of tetracoordinated silicon atoms to form eight-membered rings which are centrally capped above and below by the potassium cations (not shown). Finally, in a product 6, triaurated silicon atoms with coordination number five are linked to disilane groups to form a pentanuclear cluster anion. The potassium cation is located above the central gold atom (not shown). This work has thus provided examples based on building blocks with the coordination patterns (Si)2Si(Au)2, (Si)3Si(Au)2, and (Si)2Si(Au)3. The latter has a carbon analogue in the cation [(Me3Si)3C(AuPPh3)2]+ described previously.50,51
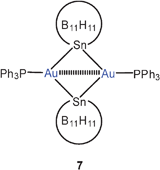
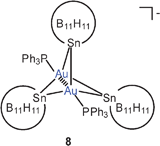
Geminal diauration of the tin atom in stanna-closo-dodecaborate clusters has yielded dinuclear complexes 7 with exceedingly small Au–Sn–Au angles (57.03°) and short Au–Au distances (2.625 Å). In the anionic complex 8, the two gold atoms (Au–Au 2.590 Å) are triply connected by the stannadodecaborate anions.52
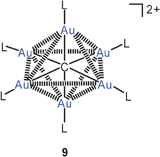
No new information on triply and quadruply aurated carbon compounds could be drawn from the recent literature, but new results have been presented on hexa-aurated methanium dications. Using diphenyl(2-pyridyl)phosphine (L) instead of triphenylphosphine ligands, the carbon-centred hexanuclear octahedralcluster 9 with six pyridine donor centers has been obtained.53 Upon treatment of this precursor with silver salts, a mixed gold/silver cluster 10 was generated which was found to still have the octahedral CAu6 core with two opposite faces capped by a silver atom. Thereby a remarkable shift of the photophysical properties is induced and manifested in the absorption and emission spectra. The structural data of the Ag2Au6C core with all Au–Au and Au–Ag distances close to 3.0 Å suggest extensive metallophilic interactions. These new results are important confirmation of all previous work on the CAu6 clusters which had not yet been reproduced in other laboratories since the first reports.54–58
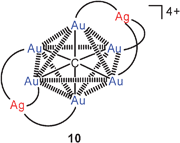
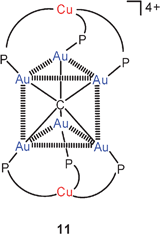
A very recent extension of this work has led to the discovery of cations with hexaaurated carbon atoms in a trigonal prismatic constitution. The reaction of 9 with [(MeCN)4Cu]+ BF−4 gave the expected product with a Cu2Au6C core, but the copper atoms are now capping the two triangular faces of the prism11 instead of two opposite triangular faces of an octahedron10. It is again assumed that metallophilic bonding (Au–Au/Cu) supports the structure of this complex. Even though a trigonal prism has only 9 Au–Au edges as compared to 12 Au–Au edges in an octahedron, there appears to be no great loss or gain in energy because for compensation all Au–Au distances in the former are found shorter (2.830 Å average) than in the latter (2.99 Å average). It requires only the introduction of a methyl substituent in the pyridyl group to convert the structure back into the octahedral form (10, Cu for Ag; 5-Me–py for py).59 It is as yet unclear if the prismatic structure of 11 is retained in solution. With a single 31P resonance in various solvents, either of the two forms (prismatic or octahedral) can prevail, or the skeleton may be fluxional on the NMR time scale. DFT calculations have shown that the energy difference between the two forms is indeed very small. The new mixed-metal clusters (10, 11) are brightly luminescent both in the solid and solution state, the emission being of triplet parentage. Clearly the extension of the core (as compared to 9) leads to an enhanced luminescence.
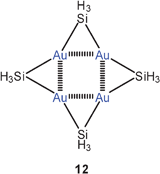
The results of theoretical calculations have shown that there may be significant aurophilic interactions in tetramers [H3EAu]4 with E = C, Si, Ge, if the metal atoms are arranged in a square with four edge-bridging E atoms (12).60 Similar Au–Au contacts have previously been assumed in the experimentally well documented oligomers [RAu]n with various organic groups R (mainly R = aryl) and with n = 4, 5.37,38,48
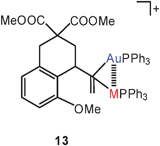
Geminal diauration of olefins has recently been in the focus of important studies with relevance to the catalytic action of gold(I) catalysts for the activation of unsaturated hydrocarbons.61a,b Mechanistic studies of the intramolecular hydroarylation of allenes have provided evidence for intermediates 13 (M = Au) with geminally diaurated olefinic units as resting states in the catalytic cycle induced by Ph3PAuN(SO2CF3)2 as a source for [(Ph3P)Au]+ electrophiles.62 In the same work, the role of silver salts as cocatalysts has been studied, and bimetallic species (13, M = Ag) have been postulated as alternative resting states of the modified catalyst. For both cations, metallophilic interactions have been ascribed a significant role in stabilizing the reactive intermediates.63
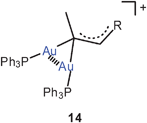
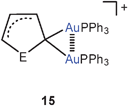
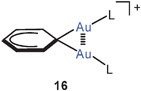
These results have been confirmed in recent work by the isolation and structural characterization of several compounds containing the diaurated olefinic cations 14 (R = OEt, cyclopropyl). In both cases narrow Au–C–Au angles and short Au–Au contacts suggest a stabilization of the cations by aurophilic bonding.64 The results are in agreement with earlier preparative and structural data65 on diaurated furans and thiophenes 15 (E = O, S) and with the related syntheses of compounds with di-aurated alkenes and arenes (16).37,66
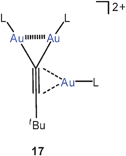
Desilylation and multiauration of the terminal alkynetBu–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–SiMe3 with tBu3PAuCl/AgSbF6 have afforded trinuclear dications 17 (L = tBu3P) in which one tBu3PAu unit is attached side-on to the triple bond, and two of these units share a terminal carbon atom. A close Au–Au contact of 2.965 Å is observed for the latter indicative of significant interactions. Using the same reagent, bis(trimethylsilyl)ethyne Me3Si–CC–SiMe3 gave a tetranuclear dication with two end-on and two side-on tBu3PAu units in which the spatial distribution of the gold atoms makes significant aurophilic interactions less likely.67 It should be noted that for 17 the bulky substituents render intermolecular interactions between gold centres impossible.
C–SiMe3 with tBu3PAuCl/AgSbF6 have afforded trinuclear dications 17 (L = tBu3P) in which one tBu3PAu unit is attached side-on to the triple bond, and two of these units share a terminal carbon atom. A close Au–Au contact of 2.965 Å is observed for the latter indicative of significant interactions. Using the same reagent, bis(trimethylsilyl)ethyne Me3Si–CC–SiMe3 gave a tetranuclear dication with two end-on and two side-on tBu3PAu units in which the spatial distribution of the gold atoms makes significant aurophilic interactions less likely.67 It should be noted that for 17 the bulky substituents render intermolecular interactions between gold centres impossible.
The situation is different in 1-carba-closo-dodecaboranyl-ethynes aurated at the alkyne terminus and at the triple bond (18). With one small end-on and one small side-on Me3PAu or Et3PAu unit (R = Me, Et), dimerization can readily occur which leads to efficient intermolecular aurophilic bonding (19). The intramolecular Au–Au contacts in the dimers 19 are 3.41 Å (R = Me) and 3.57 Å (R = Et, average), and thus at the upper limit for a significant bonding contribution, but the intermolecular contacts are found in the range of 3.04–3.14 Å for appreciable bonding.68 In the dimers a tetrahedral Au4 core is established for which there is precedent in previous work.23,58
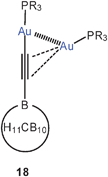
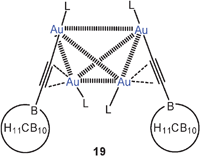
By contrast, with the larger iPr3PAu and tBu3PAu groups (18, R = iPr, tBu), the complexes are monomers in which the side-on bonded units clearly lean over to the end-on bonded one to establish an aurophilic contact of 3.29 Å (R = iPr). These results very nicely reflect the interplay of intra- and intermolecular aurophilic bonding. Moreover, the monomer–dimer equilibrium could be followed more closely for the example with R = Et and the results have provided a new reliable estimation of the energies involved (see Table 1).68
| Compound/model | Au–Au contact mode | Method | Bond energy/kcal mol−1 | Reference |
|---|---|---|---|---|
| a Non-bonding due to Coulomb repulsion; attraction verified upon addition of anionic point charges. | ||||
| (CO)AuCl | Unsupported intermolecular | MP2 | 10.0 | 235 |
| (Et3P)AuCl | Unsupported intermolecular | DFT | 9.5 | 229 |
| (NHC)AuCl | Unsupported intermolecular | DFT | 8.6 | 279 |
| [Au(CN)2]− | Unsupported inter-anionic | EH/MP2 | 7.2 | 330 |
| [Cl(AuPH3)2]+ | Unsupported inter-cationic | MP2/DFT | 19.8 | 140 |
| S(AuPH3)2 | Unsupported intermolecular | MP2/DFT | 29.8 | 109 |
| [HS(AuPH3)2]+ | Unsupported inter-cationic | MP2/DFT | a | 109 |
| [(Et3PAu)2CCB11H11] |
Unsupported inter-cationic | NMR | 8.8 | 68 |
| (dppe)Au2[S2C2(CN)2] | Fully supported intramolecular | UV/vis | 15.0 | 187 |
| [(Xantphos)2Au2]2+ | Fully supported intramolecular | NMR | 11.6 | 154 |
Very little is known about the molecular chemistry of the more extensive clustering of gold atoms at silicon. While silylation of gold atoms to produce a single Au–Si bond is well established,16 polyauration to produce species with several gold atoms bridged by a single silicon atom has not been successful in the condensed phase. Gas phase studies by mass spectrometry focusing on polynuclear anions [AunSi]− (n = 2–4) gave some first information on the reactivity pattern of such species,69 which previously were the subject of theoretical calculations and photoelectron spectroscopic investigations.70–72 In all cases the relevance of aurophilic bonding was considered and confirmed.
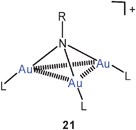
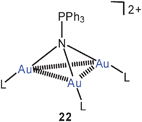
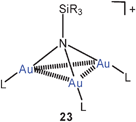
Tri-auration of amines to give cations of the type 21 is well established as summarized in earlier reviews.23,58 In a complementary study, the tri-auration of triphenylphosphinimine has been observed which affords the cation 22 with Au–Au contacts of 2.962, 3.067 and 3.219 Å length.78 Similar data had previously been reported for other phosphinimines.79 Silylamines and disilylamines can also be tri- or di-aurated, respectively, to give ammonium saltse.g. of the type 23.80
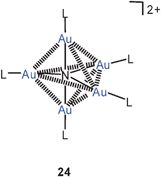
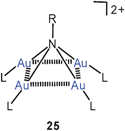
Contrary to the chemistry of poly-aurated carbon (above), work on the penta-auration of ammonia and on the tetraauration of amines which lead to dications 24, 25 has not been resumed after the early work published some years ago.81,82
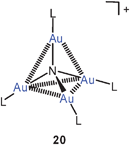
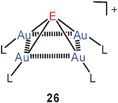
In recent theoretical work, using DFT and ab initio methods,77 the known pyramidal structure73 of {As[Au(PPh3)]4}+ (26, E = As) has been confirmed for the model with L = PH3, but a tetrahedral structure has been predicted for the P-analogue {P[Au(PH3)]4}+ (20, P for N), which appears to be a delicate borderline case. An exploration of the basis set limits for MP2 calculations has led to the conclusion that the pyramidal structure is the most likely configuration for the P-centered gas phase cation.75b However, for these cations only derivatives are known from experimental work. For simple Sb-centred species (26, E = Sb) there are also no experimental data,83 but a pyramidal structure has been calculated.73
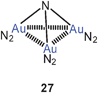
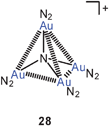
Theoretical calculations have recently also been performed on neutral and cationic nitrogen-centered polynuclear species in which molecular nitrogen N2 is introduced as the auxiliary ligand L. The models include N[Au(N2)]3 and {N[Au(N2)]4}+ with C3v and Td point group symmetry (27, 28), respectively. There is no explicit evidence for Au–Au bonding, but the short distances between the metal atoms suggest significant interactions.84 It is worth noting that still no experimental data have been provided for any trigoldamine compound N(AuL)3, while the corresponding tetragoldammonium compounds (20) are well established (above).
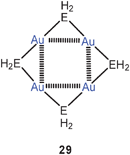
Aurophilic bonding has also been considered in theoretical calculations of the tetramers [AuNH2]4 and [AuPH2]4 with their gold atoms arranged in squares and with edges bridged by the NH2 or PH2 groups (29, E = N, P).59 These species were found to be stable, non-aromatic or even anti-aromatic molecules, but there is as yet no experimental evidence for amides or phosphides [AuER2]4 with E = N, P.
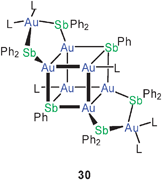
In the present context it is of great interest that from the complex reaction mixture obtained from (Et3P)AuCl and Ph2SbSiMe3/PhSb(SiMe3)2 a cluster Au8(SbPh)2(SbPh2)4(PEt3)6 could be crystallized (30). In this structure, the Sb atoms of the central heterocubic unit Au6Sb2 are part of five-membered rings with very narrow Au–Sb–Au angles (90.5°) and therefore short Au–Au contacts (3.041 Å).85
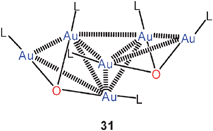
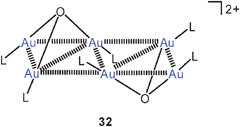
Even though the corresponding oxides (LAu)2O are still unknown because of their high affinity for [LAu]+ electrophiles, oxonium salts [O(AuL)3]+ X− can be viewed as sources of such [LAu]+ electrophiles. This has meanwhile been used extensively in homogeneous catalysis of organic transformations by gold(I) compounds.63,88–91 For this purpose, triphenylphosphine Ph3P was the ligand L of choice in almost all cases.
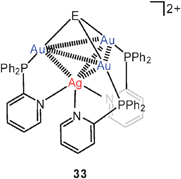
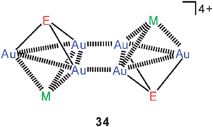
In another extensive research programme, the aryl groups in Ph3P have partially been replaced by 2-pyridyl groups in order to offer additional coordination sites for other coinage metals. In this way it was possible to tune the photophysical properties of the clusters by additional metallophilic bonding.92,93 The dication 33 (E = O) has a quasi-tetrahedral Au3Ag core unit with the Au3 face capped by the oxygen atom. All Au–Au and Au–Ag contact distances allow for metallophilic bonding (2.95–3.06 and 2.90–3.06 Å, respectively). The analogous copper compound has the same structure in one polymorph, but in a second one the dications are associated via interionic aurophilic bonding (edge–edge) into dimers (34, E = O, M = Cu). A similar arrangement in dimers is found for dications with ligands L = Ph2PCH2CH2–2–Py. As the pyridyl groups are dangling on ethylidene bridges in this case, the Cu and Ag atoms cannot be kept in the vicinity of the Au3 triangle without a significant loss of entropy, and only Au–Au contacts are possible. Analogous compounds have been prepared with M = Cu, Ag and E = S, Se (sulfonium and selenonium salts).92,93
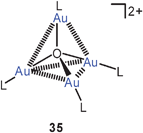
Tetra(gold)oxonium dications with a conventional tetrahedral structure 35 with triphenylphosphine ligands and tetrafluoroborate counterions were prepared for the first time in 1995, and this work had since not been followed up.94 It was only in a very recent study that the dication with the formula [O(AuL)4]2+ was observed, but associated with Keggin-type polyoxometalate anions in two different ethanol solvates and in one hydrate.95 Surprisingly, however, in both crystalline ethanol solvates the dication was shown to have a trigonal-pyramidal structure with approximately C3v symmetry (36). In this geometry, there are three short Au–Au contacts (apical–equatorial, average 2.94 Å) and three long Au–Au distances (equatorial–equatorial, average 3.60 Å). Accordingly, the average apical–equatorial and equatorial–equatorial angles Au–O–Au are <90° and 120°, respectively. In an anion-exchange resin the polyoxometalate could be converted into the known tetrafluoroborate salt with the tetrahedral structure of the dication (35). Solid-state 31P NMR investigations have confirmed the presence of two different types of phosphine ligands (1 axial, 3 equatorial) in the crystals, but solution studies (in DMSO-d6 at 25°) showed equivalence of all four ligands. The δ31P value of ca. 25 ppm is the same as that reported for the BF4 salt, suggesting a quasi-tetrahedral or a fluxional structure in solution.95
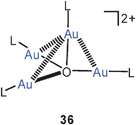
These new results complement previous findings with the corresponding isoelectronic ammonium salts 20 which are also known to have a flexible skeleton easily distorted away from the standard tetrahedral structure.10,73 The non-Le Bel-van't Hoff behaviour of these and related species (with phosphorus,83,96arsenic73,96 and sulfur centers23,58,97) is one of the most obvious consequences of aurophilic interactions. Because the combined energies of these interactions can effectively compensate the energy loss associated with the distortion of the classical tetrahedral structure, all element-centred units of the type [E(AuL)n]m+ show these structural phenomena.97,98 In the present case, it is the influence of anions very different in size and charge density (POM3−vs.BF−4) which induces the drastic folding of the tetrahedron 35 to the trigonal pyramid 36 which is otherwise unprecedented and completely unexpected in the structural chemistry of first row elements with an closed octet shell (C4−, N3−, O2−, F−).
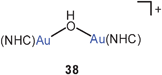
All the recent observations summarized here for oxygen-bridging of gold atoms were made with tertiary phosphines as auxiliary ligands L. Very different behaviour was encountered for units [LAu]+ with L = NHC (an N-heterocyclic carbene). Treatment of (NHC)AuCl substrates with KOH gave the mononuclear hydroxides (NHC)AuOH (37), so far unknown with L = R3P, and with a different stoichiometry of reactants the product 38 with a hydroxo-bridged dinuclear cation could be isolated. The cation in 38 has a bent structure, but the Au–O–Au angle is surprisingly large at 129.5° associated with an Au–Au distance of as much as 3.75 Å, ruling out any significant direct aurophilic interaction.99 Similar structural data have been accumulated of salts 38 with other anions [CF3SO−3, SbF−6, N(SO2CF3)−2, B(C6F5)−4] which lead to the conclusion that the wide angular structure of the cation is not induced by the nature of the anions, but is an intrinsic characteristic of the species caused by the NHC ligands.100 The authors have offered a schematic representation of (NHC)Au–Au interactions based on Au(d2z)–Au(pz) overlap, with a calculated Au–O–Au angle of 137.9°, which in the framework of second-order perturbation theory E(2) yields an energy of no less than 8.7 kcal mol−1, and with a non-negligible Mayer Bond Order of 0.14.

Because for other systems any Au–Au distances of >3.5 Å have so far been ascribed very little if any aurophilic contributions, the new structural details with NHC instead of R3P ligands may indicate that for the former cases interactions along the Au–O–Au bridge have to be considered which reach a maximum at large rather than at small bond angles. The differences may arise from the different donor/acceptor properties of NHC as compared to R3P, evident also e.g. in the different catalytic action of [(NHC)Au]+ and [(R3P)Au]+ electrophiles,61,101 but steric effects may also play a major role.
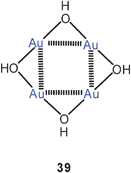
Tetrameric gold(I) hydroxide was considered in recent theoretical calculations.59 A square planar structure with edge-bridging OH groups was found to be stable (39), but neither this hydroxide nor any alkoxide/aryloxide/siloxide with this type of structure have been encountered in experimental work.
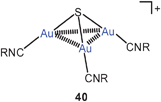
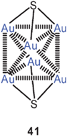
Up to 2005, all compounds of this type had tertiary phosphines as auxiliary ligands L. In one case it has then been shown that the gold clustering at sulfide anions can also successfully be accomplished with L = isocyanide, RNC. Complexes with R = cycloheptyl and with PF−6 or SbF−6 as counterions have been isolated and fully characterized (40). The trinuclear cations are further associated into dimers. In these dimers, the gold atoms form a distorted octahedron with two opposite triangular faces capped by a sulfur atom (41, ligands omitted). Intra- and inter-cationic Au–Au contacts are in the range of 3.247–3.350 and 3.321–3.460 Å, respectively. The SbF−6 salt undergoes a phase transition upon cooling which leads to drastic changes in the emissive properties in the solid state.104
Multiauration has also been demonstrated for mercaptanes RSH, which can be converted into molecules LAuSR and sulfonium salts [RS(AuL)n](n−1)+(X−)(n−1) (n = 2, 3). In all dinuclear cases (42) short intracationic Au–Au contacts have been observed, but in the trinuclear dications large quasi-tetrahedral angles, Au–S–Au angles, lead to long Au–Au distances that make strong interactions unlikely.105 Notably, attempts to diaurate a dialkylsulfide to produce sulfonium salts of the type [R2S(AuPR3)2]+ 2X− have failed.106
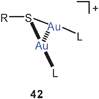
It is important to note that double auration of thiolate anions RS− can not only be achieved by the established reaction with [(R3P)Au]+ nucleophiles.102,103 Recent work has shown that oxidation of molecules R3PAuSR′ by ferrocinium tetrafluoroborate also produces salts [R′S(AuPR3)2]+ 2 BF−4, with R′SSR′ as the by-products.107
Cations [RS(AuL)2]+ (42) are often found associated into dimers, in which squares or rectangles of gold atoms are formed through unsupported Au–Au contacts (43).107
In the dication of the salt [tBuS(AuPMe3)2]2+ 2 BF−4, produced as described above, the intra- and intercationic Au–Au contacts are 3.138 and 3.112 Å, respectively, providing an example that unsupported aurophilic contacts may be shorter/stronger than supported ones even though Coulomb repulsion between the cations must be overcome. It is most interesting that ESI-mass spectrometric and DOSY measurements have shown that these tetranuclear systems are even maintained in solution and in the gas phase.108
(dppe)(AuStBu)2 has no intramolecular gold contact, probably for steric and entropic reasons, but its oxidation by ferrocinium tetrafluoroborate leads to S–S coupling of two thiolate groups and affords the salt 44 in which the four gold atoms form a Z pattern with Au–Au contacts of 2.986 (terminal) and 3.152 (central) Å.107
DFT calculations have provided structural data for the model compounds [(H3P)Au]2S and [(H3P)Au]2SH+ (42, H for R, PH3 for L) and their dimers {[(H3P)Au]2S}2 and {[(H3P)Au]2SH}22+ (43, H for R, PH3 for L) which are in good agreement—where available—with experimental dimensions for the corresponding substituted systems (R for H). The intramolecular Au–Au distances in the monomers were found to increase significantly upon protonation of the sulfur atom. For the dimers, structures with an Au4 tetrahedron are preferred over those with an Au4 rectangle. The calculated average Au–Au binding energy amounts to 31 kJ mol−1.109
In recent studies trigoldsulfonium salts {S[Au(PR3)]3}+ X− (45) have been prepared in great variety by employing tertiary phosphines with functional substituent patterns. Where bulky groups prevent intermolecular Au–Au contacts, the compact SAu3 units feature small Au–S–Au angles (<90°!) and Au–Au distances of ca. 3.25 Å. The compound with the L = mbpa ligand (46) serves as an example which was prepared because of its potential catalytic and pharmacological activities.110
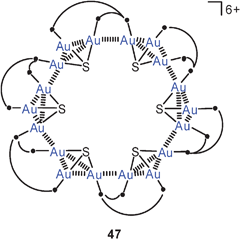
With poly(tertiary) phosphines as ligands L, the SAu3 units can be tied together to form larger aggregates. Thus with bis(diphenylphosphinoethyl)phenylphosphine, macrocyclic cations (6+) 47 have been constructed in which multiple aurophilic interactions are observed.111 The structure appears to be co-directed by the interionic Au–Au interactions which are established between pairs or triples of gold atoms. While contacts between corners and between edges (to give a pair or a square of bonded gold atoms) are quite common, contacts between a corner and an edge (to give a triangle of bonded gold atoms) are rare.97 The compound 47 (with 6 PF−6 counterions) is strongly luminescent in polar solvents, and the photophysical properties can be tuned by the addition of silver salts, which are believed to form bimetallic adducts with metallophilic interactions (Au/Ag).97,98
As already mentioned, sulfonium and selenonium analogues of the monomeric and dimeric oxonium cations 31, 33 (E = S, Se) and 34 (E = S, Se) have also been investigated in great detail with a focus on the luminescence properties of the bimetallic compounds (Au/Cu and Au/Ag). A strong dependence of the emission maxima on the nature of the clustering element E (P, S, Se) as well as of the second metal (Cu, Ag) was observed for the solid state. The structure of {[Ph2(2-Py)PAu]3Se}+ BF−4 has been determined and showed no anomalies as compared to the PPh3 analogue. The corresponding copper(I) complex has similar dimensions regarding the EAu3 unit (E = S).93,97
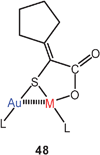
2-Cycopentylidene-2-thioglycolic acid has been shown to form both homo- and heterobimetallic complexes with [(Ph3P)Au]+ and [(Ph3P)Ag]+ units of the type 48 (M = Ag, Au). The Au–S–Au/Au–S–Ag angles are small at 80.6/79.9° owing to the typical metallophilic Au–Au/Ag contacts of 3.03/3.05 Å. The crystals are isomorphous, but there is no Au/Ag disorder, because the Ag atom is positioned near the carboxylate groups as an additional ligand. However, in CDCl3 solution at room temperature there is rapid ligand exchange on the NMR time scale which leads to coalescence phenomena in the 31P spectra. Similar fluxionality is observed for the tetranuclear silver complex. The compounds are strongly luminescent in frozen glassy solutions owing to intraligand electronic transitions, and have similar in vitro cytotoxicity.112
Ligand-free gold(I) thiolates (RSAu)n have long been a subject of special interest because of their variety of aggregation modes which have a pronounced influence on the solubility and applicability as precursors for gilding preparations (“liquid gold”).113,114 Moreover, the solubility is also relevant to some of the oldest pharmaceuticals for the chrysotherapy of rheumatoid conditions based on gold thiolates.114,115 In recent work gold(I) thiolates are of prime importance for the preparation of gold nanoparticles. In this context, the oligomers with thiopronine as the thiol have been investigated. From combined spectroscopic and powder X-ray diffraction studies and theoretical calculations it has been concluded that tetramers 49 with extensive aurophilic contacts (Au–Au 3.351 Å) are the prominent components in the precursor mixtures.116
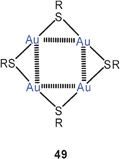
According to model calculations, the tetramers [RSAu]4 (49) are among the most important capping units of gold nanoclusters. Within this model, thiolate-protected gold nanoparticles can be formulated like Au14[(AuSMe)4]6 with aurophilic bonding in and between the tetramers and the cluster core.117,118 In other models, mononuclear anions [RSAuSR]− appear to be stapled to gold cluster surfaces. In terms of aurophilic interactions, this attachment has recently been studied by SCS-MP2 and DFT calculations.119 The phenomena observed with p-mercaptobenzoic acid as the thiol have been put into a larger context considering various types of core metals and metal mercaptides and halides.120
Trimers of the formula [RSAu]3 have also recently been suggested as precursors for quantum-sized gold nanoparticles. In model considerations an “evolution” from aurophilic bonding in these oligomers to Au–Au bonding in the clusters has been proposed.121
In a recent theoretical study, the capping of gold clusters and nanoparticles by [RSAuSR] or [RSAuS(R)AuSR] units as proposed by Häkkinen et al.122 was followed up using the superatom concept. Tetrahedral and octahedral gold clusters were provided with these capping units and the geometry optimized to reach the most stable structure. In these model systems, aurophilic contacts with distances between 2.85 and 3.05 Å prevail and contribute significantly to the overall stability.123 A nuclearity with the formula [Au12(SR)9]+ with D3 symmetry has been among the family of prominent candidates, and indeed this stoichiometry was recently discovered with RSH = N-acetyl-cysteine, the structure of which awaits to be determined.124 In the anionic particle [Au25(SCH2CH2Ph)18]− an icosahedral Au13 core has been found to be surrounded by [RS–Au–S(R)–Au–SR]− “semi-rings” (R = 2-phenylethyl) stapled through Au–S coordination and aurophilic bonding.125 It should be noted that the thiolate-capped clusters have the core gold atoms in a lower oxidation state (<1), and therefore their Au–Au contacts do not represent solely aurophilic bonding.
Anionic
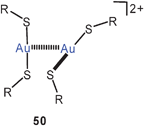
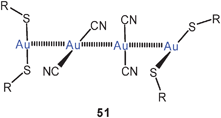
thiolato complexes of the type [Au(SR)2]− have been known for a long time, and examples with various groups R have been documented.1,126 Recent work has concentrated on a betainic thiolate ligand with a trimethylammonium function in the 4-position of thiophenol. This extra charge makes the ligand [4-Me3NC6H4S] (tab) into a neutral donor, such that the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 gold complex is a cation[(tab)2Au]+.127 Surprisingly, in the crystalline state these cations are associated into dimers 50 (R = 4-C6H4N(+)Me3) through aurophilic bonding which clearly overcomes the Coulomb repulsion between the two cations. With [Au(CN)2]− as the counterion, tetranuclear units with a chain of gold atoms are formed (51, R as for 50). Similar observations have been made for the dimerisation of anions [Au(L)2]−, e.g. for L = GeCl−3.128 For unsupported aurophilic bonding between [Au(SR)2]−anions see below.
:1 gold complex is a cation[(tab)2Au]+.127 Surprisingly, in the crystalline state these cations are associated into dimers 50 (R = 4-C6H4N(+)Me3) through aurophilic bonding which clearly overcomes the Coulomb repulsion between the two cations. With [Au(CN)2]− as the counterion, tetranuclear units with a chain of gold atoms are formed (51, R as for 50). Similar observations have been made for the dimerisation of anions [Au(L)2]−, e.g. for L = GeCl−3.128 For unsupported aurophilic bonding between [Au(SR)2]−anions see below.
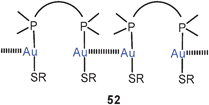
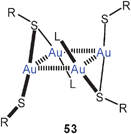
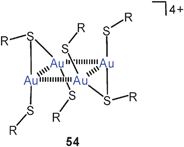
The reaction of 50 with α,ω-ditertiary phosphines leads to a variety of multinuclear cations with Ph2P(CH2)nPPh2 bridges (P–P, n = 2, 3, 4) between the metal atoms. For n = 2, 3 extensive intercationic aurophilic bonding is observed between units like [(tab)Au(P–P)Au(tab)]+ to give infinite 1D chains with only terminal tab groups (52, R as for 50), while with n = 4 the repeating units in the corresponding chains is [(tab)Au(μ-tab)Au(P–P)Au(μ-tab)Au(tab)]+ with tab in both terminal (tab) and bridging positions (μ-tab) (53, R as for 50). Ligand-free products with dications [(tab)3Au2]2+ have also been obtained, which dimerize to give tetracations 54 (R as for 50). All compounds are strongly luminescent in the solid state.129
Gold(I) thiolates are also of great relevance to the production of highly luminescent gold-containing films. To this end compounds [RSAu]n are thermolyzed in a polymer matrix. Recent work was particularly successful when using gold(I) dodecylthiolate [C12H25SAu]n in a polystyrene matrix. The thermolysis process was followed by TEM measurements and extinction, absorption and emission spectra. The results have led to the conclusion that there is a development from few-atom thiolated clusters to polynuclear thiolate aggregates stabilized by strong aurophilic interactions.130
In a study of new routes to gold nanoparticles using gold salts and long-chain mercaptanes it was observed that the precursor compounds of the general formula (RSAu)n showed strong luminescence upon UV irradiation (R = CnH2n+1; n = 2–10, 12, 14, 16). From further investigations with various analytical methods it has been concluded that aurophilic interactions within and between S(Au)m centres (m = 1, 2) play a role for the photophysical properties.131
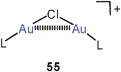
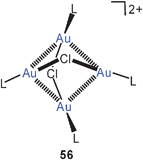
The halogenonium salts 55 have recently been shown to be excellent sources of [(R3P)Au]+ electrophiles which can function as catalysts in a variety of organic transformations.137–139 Owing to the aurophilic stabilization, the dinuclear chloronium cations are considered resting states of the homogeneous catalysts.
Quantum-chemical calculations have provided molecular dimensions for the model systems [(H3P)Au]2X+ with X = F, Cl, Br, and I and their dimers {[(H3P)Au]2X}22+ (55, 56, L = PH3). Where available (X = Cl, Br), the results are in good agreement with the experimental data for the substituted compounds (R for H). Upon dimerization of the chloronium cation, the Au–Cl–Au angle is enlarged from 79° to 102° associated with a lengthening of the Au–Au distance from 2.98–3.68 Å. Intramolecular aurophilic bonding in the monomer is thus minimized in the dimer (with D2d symmetry), but intermolecular interactions amount to 20 kcal mole−1 per Au–Au bond.140 These results complement those of earlier studies.76 For the unknown dimeric fluoronium cation (D2d symmetry), the Au–F–Au angle is predicted at 119.1°, with negligible aurophilic bonding in the monomers, but strong intermolecular interactions depending very little on the nature of the halogen atom (20.5 kcal mole−1).141
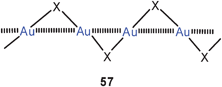
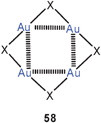
The gold(I) halides AuCl, AuBr, AuI have the well-known zigzag-structures 57 (X = Cl, Br, I) which may allow for significant Au–Au bonding.16,142a,b The structure of AuF is unknown. Recent theoretical work has considered the bonding in hypothetical tetramers [AuX]4 with X = F, Cl, Br in square-planar structures where aurophilic bonding may also contribute significantly (58).59
2.2. Semisupported aurophilic bonding between gold atoms separated by one polyatomic bridge
Short Au–Au contacts between two or more gold atoms in a molecule separated by only one multiatomic bridge have been designated as “semi-supported aurophilic bonding” as opposed to cyclic compounds with two bridging ligands (“fully supported”, Scheme 1). Classical examples for the former are the 2:1 complexes of α,ω-ditertiary phosphines (59) where both intra- (n = 1, with dppm)143 and intermolecular interactions (n = 2, with dppe)144 can be established (60).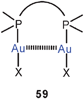
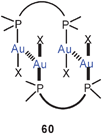
It should be noted that compounds of type 59 can have a rigid or a flexible linkage. While in the rigid case the gold atoms can be fixed in positions where Au–Au contacts are either excluded or supported, in the flexible case there is conformational freedom. If the gold atoms appear in close contacts, this conformation can be taken as evidence for aurophilic bonding which overrules the potential entropy gains that may arise in other conformations. In the following examples from the recent literature, the Au–Au contacts are maintained against possible other effects that may act against this contribution to the configurational or conformational stability.
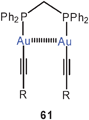
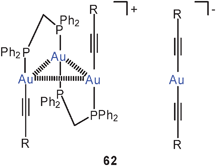
α,ω-Diphosphine complexes with gold(I) alkynyls show a variety of intra- and intermolecular (inter-ionic) Au–Au contacts depending on the length of the linker between the two P atoms. Neutral dinuclear complexes 61 [R = 4-(2,2′-bipyridyl)] with dppm as the ligand have been found to be in equilibrium with an ionic form comprised of a trinuclear cation and a homoleptic mononuclear anion (62). With dppe or dppb as ligands, only unsupported intermolecular aurophilic contacts between molecules are observed. The compounds are strongly luminescent at room temperature as solids and in CH2Cl2 or PrCN solutions.145
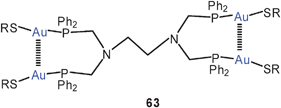
The tetranuclear complex of a tetraphosphine with gold(I) p-anisylthiolate has two intramolecular Au–Au contacts (3.148 Å) in the solid state as shown in formula 63 (R = 4-C6H4OMe). The compound is strongly luminescent in the solid state at 77 K. In solutions all four P atoms are NMR-equivalent indicating full flexibility of the skeleton.146
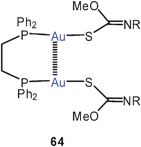
The large family of complexes of gold(I) O-methyl-N-(4-nitrophenyl)thiocarbamide with α,ω-ditertiary phosphines is also characterized by semi-supported Au–Au contacts with characteristic Au–Au contacts and the expected consequences for the luminescence properties (64, R = 4-C6H4NO2). If the P–P linker is too large, intermolecular Au–Au interactions prevail.147
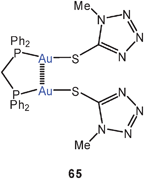
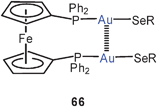
In a dinuclear dppm complex with tetrazolylthiolate ligands, a similar quasi-cyclic conformation leads to an Au–Au contact of 3.105 Å (65), but the dppe analogue again remains an open-chain molecule, which is associated through slightly slipped intermolecular Au–Au contacts into chains.148 The same association into chains has been discovered in the two selenophenolates with R = Ph or 2-(Me2NCH2)–C6H4. With the two P-atoms bridged by a ferrocene unit, the dinuclear gold(I) complex with the (2-Me2NCH2)C6H4 substituent shows the expected intramolecular Au–Au bonding (66).149
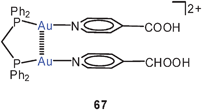
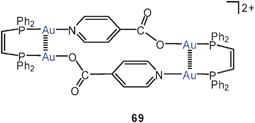
The dicationic, dinuclear dppm complex with pyridine-4-carboxylic acid (isonic acid) groups also shows the folded structure with an Au–Au lock of 3.143 Å (67), while the corresponding dppe complex remains an unlocked chain. However, the cis-dppen ligand induces again a short Au–Au contact of 3.138 Å (68). Deprotonation of 68 affords the cyclic tetranuclear complex 69 with two (“fully supported”) transannular aurophilic contacts of 3.097 Å.150
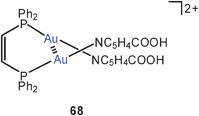
Some complementary data on intra- and intermolecular Au–Au contacts have recently also been demonstrated for the complexes of cis- and trans-bis(diphenylphosphino)ethene [Ph2P–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–PPh2] (AuI)2.151 These compounds undergo photo-induced cis/transisomerisation, and the quantum yield of this process has been shown to depend on the aurophilic interactions in the cis complex. The Au–Au interactions appear to be stronger for X = I than for X = Cl (68, X for NC6H4–4–COOH, no cationic charge), thus confirming earlier predictions that soft ligands lead to stronger interactions.28–32
CH–PPh2] (AuI)2.151 These compounds undergo photo-induced cis/transisomerisation, and the quantum yield of this process has been shown to depend on the aurophilic interactions in the cis complex. The Au–Au interactions appear to be stronger for X = I than for X = Cl (68, X for NC6H4–4–COOH, no cationic charge), thus confirming earlier predictions that soft ligands lead to stronger interactions.28–32
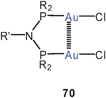
Small-bite ligands resembling dppm, like imino-diphosphites, show the expected Z-conformation bringing the Au atoms close together (3.118 Å, 70, R = 2,4-(MeO)2–C6H3O; R′ = Ph).152 A similar ligand with a 5-imino-isophthalate diester bridge forms a dinuclear complex with AuCl, in which the gold atoms are held at a bonding distance of 3.059 Å (70, R = Ph; R′ = 3,5-(MeOOC)2C6H3). With the [Au(tht)2]+ ClO−4 reagent, the cyclic complex with a fully supported transannular Au–Au contact (2.798 Å) has been obtained (71).153
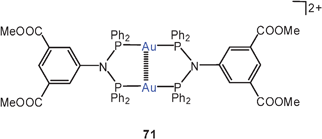
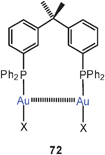
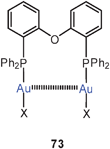
Extensive studies were carried out on dinuclear gold(I) complexes of the Xantphos type (72). A close Au–Au contact is present e.g. in the semi-supported complex with X = NO3 even though the metal atoms are separated by a chain of nine atoms.154–156 The molecule adopts a folded and twisted conformation which brings the two Au atoms as close as 2.95 Å. For 72 with X = Cl, both single crystal X-ray diffraction and EXAFS studies have given an Au–Au distance of 2.99 Å. A more flexible ligand based on diphenylether (DPEphos) (73, X = 3-Me–C6H4–S) gives similar complexes in which the Au–Au contact is 3.01 Å (as also confirmed by EXAFS studies).155–157 For the corresponding trifluoroacetate (X = F3CCO2) two polymorphs (pure and as a dichloromethane solvate) were obtained with very similar, folded conformations allowing for short Au–Au contacts of 2.944 and 2.897 Å and torsion angles O–Au–Au–O of 74.2° and 67.6°.158
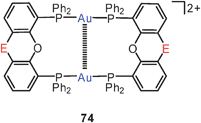
A similar folding was observed for the cyclic, fully supported complex 74 (E = CMe2) where the distance is reduced even further to 2.86 Å in an 8- or pretzel-type conformation.154 The NMR spectra of salts with dications 74 are temperature-dependent. From the coalescence of the 1H and 31P signals an enthalpy of interconversion of conformers was calculated at ca. 11.6 kcal mol−1 which probably can be ascribed to the breaking of the aurophilic contact. The difference radial distribution function calculated from solution X-ray diffraction (in nitromethane) has shown that the Au–Au distance in solution is found at 2.91 Å, only slightly longer than in the crystal.154
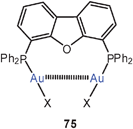
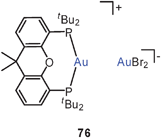
In a subsequent study the Xantphos complexes of AuBr and AuI (72, X = Br, I) were shown to have also folded conformations with aurophilic bonding, similar to that found in the complexes of the dibenzofuran-based ligand (DBFphos) 75. By introducing tBu substituents at the P-atoms of Xantphos a balance between ligand strain and aurophilic bonding has been demonstrated. With the very bulky substituents, ionic isomers 76 are formed.159
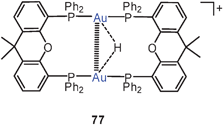
Xantphos, DBFphos and DPEphos complexes of AuCl in various stoichiometries have been employed successfully as catalysts for the dehydrogenative silylation of alcohols. Gold hydrides have been postulated as intermediates which may represent protonated Au–Au bonds, but reduction of Au(I) to Au(0) by the silanes is also involved (77).160 Similar results were obtained with bis(phosphole) analogues of Xantphos, and the proposed mechanism also includes tetra-coordinated, triangular [Au2H]+ units.161,162 This triatomic cation has recently been the subject of detailed quantum chemical calculations. It can be viewed as a protonated Au2 molecule or a gold hydride Au–H in combination with an Au+ electrophile.163a Though the Au–H–Au bridge with its intimate overlap of hydrogen and gold orbitals can be taken as the dominating bonding unit, the contribution of the underlying Au–Au contact which has been calculated to be as short as 2.60 Å is certainly very significant. There is also extensive information on the silver analogue [Ag2H]+.163b
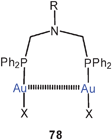
A large family of 1,5-ditertiary phosphines of the type 78 have been probed as ligands in dinuclear gold(I) complexes. Depending on the substituent pattern of the phenyl ring R at the central nitrogen atoms, the conformation of the complexes is strongly modified with significant changes in the mode of aurophilic bonding. Hydrogen bonding between carboxylic and phenolic groups attached to the aryl group R in particular determines the intra- and intermolecular interactions, but aurophilic bonding is maintained in most cases.164
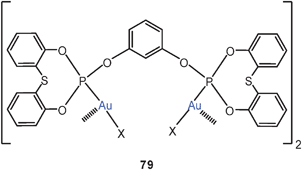
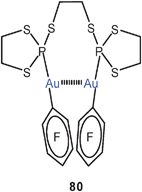
The dinuclear complex of a large-bite bis-phosphite with AuCl was found to be a cyclic dimer formed via two equivalent Au–Au contacts (79).165 In a dinuclear complex of C6F5Au with a bis(trithiophosphite) ligand (80), an Au–Au contact of only 3.467 Å is maintained in the solid state.166
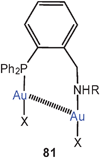
Semi-supported aurophilic bonding is also established in dinuclear complexes of P,N-difunctional donors (81). These monomers can be further aligned into chains through intermolecular Au–Au contacts.167
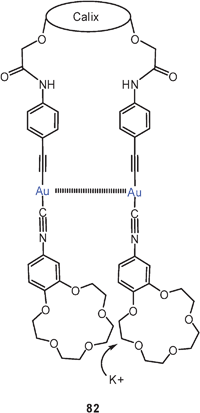
A calixarene-based dinuclear complex in which both gold atoms carry an isocyanide ligand terminated by a crown ether was found to have an open conformation in the absence of potassium cations, but to adopt a cyclic structure supported by a transannular Au–Au contact if such cations are provided (82). In this process, a low-energy emission band is switched on which has diagnostic value.168
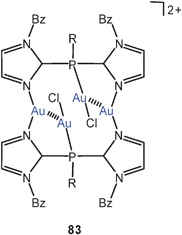
Two semi-supported Au–Au contacts in a dication 83 (Bz = benzyl; R = 1-benzylimidazol-2-yl) are formed in the reaction of [tris(1-benzylimidazole-2-yl)phosphine]gold(I) chloride with Na[AuCl4]. This dication can be associated with one AuCl−4 and one AuCl−2 anion or with two AuCl−2 anions.169
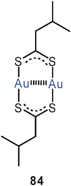
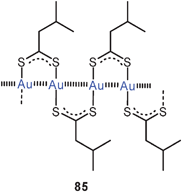
A most unusual mix of open-chain and cyclic gold(I) thiolates has been discovered as the product of the reaction of [Au(CN)2]− with isopentanedithioic acid in methanol. In the crystals obtained sinuous polymeric chains serve as a clathrate matrix for cyclic dimers of the same stoichiometry [Me2CHCH2CS2Au]n with n = 2 or ∞ (84, 85). In the polymer all gold atoms are linked into chains through Au–Au contacts (2.91–3.06 Å), and each of these contacts is bridged by a dithiocarboxylate unit. In addition, the enclosed dimers also entertain Au–Au contacts with gold atoms in the chains (3.29–3.39 Å). Crystals grown from CS2/Et2O solutions of this material gave a polymorph containing only the cyclic dimers, which are aggregated through intermolecular Au–Au contacts. An NMR and UV/vis spectroscopic study has shown that in CS2/CDCl3 solutions there is a delicate equilibrium between various oligomers in which the intra- and intermolecular aurophilic bonding is optimized depending on the temperature and the solvation conditions, which lead to a “nuclearity control”.170
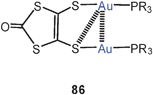
1,2-Dithiolene ligands have been shown to give dinuclear complexes of the types 86 and 87, in which the aurophilic interactions (3.198 and 3.129 Å) are very similar regardless of the nature of the five- and six-membered ring, respectively. In each case one of the two sulfur atoms is geminally coordinating two gold atoms, leaving only one gold atom in the plane of the heterocycle, while the second one is rotated away from this plane.171 The structures resemble those observed for dicyano-ethene-1,2-dithiolates and related compounds.172
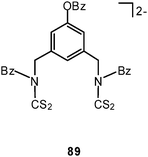
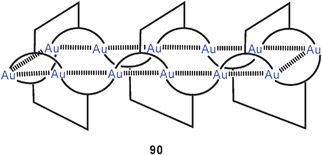
Dithiocarbamate ligands have long been known to form cyclic dinuclear gold(I) complexes with fully supported, transannular Au–Au contacts (88).16 In more recent work it has been demonstrated that with a bis(dithiocarbamate) ligand (89) a dodecanuclear macrocyclic structure 90 can be assembled with a fascinating stereochemistry (D2 symmetry). Each of the twelve Au–Au contacts in the ring (2.902–3.143 Å) is supported by a dithiocarbamate group in an undulating way above and below the macrocycle, and these groups are connected by the six corresponding 3-benzyloxy-1,5-phenylene linkers as transannular bridges. The neutral molecules are chiral and are strongly luminescent.173
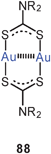
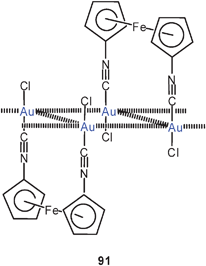
Ferrocene-1,1′-diisocyanide was found to give dinuclear complexes with AuCl and with AuNO3 (as a hydrate) in which the two substituted rings are rotated into a fully eclipsed structure (91) which allows for a close Au–Au contact (3.336 Å). In the crystals of the compound these molecules are further associated via unsupported Au–Au contacts into strings with slightly folded ribbons [Au2]n. This material has been probed as a model system for the self-assembly of ferrocene-containing monolayers on gold metal.174,175
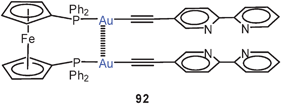
A similar organization (Au–Au 3.255 Å) has been discovered in a the dinuclear complex of 1,1′-bis(diphenylphosphino)ferrocene with a gold(I) acetylide bearing dipyridyl units for further complexation of metals (92). This eclipsed rotamer is converted into a trans-conformation (with loss of the intramolecular aurophilic interaction) upon complexation with an erbium salt, probably owing to steric reasons. The compounds are strongly luminescent with a contribution from various aurophilic contacts.176
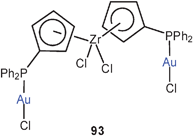
By contrast, the structure of the 1:2 complex of 1,1′-bis(diphenylphosphino)zirconocene dichloride with AuCl (93) shows in the crystal a rotamer which has the two gold atoms 7.211 Å apart ruling out any aurophilic interactions. Obviously other intra- and intermolecular forces determining the molecular packing in the crystal are more powerful and overrule the Au–Au attraction.177
2.3. Fully supported aurophilic bonding between gold atoms separated by polyatomic bridges
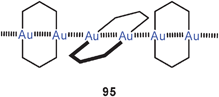
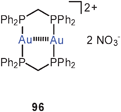
Fully supported aurophilic bonding (Scheme 1) appears in cyclic dinuclear gold(I) complexes with a large variety of ligands, from neutral bis-phosphines to anionic dithiocarboxylates (84), dithiocarbamates (88), dithiophosphates, or phosphonium-bis(methylides) (94), mainly with P, S and C donor sites.14,15,23 Transannular Au–Au contacts are most obvious in these eight-membered rings with the linearly two-coordinate gold atoms in 1,5-positions. This effect was recognized in some of the early structural work in gold chemistry1,6–9 and has intrigued theoretical chemists already at a very early stage.178–180 These studies have also included the intermolecular association of these metallacycles through semi-supported or unsupported Au–Au contacts (95). The experimental and theoretical studies carried out in the following more than three decades have provided convincing evidence that the aurophilic contacts in this type of fully supported systems are not only established out of the directional forces of the ring skeleton, but are due to a true attraction between the metals which leads to the short transannular distances. This becomes also obvious from the conformation of larger ring systems, where a folding and tilting is observed which gets the gold atoms close enough for bonding. A recent simple dicationic example of the typical eight-membered ring type is the complex [(dppm)2Au2]2+ 2 NO−3 (96) with a transannular distance of 3.025 Å.181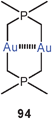
The photophysical properties associated with transannular Au–Au contacts have been considered in a review comparing the data with those of related Ag and Cu complexes. Experimental data are available for the dications with the stoichiometry [L2M2]2+ and [L3M2]2+ with L = Cy2PCH2PCy2 and Me2PCH2PMe2, and with two- and three-coordinate metal atoms, respectively. In theoretical calculations very different structures have been found for the ground and excited states, and the Raman lines for ν(Au–Au) could be correlated with the Au–Au spacing in the molecules.182
Theoretical studies (MP2)183 were also dedicated to aurophilic bonding and the related excited-state properties of the known gold(I) trithiocarbonate anions (Au–Au 2.800 Å) (97).184 A much shorter Au–Au distance was calculated for the triplet excited state responsible for the emissive properties.

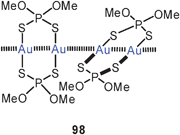
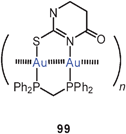
The mode of aggregation of gold(I) O,O′-dialkyldithiophosphates (98) has recently been shown to have a marked influence on the luminescence behaviour of the compounds. While the complexes are not emissive in fluid solutions, luminescence is seen upon cooling and becomes brilliant in the frozen state, similar to the behaviour of the crystalline material, for which an association of the type 95 is known from the crystal structure determination.185 Similar structural patterns were observed in gold(I) thiouracilates 99, where the reduced symmetry of the ligands induces a helical arrangement which is associated with luminescence tribochromism induced by the intermolecular aurophilic bonding.186
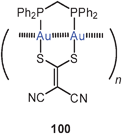
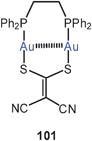
A similar helical arrangement of the gold atoms had previously been found for the cations in the crystals of the mixed-ligand complex (dppm)Au2(i-mnt) (100) [i-mnt = S2CC(CN)2], which is also emissive in the solid state. The intra- and intermolecular Au–Au contacts are 2.90 and 3.12 Å (both average). In the corresponding dppe complex (101), in which the eight-membered ring is enlarged to a nine-membered ring, a twisting of the metallacycle brings the gold atoms into close intramolecular contacts (2.850 Å).187 In this study, the association equilibria of cationic dithiocarbamates like [(dppm)Au2(dtc)]+ Y− were also studied as a function of temperature giving an estimate of the energies associated with the aggregations via aurophilic bonding. The data (up to 15 kcal mol−1) agree well with previous estimates.12,188
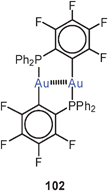
No intermolecular interactions are observed if steric bulk of the ligands prevents any close approach. The complex obtained with the 2-diphenylphosphino-perfluorophenyl ligand with C,P-donor sites in 1,3-positions is strictly monomeric with a short Au–Au contact of 2.819 Å (average for two molecules, 102).189
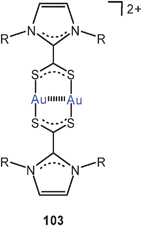
Eight-membered diauracycles are also obtained using zwitterionic imidazolium-2-dithiocarboxylate ligands, either homoleptic (103) or in combination with dppm (104). The compounds (bearing various aryl substituents at the imidazole N-atoms, and associated with PF−6 anions) are models for surface units of gold nanoparticles. If in the preparation of such nanoparticles imidazolium-dithiocarboxylates are added to the reduction mixtures, an improved yield of functionalized particles with a narrow size distribution can be achieved.190
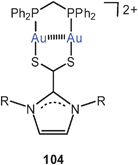
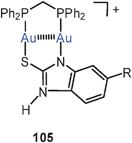
The combination of dppm with benzimidazolethiolate ligands also leads to robust neutral and cationic eight-membered rings with short transannular Au–Au contacts (ca. 2.90 Å). With a substituent R at the phenylene ring, isomers have been observed which differ in their optical properties (absorption and emission). While the cationic complexes 105 show no association in the solid state, unsupported intermolecular Au–Au contacts are established between the neutral molecules to give dimers 106. Luminescence tribochromism is observed for the trifluoroacetate salts with cations 105 with certain substituents at the benzene ring, but the origin of this phenomenon is presently unclear.191
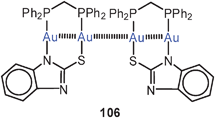
Even in macrocyclic compounds significant Au–Au contacts can be maintained. This is obvious in the conformation of a dinuclear complex prepared from dppm and 3,3′′-diethynyl-2,2′:5′,2′′-terthiophene (107), where an Au–Au distance of 3.197 Å is reached. The terthiophene ligand was introduced to provide conditions for a photochemical and/or electropolymerization which can lead to materials with photophysical and electrochemical properties co-determined by the presence of gold atoms and their clusters.192
Two transannular Au–Au contacts (3.148 Å) are established in tetranuclear macrocycles built from N,N′-bis(4-pyridyl)-oxalamide and dppm ligands (108, with perchlorate anions).193
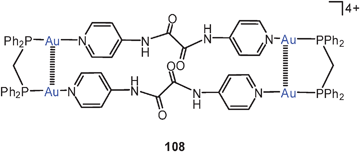
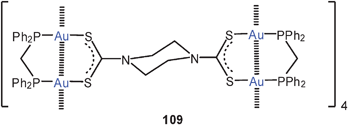
A fascination macrocyclic aggregate of eight-membered ring complexes with dppm and dithiocarbamate ligands (109) has been structurally characterized. The intramolecular Au–Au distance of 2.90 Å is complemented by intermolecular contacts of 3.12 Å length. The resulting array comprises a ring of no less than 16 gold atoms which are alternatingly fully supported or unsupported by the ligands. The aggregate is chiral owing to transannular linkages via the piperazine spacer.194
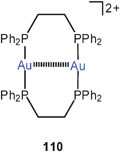
Homoleptic dicationic complexes with dppe (110) are known with a variety of counterions. In all examples the 10-membered rings are twisted to reach short Au–Au contacts, as confirmed recently for the triflate salt. It is thus obvious that the conformation of the ring cations which leads to the aurophilic contact is not determined by the packing with the anions.195
4,6-Bis(diphenylphosphino)phenoxazine (nixantphos) has been found to form a dicationic dinuclear gold(I) complex with a sixteen-membered ring (74, E = NH). In crystals of the nitrate salt and its acetonitrile solvate (1:4) the cations are fully twisted into a chiral system with extremely short transannular Au–Au contacts (2.862 and 2.856 Å, respectively). The solvent-free crystals contain only one or the other enantiomer, while those of the solvate contain both enantiomers. The molecules of the former are aligned into homochiral helices through hydrogen bonding, while in the latter a mixed hydrogen bonding occurs. The twist of the individual molecules (either left- or right-handed) is obviously also retained in solution, as proven by a low-temperature 31P NMR experiment (in CD2Cl2) which showed non-equivalent P-atoms (2:2) for both NMR-indistinguishable enantiomers: the room temperature singlet resonance is split into one AA′BB′ spin system at −80 °C.196 A complete NMR analysis of the solution (in CD2Cl2) and solid state was carried out for the nitrate salt of the dication 74 (E = CMe2). The set of parameters obtained and their temperature-dependence suggest a significant transannular aurophilic contact in a twisted conformation.197
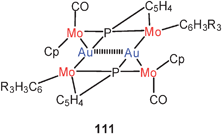
Fully supported aurophilic bonding has also been attributed to the Mo4Au2 core unit in a phosphinidene complex with the formula 111. All six metals are arranged in a centrosymmetrical, almost planar array with an Au–Au distance of 3.022 Å.198
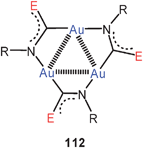
Aurophilic interactions have been considered in theoretical studies of several trinuclear gold(I) complexes with triangular symmetry. These include the classical gold(I) carbeniates 112 (E = NH, OR) with their three intramolecular Au–Au contacts.37,38,199 A recent example again has typical Au–Au distances of 3.308 Å.200
The plethora of experimental results has been reproduced by several theoretical calculations. The metallacycle [Au3(MeN = COMe)3] has been shown to have significant aurophilic bonding and an antiaromatic character.201 Other trinuclear monomers of this and related types were also the subject of MP2 and DFT studies, which provided molecular parameters comparable to the experimental data.202
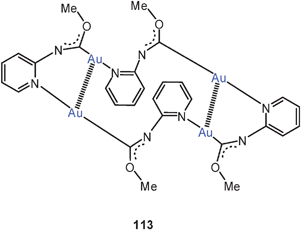
The triangular structures with the nine-membered ring may be supported in their periphery by hydrogen bonding, as found in compounds [(2-PyN = CNHMe)Au]3 (112, E = NHMe, R = 2-pyridyl).200 In the oxygen analogue [(2-PyN = COMe)Au]4 the four stoichiometric units form a 20-membered ring with two transannular Au–Au contacts (3.085 Å) (113).
It should be noted that compounds of type 114 show a pronounced tendency towards aggregation into columnar structures (95) via unsupported aurophilic bonding (ref. 200 and below).

Recent work with ligands featuring nitrogen donors in 1,3-positions has led to numerous new compounds, where the eight-membered ring structure (114) is given up in favour of an organization with a 16-membered ring (115). These rings are folded in such a way that a square, rectangle or rhombus of gold atoms is formed which allows for four intimate Au–Au contacts. The supporting 1,3-difunctional ligand can be e.g. an amidinate or a guanidinate anion (116, 117), and the Au–Au distances are all in the shorter range, often well below 3.00 Å.203–205 It should be noted that arrangements as in formula 115 have been known also from very early work on classical examples with other anionic 1,3-difunctional ligands.16

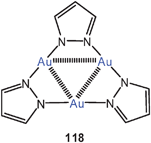
Trimeric gold(I) pyrazolates in which only the N-atoms are the donor sites (118) have the usual set of internal Au–Au interactions. There is also a fascinating aggregation with a complexity of intermolecular aurophilic contacts.206
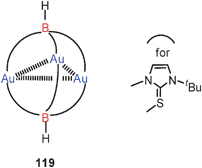
A triangle of gold atoms with three medium to very long Au–Au contacts (3.393, 3.664 and 3.868 Å) is formed in a cationic complex with the tridentate tris(mercaptoimidazolyl)borate ligand (119). The gold atoms are linearly two-coordinate by sulfur atoms, i.e. the ligands orientate their S-atoms to the gold atoms and their N-atoms to the boron atoms.207 Much tighter aurophilic bonding had previously been found for P–Au–P-coordinated trinuclear units with the tripodal tris(diphenylphosphinito)fluoroborate ligand.208
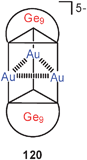
A triangle of gold atoms is found sandwiched between polyhedral Ge9 clusters in a fascinating complex 120 where the gold atoms are exclusively germanium-bound. The three Au–Au distances are in the range of 2.900–3.095 Å.209
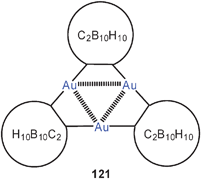
In work on gold-substituted carboranes, the trinuclear complex 121 has also been the subject of theoretical calculations.210 There are as yet no experimental data on this prototype, and the results therefore have been correlated with the data of an analogous mercury(II) compound.211
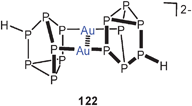
In a very recent study, HP−7 cages were employed as bridging ligands for gold(I) centers. Dinuclear dications 122 have been structurally characterized in a [(2,2,2-crypt)K]+ salt.212 The Au–Au contact of 3.077 Å is slightly longer than the Ag–Ag distance of 2.947 Å in the analogous silver compound. Because the crystals of the two compounds are isomorphous, this result is very important because it again provides evidence that at least with specific ligands argentophilic bonding may indeed be stronger than aurophilic bonding, as recently also deduced from theoretical calculations.35,36 This is particularly relevant since in several series of isomorphous mononuclear complexes the radius of Ag+ appears to be clearly larger than the radius of Au+ as also predicted on the basis of relativistic effects.213,214
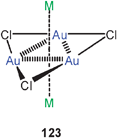
Theoretical studies (MP2) have also been focused on trinuclear units [Au3Cl3] (123) in combination with the alkali metal atoms Li, Na, K, Rb, and Cs. These systems can be used as models for the situation found in the sandwich structure with two tropylium cations. Strong aurophilic bonding has been proposed for both systems. Note that in these models systems there must be a mixed oxidation state (<+1) for the metal atoms.215,216
3. Unsupported intermolecular aurophilic bonding between gold(I) compounds
Unsupported Au–Au contacts are not only observed between simple mononuclear gold(I) complexes, but also between di- or polynuclear complexes with bridging ligands between their gold atoms, which may or may not have intramolecular aurophilic interactions. There is ample experimental evidence that aurophilic interactions of a given gold(I) centre are neither particularly directional nor limited in the number of contacts if the mutual approach of the metal atoms is not sterically hindered. Therefore, the interactions do not only lead to dimers, but also to larger aggregates and polymers (Schemes 1 and 2). It should be noted that aurophilicity has these characteristics in common with conventional van der Waals bonding, from which it is mainly distinguished by its exceptional strength.3.1. Theoretical studies
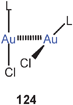
Theoretical studies of aurophilic bonding have concentrated mainly on the dimers of simple mononuclear complexes of the type L–Au–X with L and X being small neutral and anionic ligands, respectively. Over the last two decades, the results of these calculations have been summarized at various stages, reflecting the refinement of the theoretical approach to this new type of chemical bonding,29–31,217 and taking into account the latest experimental data.14,15,23,24,58,97,218–221 Several analyses of published geometrical data have also indicated certain structural preferences regarding the steric and electronic influence of L and X on the mode of interaction.26,58The most recent review—not meant to be comprehensive—was published by Muñiz, Wang and Pyykkö, and for a hypothetical dimer [L–Au–Cl]2 with “crossed” monomers (dihedral angle L–Au–Au–L = 90°, 124) the results according to the state of the art may be briefly summarized as follows:219
1. Following an early hybridization model for the bonding between closed-shell metal atoms,178,179 Hartree–Fock (HF) calculations have shown that no attraction is found at this level. By contrast, with Møller–Plasset (MP2) calculations an attraction V(R) has been verified which showed an R−6 dependence on the distance R between the metal atoms.222,223 This suggested a dispersion (London-type) interaction at this and higher levels of theory.142b,224,225 For meaningful results, relativistic effects had to be included. The dispersion mechanism is viewed as a virtual or an ionic charge transfer (A → A′, B → B′). Polarization-type mechanisms are not sufficient to yield attraction. With increasing levels of theory, the results gradually converge.75b,226,227 Calculated Au–Au equilibrium distances are in the range from 3.17 to ca. 3.40 Å, and thus only marginally larger than the experimental values.
The mechanism of the aurophilic interactions can be further broken down into (a) dispersion (London-type) interactions, (b) induction interactions, (c) electrostatic interactions, and (d) di- or multipole interactions. The results depend on the nature and symmetry of the ligands and the shifting of the dipole origins in models treated as rigid bodies.219 A detailed consideration is beyond the scope of this review.
2. The influence of the nature of both the ligands X and L on the dimerization of monomers L–Au–X has been followed in earlier and in more recent calculations. Polarizable, “soft” anions X lead to more efficient aurophilic bonding (e.g. Cl < Br < I),31,32 but for polyatomic anions (CN, SCN, SR, CCRetc.) other effects may have a marked influence, either electronic or steric. Regarding the neutral ligands L, a more comprehensive series was investigated, and the following sequence of increasing aurophilicity was found: NF3 < CO < MeCN < PF3 < SH2 < NH3 < PH3 < NHC. For carbenes (NHCs) and other “flat” ligands (triazine, pyridineetc.), the relative orientation of their planes in the dimer has a marked influence on the stability of the aggregate.
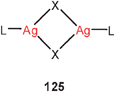
3. A comparison of aurophilicity with the bonding in the corresponding systems with copper (cupro-) and silver (argentophilicity) has shown an increase in the order of Cu < Ag < Au at the MP2 level,226,227 ascribed mainly to relativistic effects. However, at higher levels the interaction energy V(R) was found to weaken monotonously from Ag to the heavier congeners (Au and Rg), with some uncertainty for copper.35,36 It should be noted that there are as yet very few experimental data which could allow an estimation of the energy associated with unsupported argentophilic interactions of the type considered as models for the aurophilic interactions (124, Ag for Au). In contrast to the abundant L–Au–X monomers, no such monomers L–Ag–X are known except for cases with extremely bulky ligands which anyhow rule out any intimate approach of two metal atoms in the corresponding hypothetical dimers. With sterically undemanding groups L and X, the silver complexes form oligomers or polymers mainly through short Ag–X contacts (e.g. 125, 126).
4. For systems with the aggregations extended beyond dimers, with the gold atoms engaged in more than one Au–Au contact, the energy per contact may be a little less than additive, but there is no definite answer to this open question. Among the large number of examples with one-dimensional chains of gold atoms built from L–Au–X molecules (see below) there are cases with Au–Au contacts alternating in distance (–short–long–short–long–), and also with equidistant gold atoms, suggesting that the differences in energy may be very small indeed. With supporting ligands, trimers and tetramers are also common (above), but unsupported cases are rare.
In other more recent theoretical studies, the effect of aurophilic bonding on the physical properties has been investigated. At the MP2 level it has been found that the Nonlinear Optical Properties (NLO) of molecules H3P–M–X (M = Cu, Ag, Au; X = F, Cl;) are altered significantly upon the aggregation to dimers with the crossed (staggered) conformation (124, L = PH3, M for Au, X for Cl) considered also in other studies (above). Polarizability (α) and second-order hyperpolarizability tensors (γ) of the components are larger in the dimers than in the monomers. The charge transfer along the M–M contact appears to play the key role in increasing the parameters α and γ, while the first-order hyperpolarizability tensor β is mainly affected by the nature of the ligands and only slightly influenced by the M–M contact.228
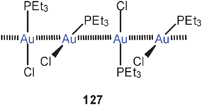
The association of (Et3P)AuCl into dimers and its consequences for the luminescence properties has been analyzed by TD-DFT calculations.229 An Au–Au distance of 3.6 Å was calculated for the dimer, in agreement with the alternating distances found in the crystals of the compound (3.568 and 3.615 Å),230 where it is associated into a chain polymer (127). The energy associated with the dimerisation was calculated to be −9.5 kcal mol−1. The low-temperature luminescence of the solid appears to be strongly influenced by the Au–Au interactions. The emission disappears at higher temperatures and in solution.
The photophysical properties of the compounds with ditertiary phosphine ligands have been investigated by XEOS and XAFS methods. For powder samples it has been demonstrated by EXAFS that aurophilic bonding (Au–Au ca. 3.10 Å) is present in almost all cases. The optical emission observed with these complexes has been assigned to various transitions within the molecules and their aggregates, with significant contributions from the Au–Au interactions.231
3.2. (Carbonyl)gold(I) halides
Molecules (CO)AuCl have long been known to be associated in the crystal into infinite puckered layers through aurophilic contacts of 3.38 Å.232–234 Recent photophysical and theoretical studies (MP2) have demonstrated that oligomerisation of (CO)AuCl molecules can also be demonstrated in acetonitrile solutions. The absorption spectra show concentration dependent deviations from Beer's law, which suggests the formation of at least dimeric species 128. The temperature dependence of the luminescence behaviour leads to similar conclusions. Calculated electronic states indicate a good agreement with the experimental findings. The distances computed for the ground states (ca. 3.2 Å) are contracted in the phosphorescent excited states (to ca. 2.6 Å) extended beyond two adjacent molecules. From the calculated values, the aurophilic binding energy has been estimated to be ca. 10 kcal mol−1.235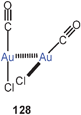
3.3. (Phosphine)gold(I) halides
The significance of unsupported aurophilic bonding in crystals of compounds (R3E)AuX with R = alkyl, aryl; E = P, As and X = Cl, Br, I has been probed in extensive preparative and structural studies with a large variety of tertiary phosphines and several tertiary arsines, including also data from the literature. For the same purpose, the 1:1 adducts of the type (R3E)X2 have also been included, where the diatomic molecules Au–X are replaced by the molecular halogens X–X. In addition, several complexes (R3E)AuR′ with R′ = aryl were also investigated.236–238 Only very few cases were found where aurophilic bonding was actually observed. From this work, (p-tolyl)3PAuCl is now known to appear in two polymorphic forms, one with and one without aurophilic contacts, depending on the mode of crystallization, indicating a delicate balance of intermolecular forces.239,240 In all other cases this interaction was overruled by steric effects and the prevalence of other secondary bonding, mainly several kinds of hydrogen bonding. 2-Acylphenyl(diphenylphosphine)gold chloride is also not associated in the solid state. With the acetyl substituent larger than the methyl group in the corresponding diphenyl(2-methylphenyl)phosphine complex, the aggregation through Au–Au bonding is energetically disfavoured.241
These results have shown that generally the shapes and volumes of the molecules under consideration determine the mode of aggregation and the packing in crystals through the various specific hydrogen bonding and van der Waals interactions. With bulky ligands, or with a preference for concerted hydrogen bonding between the substituents (phenyl or tolyl “embrace” etc.),242 no aurophilic contacts can be established.
The AuCl complexes of trithiophosphites, (RS)3PAuCl, are also aggregated in the solid state through aurophilic bonding. For R = Me, chains of molecules are formed with Au–Au contacts of 3.294 Å (129). The compound is isomorphous with the corresponding phosphite complex (MeO)3PAuCl (Au–Au 3.163 Å). It is remarkable that the aggregation is not instead established via common Au–S contacts as suggested by the thiophilic properties of gold(I).166
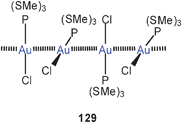
Surprisingly, the tetranuclear complexes of gold(I) halides with a tripodal tetraphosphine have been found to form exclusively unsupported intermolecular aurophilic interactions in the solid state (130; X = Br, Au–Au 3.270 Å; X = I, Au–Au 3.184 Å; phenyl groups and dangling (CH2)2PPh2AuX groups omitted). The gold atoms within one molecule are too far apart and an entropy-costly folding would be required to reach a conformation for any intramolecular alternatives.243 The structure of the corresponding chloride (X = Cl) is very similar.244 All compounds of this type show strong luminescence.
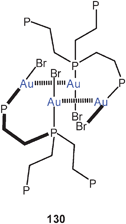
3.4. (Phosphine)gold(I) thiolates, amides and imides
Aggregation of complexes (R3P)AuSR′ was observed in much of the earlier work,16,23,58 but recent studies have revealed specific interactions in a number of functional variations. The interplay of hydrogen bonding and aurophilic interactions became obvious in derivatives of 4-sulfanyl-benzoic acid which can form cyclic tetramers with two Au–Au contacts (131). With other phosphines than Ph3P, chains are also formed, and the intermolecular interactions occur between P–Au–S units with an extended Au–Au contact owing to an intriguing slippage of these units (132 shows only the connecting unit for the Et3P complex).245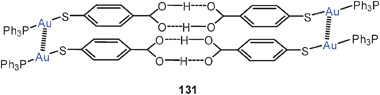
[Diphenyl(2-pyridyl)phosphine]gold(I) pyridine-2-thiolate is not associated, probably for steric reasons, but the dication generated upon protonation shows a short Au–Au contact of 2.978 Å clearly supported by two N–H–N hydrogen bonds (133; the twist of the 8-membered ring is not shown). The PF−6 salt shows strong photoemission in dichloromethane solution.246
Carbonimidothioates have been found to be associated into dimers only with a substituent pattern which minimizes steric hindrances, as in 134 (R = p-tolyl) with Au–Au 3.087 Å.247
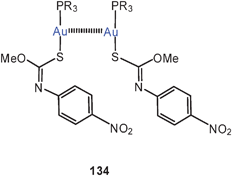
A number of (phosphine)gold complexes with purinate and purinylaminate ligands have been structurally characterized. Unsupported Au–Au contacts of 3.231 Å were found in the dimer of (tributylphosphine)gold(I) 9H-purin-9-ate (135), but also in related dinuclear complexes with dppe and dppp.248Gold(I) O-methyl-N-(4-nitrophenyl)thiocarbamide forms several complexes with tertiary phosphines which are associated into dimers or “slipped” dimers (136), in which the Au–Au and Au–S contacts appear to be very flexible, a rather common phenomenon in gold thiolates.147
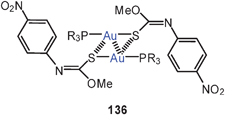
3.5. (Phosphine)gold alkynyls and aryls
Many compounds of the type (R3P)AuCCR′ have been documented in the literature,37,38,48 and most of the structurally characterized examples feature the expected intermolecular Au–Au contacts. Even molecules with an extended framework of ligands are found to be assembled in the solid state in such a way that this bonding can contribute to the overall stability. The dinuclear monomers 137 form “staircase-like” chains with short contacts Au–Au 2.990 Å, supported by face-to-face stacking of (hetero)aromatic rings. Both the solid and its solutions in dichloromethane are strongly photoluminescent. A large number of complexes with the same set of ligands was prepared with mixed coinage metals (mainly Au–Ag). In all cases an extended network of metallophilic bonding supports their complex molecular frameworks.249a–d
(Tetracenyl)ethynyl complexes (138) have recently been investigated in order to further probe the nature and origin of the photoemissive properties of this class of compounds. While the Ph3P complex is not associated in the solid state, the Me3P complex is aggregated into stacks with crystallographic 31 and 32 screw axes. The gold atoms are equally spaced (3.26 Å) and form undistorted right- and left-handed helices (Au–Au–Au 134.2°). Surprisingly, the crystals and solutions of the monomeric Ph3P complex are emissive, while the crystals of the polymeric Me3P complex are silent. However, solutions of the latter are strongly luminescent, which suggests that in this case the helical assembly quenches the emission. The effect has tentatively been ascribed to the relative orientation of the tetracene groups, because the fluorescence occurs from the excited state which is polarized along the long axis of the arene leading to exciton delocalization.250
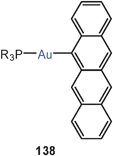
Two complexes with an octanuclear gold core assembled through aurophilic contacts have been obtained from (tBuCCAu)n and [(Ph2P-X-PPh2)2Au2]2+ 2 PF−6 salts (X = –CC–, p-C6H4–). The cations have the stoichiometry [Au8(C2tBu)6(Ph2PXPPh2)2]2+ with an extensive network of π-(CC)–Au and Au–Au interactions (not shown here in the diagram owing to the complexity of the system). The crystals of both compounds are strongly photoemissive, and a similar luminescence is also observed for the solution of the former, while that of the latter is different owing to a fluxionality which is also obvious from NMR studies. The crystals of the compound with the p-phenylene bridge show thermochromism, and those of the alkynediyl-bridged compound change their emission after grinding. The effects are all ascribed to the rupture and formation of aurophilic contacts under thermal or mechanical stress induced by different packing patterns of the dications.251
The aggregation of a series of (triphenylphosphine)gold(I) aryls with different substitution patterns has also been investigated. The compounds with 4-methoxy- (p-anisyl-), 3-fluoro- and 3,5-difluorophenyl groups have been shown to be dimers in the solid state with Au–Au distances of 3.135, 3.120 and 3.108 Å, respectively. The distances thus show no significant dependence on the electronic effect of the substituents. In all cases, a crossed conformation (139) minimizes the steric repulsion of the ligands and probably optimizes the interaction energy.252
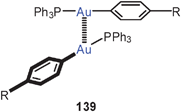
3.6. Gold(I) complexes L–Au–X with sulfur-functional ligands L.
Complexes of the type (R2S)AuX have long been known and are used extensively in gold chemistry as convenient reagents.1,6–10,16,253 With simple thioethers, the molecules are mostly assembled into 1-dimensional aggregates centred by a chain of gold atoms. Similar arrangements have recently been found with the (trithiane)AuCl complex which can be crystallized in two polymorphs with a remarkably different array of the monomers. One of the polymorphs features an only slightly folded chain with equidistant gold atoms (140, Au–Au 3.558 Å), while the chain of the second polymorph contains centrosymmetrical tetramers (141) with much shorter distances [Au–Au 3.136 (central) and 3.240 Å (peripheral)] and a zigzag conformation. These tetramers are connected into chains through significantly longer Au–Au contacts of 3.514 Å. In both polymorphs the S–Au–Cl axes of neighbouring monomers have the staggered, or “crossed swords” orientation typical for this class of complexes, but with different dihedral angles Cl–Au–Au–Cl. Both materials show almost the same intense red luminescence in the solid state at room temperature, which undergoes a shift to longer wavelengths at low temperature.254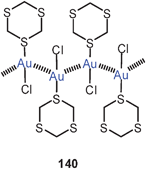
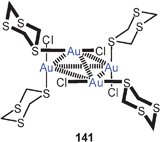
The structure of dithioether complexes follows the same pattern as recently demonstrated for ligands like bis(methylthio)methane and 1,2-bis(phenylthio)ethane and their homologues.255 In crystals of the 1:1 complexes like (MeSCH2SMe)AuCl the molecules are arranged in chains with a zigzag string of gold atoms (142, Au–Au 3.166 Å). By contrast, in 1:2 complexes like (PhSCH2CH2SPh)(AuCl)2, sheets are generated with the gold atoms arranged in parallel strings (143, Au–Au 3.120 and 3.150 Å). The network of the sheets contains 14-membered rings Au6S4C4. This structure was determined at two different temperatures, and a significant shrinkage of the unit cell was observed at low temperature. This shrinkage concerned mainly the direction involving the aurophilic interactions which clearly are most sensitive, probably owing to a flat energy profile of the lengthening and shortening of the Au–Au contacts. For (PhSCH2SPh)(AuCl)2 and (PhS(CH2)3SPh)(AuCl)2 (two polymorphs), chain structures established via Au–Au contacts (Au–Au 3.035, 3.210, 3.192 Å, respectively) were observed. The compounds show strong solid state luminescence at low temperatures.255,256
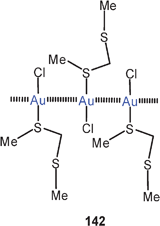
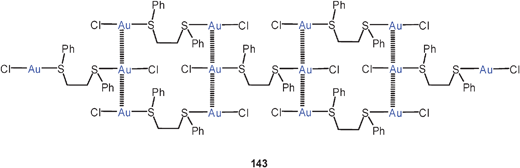
(Tetrahydrothiophene)(pentafluorophenyl)gold(I), (tht)AuC6F5, was found to have a chain structure in the solid state with the gold atoms in an almost linear array, with the Au–Au contacts alternating between 3.128, 3.191 and 3.306 Å, and with a variety of torsional angles S–Au–Au–S (144). Two neighbouring pentafluorophenyl groups are facing each other.257 A chain structure was also found for (tetrahydrothiophene)gold(I) tetrafluorophthalimide. The molecules are stacked establishing Au–Au contacts of 3.322 Å with only minor deviations of the resulting gold chain from linearity (Au–Au–Au 172.6°) and with a minimum of overlap between the phthalimido ligands. The aggregation is thus not governed by common π–π stacking, but appears to be determined by the aurophilic interactions (145).258
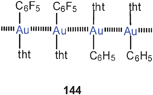
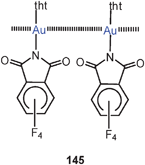
4,5-Bis[(2-hydroxyethyl)thio]-1,3-dithiole-2-thione gives a 1:1 complex with AuCl, which is a dimer in the solid state (146, R = CH2CH2OH).171
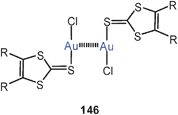
The 2:1 complexes of AuCl with 2-pyridine-thiol (147) show only very weak aurophilic interactions between the cations in the solid state (Au–Au 3.50 Å). In the crystals of these compounds and of their deprotonated forms a set of hydrogen bonds involving also the counterions appears to make the dominating contributions to the overall stability.259
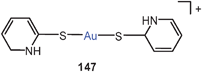
3.7. (Amine)- and (pyridine)gold(I) complexes
Complexes of the simple composition (NH3)AuX are not known, and their analogues with primary and secondary amines are represented only by rare examples.16 However, recent work has provided evidence that compounds (RNH2)AuCl with long-chain amines can readily be obtained and appear to aggregate in hexane solution through extended aurophilic bonding (148). This assembly induces the formation of gold nanoparticles and nanowires in the reduction or thermal decomposition of such solutions. With (oleylamine)AuCl the polymer has been isolated and characterized by analytical and spectroscopic methods, including low-angle X-ray diffraction.260,261
In a theoretical study (MP2)262 of the model dimers (H3NAuCl)2 and (Me2NHAuCl)2, efficient aurophilic interactions supported by hydrogen bonding have been diagnosed with Au–Au equilibrium distances near 3.20 Å (149, R = H, Me). A model of a known phosphonite complex (150) has also been included in this investigation, confirming the experimental data.263
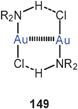
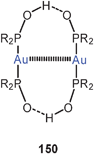
The most simple case of aurophilic bonding between cationic species with gold(I) atoms coordinated homoleptically by nitrogen ligands are the ammonia complexes in salts [Au(NH3)2]+ X−. For X = Br the cations have an Au–Au distance of 3.414 Å indicating weak interactions which complement a network of Br–H–N hydrogen bonds (151).264
Cationic, cyclic bis(imidazolyl)gold(I) complexes with different anions (152) have been shown to be aggregated via inter-cationic Au–Au contacts into strings with Au–Au distances of 3.289 and 3.359 Å for BF4 and CF3SO3 anions, respectively. There is no transannular aurophilic contact within the cations.265
Pyridine complexes (py)AuX were among the first families of compounds for which extended aurophilic bonding in chain-like assemblies of the molecules in crystals has been demonstrated.16 Compounds of this composition may appear as ionic isomers [(py)2Au]+ [AuX2]−, but this does not preclude aurophilic bonding between the components.266
In recent work it has been demonstrated that (3-bromopyridine)gold(I) chloride crystallizes in tetranuclear units with a central Au–Au contact between the cations of 3.311 and two cation–anion contacts of 3.268 Å (153). The array may be supported by π–π stacking of the rings and by C–H–Cl hydrogen bonding, but it is nevertheless remarkable that the intermetallic contact between two cations is only slightly longer than that within each cation–anion pair.267
The photophysical properties of the compounds with bipyridyl and α,ω-dipyridyl ligands have been investigated by XEOS and XAFS methods. For powder samples it has been shown that aurophilic bonding (Au–Au ca. 3.10 Å) is present in almost all cases. The optical emission observed with these complexes has been assigned to various transitions within the molecules and their aggregates, with significant contributions from the Au–Au interactions.231
Recent structural studies of a series of (pyridine)gold(I) alkynyls have provided only very few examples with aurophilic contacts in the solid state. The compound (p-Me2N–C6H4N)AuCC–Ph, which is strongly luminescent in the solid state, forms dimers with Au–Au 3.190 Å (154). Most other compounds of this type also appear as dimers, but show a slippage of the parallel orientated monomers which lengthens the Au–Au contact beyond the usual 3.5 Å considered as the upper limit for significant bonding, in favour of other stacking interactions.268
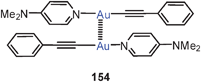
Polyvinylpyridine has been used as a matrix for the complexation of gold in a new approach to the preparation of metallopolymers. Through a reaction with (tht)AuC6F5 a strongly luminescent material was obtained (155). Its properties are assigned to intra- and interchain aurophilic interactions. An application of the polymer in PLEDs has been considered.269
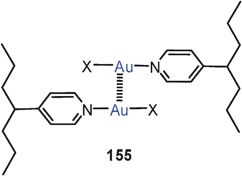
3.8. (Isocyanide)gold(I) complexes
Complexes of the type (RNC)AuX have been investigated extensively over more than three decades. A large variety of examples with different substituents R and ligands X are known, and many of these were shown to be associated in the solid state via aurophilic interactions.16–18,37,38 Apart from experimental studies, several theoretical investigations were carried out which provided a more detailed picture of the mechanisms of the bonding involved.270In most of the more recent work it has been tried to relate the remarkable photophysical properties of these compounds to the structure and bonding systems with selected substitution patterns (R and X). In this context the known model CyNCAuCl and the new, sterically more crowded example Me3CCH2CMe2NCAuCl (156) have been investigated. The structural patterns are very similar. In crystals of 156, the molecules also form dimers with a parallel head-to-tail arrangement and long Au–Au contacts of 3.418 Å, and yet the material is strongly luminescent with a large Stokes shift, probably owing to excimeric interactions between the dimers which are aligned into chains in the crystal. The long ground-state distances in model dimers have been shown by MP2 calculations to be shortened very significantly (to ca. 2.6 Å) in the phosphorescent excited state. By contrast, discrete dimers such as (tBuNCAuCl)2 feature different emissive behaviour with a much smaller Stokes shift.235
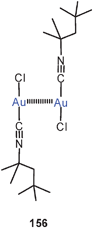
The solid-state structures of the three halide complexes p-Me–C6H4SO2CH2NCAuX (X = Cl, Br, I) show a slight, but progressive influence of the halogen atom, which gives rise to a significant lengthening of the short aurophilic contacts in crossed-sword dimers (157). The crystals exhibit blue-green emission, while the emission of known examples RNCAuX with the more common parallel head-to-tail configuration is shifted to orange-red. In DFT calculations of (MeNCAuCl)n models, some of the variations in the photophysical properties could be reproduced and rationalized.271,272
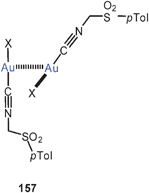
In related work it has been demonstrated that the aggregation of RCNAuCl molecules caused by photoexcitation leads to dimers and trimers with exceedingly high formation constants (in dichloromethane solutions). The photoproduct quantum yield is concentration-dependent and varies with the nature of the substituent R. With additives in the solutions, the photochemical formation of gold nanoparticles can be initiated and controlled.273
Different conformations were observed in the dimeric complexes with 2,6-dimethylphenylisocyanide upon substitution of chloride by thiophenolate: the former has the crossed-swords structure (Au–Au 3.54 Å), while the latter exhibits an antiparallel alignment (3.65 Å) (158, 159).274 On the other hand, with an isocyanide bearing a long and rigid 4′-ferrocenyl-4-diphenylyl-isocyanide, both the chloride and the thiophenolate have the structure of head-to-tail dimers with Au–Au distances of 3.376 and 3.333 Å, respectively (160, Fc = ferrocenyl). Clearly the packing of long rods is much more efficient in parallel orientation, such that other conformations are disfavoured.275
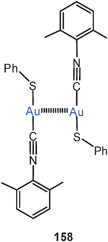
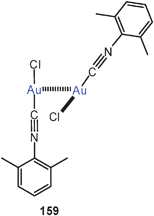
The four new examples274,275 confirm the general observation that gold(I) isocyanide complexes have only weak aurophilic bonding with Au–Au distances at the long end of the range considered for significant interactions, regardless of the conformation of the rotamers specified by their C–Au–Au–C dihedral angle. Therefore there are also many cases, where no aurophilic bonding is observed at all, such as in crystals of 1,4-(C6F5–Au–CN)2–C6H4 which has Au–Au distances of 5.19 Å. However, this material shows an interesting mechanochromical luminescence, which may be ascribed to slippages of the rods which allow for shorter Au–Au contacts responsible for the emission behaviour. Upon annealing with solvent, the original crystal structure is restored (161).276
A (phenylisocyanide)gold(I) probe has been attached to guanosine oligomers viaalkynyl functions in order to obtain sensor capabilities for potassium cations. In the presence or absence of these cations, a switchable emission is observed which is believed to be based on aurophilic interactions. A model tetramer with four gold atoms has been considered, which can sandwich a potassium cation, thereby generating four pairs of gold atoms with reversible aurophilic bonding. The formation of oligomers is based on extensive hydrogen bonding between the guanosine groups. Remarkable changes in the circular dichroism are observed upon complexation, and the absorption and emission properties are modified such that an on/off switch behaviour is obtained (162).277
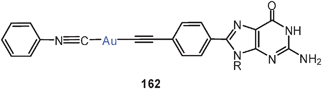
It should finally be remembered that 1,1′-diisocyanoferrocene forms a dinuclear complex with AuCl which is aggregated in the crystal into a string with an undulated double-string of gold atoms (91). This remarkable structure is a unique case of multiple aurophilic bonding (supported and unsupported). It should be noted, however, that even in this intimate arrangement the Au–Au distances are all larger than 3.30 Å (3.336, 3.354, 3.484 Å).174,175
3.9. (Carbene)- and (carbeniate)gold(I) complexes
Gold(I) complexes of carbenes [CY2] with Y = RO, RS, NHR, NR2etc. and the corresponding carbeniate anions [CYZ]− with Z = NRetc. have been known from the early 1970s.16–18,37,38 The examples include neutral molecules (Y2C)AuX with X = halide, thiolate etc., and (YZC)AuL with L = R3Petc., but also cations [(Y2C)2Au]+ and [(Y2C)AuL]+ with L = R3P, and finally anions with [(YZC)2Au]−. Many of these species have been found to be associated in the solid state to form either dimers or chain polymersvia aurophilic interactions. However, mainly owing to steric effects, the aggregation may also be steered by hydrogen bonding and other specific binding leaving Au–Au interactions ineffective.278While some work has been continued on complexes with open-chain carbenes (Fischer carbenesetc.), much of the recent investigations have focused mainly on complexes with N-heterocyclic carbenes (NHCs) which have appeared to be extremely valuable homogeneous catalysts for organic transformations.61,63
In the course of these studies, the solid-state structures of a variety of prototypes have been determined. The results have shown that aurophilic interactions play a major role in the aggregation of the molecules and ions in the crystals, and that these interactions may also be not negligible in solution.279,280 Thus, (NHC)AuCl complexes with the imidazol-2-ylidene group bearing methyl, i-butyl, cyclohexyl, or benzyl/N-tert-butyl-acetamido groups were all shown to be dimers with Au–Au distances between 3.05 and 3.20 Å (163). Therein, the monomers are either eclipsed or staggered (C–Au–Au–X dihedral angles between 0 and 180°). The new data on these complexes with imidazol- and benzimidazol-based carbenes confirm earlier results regarding not only the distances, but also the crossed-swords conformation in the dimers.280–285
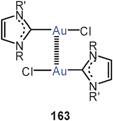
The bond energy of the latter example (Au–Au 3.2042 Å) has been calculated by DFT methods to be 8.6 kcal mol−1. Surprisingly, the Ag–Ag distance in the crystallographically isomorphous silver analog was found to be slightly shorter (at 3.1970 Å) and its bond energy much higher (at 12.8 kcal mol−1).279 These observations lend support to the conclusions drawn from calculations on simple [(H3P)MX]2 molecules, which have suggested stronger metallophilic bonding for M = Ag than for M = Au, even though relativistic effects might favour the bonding between the heavier atoms.25,35
In very recent work, dinuclear complexes of a bidentate carbene complex with an isomannide (dehydrohexitole) bridge have been described (164). The molecules are aggregated into chains via intermolecular Au–Au contacts of 3.295 Å in length. The crystals are strongly emissive.286
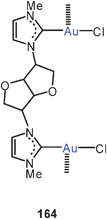
The combination of an NHC with a pyridine ligand leads to cationic complexes [(NHC)Au(py)]+ X− (165). In crystals of ten representatives of this class of compounds, the cations are all stacked in such a way that zigzag chains of gold atoms are assembled along a crystallographic axis, but the Au–Au distances depend strongly on the nature of the substituents of the pyr ligand (4-Me–, 4-Me2N–, 4-Ph, 4-CNetc.). With 4-Me2N–py, with the N,N′-dimethylated or -diethylated carbene and with X = PF−6, e.g., the Au–Au contacts are 3.473 and 3.436 Å, and therefore still to be considered as bonding, but with other substituent patterns a slippage of the molecules can lead to much larger distances, well beyond the commonly set limit of 3.5 Å. The luminescence properties of the solids have been investigated and the results rationalized by a comparison with theoretical results (DFT and time-dependent DFT). The LUMO of the transition state is assumed to be determined mainly by the aurophilic contacts.287
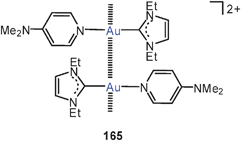
An N-unsubstituted and N′-CH(OEt)2 substituted imidazol-2-ylidene, a rare case of a “protic NHC”, forms a 2:1 complex with AuCl (166), which is associated in the crystal only via extensive hydrogen bonding. This result again illustrates the delicate interplay between various weak forces in the assembly of the components in crystals.288
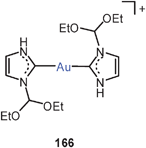
The reaction of an N-alkenyl-, N′-alkynyl-imidazolium salt with an organolithium reagent and Me3PAuCl resulted in a product containing a trinuclear cation in which all three equivalent gold atoms bear each an NHC and an alkynyl ligand, together with a bis(trimethylphosphine)gold(I) cation and one counterion. As a most remarkable case of polynuclear aurophilic bonding in a star-type Au3Au core, the [(Me3P)2Au]+ cation was found to be inserted like an axle into the hub of the tri-spoked wheel, establishing three Au–Au contacts of 3.25–3.30 Å (167). This array is the first example of a rotaxane supported by aurophilic bonding.289 In this context it should be noted that catenanes (168) supported by aurophilic bonding are well established from earlier work,290,291 but there were no more recent contributions to this area.
The mode of aggregation of complexes with non-cyclic carbenes has long been known to be also strongly influenced by the substitution pattern, by the counterions, and by solvate components.292 Recent work with pyridine-functionalized carbenes has afforded dimers and polymers featuring both intra- and interligand hydrogen bonding and aurophilic interactions with Au–Au distances of 3.255 and 3.311 Å (169, 170). Even subtle changes in the arrangement of the substituents or the inclusion of solvate molecules may cancel the aurophilic contact.293
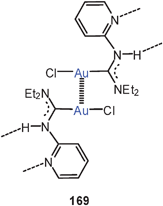
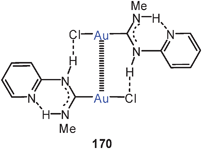
Surprisingly, the assembly of salts with cationic bis(carbene)gold(I) complexes in the solid state is far less sensitive to the nature of the substituents. Compounds of the type {[(RHN)2C]2Au}+ X− (171) are generally found to be stacked to form a linear or quasi-linear string of equidistant gold atoms. For R = Me and X = PF6, AsF6, SbF6 the stacking is modified by the nature of the counterion and by solvate molecules included in the crystals upon different conditions of crystal growth. The Au–Au contacts are generally rather long, in the range from 3.489 to 3.538 Å. As expected, these structural changes have a marked influence on the photophysical properties.294,295
Similar results have been found for the corresponding methoxy-methylamino-carbene complexes with counterions PF−6, ClO−4, CF3SO−3, CF3CO−2, and I− (172). With Au–Au distances in the range of 3.263–3.335 Å in linear or slightly kinked chains of gold atoms, the crystals are photoemissive with high quantum efficiencies, while solutions show no appreciable emission. The color of the emission can be tuned from blue to yellow by the choice of the counterion.296
Oligomeric gold(I) carbeniate complexes (173, R = alkyl, aryl;  , OR′ etc.) have been an active area of research since their discovery in 1972 and 1974,297,298 mainly owing to their fascinating structure-dependent photophysical properties. The results have been reviewed at various stages,299–302 and research has continued at a high level of activity.
, OR′ etc.) have been an active area of research since their discovery in 1972 and 1974,297,298 mainly owing to their fascinating structure-dependent photophysical properties. The results have been reviewed at various stages,299–302 and research has continued at a high level of activity.
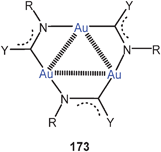
As already mentioned above, in a recent theoretical study (MP2 and DFT) the aurophilic attractions in and between molecules {[(MeO)(MeN)C]Au}3 have been reconsidered (174). With trigonal prismatic models (D3h symmetry), the experimental structural parameters could be reproduced and the photophysical properties (MMCT, MLCT) related to intermolecular Au–Au contacts in hexanuclear dimers-of-trimers and larger stacked aggregates.202Experimental data have also become available for [(2-PyNCNHMe)Au]3 which forms a dimer with a trigonal prism of gold atoms which has three vertical Au–Au distances of 3.264 Å. The compound is intensely luminescent in both the crystalline and solution (dichloromethane) state, and this property has been related to the aurophilic bonding.200
The trinuclear gold(I) triazolates 175 (for clarity, five-membered rings for 3,5-dialkyl-1,2,4-triazoles) have also been shown to form dimers-of-trimers through distinct aurophilic bonding. The conformation was found to be temperature-dependent with a change from C2 to approximately D3 symmetry upon warming from 95 K to room temperature. The low-temperature form has one particularly short Au–Au contact of 3.193 Å, while at room temperature all three Au–Au bonds are near 3.42 Å. These changes lead to gradual shifts in the emission spectra of the crystals. The aggregates are also present in dichloromethane solutions which show isoemissive and isosbestic points in the concentration-dependent spectra. Upon addition of acid, the aggregation of the dimers breaks down and the emission is quenched, but can be regenerated upon neutralization.303
Trinuclear gold(I) imidazolates form dimers-of-trimers with C2 symmetry (175, five-membered rings for N-benzyl-imidazoles), intermolecular Au–Au distances of 3.346 (2×) and 3.558 (1×) Å and a torsion angle of the two rings of only 17°. The crystals show green luminescence even at room temperature, with an enhancement of intensity and lifetime at low temperatures. This is changed in addition compounds with perfluorinated fluorophores like octafluoronaphthaline. In crystals of this adduct the trinuclear complexes and the fluorocarbons are stacked exactly on top of each other. The modified emission is observed both for the crystals and for frozen solutions in tetrahydrofuran.304
The same type of trinuclear gold(I) imidazolates (175) with different substitution patterns were introduced in properly designed scaffolds of trigonal symmetry that can hold two or three of the trinuclear units. Scaffolds of D3h symmetry (176) were built from linear 1,4-di(4-pyridyl)benzene, diazine and tris(4-pyridyl)triazine ligands connected through (en)Pd(II) linkers. In the cage with three eclipsed trinuclear units, the Au–Au contacts are in the range of 3.206–3.230 Å. The inclusion compounds are non-emissive owing to an efficient quenching by host–guest interactions. The solution absorption spectra show drastic changes upon addition of silver ions suggesting that silver ions are included between the stacked components establishing additional metallophilic interactions.305
Trinuclear gold(I) pyrazolate complexes have been provided with three large substituents carrying each two very long alkyl chains (177, R = octadecyl). As a sol in various solvents, this compound is non-emissive, but upon gentle heating (40–50 °C) in hexane and recooling it forms a red-luminescent organo-gel in hexane. There is X-ray diffraction and other experimental evidence that in this gel the trinuclear complex units are stacked by establishing aurophilic bonding which gives rise to fundamentally different photophysical properties. Heating of the gel leads to a reversible loss of the emission. The colour of the luminescence can be altered from red to blue by the addition of traces of silver cations, and this process is also reversible (upon addition of chloride anion). These phenomena have again been rationalized by assuming an insertion and metallophilic binding of the silver ions between the gold pyrazolate trimers.306
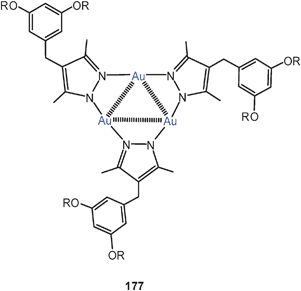
The aggregation of trinuclear gold(I) pyrazolates with small innocent substituents follows a more complicated pattern. Only two of the three gold atoms are involved in the stacking into dimers, while the third one entertains Au–Au contacts with a neighbouring dimer (178, for clarity, only dimers are shown, five-membered rings omitted).206 This peculiar mode of stacking has been confirmed by the solid state structural characterization of the 3,5-bis(trifluoromethyl)-substituted compound, but its intermolecular distances are much longer (3.885 Å) indicating only weak if any interactions. In a thorough treatment of the electronic structure of the pyrazolates with different substituents (H, Me, CF3) by DFT and MP2 techniques, the effects of the substituents on the structure and the photophysical properties have been analyzed. The CF3-substituted case was shown to have high acidity (acceptor properties), which materialized in the formation of a toluene solvate. In crystals of this compound, the stacking of the pyrazolate trimers is abandoned by an insertion of the toluene molecules between the trimers.307
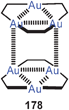
Crystals of the copper(I) and silver(I) analogues of 178 (Cu or Ag for Au; various decorative substituents) are isomorphous, but show a variation in the intra- and intermolecular M–M distances and in small details of the stacking. Shorter Ag–Ag distances suggest that the silver compound has the strongest metallophilic bonding within the series.307
3.10. Homoleptic anionic gold(I) complexes [AuX2]− with halide, pseudohalide and sulfurous ligands X
It has been observed already in earlier work that anions of the type [AuX2]− have a tendency to aggregate through close contacts between the gold centres.16 This was a surprising phenomenon since the approach clearly takes place against Coulomb repulsion between two or more units of negative charge. The aggregation is not only relevant to the crystalline state, but also to solutions of dicyanoaurates(I) and the absorption of the anions out of solutions on surfaces. Since [Au(CN)2]− is among the candidates, this behaviour is particularly important for technical processes such as cyanide leaching of ores, for the recovery of gold, for galvanic and electroless gold plating etc. Therefore much work has been dedicated to studies of the deposition of the complex in the carbon-in-pulp process, where the efficient uptake from solutions clearly relies on some specific interactions.308,309 In more recent studies, the ambiphilic role of [Au(CN)2]− anions has been exploited in new concepts of crystal engineering, where the anions can be coordinated end-on to other metal atoms and linked to each other through aurophilic bonding (below). Besides the cyanide complexes, current studies in this area have focused on analogues with halide, pseudohalide and sulfurous ligands.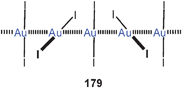
Stacking of [AuX2]− anions with complex gold(I) cations through aurophilic bonding into dimers or polymers with alternating distances between gold atoms is very common.16,311,312 In recent work the first examples with complex gold(III) cations have been discovered (180). The AuI–AuIII distance of 3.34 Å is very similar to that between two AuI centres. The Cl–Au–Cl axis of the anion is parallel to the N–Au–N unit in the cation, as expected based on electrostatic considerations.313 In most previous examples of AuI–AuIII aurophilicity, the bonding is supported by a ligand (see below).
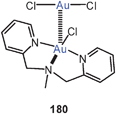
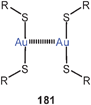
The structures and photophysical characteristics of di(thiocyanato)gold(I) salts M+ [Au(SCN)2]− have been investigated in great detail in an attempt to correlate the emissive properties with the Au–Au distances found in the crystalline state. Surprisingly, the mode of aggregation of the anions depends very strongly on the nature of the cation: isolated monomers with the cyano groups in trans-conformation are found with the bulky and rigid Ph4P+ cations, isolated dimers with the cyano groups in cis-conformation at each gold atom are observed with the bulky, but more flexible nBu4N+ cations (Au–Au 3.070 Å), while infinite chains of anions are obtained with M = NH4, K, Rb, Cs. Along these chains, the Au–Au distances alternate (short–long–short–long) with distances in the range of 3.006–3.240 Å (180). The salt with Me4N+ cations features trimers loosely aggregated into kinked chains (3.179 and 3.265 Å). While the phosphonium salt with its monomeric anion is photo-silent, the other crystalline solids are strongly luminescent, and their emission maxima appear to be shifted to lower energy (from 19379 to 14925 cm−1) as the aurophilic contacts are increased (from 3.006 to 3.240 Å),314,315 confirming earlier suggestions at least for this family of compounds.316,317
In a subsequent study, very detailed experimental and theoretical work on K[Au(SCN)2] was carried out which allowed a further description of the phenomena involved. Temperature-dependent photoluminescence studies of the crystals have revealed a green phosphorescence and a blue fluorescence, both becoming well resolved at 4 K. The calculations (MP2, DFT) of a dianionic, crossed model dimer (182) gave a S0 ground state geometry with an Au–Au distance of 2.95 Å, which shrinks to only 2.62 Å in the excited lowest triplet state T1. The blue-shift in the phosphorescence energies and the reduced Stokes shifts with the shortening of the Au–Au distances induced by cations of different sizes were confirmed. Moreover, frozen solutions of K[Au(SCN)2] with different concentrations in acetonitrile (77 K) show a red-shift of the phosphorescence and larger Stokes shifts as compared to the crystals, indicating aurophilic bonding in the glass state. The calculations have also shown that the ground state structure of the dimeric anion is changed drastically in the excited state, which is accepted more readily in a more spatial and flexible environment. This may explain the strong dependence of the emission on the nature of the cations and on the temperature.318
The crystal structures of sodium bis(thiosulfato)gold(I) dihydrate were determined already in the 1970s in two independent studies, but only a short remark refers to the observation of interanionic Au–Au contacts of 3.302 Å in dimeric units (183).319,320 At this time, only very few structural data exhibiting similar phenomena were available.321
In a very recent investigation, a similar organizational pattern has been found in the structure of tetraethylammonium bis(methylthiosulfonato)gold(I) (182). Within the centrosymmetrical pair of anions there is an Au–Au contact of 3.107 Å. The dihedral angle S–Au–Au–S of 21.8° in the thiosulfonate 184 is to be compared with a corresponding angle of 66.8° in the thiosulfate 183. The two complexes have in common that the gold atoms are strictly sulfur-bonded with quasi-linear S–Au–S bonds complemented by perpendicular aurophilic bonding to give a T-shaped coordination. The conformational differences (staggered vs. eclipsed) and the different Au–Au distances can be ascribed to the different charges of the thiosulf(on)ate anions (−2 vs. −1) and some efficient hydrogen bonding in the hydrate.322(Phosphine)gold(I) thiosulfonates had also been shown to be associated via aurophilic interactions (185; R = Me, Au–Au 2.996; R = Ph, 3.051 Å).323 Bis(organosulfinato)gold(I) anions are known to form dinuclear units with bis(triphenylphosphine)gold(I) cations through an Au–Au contact of 2.943 Å, in this case supported by electrostatic attraction (186).324
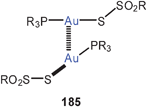
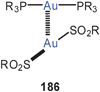
In crystals of some of the simple alkali dicyanoaurates(I) the anions are only loosely aggregated, such as in K[Au(CN)2] with Au–Au contacts of as much as 3.64 Å.327 However, the crystal structure analysis of Ba2[Au(CN)2]4 (diaza-18-crown-6)2 (H2O)3 has provided data for mono-, di- and trimeric anions in the same crystal. The Au–Au distances in the dimer and trimer are 3.267 (1×) and 3.565 (2×) Å, respectively.328 By contrast, only monomeric anions are present in nBu4N+ [Au(CN)2].329
Crystals of K[Au(CN)2] are luminescent, and the solutions in water or methanol also have comparable photoemissive properties. These can be tuned by the choice of solvent, concentration, temperature, and excitation wavelength, and the observed bands can be assigned to excimers and exciplexes [Au(CN)2]n−n (n = 2, 3). This assignment has been confirmed by theoretical calculations (MP2), which showed an equilibrium distance of 2.90 Å for a staggered ground state dimer (187) and of 2.664 Å for the first triplet excited state. The energy calculated for the aurophilic bonding in the ground state dimer amounts to 32 kJ mol−1, notably higher than for the silver analog (25 kJ mol−1).330
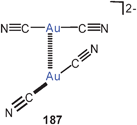
Dimers 187 (Au–Au 3.262 Å) are also present in crystals of the formula [L2Hg]2+ 2 [Au(CN)2]− where L represents the zwitterionic ligand 4-Me3N(+)–C6H4–S(−). This dimer occurs—flanked by two [L2Au]+ cations—in the compound 188 [Au–Au 3.069 (centre), 3.024 and 3.031 Å], with a linear chain of four gold atoms.127,129
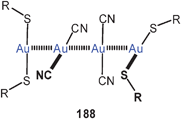
Crystals of Tl[Au(CN)2] were investigated by neutron diffraction and shown to have a complex arrangement of the components with both Au–Au and Au–Tl metallophilic contacts. The former vary with the sites in the crystal within the range from 3.037 to 3.560 Å (189).331 The structure and bonding has recently been reinvestigated by multinuclear NMR spectroscopy on isotopically enriched samples providing important benchmark data for further studies.326
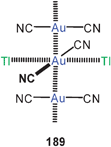
In this context the NMR characteristics of AuCN powders have also been reconsidered. Depending on the method of preparation and mechanical deformation, high degrees of disorder of the known ideally linear structure have been discovered which indicate extensive chain slippage and misalignment (190).326 This result suggests that the perfect hexagonal symmetry of layers of gold atoms (Au–Au 3.396 Å) separated by cyanide spacers (191, vertical lines for CN linkers to the next sheet of gold atoms)325,332 may be only an idealized picture not very close to reality when dealing e.g. with commercially available ochre-coloured samples.
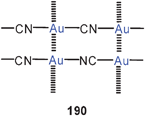

Lead(II) cyanoaurates(I) have been studied in some detail in attempts to interpret the photophysical properties and to trace the lone pair effect of Pb2+, by detailed structural, UV and NMR spectroscopic and theoretical investigations. Depending on the nature of auxiliary ligands, the structures reflect an interplay between various weak forces (hydrogen bonding, π–π stacking, lone pair repulsion, aurophilic bonding). While in crystals of the phenanthroline complex [Pb(L)2]2+ 2 [AuCN)2]− with L = phen no significant Au–Au contacts are discernible, and in the bipyridyl complex these contacts are still long at 3.592 Å, in the ethylenediamine complexes they are reduced to 3.249 Å (192). The emission properties of the three compounds are complex owing to the various modes of excitation possible for two heavy metals and very different ligand systems, but for cases with a clear structural basis the influence of aurophilic bonding could be traced.333 In previous work, crystals of the related aquo complex Pb(H2O)[Au(CN)2]2 were found to be highly birefringent due to similar bonding situations. With only water molecules as ligands, six cyanoaurate anions are coordinated to the octacoordinated lead atom and are grouped in units of three with weak Au–Au bonding (3.506 Å). The alignment of these units leads to additional, somewhat shorter Au–Au contacts (3.488 Å). The hydrate had been known from previous work to be luminescent in the solid state.334 From powder X-ray and NMR data, the anhydrous compound has been tentatively assigned a homoleptic octacoordination by cyanoaurate anions of the lead(II) cation, where two units of four form squares of gold atoms (193, 194).335
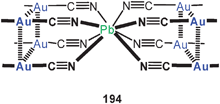
The combination of dicyanoaurate(I) anions with first row transition metal cations is a particularly popular field of investigation, mainly owing to the multifaceted structural motifs—representing one of the active areas of supramolecular chemistry and crystal engineering—and to the variety of cooperative magnetic phenomena observed therein. Aurophilic bonding contributes to the mode of assembly of the components in almost all cases. Some of the compositions are exceedingly complex, but can be broken down to simple free-standing, interwoven or interpenetrating networks.
Thus the crystalline material of the compositions {Mn[Au(CN)2]2 (H2O)4} [Au(CN)2]2(4,4′-H2bipy)(H2O), where H2bipy is diprotonated 4,4′-bipyridyl, shows an aggregation determined by an interplay of coordinative, aurophilic and hydrogen bonding, complemented by π–π stacking of the heteroarenes. It contains both end-on coordinated (to MnII) and free [Au(CN)2]− anions, but both of these are connected to each other through a network of aurophilic bonding via Au–Au contacts of ca. 3.444 Å in a common layer (195).336
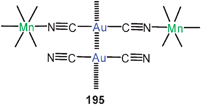
Similar principles of structure were found in the compound {Mn[Au(CN)2]2(nitpy)2(H2O)2]}n, where nitpy is the permanent organic radical 2-(4′-pyridyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide. The two [Au(CN)2]− anions are attached trans and end-on to the MnII centre, but entertain short Au–Au contacts with the corresponding units of neighbouring complexes (3.331 Å). Note that this alignment also allows for hydrogen bonding with coordinated water molecules (196).337
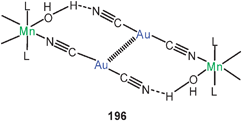
In a previous study,338nickel(II) and zinc(II) complexes of another permanent organic radical (4,5-dihydro-4,4,5,5-tetramethyl-2-(pyridin-2-yl)-1H-imidazol-1-yloxyl) with the general formula {M(tpyimo)2[Au(CN)2]2}n had been prepared in which the dicyanoaurate anions form chains with slightly alternating or identical Au–Au contacts (3.264 and 3.260 Å for M = Ni, 3.339 Å for M = Zn). One CN group of each anion is coordinated to a Ni/Zn centre, while the other one remains uncoordinated (197).
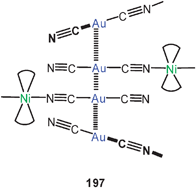
Two-dimensional networks co-determined by aurophilic bonding were observed in iron complexes of the type{Fe(3-Xpy)2[Au(CN)2]2}, where 3-Xpy represents 3-F–, 3-Cl–, 3-Br–, and 3-I–pyridine. The four homologues have closely related structures, the latter three being even isomorphous. Each FeII centre is connected to four other FeII centres via [Au(CN)2]− anions, forming a 2D grid. Two of these grids form a double layer in which the gold atoms are placed on top of each other with short Au–Au contacts of 3.082 and 3.124 Å (X = F) and of average 3.05 Å (X = Cl, Br, I) (198). Similar results were found for {Fe(3-Ipy)2[Au(CN)2]2] (3-Ipy)0.5, where the extra 3-Ipy molecules are intercalated between the double layers.339
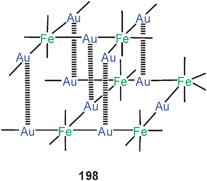
Square grids built from dinuclear complexes of bipyrimidine with trans-di(amino)metal(II) nodes and [Au(CN)2]− spacers are also present in crystals of {[M(NH3)2]2(bpym)[Au(CN)2]2}[Au(CN)2]2 with M = Ni, Co, Cu. As expressed by the formula, in addition to the grid-building [Au(CN)2]− units there are two additional such units which are not connected to the M centres, but form aurophilic bonds between the former thus extending the connectivity (Au–Au 3.138, 3.123, 3.144 Å). Zigzag chains of gold atoms are thereby connected to form a honeycomb array.340
Almost the same honeycomb array of gold atoms was detected in copper(II) complexes with pyrimidine-2-carboxylate (pymca) ligands. The specific phase K{[Cu(NH3)2]2(pymca)[Au(CN)2]2}[Au(CN)2]2 has again bridging and uncoordinated [Au(CN)2]− anions. The former connect the dinuclear copper(II) complex to form a square grid, similar to the one shown as 198, and these grids are linked through aurophilic bonding between the remainder anions to give the connectivity pattern 199 for the gold atoms.341
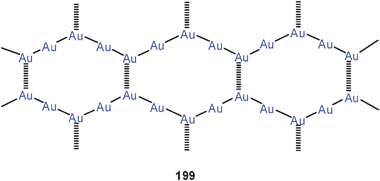
Complexes of Mn(II) and Cd(II) with bipyridyl and phenanthroline ligand have also been aggregated viadicyanoaurate anions into networks supported by aurophilic bonding.
In the example {Mn(phen)2(H2O)[Au(CN)2]}2[Au(CN)2]2(H2O)4 the four anions of the formula unit form a string of four gold atoms (Au–Au 3.101 and 3.176 Å), but only two of the eight CN groups are coordinated to a MnII centre (200). In {Cd(bipy)2(H2O)[Au(CN)2]}[Au(CN)2] also only every second anion in a chain of anions (Au–Au 3.179 Å) is Cd-coordinated.342 In a corresponding Cd complex with only one phenanthroline ligand, {Cd(phen)(H2O)[Au(CN)2]2}, both anions are attached to a Cd centre and bonded aurophilically to neighbouring anions (Au–Au 3.190 Å) to form chains, which are further connected (Au–Au 3.415 Å) into layers.343
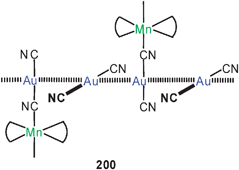
An [Au(CN)2]− unit also has the role of the connector between the two copper(II) centres in a complex of the formula {[Cu(acac)(bipy)]2[Au(CN)2]}+ ClO−4. These dinuclear copper complexes pair up in a crossed orientation and form a central Au–Au contact of 3.295 Å (201, L = bipy).344
Aurophilic contacts (3. 547 Å) obviously are only of marginal relevance to the structure of the complexes {M(pyz)[Au(CN)2]2}n with [Au(CN)2]− anions and pyz = pyrazine molecules as spacers between octahedrally coordinated dications M = Co, Zn. The compounds are not photoluminescent in the solid state. By contrast, in the related zinc complex {Zn(bipy)[Au(CN)2]}[Br/CNAuCN] (with bipy = 4,4′-bipyridyl and a mixed occupancy and a Br/CN disorder in an anion) the components are more firmly bonded via short Au–Au contacts of 3.118 and 3.126 Å, and the crystals are strongly emissive. Both types of compounds were prepared through a new thermal reductive elimination route from the corresponding gold(III) compounds.345
In a coordination polymer of the formula K2{Tb[Pt(CN)4]2(H2O)4}[Au(CN)2](H2O)2 there are both Au–Au and Au–Pt metallophilic interactions with distances of 3.195 and 3.266 Å, respectively. The neighbouring [Au(CN)2]− units appear in both an eclipsed and a staggered conformation (202, 203). The compound is strongly luminescent showing a Tb3+ emission in the solid state.346
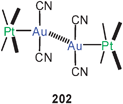
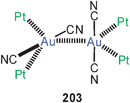
4. Aurophilic interactions between gold(I) and gold(III) centres
Two reviews have been published on metallophilic bonding including also some aurophilic bonding between Au(III) centres.220,221 It appears that there are still only few examples of Au(I)–Au(III) and Au(III)–Au(III) interactions in molecular systems. Early cases have been the fully supported bonding in dinuclear 2-phosphino-arylgold complexes with an either heteroleptic (204) or homoleptic configuration (205) (Au–Au 2.887 and 2.931 Å, respectively), complemented by one semi-supported form (206).347 A similar situation had been described for the cyclic ylide complex 207 (Au–Au 3.061 Å).348 In tri(gold)selenium compounds with Au(I) and Au(III) present in the ratio 2:1 or 1:2, only the distance between the two Au(I) centers in the former suggests aurophilic interactions (2.946 Å) (208, L = PR3; X = C6F5). The AuI–AuIII distances are all much longer, the shortest at 3.412 Å.349
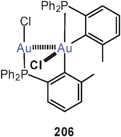
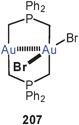
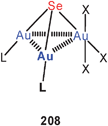
An unsupported case was found, as mentioned above, in the stacking of the cations and anions of chloro[bis(2-pyridylmethyl)amido]gold(III) dichloroaurate(I) with AuI–AuIII 3.34 Å (180).313
The stacking of the tetraazidoaurate(III) anions in the tetramethylammonium salt has been assigned to AuIII–AuIII interactions, because the slightly alternating distances of 3.507 and 3.584 Å are short enough to allow for at least weak contributions. Preliminary DFT calculations showed virtually no bonding in a model gas phase dimer of anions in the absence of support from the cations (209).350
Several theoretical studies (ab initio, MP2) of the underlying d10–d8 and d8–d8 interactions have been published. The earlier work351 has recently been summarized and complemented using various intramolecular (supported) and intermolecular (unsupported) model systems (e.g. 210). The dispersion, induction and electrostatic multipole components of the interactions have been analyzed, and a satisfactory agreement with experimental data has been reached. Aurophilic bonding with significant interaction energies was found in all cases at least for staggered conformations avoiding steric and electrostatic interference.352
5. Summary and conclusions
In this review it has been tried to screen the results of investigations on aurophilic bonding published in the last five years (up to mid-2011), and to illustrate the impact that the aurophilicity concept has had on the progress and current interest in all major areas of gold chemistry. It is obvious that aurophilicity phenomena can be recognized in virtually all classes of molecular low-valent gold compounds. The same is true for general solid-state chemistry of gold, including alloys and polynary phases of gold with metalloids and main group elements not considered in this review.The typical range of aurophilic Au–Au distances is now well established and recognized to be exceptionally broad, ca. 2.85–3.50 Å. The influence of the intrinsic properties of ligands and substituents on these distances has been estimated, and the results can be used as a guidance in a search for specific aggregations and structures. It is also well documented that aurophilic interactions are not very sensitive to variations in conformational angles like e.g. L-Au–Au or X-Au–Au for associated molecules L–Au–X, although angles near 90° (for staggered conformations) prevail. Both the lengthening of the Au–Au contact and the widening of these angles appear to have a flat energy profile making the systems very flexible. Moreover, each gold atom can entertain more than one Au–Au contact without significant loss in binding energy for each contact. Therefore, mononuclear or polynuclear gold compounds may not only dimerize, but form also oligomers and polymers with one- or two-dimensional arrays of gold atoms. The frequently observed existence of polymorphs of a given system indicates that the differences in energy between the various aggregates must indeed be quite small.
Earlier estimates or measurements of the Au–Au binding energy (5–15 kcal mol−1),23 which have placed aurophilic bonding in this regard next to hydrogen bonding, have been confirmed in several new studies which are listed in Table 1. This range of binding energies is clearly enough to keep some of the stronger Au–Au contacts intact also in solution, in particular at high concentrations and if too powerful donor solvents are excluded. It is therefore of no surprise that evidence is accumulating for aurophilic bonding in states of aggregation other than the solid state, where the early observations had been made almost exclusively.
State-of-the-art theoretical chemistry is able to explain the basic phenomenon, and experimental data have been well reproduced even for some larger multi-atom systems. The role of relativistic effects, which was strongly emphasized in the earlier work in order to explain the specific observations with gold, is now better defined. This, however, has led to uncertainties regarding the relative importance of relativistic effects in metallophilic bonding in general, and with the remainder coinage metals in particular. Since cases have become known where in a given family of complexes the Ag–Ag contacts are actually shorter than the Au–Au contacts, it appears that the significance of relativity-related contributions is less than initially surmised. On the other hand, it is probably not simply a coincidence that other classics of metallophilic bonding were all observed with the heavy metals neighbouring gold in the Periodic Table (Ir, Pt, Hg, Tl), where relativistic effects reach their maximum.
Aurophilic bonding thus should be taken as a prominent case of closed-shell interactions of metal atoms, which may be summarized as metallophilic interactions. These interactions are more obvious with gold because of its peculiar general bonding characteristics, which include the lowest coordination number of all metals with highly directional covalent bonding, its high electronegativity, and its small, relativistically contracted atomic radius.
These unusual features of aurophilic bonding give many opportunities for the design of novel compounds, for supramolecular architectures and for crystal engineering of gold-containing materials. In the new structures, the aurophilic contacts have specific consequences for the physical properties of the materials which may be of great interest for various applications, including also the preparation and functionalization of gold nanoparticles and the engineering of gold surfaces. Presently the main focus is on the photophysical effects which are probed as photocatalysts, vapochromic or solvatochromic sensors, light-emitting diodes, and other devices.
The authors would like to conclude this dry summary by highlighting one of the latest contributions which exhibits all the intriguing facets of metallophilicity, published under the headline of “molecular amalgamation”: bis(pentafluorophenyl)mercury(II) and pentafluorophenyl(trimethylphosphine)gold(I) have been shown to form stacks of the components in a 1:2 molar ratio, in which dimers [(Me3P)AuC6F5]2 alternate with single molecules [(C6F5)2Hg] (211). The string of metal atoms of this molecular amalgam has distances Au–Au and Au–Hg of 3.0504(5) and 3.1860(2) Å, and angles Au–Hg–Au and Au–Au–Hg of 147.85(1)° and 180.0°, respectively. Neighbouring molecules show staggered conformations which indicate the absence of any significant π–π stacking and therefore suggest that metallophilic bonding is the main force determining the alignment of the components with their closed-shell metal centres (5d10). Theoretical calculations (MP2) of gas phase dinuclear model units [(Me3P)AuC6F5]2 and [(Me3P)AuC6F5·(C6F5)2Hg] in staggered conformations have given binding energies of −13.60 and −17.5 kcal mol−1, respectively, mainly based on dispersion forces and with very significant contributions from relativistic effects. The crystals are strongly luminescent, but silent in solution indicating extensive degradation of the stacks in solution. This report thus shows in a nutshell how the wealth of information provided in recent years is now giving a consistent picture of the origin and consequences of metallophilic bonding which can assist in the design and construction of many more novel aggregates with interesting properties.353
Acknowledgements
The work on this review was generously supported by Fonds der Chemischen Industrie, Frankfurt am Main, Germany.References
- R. J. Puddephatt, The Chemistry of Gold, Elsevier, Amsterdam, 1978 Search PubMed.
- S. Åkerstrom, Ark. Kemi, 1959, 14, 387 Search PubMed.
- S. Esperås, Acta Chem. Scand., Ser. A, 1976, 30, 527 Search PubMed.
- H. Schmidbaur, Acc. Chem. Res., 1975, 8, 62 CrossRef CAS.
- H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1976, 15, 728 CrossRef.
- P. G. Jones, Gold Bull., 1981, 14, 102 CrossRef CAS.
- P. G. Jones, Gold Bull., 1981, 14, 159 CrossRef CAS.
- P. G. Jones, Gold Bull., 1983, 16, 114 CrossRef CAS.
- P. G. Jones, Gold Bull., 1986, 19, 46 CrossRef CAS.
- A. N. Nesmeyanov, E. G. Perevalova, Y. T. Struchkov, M. Y. Antipin, K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1980, 201, 343 CrossRef CAS.
- F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1988, 27, 1544 CrossRef.
- H. Schmidbaur, W. Graf and G. Müller, Angew. Chem., Int. Ed. Engl., 1988, 27, 417 CrossRef.
- W. S. Rapson, Gold Bull., 1989, 22, 19 CrossRef.
- H. Schmidbaur, Gold Bull., 1990, 23, 11 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 2000, 33, 3 CrossRef CAS.
- Gold, Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, Wiley, Chichester, 1999 Search PubMed.
- Gold Chemistry, Applications and Future Directions in the Life Sciences, ed. F. Mohr, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Modern Supramolecular Gold Chemistry, ed. A. Laguna, Wiley-VCH, Weinheim, 2009 Search PubMed.
- C.-M. Che, S.-W. Lai, in ref. 17, p. 250.
- S. Coco, P. Espinet, in ref. 17, p. 357.
- V. W.-W. Yam and E. C.-C. Cheng, Top. Curr. Chem., 2007, 281, 269 CrossRef CAS.
- M. Bardaji and A. Laguna, J. Chem. Educ., 1999, 76, 201 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC.
- S. S. Pathaneni and G. R. Desiraju, J. Chem. Soc., Dalton Trans., 1993, 319 RSC.
- L. Magnko, M. Schweizer, G. Rauhut, M. Schütz, H. Stoll and H.-J. Werner, PhysChemChemPhys, 2002, 4, 1006 RSC.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Inorg. Chem., 2007, 46, 6444 CrossRef CAS.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741 RSC.
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef.
- P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412 CrossRef.
- P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113 CrossRef.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967 RSC.
- P. Schwerdtfeger, M. Lein, in ref. 18, p. 183.
- P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578 CrossRef CAS.
- E. O'Grady and N. Kaltsoyannis, PhysChemChemPhys, 2004, 6, 680 RSC.
- L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 230 CrossRef CAS.
- H. Schmidbaur, Gmelin Handbook of Inorganic Chemistry: Gold, Organic Compounds, Springer-Verlag, Berlin, 1980 Search PubMed.
- H. Schmidbaur and A. Schier, “Organogold Chemistry”, in “Comprehensive Organometallic Chemistry III”, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2006, vol. 2, p. 251 Search PubMed.
- H. Schmidbaur, Nachr. Chem., Tech. Lab., 1979, 27, 620 CrossRef CAS.
- H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1983, 22, 907 CrossRef.
- H. Schmidbaur, Angew. Chem., Int. Ed., 2007, 46, 2984 CrossRef CAS.
- H. Schmidbaur and O. Gasser, Angew. Chem., Int. Ed. Engl., 1976, 15, 502 CrossRef.
- J. Vicente, A. R. Singhal and P. G. Jones, Organometallics, 2002, 21, 5887 CrossRef CAS.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038 CrossRef CAS.
- M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295 CrossRef CAS.
- E. I Smyslova, E. G. Perevalova, V. P. Dyadchenko, K. I. Grandberg, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1981, 215, 269 CrossRef.
- M. C. Gimeno, A. Laguna, M. Laguna, F. Sanmartin and P. G. Jones, Organometallics, 1993, 12, 3984 CrossRef CAS.
- H. Schmidbaur, A. Grohmann, M. E. Olmos and A. Schier, in The Chemistry of Organic Derivatives of Gold and Silver, ed. S. Patai and Z. Rappoport, J. Wiley & Sons, Chichester, 1999, p. 244 Search PubMed.
- M. Wilfling and K. W. Klinkhammer, Angew. Chem., Int. Ed., 2010, 49, 3219 CrossRef CAS.
- S. Bommers, H. Beruda, N. Dufour, M. Paul, A. Schier and H. Schmidbaur, Chem. Ber., 1995, 128, 137 CrossRef CAS.
- N. Dufour, A. Schier and H. Schmidbaur, Organometallics, 1993, 12, 2408 CrossRef CAS.
- S. Hagen, I. Pantenburg, F. Weigend, C. Wickleder and L. Wesemann, Angew. Chem., Int. Ed., 2003, 42, 1501 CrossRef CAS.
- J.-H. Jia and Q.-M. Wang, J. Am. Chem. Soc., 2009, 131, 16634 CrossRef CAS.
- H. Schmidbaur, B. Brachthäuser, O. Steigelmann and H. Beruda, Chem. Ber., 1992, 125, 2705 CrossRef CAS.
- F. P. Gabbaï, A. Schier, J. Riede and H. Schmidbaur, Chem. Ber., 1997, 130, 111 CrossRef.
- H. Schmidbaur, B. Brachthäuser and O. Steigelmann, Angew. Chem., Int. Ed. Engl., 1991, 30, 1488 CrossRef.
- O. Steigelmann, P. Bissinger and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1990, 29, 1399 CrossRef.
- H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391 RSC.
- (a) J.-H. Jia, J.-X. Liang, Z. Lei, Z.-X. Cao and Q.-M. Wang, Chem. Commun., 2011, 47, 4739 RSC; (b) K. Doll, P. Pyykkö and H. Stoll, J. Chem. Phys., 1998, 109, 2339 CrossRef CAS.
- E. E. Karagiannis and C. A. Tsipis, Organometallics, 2010, 29, 849 CrossRef.
- (a) H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2 CrossRef CAS; (b) H. G. Raubenheimer and H. Schmidbaur, S. Afr. J. Sci., 2011, 107, 31 CrossRef CAS.
- D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS.
- H. Schmidbaur and A. Schier, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 329 CAS.
- G. Seidel, C. W. Lehmann and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 8466 CrossRef CAS.
- K. A. Porter, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 4922 CrossRef CAS.
- A. N. Nesmeyanov, E. G. Perevalova, K. I. Grandberg, D. A. Lemenovskii, T. V. Baukova and A. B. Afanasova, J. Organomet. Chem., 1974, 65, 131 CrossRef CAS.
- T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313 RSC.
- A. Himmelspach, M. Finze and S. Raub, Angew. Chem., Int. Ed., 2011, 50, 2628 CrossRef CAS.
- Y. Cao, R. F. Höckendorf and M. K. Bayer, ChemPhysChem, 2008, 9, 1383 CrossRef CAS.
- X. Li, B. Kiran and L. S. Wang, J. Phys. Chem., 2005, A 109, 4366 Search PubMed.
- B. Kiran, X. Li, H. J. Zhai, L. F. Cui and L. S. Wang, Angew. Chem., Int. Ed., 2004, 43, 2125 CrossRef CAS.
- B. Kiran, X. Li, L. S. Zhai and L. S. Wang, J. Chem. Phys., 2006, 125, 133204 CrossRef.
- E. Zeller, H. Beruda, A. Kolb, P. Bissinger, J. Riede and H. Schmidbaur, Nature, 1991, 352, 141 CrossRef CAS.
- Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1984, 277, 143 CrossRef CAS.
- (a) J. Li and P. Pyykkö, Inorg. Chem., 1993, 32, 2630 CrossRef CAS; (b) P. Pyykkö and P. Zaleski-Ejgierd, J. Chem. Phys., 2008, 128, 124309 CrossRef.
- P. Pyykkö and T. Tamm, Organometallics, 1998, 17, 4842 CrossRef.
- H. Fang and S.-G. Wang, J. Phys. Chem. A, 2007, 111, 1562 CrossRef CAS.
- G. Pivoriunas, C. Maichle-Mössmer, S. Schwarz and J. Strähle, Z. Anorg. Allg. Chem., 2005, 631, 1743 CrossRef CAS.
- A. Bauer, N. W. Mitzel, A. Schier, D. W. H. Rankin and H. Schmidbaur, Chem. Ber./Recl., 1997, 130, 323 CrossRef CAS.
- K. Angermaier and H. Schmidbaur, Chem. Ber., 1995, 128, 817 CrossRef CAS.
- K. Angermaier and H. Schmidbaur, Inorg. Chem., 1995, 34, 3120 CrossRef CAS.
- A. Schier, A. Grohmann, J. M. Lopez-de-Luzuriaga and H. Schmidbaur, Inorg. Chem., 2000, 39, 547 CrossRef CAS.
- H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 853–859 CAS.
- X. Liang, X. Wu, T. Dong, Zh. Qin, K. Tan, X. Lu and Z. Tang, Angew. Chem., Int. Ed., 2011, 50, 2166 CrossRef CAS.
- D. Fenske, A. Rotheberger and S. Wieber, Eur. J. Inorg. Chem., 2007, 3469 CrossRef CAS.
- K. Angermaier and H. Schmidbaur, Inorg. Chem., 1994, 33, 2069 CrossRef CAS.
- S.-C. Chung, S. Krüger, H. Schmidbaur and N. Rösch, Inorg. Chem., 1996, 35, 5387 CrossRef CAS.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415 CrossRef CAS.
- B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978 CrossRef CAS.
- B. D. Sherry, L. Maus, B. N. Laforteza and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 8132 CrossRef CAS.
- G. E. Veitch, E. Beckmann, B. J. Burke, A. Boyer, S. L. Maslen and S. V. Ley, Angew. Chem., Int. Ed., 2007, 46, 7629 CrossRef CAS.
- Q.-M. Wang, Y.-A. Lee, O. Crespo, J. Deaton, C. Tang, H. J. Gysling, M. C. Gimeno, C. Larraz, M. D. Villacampa, A. Laguna and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 9488 CrossRef CAS.
- O. Crespo, M. C. Gimeno, A. Laguna, C. Larraz and M. D. Villacampa, Chem.–Eur. J., 2007, 13, 235 CrossRef.
- H. Schmidbaur, S. Hofreiter and M. Paul, Nature, 1995, 377, 503 CrossRef CAS.
- K. Nomiya, T. Yoshida, Y. Sakai, A. Nanba and S. Truruta, Inorg. Chem., 2010, 49, 8247 CrossRef CAS.
- H. Fang and S.-G. Wang, J. Phys. Chem. A, 2007, 111, 1562 CrossRef CAS.
- M. C. Gimeno and A. Laguna, Chem. Soc. Rev., 2008, 37, 1952 RSC.
- M. Bardaji , in ref. 18, p. 403.
- S. Gaillard, J. Bosson, R. S. Ramon, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 13729 CrossRef CAS.
- R. S. Ramon, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2011, 17, 1238 CrossRef CAS.
- N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC.
- K. Angermaier and H. Schmidbaur, Chem. Ber., 1994, 127, 2387 CrossRef CAS.
- K. Angermaier and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 879 CAS.
- E. M. Gussenhoven, J. C. Fettinger, D. M. Pham, M. M. Malwitz and A. L. Balch, J. Am. Chem. Soc., 2005, 127, 10838 CrossRef CAS.
- A. Sladek, K. Angermaier and H. Schmidbaur, Chem. Commun., 1996, 1959 RSC.
- A. Sladek and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 1207 CAS.
- A. Battisti, O. Bellina, P. Diversi, S. Losi, F. Marchetti and P. Zanello, Eur. J. Inorg. Chem., 2007, 865 CrossRef CAS.
- F. Balzano, A. Cuzzola, P. Diversi, F. Ghiotto and G. Uccello-Barretta, Eur. J. Inorg. Chem., 2007, 5556 CrossRef CAS.
- H. Fang, X.-G. Zhang and S.-G. Wang, J. Mol. Model, 2009, 15, 461 CrossRef CAS.
- J.-C. Shi, B.-S. Kang and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1997, 2171 RSC.
- T. K.-M. Lee, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 17646 CrossRef CAS.
- E. Barreiro, J. S. Casas, M. D. Couce, A. Laguna, J. M. López-de Luzuriaga, M. Monge, A. Sánchez, J. Sordo, J. M. Varela and E. M. V. López, Eur. J. Inorg. Chem., 2011, 1322 CrossRef CAS.
- G. Landgraf , in ref. 16, p. 143.
- C. Corti and R. Holliday, Gold: Science and Applications, CRC Press, 2010 Search PubMed.
- C. F. Shaw III , in ref. 16, p. 259.
- C. A. Simpson, C. L. Farrow, P. Tian, S. J. L. Billinge, B. J. Hoffmann, K. M. Karkness and D. E. Cliffel, Inorg. Chem., 2010, 49, 10858 CrossRef CAS.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem., 2006, 110, 9927 Search PubMed.
- H. Grönbeck, M. Walter and H. Häkkinen, J. Am. Chem. Soc., 2006, 128, 10268 CrossRef.
- P. Pyykkö, X.-G. Xiong and J. Li, Faraday Discuss., 2011 10.1039/C1FD00018G.
- H. Schnöckel, A. Schnepf, R. L. Whetten, C. Schenk and P. Henkel, Z. Anorg. Allg. Chem., 2011, 637, 15 CrossRef.
- R. Jin, Y. Zhu and H. Qian, Chem.–Eur. J., 2011, 17, 6584 CrossRef CAS.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. J. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756 CrossRef CAS.
- D. Jiang, R. L. Whetten, W. Luoi and S. Dai, J. Phys. Chem. C, 2009, 113, 17291 CAS.
- Y. Zhang, S. Shuang, C. Dong, C. K. Lo, M. C. Paau and M. M. F. Choi, Anal. Chem., 2009, 81, 1676 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754 CrossRef CAS.
- J. P. Fackler Jr, W. E. van Zyl, B. A. Prihoda, in ref. 16, p. 795.
- J. Chen, W.-H. Zhang, X.-Y. Tang, Z.-G. Ren, H.-X. Li, Y. Zhang and J.-P. Lang, Inorg. Chem., 2006, 45, 7671 CrossRef CAS.
- A. Bauer and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 5324 CrossRef CAS.
- A.-X. Theng, Z.-G. Ren, L.-L. Li, H. Shang, H.-X. Li and J.-P. Lang, Dalton Trans., 2011, 589 Search PubMed.
- A. S. Susha, M. Ringler, A. Ohlinger, M. Paderi, N. LiPira, G. Carotenuto, A. L. Rogach and J. Feldmann, Chem. Mater., 2008, 20, 6169 CrossRef CAS.
- S.-H. Cha, J.-U. Kim, K.-H. Kim and J.-C. Lee, Chem. Mater., 2007, 19, 6297 CrossRef CAS.
- P. G. Jones, G. M. Sheldrick, R. Usón and A. Laguna, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1486 CrossRef.
- A. Bayler, A. Bauer and H. Schmidbaur, Chem. Ber./Recl., 1997, 130, 115 CrossRef CAS.
- S. Hofreiter, M. Paul and H. Schmidbaur, Chem. Ber., 1995, 128, 901 CrossRef CAS.
- A. Hamel, N. W. Mitzel and H. Schmidbaur, J. Am. Chem. Soc., 2001, 123, 5106 CrossRef CAS.
- H. Schmidbaur, A. Hamel, N. W. Mitzel, A. Schier and S. D. Nogai, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4916 CrossRef CAS.
- A. S. K. Hashmi, E. Kurpejovic, W. Frey and J. W. Bats, Tetrahedron, 2007, 63, 5879 CrossRef CAS.
- A. S. K. Hashmi, R. Salathe and W. Frey, Eur. J. Org. Chem., 2006, 4340 CrossRef CAS.
- A. S. K. Hashmi, S. Schäfer, M. Wölfle, C. D. Gil, P. Fischer, A. Laguna, M. C. Blanco and M. C. Gimeno, Angew. Chem., Int. Ed., 2007, 46, 6184 CrossRef CAS.
- H. Fang, S.-G. Wang and X.-G. Zhang, Int. J. Quantum Chem., 2009, 109, 526 CrossRef CAS.
- H. Fang, X.-G. Zhang and S.-G. Wang, PhysChemChemPhys, 2009, 11, 5796 RSC.
- (a) H. Raubenheimer, S. Cronje, in ref. 16, p. 557; (b) K. Doll, P. Pyykkö and H. Stoll, J. Chem. Phys., 1998, 109, 2339 CrossRef CAS.
- H. Schmidbaur, A. Wohlleben, F. Wagner, O. Orama and G. Huttner, Chem. Ber., 1977, 110, 1748 CrossRef CAS.
- P. A. Bates and J. M. Waters, Inorg. Chim. Acta, 1985, 98, 125 CrossRef CAS.
- J. Vicente, J. Gil-Rubio, N. Barquero, P. G. Jones and D. Bautista, Organometallics, 2008, 27, 646 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, R. C. Puelles and E. Sánchez-Forcada, Eur. J. Inorg. Chem., 2007, 4001 CrossRef.
- S. Y. Ho, E. C.-C. Cheng, E. R. L. Tiekink and C. W.-W. Yam, Inorg. Chem., 2006, 45, 8165 CrossRef CAS.
- A. Ilie, C. I. Rat, S. Scheutzow, C. Kiske, K. Klux, T. M. Klapötke, C. Silvestru and K. Karaghiosoff, Inorg. Chem., 2011, 50, 2675 CrossRef CAS.
- O. Crespo, M. C. Gimeno, A. Laguna, M. Kulcsar and C. Silvestru, Inorg. Chem., 2009, 48, 4134 CrossRef CAS.
- P. Teo, J. Wang, L. L. Koh and T. S. A. Hor, Dalton Trans., 2009, 5009 RSC.
- J. B. Foley, S. E. Gay, M. J. Vela, B. M. Foxman, A. E. Bruce and M. R. M. Bruce, Eur. J. Inorg. Chem., 2007, 4946 CrossRef CAS.
- M. S. Balakrishna, S. Naik and S. M. Mobin, Inorg. Chim. Acta, 2010, 363, 3010 CrossRef CAS.
- D. Wiedemann, M. T. Gamer and P. W. Roesky, Z. Anorg. Allg. Chem., 2009, 635, 125 CrossRef CAS.
- A. Deák, T. Megyes, G. Tárkány, P. Király, L. Biscók, G. Pálinkás and P. J. Stang, J. Am. Chem. Soc., 2006, 128, 12668 CrossRef.
- H. de la Riva, A. Pintado-Alba, M. Nieuwenhuyzen, C. Hardacre and M. C. Lagunas, Chem. Commun., 2005, 4970 RSC.
- A. Pintado-Alba, H. de la Riva, M. Nieuhuyzen, D. Bautista, P. R. Raithby, H. A. Sparkes, S. J. Teat, J. M. Lopez-de-Luzuriaga and M. C. Lagunas, Dalton Trans., 2004, 3459 RSC.
- T. Tunyogi and A. Deák, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m133 Search PubMed.
- M. C. Lagunas, C. M. Fierro, A. Pintado-Alba, H. de la Riva and S. Betanzos-Lara, Gold Bull., 2007, 40, 135 CrossRef CAS.
- D. V. Partiyka, J. B. Undegraff III, M. Teller, A. D. Hunter and T. G. Gray, Dalton Trans., 2010, 5388 RSC.
- H. Ito, T. Saito, T. Miyahara, Ch. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829 CrossRef CAS.
- A. Escalle, G. Mora, F. Gagosz, N. Mézailles, X. F. Le Goff, Y. Jean and P. Le Floch, Inorg. Chem., 2009, 48, 8415 CrossRef CAS.
- S. Labouille, A. Escalle-Lewis, Y. Jean, N. Mézailles and P. Le Floch, Chem.–Eur. J., 2011, 17, 2256 CrossRef CAS.
- (a) R. J. F. Berger, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 388 CAS; (b) R. Mitrić, J. Petersen, A. Kulesza, M. I. S. Röhr, V. Bonačić-Koutecký, C. Brunet, R. Antoine, P. Dugourd, M. Broyer and R. A. J. O'Hair, J. Phys. Chem. Lett., 2011, 2, 548 CrossRef.
- M. B. Smith, S. H. Dale, S. J. Coles, T. Gelbrich, M. B. Hursthouse and M. E. Light, CrystEngComm, 2006, 8, 140 RSC.
- M. S. Balakrishna, P. Kumar, B. Punji and J. T. Mague, J. Organomet. Chem., 2010, 695, 981 CrossRef CAS.
- C. E. Strasser, S. Cronje, H. Schmidbaur and H. G. Raubenheimer, J. Organomet. Chem., 2006, 691, 4788 CrossRef CAS.
- D. B. G. Williams, T. Traut, F. H. Kriel and W. E. van Zyl, Inorg. Chem. Commun., 2007, 10, 538 CrossRef CAS.
- X. He, W. H. Lam, N. Zhu and V. W.-W. Yam, Chem.–Eur. J., 2009, 15, 8842 CrossRef CAS.
- A. Burini, R. Galassi, S. Ricci, F. Bachechi, A. A. Mohamed and J. P. Fackler Jr, Inorg. Chem., 2010, 49, 513 CrossRef CAS.
- M. L. Gallego, A. Guijarro, O. Castillo, T. Parella, R. Mas-Balleste and F. Zamora, CrystEngComm, 2010, 12, 2332 RSC.
- F. Guyon, A. Hameau, A. Khatyr, M. Knorr, H. Amrouche, D. Fortin, P. D. Harvey, C. Strohmann, A. L. Ndiaye, V. Huch, M. Veith and N. Avarvari, Inorg. Chem., 2008, 47, 7483 CrossRef CAS.
- R. M. Davilla, A. Elduque, T. Grant, R. J. Staplesand and J. P. Fackler Jr, Inorg. Chem., 1993, 32, 1749 CrossRef.
- Q.-F. Sun, T. K.-M. Lee, P.-Z. Li, L.-Y. Yao, J.-J. Huang, J. Huang, S.-Y. Yu, Y.-Z. Li, E. C.-C. Cheng and V. W.-W. Yam, Chem. Commun., 2008, 5514 RSC.
- U. Siemeling, D. Rother, C. Bruhn, H. Fink, T. Weidner, F. Träger, A. Rothenberger, D. Fenske, A. Priebe, J. Maurer and R. Winter, J. Am. Chem. Soc., 2005, 127, 1102 CrossRef CAS.
- T. Weidner, N. Ballay, M. Zharnikov, A. Priebe, N. J. Long, J. Maurer, R. Winter, A. Rothenberger, D. Fenske, D. Rother, C. Bruhn, H. Fink and U. Siemeling, Chem.–Eur. J., 2008, 14, 4346 CrossRef CAS.
- H.-B. Xu, L.-Y. Zhang, J. Ni, H.-Y. Chao and Z.-N. Chen, Inorg. Chem., 2008, 47, 10744 CrossRef CAS.
- M. Moriya, R. Fröhlich, G. Kehr, G. Erker and S. Grimme, Chem.–Asian J., 2008, 3, 753 CrossRef CAS.
- P. K. Mehrotra and R. Hoffmann, Inorg. Chem., 1978, 17, 2187 CrossRef CAS.
- A. Dedieu and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 2074 CrossRef CAS.
- Y. Jiang, S. Alvarez and R. Hoffmann, Inorg. Chem., 1985, 24, 749 CrossRef CAS.
- L.-A. de Jongh, C. E. Strasser, S. Cronje and H. G. Raubenheimer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2137 Search PubMed.
- D. L. Philips, C.-M. Che, K. H. Leung, Z. Mao and M.-C. Tse, Coord. Chem. Rev., 2005, 249, 1476 CrossRef.
- Y.-R. Guo, Q.-J. Pan, G.-Z. Fang and Z.-M. Liu, Chem. Phys. Lett., 2005, 413, 59 CrossRef CAS.
- J. Vicente, M. Chicote, P. González-Herrero and P. G. Jones, J. Chem. Soc., Chem. Commun., 1995, 745 RSC.
- Y.-A. Lee, J. E. McGarrah, R. J. Lachicotte and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 10662 CrossRef CAS.
- Y.-A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778 CrossRef CAS.
- S. S. Tang, C.-P. Chang, I. J. B. Lin, L.-S. Liou and J.-C. Wang, Inorg. Chem., 1997, 36, 2294 CrossRef CAS.
- D. E. Harwell, M. D. Mortimer, C. B. Knobler, F. A. L. Anet and M. F. Hawthorne, J. Am. Chem. Soc., 1996, 118, 2679 CrossRef CAS.
- M. A. Bennett, S. K. Barghava, N. Mitzadeh, S. H. Privér, J. Wagler and A. C. Willis, Dalton Trans., 2009, 7537 RSC.
- S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Inorg. Chem., 2010, 49, 1784 CrossRef CAS.
- J. Schneider, Y.-A. Lee, J. Pérez, W. W. Brennessel, C. Flaschenriem and R. Eisenberg, Inorg. Chem., 2008, 47, 957 CrossRef CAS.
- A. M. Kuchison, M. O. Wolf and B. O. Patrick, Inorg. Chem., 2010, 49, 8802 CrossRef CAS.
- B.-C. Tzeng, H.-T. Yeh, Y.-L. Wu, J.-H. Kuo, G.-H. Lee and S.-M. Peng, Inorg. Chem., 2006, 45, 591 CrossRef CAS.
- S.-Y. Yu, Z.-X. Zhang, E. C.-C. Cheng, Y.-Z. Li, V. W. W. Yam, H.-P. Huang and R. Zhang, J. Am. Chem. Soc., 2005, 127, 17994 CrossRef CAS.
- C. E. Strasser, S. Cronje and H. G. Raubenheimer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m914 Search PubMed.
- T. Tunyogi, A. Deák, G. Tárkányi, P. Király and G. Pálinkás, Inorg. Chem., 2008, 47, 2049 CrossRef CAS.
- G. Tárkány, P. Király, G. Pálinkás and A. Deák, Magn. Reson. Chem., 2007, 45, 917 CrossRef.
- M. A. Alvarez, I. Amor, M. E. Garcia and M. A. Ruiz, Inorg. Chem., 2008, 47, 7963 CrossRef.
- J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1179 CrossRef CAS.
- C. Bartolomé, M. Carrasco-Rando, S. Coco, C. Cordovilla, P. Espinet and J. M. Martin-Alvarez, Organometallics, 2006, 25, 2700 CrossRef.
- A. Muñoz-Castro, D. M.-L. Carey and R. Arratia-Pérez, Chem. Phys. Lett., 2009, 474, 290 CrossRef.
- F. Mendizabal, B. Aguilera and C. Olea-Azar, Chem. Phys. Lett., 2007, 447, 345 CrossRef CAS.
- M. D. Irwin, H. E. Abdou, A. A. Mohamed and J. P. Fackler Jr, Chem. Commun., 2003, 2882 RSC.
- H. E. Abdou, A. A. Mohamed, J. M. Lopez-de-Luzuriaga and J. P. Fackler Jr, J. Cluster Sci., 2004, 15, 397 CrossRef CAS.
- A. A. Mohamed, Coord. Chem. Rev., 2010, 254, 1918 CrossRef CAS.
- G. Yang and R. G. Raptis, Inorg. Chem., 2003, 42, 262 Search PubMed.
- D. V. Patel, K. A. Kreisel, G. P. A. Yap and D. Rabinovich, Inorg. Chem. Commun., 2006, 9, 748 CrossRef CAS.
- C. Hollatz, A. Schier and H. Schmidbaur, Inorg. Chem. Commun., 1998, 1, 115 CrossRef CAS.
- A. Spiekermann, S. D. Hoffmann, F. Kraus and T. F. Fässler, Angew. Chem., Int. Ed., 2007, 46, 1638 CrossRef.
- O. Crespo, M. C. Gimeno, A. Laguna, I. Ospino, G. Aullón and J. M. Oliva, Dalton Trans., 2009, 3807 RSC.
- X. Hong, K.-K. Cheng, C.-X. Guo and C.-M. Che, J. Chem. Soc., Dalton Trans., 1994, 1867 RSC.
- C. M. Knapp, C. S. Jackson, J. S. Large, A. L. Thompson and J. M. Goicoechea, Inorg. Chem., 2011, 50, 4021 CrossRef CAS.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006 CrossRef CAS.
- U. M. Tripathy, A. Bauer and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1997, 2865 RSC.
- J. Muñiz, L. E. Sansores, P. Pyykkö, A. Martínez and R. Salcedo, J. Mol. Model., 2009, 15, 1165 CrossRef.
- J. Muñiz, L. E. Sansores, A. Martinez and R. Salcedo, J. Mol. Model., 2008, 14, 417 CrossRef.
- P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133 CrossRef.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151 CrossRef CAS.
- J. Muniz, C. Wang and P. Pyykkö, Chem.–Eur. J., 2011, 17, 368 CrossRef CAS.
- L. H. Doerrer, Comments Inorg. Chem., 2008, 29, 93 CrossRef CAS.
- L. H. Doerrer, Dalton Trans., 2010, 39, 3543 RSC.
- J. Li and P. Pyykkö, Chem. Phys. Lett., 1992, 197, 586 CrossRef CAS.
- P. Pyykkö and F. Mendizabal, Chem.–Eur. J., 1997, 3, 1458 CrossRef.
- N. Runeberg, M. Schütz and H.-J. Werner, J. Chem. Phys., 1999, 110, 7210 CrossRef CAS.
- P. Pyykkö and Y.-F. Zhao, Angew. Chem., Int. Ed. Engl., 1991, 30, 604 CrossRef.
- P. Pyykkö, N. Runeberg and F. Mendezabal, Chem.–Eur. J., 1997, 3, 1451 CrossRef.
- P. Pyykkö and M. Straka, PhysChemChemPhys, 2000, 2, 2489 RSC.
- S.-L. Sun, G.-C. Yang, C.-S. Qin, Y.-Q. Qiu, L.-K. Yan, Z.-M. Su and R.-S. Wang, Int. J. Quantum Chem., 2010, 110, 865 CAS.
- J.-G. Kang, Y.-K. Jeong, S.-I. Oh, H.-J. Kim, C. Park and E. R. R. Tiekink, Bull. Korean Chem. Soc., 2010, 31, 2151 CrossRef CAS.
- E. R. R. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1233 CrossRef.
- P.-S. G. Kim, Y. Hu, M.-C. Brandys, T. J. Burchell, R. J. Puddephatt and T. K. Sham, Inorg. Chem., 2007, 46, 949 CrossRef CAS.
- P. G. Jones, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 823 Search PubMed.
- F. Calderazzo, J. Organomet. Chem., 1990, 400, 303 CrossRef CAS.
- W. Manchot and H. Gall, Ber. Bunsenges. Phys. Chem., 1925, 58, 2175 Search PubMed.
- O. Elbjeirami, S. Yockel, C. F. Campana, A. K. Wilson and M. A. Omary, Organometallics, 2007, 26, 2550 CrossRef CAS.
- N. A. Barnes, K. R. Flower, S. A. Fyyaz, S. M. Godfrey, A. T. McGown, P. J. Miles, R. G. Pritchard and J. E. Warren, CrystEngComm, 2010, 12, 784 RSC.
- N. A. Barnes, K. R. Flower, S. M. Godfrey, P. A. Hurst, R. Z. Khan and R. G. Pritchard, CrystEngComm, 2010, 12, 4240 RSC.
- K. R. Flower, A. T. McGown, P. J. Miles, R. G. Pritchard and J. E. Warren, Dalton Trans., 2010, 39, 3509 RSC.
- R. C. Bott, P. C. Healy and G. Smith, Aust. J. Chem., 2004, 57, 213 CrossRef CAS.
- P. D. Cookson and E. R. T. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1896 CrossRef.
- U. Monkowius and M. Zabel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m196 Search PubMed.
- I. Dance, Mol. Cryst. Liq. Cryst., 2005, 440, 265 CAS.
- D. Fernández, M. I. García-Seijo, M. Bardaji, A. Laguna and M. E. Garcia-Fernández, Dalton Trans., 2008, 2633 RSC.
- A. L. Balch and E. Y. Fung, Inorg. Chem., 1990, 29, 4764 CrossRef CAS.
- J. D. E. T. Wilton-Ely, A. Schier, N. W. Mitzel and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 2001, 1058 RSC.
- L. Hao, A. Mansour, R. J. Lachicotte, H. J. Gysling and R. Eisenberg, Inorg. Chem., 2000, 39, 5520 CrossRef CAS.
- F. S. Kuan, S. Y. Ho, P. P. Tadbuppa and E. R. T. Tiekink, CrystEngComm, 2008, 10, 548 RSC.
- U. E. I. Horvath, S. Cronje, J. M. McKenzie, L. J. Barbour and H. G. Raubenheimer, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1605 CAS.
- (a) I. O. Koshevoy, L. Koskinen, E. S. Smirnova, M. Haukka, T. A. Pakkanen, A. S. Melnikov and S. P. Tunik, Z. Anorg. Allg. Chem., 2010, 636, 795 CrossRef CAS; (b) I. O. Koshevoy, L. Koskinen, M. Haukka, S. P. Tunik, P. Y. Serdobintsev, A. S. Melnikov and T. A. Pakkanen, Angew. Chem., Int. Ed., 2008, 47, 3942 CrossRef CAS; (c) I. O. Koshevoy, A. J. Karttunen, S. P. Tunik, M. Haukka, S. I. Selivanov, A. S. Melnikov, P. Y. Serdobintsev, M. A. Khodorkovsky and T. A. Pakkanen, Inorg. Chem., 2008, 47, 9478 CrossRef CAS; (d) I. O. Koshevoy, Y.-C. Lin, A. J. Karttunen, M. Haukka, P.-T. Chou, S. P. Tunik and T. A. Pakkanen, Chem. Commun., 2009, 2860 RSC.
- M.-H. Nguyen and J. H. K. Yip, Organometallics, 2010, 29, 2422 CrossRef CAS.
- I. O. Koshevoy, C.-L. Lin, A. J. Karttunen, M. Haukka, C.-W. Shih, P.-T. Chou, S. P. Tunik and T. A. Pakkanen, Chem. Commun., 2011, 47, 5533 RSC.
- C. Croix, A. Balland-Longeau, H. Allouchi, M. Giorgi, A. Duchene and J. Thibonnet, J. Organomet. Chem., 2005, 690, 4835 CrossRef CAS.
- S. Ahrland, K. Dreisch, B. Norén and Å. Osklarsson, Mat. Chem. Phys., 1993, 35, 281 CrossRef CAS.
- C.-Y. Yue, W.-D. Chen, F.-L. Jiang, R. Feng and M.-C. Hong, Inorg. Chem. Commun., 2010, 13, 105 CrossRef CAS.
- M. O. Awaleh, F. Baril-Robert, C. Reber, A. Badia and F. Brisse, Inorg. Chem., 2008, 47, 2964 CrossRef CAS.
- M. G. B. Drew and M. J. Riedl, J. Chem. Soc., Dalton Trans., 1973, 52 RSC.
- J. Coetzee, W. F. Gabrielli, K. Coetzee, O. Schuster, S. D. Nogai, S. Cronje and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2007, 46, 2497 CrossRef CAS.
- N. Savjani, S. J. Lancaster, S. Bew, D. L. Hughes and M. Bochmann, Dalton Trans., 2011, 1079 RSC.
- M. T. Räisänen, N. Runeberg, M. Klinga, M. Nieger, M. Bolte, P. Pyykkö, M. Leskelä and T. Repo, Inorg. Chem., 2007, 46, 9954 CrossRef.
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Kogel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900 CrossRef CAS.
- J. Zeng, Y. Ma, U. Jeong and Y. Xia, J. Mater. Chem., 2010, 20, 2290 RSC.
- F. Mendizabal, D. Reyes and C. Olea-Azar, Int. J. Quantum Chem., 2006, 106, 906 CrossRef CAS.
- C. Hollatz, A. Schier and H. Schmidbaur, J. Am. Chem. Soc., 1997, 119, 8115 CrossRef CAS.
- D. M. P. Mingos, J. Yau, S. Menzer and D. J. Williams, J. Chem. Soc., Dalton Trans., 1995, 319 RSC.
- C. E. Strasser, L. Dobrzanska, H. Schmidbaur, S. Cronje and H. G. Raubenheimer, J. Mol. Struct., 2010, 977, 214 CrossRef CAS.
- H.-N. Adams, W. Hiller and J. Strähle, Z. Anorg. Allg. Chem., 1982, 485, 81 CrossRef CAS.
- M. Freytag and P. G. Jones, Chem. Commun., 2000, 277 RSC.
- K. J. Kilpin, R. Horvath, G. B. Jameson, S. G. Telfer, K. C. Gordon and J. D. Crowley, Organometallics, 2010, 29, 6186 CrossRef CAS.
- M. A. Rawashdeh-Omary, J. M. López-de-Luzuriaga, M. D. Rashdan, O. Elbjeirami, M. Monge, M. Rodrígez-Castillo and A. Laguna, J. Am. Chem. Soc., 2009, 131, 3824 CrossRef CAS.
- R.-Y. Liau, T. Mathieson, A. Schier, R. J. F. Berger, N. Runeberg and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 881 CAS.
- O. Elbjeirami, M. W. A. Gonser, B. N. Stewart, A. E. Bruce, M. R. M. Bruce, T. R. Cundari and M. A. Omary, Dalton Trans., 2009, 1 Search PubMed.
- O. Elbjeirami, S. Yockel, C. F. Campana, A. K. Wilson and M. A. Omary, Organometallics, 2007, 26, 2550 CrossRef CAS.
- O. Elbjeirami and M. A. Omary, J. Am. Chem. Soc., 2007, 129, 11384 CrossRef CAS.
- V. P. Dyadchenko, N. M. Belov, M. A. Dyadchenko, Y. L. Slovokhotov, A. M. Banaru and D. A. Lemenovskii, Russ. Chem. Bull., Int. Ed., 2010, 59, 539 CrossRef CAS.
- V. P. Dyadchenko, N. M. Belov, D. A. Lemenovskii, M. Y. Antipin, K. A. Lyssenko, A. E. Bruce and M. R. M. Bruce, J. Organomet. Chem., 2010, 695, 304 CrossRef CAS.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef.
- X. Meng, T. Moriuchi, M. Kawahata, K. Yamaguchi and T. Hirao, Chem. Commun., 2011, 47, 4682 RSC.
- I. J. B. Lin and C. S. Vasam, Can. J. Chem., 2005, 83, 812 CrossRef CAS.
- L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 230 CrossRef CAS.
- A. John and P. Ghosh, Dalton Trans., 2010, 39, 7183 RSC.
- M. V. Baker, P. J. Bernard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625 CrossRef CAS.
- M. K. Samantaray, K. Pang, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 4135 CrossRef.
- C. Dash, M. M. Shaikh, R. J. Butcher and P. Ghosh, Inorg. Chem., 2010, 49, 4972 CrossRef CAS.
- H. M. J. Wang, C. Y. L. Chen and I. J. B. Lin, Organometallics, 1999, 18, 1216 CrossRef CAS.
- P. D. Fremont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411 CrossRef.
- C. Carcedo, J. C. Knight, S. J. A. Pople, I. A. Fallis and A. Dervisi, Organometallics, 2011, 30, 2553 CrossRef CAS.
- J. Y. Z. Chiou, S. C. Luo, W. C. You, A. Bhattacharyya, C. S. Vasam, C. H. Huang and I. J. B. Lin, Eur. J. Inorg. Chem., 2009, 1950 CrossRef CAS.
- P. C. Kunz, C. Wetzel, S. Kögel, M. U. Kassack and B. Spingler, Dalton Trans., 2011, 40, 35 RSC.
- U. E. I. Horvath, J. M. Mckenzie, S. Cronje, H. G. Raubenheimer and L. J. Barbour, Chem. Commun., 2009, 6598 RSC.
- D. M. P. Mingos, J. Yau, S. Menzer and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1995, 34, 1894 CrossRef CAS.
- R. J. Puddephatt, Coord. Chem. Rev., 2001, 216, 313 CrossRef.
- R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327 CrossRef CAS.
- C. Bartolomé, M. Carrasco-Rando, S. Coco, C. Cordivilla, J. M. Martín-Alvarez and P. Espinet, Inorg. Chem., 2008, 47, 1616 CrossRef.
- R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327 CrossRef CAS.
- D. Rios, D. M. Pham, J. C. Fettinger, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2008, 47, 3442 CrossRef CAS.
- M. Saitoh, A. L. Balch, J. Yuasa and T. Kawai, Inorg. Chem., 2010, 49, 7129 CrossRef CAS.
- G. Minghetti and F. Bonati, Angew. Chem., Int. Ed. Engl., 1972, 11, 429 CrossRef CAS.
- J. E. Parks and A. L. Balch, J. Organomet. Chem., 1974, 71, 453 CrossRef CAS.
- E. Y. Fang, M. M. Olmstead, J. C. Vickery and A. L. Balch, Coord. Chem. Rev., 1998, 171, 151 CrossRef.
- A. Burini, A. A. Mohamed and J. P. Fackler Jr, Comments Inorg. Chem., 2003, 24, 253 CrossRef CAS.
- H. E. Abdou, A. A. Mohamed, J. P. Fackler Jr, in ref. 17, p. 24 ff.
- A. Omary, A. A. Mohamed, M. A. Rawashdeh-Omary and J. P. Fackler Jr, Coord. Chem. Rev., 2005, 249, 1372 CrossRef.
- C. Yang, M. Messerschmidt, P. Coppens and M. A. Omary, Inorg. Chem., 2006, 45, 6592 CrossRef CAS.
- O. Elbjeirami, M. D. Rashdan, V. Nesterov and M. A. Rawashdeh-Omary, Dalton. Trans., 2010, 39, 9465 RSC.
- T. Osuga, T. Murase, K. Ono, Y. Yamauchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 15553 CrossRef CAS.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS.
- M. A. Omary, M. A. Rawashdeh-Omary, M. W. A. Gonser, O. Elbjeirami, T. Grimes, T. R. Cundari, H. V. K. Diyabalanage, C. S. P. Gamage and H. V. Rasika Dias, Inorg. Chem., 2005, 44, 8200 CrossRef CAS.
- M. D. Adams, M. W. Johns, D. W. Drew, in ref. 16, p. 65.
- J. Marsden and I. House, The Chemistry of Gold Extraction, Ellis Howood, New York, 1992 Search PubMed.
- Z. Tang, A. P. Litvinchuk, H.-G. Lee and A. M. Guloy, Inorg. Chem., 1998, 37, 4752 CrossRef CAS.
- A. Bauer, W. Schneider and H. Schmidbaur, Inorg. Chem., 1997, 36, 2225 CrossRef CAS.
- P. Pyykkö, W. Schneider, A. Bauer, A. Bayler and H. Schmidbaur, J. Chem. Soc., Chem. Commun., 1997, 1111 RSC.
- L. Cao, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2007, 46, 1361 CrossRef CAS.
- N. L. Coker, J. A. Krause Bauer and R. C. Elder, J. Am. Chem. Soc., 2004, 126, 12 CrossRef CAS.
- N. L. Coker, C. E. Bedel, J. A. Krause and R. C. Elder, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m319 Search PubMed.
- J. M. Forward, D. Bohmann, J. P. Fackler Jr and R. Staples, Inorg. Chem., 1995, 34, 6330 CrossRef CAS.
- Y.-A. Lee, J. E. Mcgrath, R. J. Lachicotte and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 10662 CrossRef CAS.
- R. K. Arvapally, P. Sinha, S. R. Hettiarachchi, N. L. Coker, C. E. Bedel, H. H. Patterson, R. C. Elder, A. K. Wilson and M. A. Omary, J. Phys. Chem. C, 2007, 111, 10689 CAS.
- H. Ruben, A. Zalkin, M. O. Faltens and D. H. Templeton, Inorg. Chem., 1974, 13, 1836 CrossRef CAS.
- R. Baggio and S. Baggio, J. Inorg. Nucl. Chem., 1973, 35, 3191 CrossRef CAS.
- S. L. Lawton, W. J. Rohrbaugh and G. T. Kokotailo, Inorg. Chem., 1972, 11, 2227 CrossRef CAS.
- A. J. Fischmann and L. Spiccia, Dalton Trans., 2011, 40, 4803 RSC.
- P. Römbke, A. Schier, F. Wiesbrock and H. Schmidbaur, Inorg. Chim. Acta, 2003, 347, 123 CrossRef.
- P. Römbke, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 2001, 2482 RSC.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1747 RSC.
- K. J. Harris and R. E. Wasylishen, Inorg. Chem., 2009, 48, 2316 CrossRef CAS.
- A. Rosenzweig and D. T. Cromer, Acta Crystallogr., 1959, 12, 709 CrossRef CAS.
- C. M. Beavers, L. Paw and M. M. Olmstead, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m300 Search PubMed.
- R. J. Schubert and K.-J. Range, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1118 CAS.
- M. A. Rawashdeh-Omary, M. A. Omary, H. H. Patterson and J. P. Fackler Jr, J. Am. Chem. Soc., 2001, 123, 11237 CrossRef CAS.
- N. Blom, A. Ludi, H.-B. Bürgi and K. Richy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1767 CrossRef.
- G. A. Bowmaker, B. J. Kennedy and J. C. Reid, Inorg. Chem., 1998, 37, 3968 CrossRef CAS.
- M. J. Katz, V. K. Michaelis, P. M. Aguiar, R. Yson, H. Lu, H. Kalurachchi, R. J. Batchelor, G. Schreckenbach, S. Kroeker, H. H. Patterson and D. B. Leznoff, Inorg. Chem., 2008, 47, 6353 CrossRef CAS.
- H. H. Patterson, J. Bourassa and G. Shankle, Inorg. Chim. Acta, 1994, 226, 345 CrossRef CAS.
- M. J. Katz, P. M. Aguiar, R. J. Batchelor, A. A. Bokov, Z. G. Ye, S. Kroeker and D. B. Leznoff, J. Am. Chem. Soc., 2006, 128, 3669 CrossRef CAS.
- W. Dong, Y. Ouyang, L.-N. Zhu, D.-Z. Liao, Z.-H. Jiang, S.-P. Yan and P. Cheng, Inorg. Chem. Commun., 2007, 10, 779 CrossRef CAS.
- H.-B. Zhou, S.-P. Wang, Z.-Q. Liu, D.-Z. Liao, Z.-H. Jiang, S.-P. Yan and P. Cheng, Inorg. Chim. Acta, 2006, 359, 533 CrossRef CAS.
- S.-P. Wang, Y. Song, D.-Z. Gao, L.-C. Li, Q.-M. Wang, D.-Z. Liao, Z.-H. Jiang and S.-P. Yan, Helv. Chim. Acta, 2005, 88, 3000 CrossRef CAS.
- G. Agusti, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2008, 47, 2552 CrossRef CAS.
- J. Suárez-Varela, H. Sakiyama and E. Colacio, Dalton Trans., 2007, 249 RSC.
- J. Suárez-Varela, A. J. Mota, H. Aouryaghal, J. Cano, A. Rodríguez-Diéguez, D. Luneau and E. Colacio, Inorg. Chem., 2008, 47, 8143 CrossRef.
- Y. Guo, Y. Ma, N. Zhou, Z.-Q. Liu, Q.-L. Wang, A.-P. Yan and D.-Z. Liao, Z. Anorg. Allg. Chem., 2010, 636, 865 CrossRef CAS.
- Y. Guo, Z.-Q. Liu, B. Zhao, Y.-H. Feng, G.-F. Xu, S.-P. Yan, P. Cheng, Q.-L. Wang and D.-Z. Liao, CrystEngComm, 2009, 11, 61 RSC.
- A. M. Madalan, N. Avarvari and M. Andruh, Cryst. Growth Des., 2006, 6, 1671 CAS.
- J. S. Ovens and D. B. Leznoff, Dalton Trans., 2011, 40, 4140 RSC.
- L. Ladner, T. Ngo, C. Crawford, Z. Assefa and R. E. Sykora, Inorg. Chem., 2011, 50, 2199 CrossRef CAS.
- M. A. Bennett, D. C. R. Hockless, L. L. Welling and A. C. Willis, Organometallics, 2001, 20, 79 CrossRef CAS.
- R. G. Raptis, L. C. Porter, R. J. Emrich, H. H. Murray and J. P. Fackler Jr, Inorg. Chem., 1990, 29, 4408 CrossRef CAS.
- S. Canales, O. Crespo, M. C. Gimeno, P. G. Jones, A. Laguna and F. Mendizabal, Organometallics, 2001, 20, 4812 CrossRef CAS.
- T. M. Klapötke, B. Krumm, J.-C. Galvez-Ruiz and H. Nöth, Inorg. Chem., 2005, 44, 9625 CrossRef.
- F. Mendizabal, G. Zapatero-Torres and C. Oleo-Azar, Chem. Phys. Lett., 2003, 382, 92 CrossRef CAS.
- F. Mendizabal and P. Pyykkö, PhysChemChemPhys, 2004, 6, 900 RSC.
- J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and T. Lasanta, Chem. Commun., 2011, 47, 6795 RSC.
| This journal is © The Royal Society of Chemistry 2012 |