Update on selective oxidation using gold
Cristina Della
Pina
,
Ermelinda
Falletta
and
Michele
Rossi
*
Dipartimento di Chimica Inorganica, Metallorganica e Analitica “L. Malatesta”, Università degli Studi di Milano, CNR-ISTM, via Venezian, 21, 20133 Milano, Italy. E-mail: michele.rossi@unimi.it; Fax: +39 02 50314405; Tel: +39 02 50314397
First published on 5th July 2011
Abstract
This critical review aims to update the recent development in the selective oxidation of organic compounds by gold catalysis, highlighting the progress in the last three years. Following the impressive developments in the last decades, several protocols for catalytic oxidation are today available, which are based on the extraordinary properties of gold in terms of catalytic activity, selectivity, reusability and resistance to poisons. Beside many other applications, gold can be recommended for green processes dedicated to fine chemicals, pharmaceuticals and the food industry owing to its recognized bio-compatibility. The collected literature is focused on experiments concerning the oxidation of different chemical groups and could be of interest, in the wide area of organic chemistry, for improving previous processes or for exploring new catalytic pathways (174 references).
 Cristina Della Pina | Cristina Della Pina received the Master’s degree and awarded the PhD degree in industrial chemistry at the Università degli Studi Milano under the supervision of Prof. Michele Rossi. She is presently Assistant Professor of General Chemistry and her research work focuses on the development of heterogeneous catalysts for the selective oxidation of organic compounds. |
 Ermelinda Falletta | Ermelinda Falletta got her Master’s degree in organic chemistry (2004) at the University of Palermo. In 2005 she joined Prof. Rossi's research group by a scholarship of the Centre of Excellence CIMAINA at the University of Milan and then she was involved in the research of gold catalysis as a graduate technician at the Department of Inorganic Chemistry. She is working on her PhD thesis concerning new environmentally friendly synthetic routes for conducting organic polymers and their technological applications. |
 Michele Rossi | Michele Rossi obtained his master degree in Industrial Chemistry at the Università degli Studi di Milano. After two decades at the University of Bari, where he early became full professor of chemistry, he transferred to Milano where he held the Chair of General and Inorganic Chemistry up to his retirement. His research has been mainly devoted to the activation of small molecules as H2, N2 and O2. His research group has been pioneering in the application of gold catalysis to the liquid phase oxidation of alcohols and sugars. |
1. Introduction
One of the most exciting contributions to catalysis science has been the discovery of the catalytic properties of gold nanoparticles, which has led to the renaissance of gold chemistry. Basic reports in the last ten years concern many important applications such as low-temperature CO oxidation,1–13 H2O2 synthesis from the elements,14–16 water gas shift,17,18 C–C coupling reactions,19–21 synthesis of N and O-heterocycles22 and hydrogenation of organic compounds.23–27Oxidation by gold in organic chemistry is of relevant interest in supporting green processes based on the use of stable, selective and non-toxic heterogeneous catalysts, as well as air or pure O2 as eco-friendly oxidants. In many processes, in fact, gold catalysts fulfil all the above features.
Considering the organic substrates, strategic applications of gold catalysis concern the transformation of renewable biological resources, a pressing requirement for balancing the natural CO2 cycle.
The selective oxidation using gold has been previously reviewed until ca. 2007,28 and later general aspects of gold catalysis, including selective oxidation, have been reported.14,29–35
The present review aims to outline the recent progress in the selective oxidation applied to chemical synthesis, assembling the literature according to the typology of the organic reagents, as adopted in the previous paper.28
2. Selective oxidation of hydrocarbons
Selective oxidation of hydrocarbons with oxygen is of crucial importance for the sustainable exploitation of conventional fossil feedstock and over recent years the aerobic functionalization of aliphatic compounds has attracted new experimental research producing important results particularly in the C–H bond activation.2.1 Methane oxidation
Basic research of Shilov et al. shows the fundamental role of the bioinorganic catalysis in the aerobic oxidation of methane mimicking enzymatic catalysis.36 According to recent developments,37 gold complexes with the bioflavonoid rutin ligand in water are able to catalyze the selective oxidation of methane to methanol by air at room temperature and atmospheric pressure. The oxidative activation of the aliphatic C–H bond, which includes methane, ethane and propane, is mediated by Au(I) in the presence of NADH as a reducing agent.The TON per mole of Au over a period of time of 48 h is 0.6 h−1 for methane and increases to 4.2 h−1 for ethane and 4.6 h−1 for propane, thus following the drop in the C–H bond cleavage energy of the hydrocarbons. Although far from practical application, these experiments on alkane oxidation in solution allow speculation on the reaction mechanism, a basic step for progress in hydrocarbon oxidation. According to a suggested molecular model, the activation of the aliphatic C–H bond occurs upon oxidative addition to the gold complex. Considering that gold is not an oxophilic coordination site and methylgold complexes are rather resistant to hydrolysis, it has been assumed that the C–O bond formation is caused by hydrolysis of the C–H bond rather than molecular oxygen insertion, the role of this latter being restricted to the dehydrogenation activity producing hydrogen peroxide as the by-product (Scheme 1).
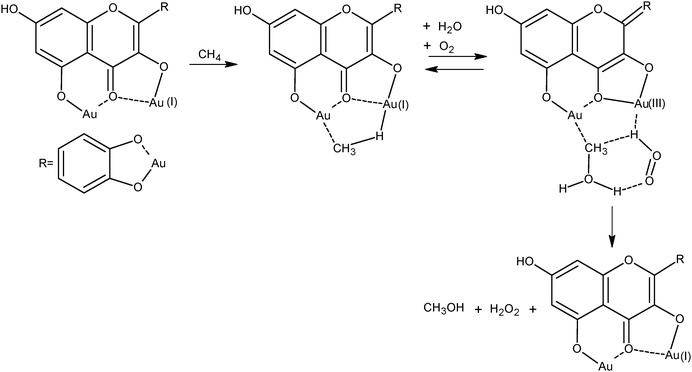 | ||
| Scheme 1 Activation of methane and oxygen by gold complex. | ||
This is an important point, and also in catalytic processes by gold nanoparticles, because many experiments show that the aerobic oxidation of oxygenated hydrocarbons (alcohols and carbonyls) occurs by a similar dehydrogenation mechanism producing H2O2 through a 2-elecron reduction of O2,38 as noted in the following sections.
Despite the great potential interest, the selective oxidation of CH4 by gold, in the gas phase, has been scarcely investigated. Recently, Llorca et al. have reported that plasma-activated gold nanoparticles catalyze the oxidation of methane to formic acid by oxygen in the presence of hydrogen.39 Catalytic tests were performed at atmospheric pressure over core–shell Au-dodecanethiol protected particle films deposited over cordierite monoliths. The peculiar plasma treatment was accomplished by a glow discharge ensuring the removal of the capping thiol and the formation of the active gold aggregates (3–4 nm). On the contrary, a conventional thermal treatment led to inactive material.
At 200 °C, the specific yield towards HCOOH was 2.9 mmol h−1 gAu−1. In this reaction H2 is fed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio with O2. Thus, considering that Au nanoparticles are active for the reaction of H2 and O2 to form H2O2,15 this result suggests that active oxygen species, as surface hydroperoxides, are involved in methane activation by O2–H2 over heterogeneous Au catalyst, in analogy to the above reported activation in liquid phase by Shilov et al. and to the use of sacrificial H2 in propene epoxidation (see section 2.8).
:1 molar ratio with O2. Thus, considering that Au nanoparticles are active for the reaction of H2 and O2 to form H2O2,15 this result suggests that active oxygen species, as surface hydroperoxides, are involved in methane activation by O2–H2 over heterogeneous Au catalyst, in analogy to the above reported activation in liquid phase by Shilov et al. and to the use of sacrificial H2 in propene epoxidation (see section 2.8).
2.2 Cyclohexane oxidation
Compared to methane and linear alkanes, the selective oxidation of cyclohexane is a smooth reaction having industrial interest as an alternative route to cyclohexanol-cyclohexanone (KA oil) intermediates for nylon industry (Scheme 2).40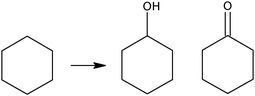 | ||
| Scheme 2 The selective oxidation of cyclohexane to KA oil | ||
Two commercial processes are presently operated in liquid phase, the noncatalytic autoxidation process and the catalyzed process, the latter making use of dissolved cobalt(II) naphthenate.
In recent years, heterogeneous gold catalysts have been experimented with to support a cleaner process. Following previous work on the conversion of cyclohexane by Au on Al2O3 catalyst,41 Xu et al. have investigated new more efficient catalytic systems.42
Accordingly, gold nanoparticles were deposited on alumina pre-treated by silica via a surface sol–gel post-modification method and applied for cyclohexane oxidation using oxygen in the absence of any solvent and initiator. The results show that the surface doping of alumina support with silica is effective in stabilizing gold nanoparticles after calcination at 500 °C for 3 h. In fact, the catalytic activity in terms of specific reaction rate and the selectivity to cyclohexanone are appreciably enhanced up to 13.9 mol h−1 gAu−1 with total selectivity (cyclohexanol + cyclohexanone) above 90%. This effect has been ascribed to the decreased acidity and hydrophilicity caused by surface substitution of alumina with silica.
However, TOF and selectivity are quite high only at the early stage of the reaction (8%–10% conversion) owing to the marked deterioration of the catalytic performance. No conversion was observed in the absence of gold, thus confirming its catalytic role.
The elegant paper of Hereijgersa and Weckhuysen questions the true catalytic role of gold in cyclohexane oxidation.43 Comparing the aerobic oxidation of cyclohexane to cyclohexanone and cyclohexanol over Au catalysts (Au/Al2O3, Au/TiO2, and Au/SBA-15) with the industrial autoxidation process, the authors found that, in contradiction with the literature results, the Au-based catalysts did not exhibit excellent catalytic performance and the combined selectivity decreases below 70% with increasing conversion above 5%. In addition, the observed product and by-product evolution was typical for the autoxidation process, although a significant increase in adipic acid and CO2 formation was observed. Further oxidation experiments containing a radical scavenger completely inhibited the reaction and provided proof that the oxidation follows a radical-chain mechanism. This explains the low selectivity at increasing conversion.
However, the catalytic role of gold in cyclohexane oxidation has been reaffirmed in a following research where gold clusters, Aun (n = 10, 18, 25, 39), on hydroxyapatite, with atomically controlled sizes, show remarkable activity (up to TOF = 18500 h−1) and high selectivity (>90%) in the solvent-free oxidation of cyclohexane at a moderate temperature (150 °C) and atmospheric pressure of oxygen.44
No conversion was observed in the absence of gold, indicating the catalytic role of the metal. The addition of a small amount of TBHP was essential to initiate the reaction, suggesting that this oxidation proceeds via a complex radical chain mechanism. In all the cases, the selectivity towards cyclohexanol + cyclohexanone was very high, reaching 99% for smaller Au clusters, the ketone/alcohol ratio being close to unity. It has been pointed out that the TOFs of the Aun clusters are related to their dimension according to a volcano-type curve showing a maximum at n = 39 (Fig. 1).
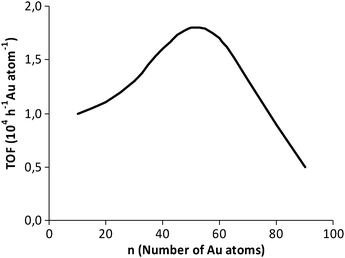 | ||
| Fig. 1 The dependence of the catalytic activity on the cluster size in the cyclohexane conversion. | ||
In conclusion, the question about the true catalytic role of gold in liquid phase oxidation of cyclohexane is still open and a table of discussion about this intriguing argument would be of great interest.
2.3 Oxidation of toluene
Toluene is a basic commodity in organic chemistry which can be functionalized by oxygen to benzyl alcohol, benzaldehyde, benzoic acid, and benzyl benzoate. These products are versatile intermediates in the manufacture of solvents, dyes, pharmaceuticals, perfumes, plasticizers, food preservatives and flame retardants.While benzaldehyde is produced by the chlorination of toluene followed by saponification, direct oxidation of toluene is presently performed to produce benzoic acid by the liquid-phase reaction using oxygen under severe conditions.40 Although many attempts have been devoted to the optimization of this reaction using cobalt45 and other different transition metals as copper, manganese and chromium,46–49 the conversion is still poor (15%) in order to ensure a convenient selectivity.50–55
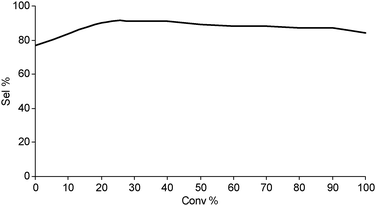
New fundamental experiments on the heterogeneous oxidation of toluene have been presented by Hutchings' group, highlighting the ability of bimetallic Au–Pd in promoting the aerobic oxidation at the CH3 group.56 Under moderate conditions (160 °C, 0.1 MPa oxygen pressure), 100% conversion of toluene has been observed leading to benzyl benzoate in very high yield (around 90%). It has been considered that the high selectivity to benzyl benzoate results from consecutive reactions producing benzyl alcohol, benzaldehyde, the coupling of these latter compounds to give the hemiacetal, followed by further oxidation to the ester (Scheme 3). This hypothesis has been demonstrated by several control experiments. The impressive selectivity to benzylbenzoate up to the total conversion of toluene is noted in Fig. 2.
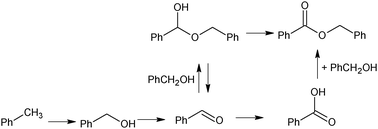 | ||
| Scheme 3 Mechanism of toluene oxidation–condensation to benzyl benzoate with Au–Pd catalyst. | ||
Applying the Au–Pd catalyst to the oxidation of 2-, 3-, and 4-methoxytoluene and 2-, 3-, and 4-nitrotoluene, the authors found this method to be of general application for the oxidation of substituted toluenes, thus opening great interest in organic synthesis.
A strong effect of the material chosen as the metal support has been detected by comparing two different catalytic systems, AuPd/C and AuPd/TiO2: the first had a double catalytic activity with respect to the second one, despite both catalysts having a very similar size distribution of Au–Pd particles. It has been supposed that the difference in the catalytic activity may be related to the morphology of gold nanoparticles, in particular the different number of low coordination sites in the two catalysts.
2.4 α-Pinene oxidation
Oxygen addition to α-pinene represents an interesting target for new synthetic route to commercial products, in particular verbenol and verbenone, which are key intermediates for flavors, fragrances and therapeutic agents57–60 (Scheme 4). A second important aspect of α-pinene oxidation is related to mechanistic aspects of selectivity owing to the complexity of this molecule. Therefore, many catalytic systems have been applied to perform the selective oxidation of α-pinene, including homogeneous and heterogeneous catalysts, but with doubtful results.61–70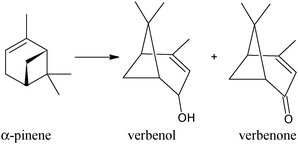
Recently, gold catalysis has been applied to α-pinene oxidation resulting in a remarkable activity.71 Thus, a series of bimetallic catalysts Au–M (where M = Cu, Co and Ru) were supported on TiO2via deposition–precipitation method. The alloyed particles having an average particle size in the range of 3–5 nm were able to convert α-pinene according to the order: AuCu/TiO2 > AuCo/TiO2 > Cu/TiO2 > Au/TiO2 > AuRu/TiO2. In spite of the low selectivity (no single compound exceeds 50% at 70–94% conversion), the reacted mixture contains commercially interesting compounds as verbenone, verbenol, α-pinene oxide and alkyl-pinene peroxide which can be separated upon distillation. By comparing various catalysts, Au–Cu/TiO2 was found to be the most active and selective catalyst towards the formation of verbenone which can be obtained, however, with a modest 48% selectivity at 97% conversion.
Comparing the kinetics of the uncatalyzed and the metal-catalyzed reaction (Fig. 3), as well as the relative product distribution (Table 1), the beneficial role of the bimetallic catalyst is evident in increasing the reaction rate and selectivity. A radical mechanism has been proposed for the metal mediated oxidation of α-pinene, where alkoxy and alkyl-peroxy radicals are formed at the perimeter interface between the metal particles and the support via the adsorption of alkyl peroxide.
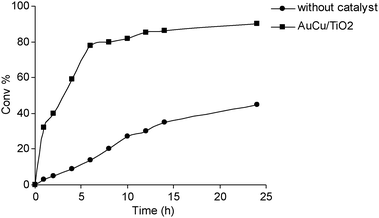 | ||
| Fig. 3 Catalyzed and uncatalyzed oxidation of α-pinene. | ||
2.5 Oxidation of styrene
Beside the interest for organic synthesis, styrene has become a selected molecular probe for testing the activation of molecular oxygen by gold. It is known that gold catalysts promote the aerobic oxidation of styrene which proceeds smoothly in the presence of a small amount of hydrogen peroxide or organic hydroperoxides as an oxygen chain initiator.72 Recent studies have demonstrated that very small gold entities (ca. 1.4 nm) derived from Au55(PPh3)12Cl6 cluster, supported on inert materials, are also robust catalysts for the selective oxidation of styrene by dioxygen in toluene solution in the absence of peroxidic species. In any case, low activity (TOF ca. 0.02 s−1) and poor selectivity towards styrene epoxide (14.0%) were observed, benzaldehyde (82.3%) being the main product, beside small quantities of acetophenone (3.9%) (Scheme 5).73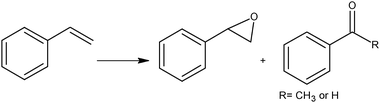
By comparing gold nanoparticles of increasing diameters, a size threshold in catalytic activity has been found resulting in complete inactivity for particles with diameters >∼2 nm. This experimental result suggests that O2 activation arises from the altered electronic structure intrinsic to small gold nanoparticles. Following research has confirmed the strong effect of the particles dimension on the oxygen activation during the oxidation of styrene mediated by gold nanoparticles, as well as the role of peroxidic species.
According to Jin et al.,74 the very small colloidal clusters of gold, Au25, Au38, and Au144, capped with phenylethanethiolate, e.g. Au25 (SC2H4Ph)18, were supported on hydroxyapatite (HAP) and SiO2 and applied in styrene oxidation. Comparing three different oxidation processes, the first using t-butylhydroperoxide (TBHP) as the sole oxidant, the second employing O2 as the oxidant in the presence of a small amount of TBHP as initiator, and the third using O2 as the sole oxidant, the authors found that, in the case of TBHP oxidant, the catalytic activity of Aun was almost independent on the size of the cluster. On the contrary, with O2 as the oxidant, the following order of activity Au25 > Au38 > Au144 was observed, in line with the size-dependent activity reported in ref. 75. The catalytic activity of the thiolate-capped cluster is surprising, considering the expected poisoning effect of sulfur compounds as, for example, detected at very low levels during glucose oxidation with molecular oxygen.75
After removing the capping thiolate by calcination, the catalytic activity of gold particles is still present. However, aggregation of gold was observed which may contrast the expected benefit of de-capping the catalytic centers.74 The decrease of the catalytic activity due to sintering is important only in the case of the oxidation by sole O2 (Fig. 4). Considering the kinetic data obtained under similar reaction conditions with Au25-HAP capped catalyst at 80 °C in toluene solution, TBHP was much more effective (ca. 90% conv.) than O2 (ca. 24% conv.), while TBHP as activator plus O2 as the oxidant showed an intermediate behavior (ca. 47% conv.). Detailed experiments dealing with oxidation by O2 are also reported elsewhere by the same research team.76
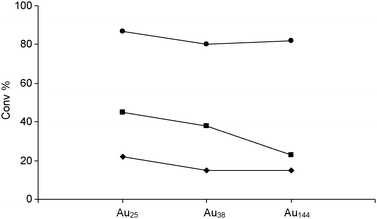 | ||
| Fig. 4 Catalytic activity of uncalcined Aun(SR)m/HAP catalysts for styrene oxidation. TBHP as the oxidant (●); TBHP as an initiator and O2 as the main oxidant (■); O2 as the sole oxidant (✦). Reaction conditions: 80 °C for 12 h in toluene. | ||
It is important to note that, according to the above experiments, the oxidation of styrene produces benzaldehyde with high selectivity, ca. 100% in the case of TBHP and (O2 + TBHP) oxidants, and ca. 80% selectivity with sole O2 oxidant. Acetophenone and styrene oxide (Scheme 5) were formed only as minor reaction products.
A mechanism of selective oxidation of styrene by the three different oxidation systems has been proposed, highlighting the activation of O2 on small gold clusters and the role of peroxidic species.74
Almost at the same time, another research team has reported styrene oxidation mediated by gold clusters, but with a surprisingly different result concerning the selectivity. Thus, according to Tsukuda et al., Au25 cluster supported on hydroxyapatite is able to oxidize styrene with 100% conversion using tert-butylhydroperoxide in toluene solution at 80 °C with high selectivity, 92%, which is, however, directed towards styrene oxide instead of benzaldehyde.77
Considering that in these experiments the solvent (toluene), reaction temperature (80 °C) and oxidant (TBHP) are the same as those used in ref. 76, while the cluster precursor Au25(SG)18 (SG = glutathione ligand) (I) has a close similarity with precursor Au25(SC2H4Ph)18 (II), it is hard to rationalize why the oxidation of styrene leads to 92% styrene oxide starting from II and 100% benzaldehyde starting from I. It is worth noting that both the capped precursors were deposited on the same support, hydroxyapatite, and used after calcination at 200–300 °C. According to Tsukuda and coworkers,77 the properties of the Au/HPA catalyst are quite sensitive to poorly clarified morphological aspects. In particular, the catalyst prepared from the Au25(SG)18 precursor displays much higher selectivity to styrene oxide (>90%) than the Au/HPAads catalyst prepared from HAuCl4 by adsorption technique (ca. 60% selectivity) despite a similar activity and dimension of the supported gold particles (ca. 1.4 nm and ca. 1.7 nm, respectively) (Fig. 5).
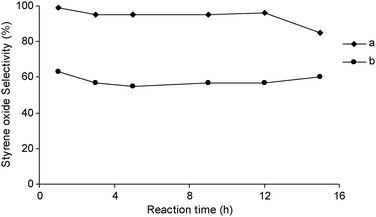 | ||
| Fig. 5 Selectivity of Au25-HAP (a) and Au-HAPAd (b) to styrene oxide. | ||
The electronic spectra of the two catalysts, however, differ owing to the absence of the plasmon band in the cluster-derived catalyst and its presence in the other one, indicating the less and more metallic character of the two gold entities.
Kinetic experiments have been used for interpreting the different chemical behavior: it has been demonstrated that the less selective catalyst is more effective in decomposing styrene oxide into a variety of products, thus lowering the selectivity.
The versatility of styrene as intermediate for the production of oxygenated products by gold catalysis is emphasized in a study describing a new selective route to acetophenone.78 Thus, a supported Pd–Au catalyst is efficient for the selective oxidation of styrene to acetophenone with H2O2 in supercritical CO2. The presence of the CO2 medium allows high conversion of styrene and enhances the selectivity to acetophenone by inhibiting the formation of different by-products such as benzaldehyde and benzoic acid. The nature of the support has great influence on the catalytic performance of Pd–Au catalysts. Al2O3 was the best support allowing a relevant 87% selectivity at 68% conversion achieved at 9 MPa and 120 °C. In order to make the reaction of general synthetic interest, substituted styrenes have been reacted under similar conditions. The result was that electron-donating groups at the para position increased the conversion of styrene, and electron-withdrawing groups at the meta position decreased both the conversion and the selectivity to acetophenone.
Beside synthetic applications, the ligand-covered gold nanoparticles have been investigated in order to derive useful models. The ab initio investigation of structural, electronic, catalytic and selective properties of Au55(PPh3)12Cl6 has been reported by Zeng et al.79 It has been proposed that the activation of O2 derives from a combined effect of triphenylphosphine ligands and surface structure of the “magic-number” quasi-icosahedral Au55 core, where ligand-encompassed triangular Au6 faces act as the active sites. Under the Eley-Rideal mechanism, the active sites accommodate styrene and the O2 molecules, these latter being activated towards the formation of superoxo species. The negatively charged and low-coordinated Au atoms in the active site facilitate O2 activation leading to a high yield of benzaldehyde. However, as pointed out by Turner et al.,73 the removal of ligands by heat treatment can significantly increase the activity and selectivity of Au nanoparticles. Further DFT calculations showed that catalytic activity of gold cluster is related to the size and the structure of the cluster.80
2.6 Oxidation of phenols
Quinones represent an important class of chemical intermediates for drugs, fragrances and vitamins production. Recently, heterogeneous catalytic routes have been reported to produce quinones from the corresponding phenols in the liquid phase, using TBHP, hydrogen peroxide or molecular oxygen as the oxidants.81 In this context, a new study on the performance of gold nanoparticles in the oxidation of substituted phenols using hydrogen peroxide has been reported.82 By investigating the oxidation of various substituted phenols (2,6-di-tert-butyl phenol and 2,3,6-trimethyl phenol) with aqueous hydrogen peroxide, it has been found gold deposited on inorganic supports behaves as an efficient catalyst. Differently from conventional catalysts, such as Ti-containing mesoporous silicas, which convert phenols to the corresponding benzoquinones, gold nanoparticles displayed a very selective production of biaryl compounds (3,3′,5,5′-tetra-tert-butyl diphenoquinone and 2,2′,3,3′,5,5′-hexamethyl-4,4′-biphenol, respectively, Scheme 6). Product yield and selectivity depend on the solvent used, the best results being obtainable in methanol with yields >98%. It has been suggested that the low stability of Au–H2O2 adducts, compared to high stability of Ti–OOH species, favours the formation of phenoxy radicals and their C–C coupling reactions. This unusual behavior of the gold catalyst opens up interesting perspectives in the clean synthesis of biaryl compounds for fine chemicals.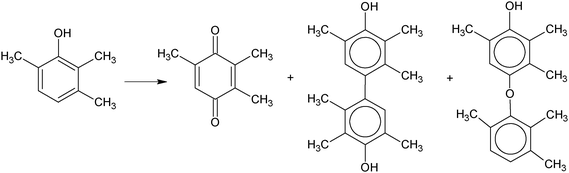
2.7 Oxidation of benzylic compounds
Benzylic compounds undergo a smooth oxidation to ketones (Scheme 7) by means of supported gold catalyst, Au/TiO2, without any organic solvent under mild reaction conditions (≤100 °C, 1 atm O2). For instance, indane was oxidized outside the aromatic ring with high selectivity 90% to 1-indanone at 46% conversion. The catalyst can be recycled without loss of activity.83 The result of this study confirms the inertness of the aromatic ring towards oxygen addition already detected in the case of toluene oxidation (at the methyl group), styrene oxidation (at the olefinic carbons) and phenols oxidation (C–C coupling).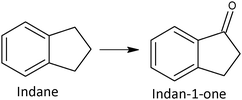
2.8 Oxidation of propene
The pioneering research on the selective oxidation of propene to propene oxide (PO) by gold catalysis started by Haruta in 19971 leading, in lab-scale experiments, to important results of commercial application exceeding 300 gPO gAu−1 h−1.84 It has been previously recognized that oxygen activation in the epoxidation reactions requires the use of sacrificial hydrogen in order to generate active hydroperoxidic species.72Now, academic studies and patent claims have presented strategic results for further improvements, which can open concrete perspectives for the realization of a gold-catalyzed process to PO avoiding, or limiting, the use of H2 in the reagents.
As reported by Lee et al., subnanometric gold clusters (Au6–Au10) are able to catalyze propene epoxidation in the absence of molecular hydrogen, this latter being advantageously replaced by the safer water vapor.85 According to the authors, the role of water as a substitute of H2 is to ensure the hydroxy equilibrium at the surface of the support, a key step for O2 activation. A second important point outlined in this investigation is that alumina, instead of titania, has been applied as the support of gold. Titania was previously considered as an essential aid for assisting the formation of peroxidic species.
In a parallel study, Ojeda and Iglesia also describe the epoxidation of propene in the absence of sacrificial hydrogen, using the classic Au/TiO2 catalyst in the presence of water.86 According to the proposed mechanism, the crucial step of the reaction is the formation of hydroperoxidic species from H2O–O2 interaction at the surface of nanometric gold particles. Compared to the reacting system O2 + H2, the rate of propene conversion to PO with H2O + O2 is significantly lower, but the latter system is safer and avoids significant losses of the costly H2 co-reactant via its unproductive pathways to H2O. According to the report, the thermodynamically unfavorable HOO species can be formed as a reaction intermediate because of the their kinetic coupling with propene epoxidation that splits the HOO intermediates to form PO.
The target of avoiding the presence of a relevant amount of H2 in the selective oxidation of propene has been also fulfilled by Haruta et al.87 In particular, the process has been proposed which is characterized by the use of oxygen in the presence of water and a catalytic amount of hydrogen. The gas phase process occurs in the presence of gold nanoparticles supported on an alkali-treated titanosilicalite or mesoporous titanosilicate. The key factor described in the patent is the use of a catalytic amount of hydrogen, the role of H2 being substituted by water present in the reacting gas.
According to this combined process, it is possible to produce propene oxide at a much higher rate with respect to the H2-free methods by Lee et al.85 and Ojeda and Iglesia.86
3. Oxidation of alcohols
The aerobic oxidation of alcohols by gold has been widely investigated owing to interest in organic synthesis, principally to carbonylic and carboxylic compounds, as a model reaction for oxygen activation. Among many different substrates, benzyl alcohol and glycerol were the most investigated ones.The liquid phase oxidation of benzyl alcohol is catalyzed by several transition metal catalysts and occurs smoothly due to activation of the aromatic ring. It has been often used as a reference test of the activity of gold catalysts. From the synthetic point of view, the chlorine-free route to benzaldehyde represents a concrete synthetic goal,28 while scientific interest is reserved to the effect of the ring substitution on the reactivity.
The importance of the supporting material in promoting gold catalysis has been outlined by several research groups moving from the former conventional materials, as C, SiO2, Al2O3, TiO2, to more sophisticated materials. As we will see, however, good results in terms of activity and selectivity can be achieved independently from the support. Thus, carbon nanotubes and nanofibers have been used as an alternative to activated carbon.88 In order to achieve a better metal distribution, these materials were previously functionalized by oxidation with nitric acid followed by amination with gaseous NH3. TEM analysis revealed that the introduction of nitrogen functionalities improved the dispersion of the metal nanoparticles on the surface of the support. This led to improved activity of the catalysts with respect to pristine carbon nanostructures when tested in the liquid phase oxidation of benzyl alcohol and cinnamyl alcohol. The bimetallic Au–Pd catalysts showed good activity (max. TOF 1099 h−1) and selectivity to the aldehyde (max. 96%).
Metal–organic frameworks (MOFs) are emerging porous materials obtained from metal ions and organic linkers, e.g. Cr3(F,OH)O[(O2C)–C6H4–(CO2)]3, which show spatial arrangement and nanosized channels similar to zeolites. According to Liu et al.,89 gold nanoparticles, deposited on MOF from colloidal precursors, has led to the preparation of active heterogeneous catalysts for a variety of reactions.
In particular, benzylic alcohols, either primary or secondary, have been converted to the corresponding benzylic aldehydes or ketones in quantitative yield. Substituted benzyl alcohols containing electron-donating groups such as CH3, OCH3, or OH, were more easily oxidized than those containing electron withdrawing groups.
A simple and efficient procedure for the preparation of gold(0) nanoclusters stabilized by zeolite framework has been presented, which includes the ion-exchange-impregnation of Au3+ ions with zeolite-Y followed by reduction using sodium borohydride.90 The resulting Au(0)–NaY catalysts were found to be highly active, selective and reusable catalyst in the aerobic oxidation of benzyl alcohol which was carried out in toluene at 80 °C in the presence of stoichiometric amounts of Na2CO3. No explanation is given by the authors on the role of the basic carbonate which was used in the absence of water. In any case, the reaction leads to benzaldehyde in high yield (99% selectivity at 95% conversion) and the robust catalyst can be reused. The reaction rate is, however, low (TOF 55 h−1).
The preparation method and the catalytic properties of gold deposited on structured mesoporous silica materials have been reported.91 Au/SBA-15 and Au–Pd/SBA-15 catalysts were obtained by the grafting methods using 3-(mercaptopropyl)trimethoxylsilane as the grafting agent. It was found that the Au–Pd/SBA-15 catalyst could highly disperse metal nanoparticles in the mesoporous channels and exhibits excellent catalytic performances (TOFs up to 990 h−1) for the selective oxidation of benzyl alcohol to benzaldehyde (max. selectivity 98% at 34% conversion) under mild reaction conditions (aqueous Na2CO3 solution, air at atmospheric pressure, 80 °C). According to the authors, a high stability of the catalyst has been obtained by preventing agglomeration and leaching of metal nanoparticles restricted inside the mesopores. The addition of Pd to Au/SBA-15 catalysts decreased the size of gold nanoparticles, thus improving the activity.
Another simple, effective method has been utilized to immobilize very small, ca. 1 nm, Au clusters within mesoporous silicas (SBA-15, MCF, HMS) using triphenylphosphine-protected clusters as precursors, which were deposited on the silica surface in an organic medium.92 A feature of this method is the ability to disperse Au clusters homogeneously over a large surface area by optimizing the solvent-mediated interaction. The composite material was calcined to remove the protecting ligands producing the active catalysts.
In order to evaluate the catalytic activity of the SBA-supported Au clusters, the authors have developed an unusual testing method: benzyl alcohol was oxidized in aqueous solution using H2O2 as the oxidant and microwave irradiation was adopted as an alternative heating system. Under these conditions, the selectivity privileged the unusual formation of benzoic acid (max. 96% at total conversion) while benzaldehyde and benzyl benzoate were co-products. Other substituted benzylic alcohols were readily oxidized to produce carboxylic acids with excellent yields.
New inorganic composites containing Ga–Al oxides and Au have been prepared by Cao et al.93
A series of binary mesostructured Ga–Al mixed-oxide supports (GaxAl6−xO9; x = 2, 3, 4), along with unitary oxides of γ-Ga2O3 and γ-Al2O3, was firstly synthesized through an alcoholic sol–gel method. The X-ray diffraction patterns of the binary substrates are consistent with solid solutions with a spinel-type structure. Onto these mixed oxides, gold was deposited from a HAuCl4-urea solution producing dispersed nanoparticles (<6 nm).
One particular catalyst having the composition Au/Ga3Al3O9 was so active to allow the aerobic oxidation of benzyl alcohol at ambient temperature. Under extreme conditions (at 160 °C, solvent free), Au/Ga3Al3O9 catalyst produced the very high TOF of 25000 h−1, thus showing great potential for practical applications. We should remember the historical TOFs (up to 270000 h−1) reported in 2006 by Hutchings' group using Au–Pd nanoparticles on conventional TiO2 support.94
Underpinned by parallel experiments, a dehydrogenation mechanism has been confirmed for the conversion of alcohols to aldehydes. In fact, the Au–Ga2O3–Al2O3 system showed a high activity towards the evolution of molecular hydrogen during the desorption of 2-propanol. A maximum effect has been observed just in the case of the most active Ga3Al3O9 solid solution (Fig. 6).
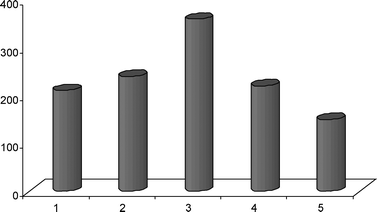 | ||
| Fig. 6 Dependence of the catalytic activity on catalyst composition (1 = Au/Ga2O3; 2 = Au/Ga4Al2O9; 3 = Au/Ga3Al3O9; 4 = Au/Ga2Al4O6; 5 = Au/Al2O3). | ||
Ceria prepared using supercritical conditions has been reported as an innovative material for supporting gold-palladium nanoparticles, which was a quite effective catalyst for the aerobic oxidation of benzyl alcohol in solvent-free conditions.95 The monometallic Au and Pd and bimetallic Au–Pd catalysts, prepared from the scCeO2 support, were evaluated at 140 °C with O2 in the absence of solvent appearing highly active and selective for benzaldehyde (ca. 91% selectivity). The other products were mainly benzyl benzoate (3%), benzoic acid (2%) and toluene (3%). The Au–Pd supported on supercritical CeO2 catalyst was approximately 4 times more active than Au–Pd supported on conventional CeO2. The morphology of the catalysts (HAADF imaging, STEM–XEDS mapping and XPS spectroscopy) showed that both Au and Pd, in form of Au(0) and Pd(II) entities, were intimately mixed and uniformly dispersed over the supercritical nanocrystalline CeO2 support.
The unusual catalytic system, NaAuCl4/Cs2CO3 was found to be a simple, recyclable, and efficient catalyst for the oxidation of benzylic alcohols by O2 at room temperature under normal air atmosphere without the need for any further support.96 Of particular relevance, this system gave excellent selectivity to aldehydes for a wide range of primary alcohols. Atomic force microscopy, TEM, XPS and diffuse reflectance ultraviolet spectroscopy showed in the catalyst the presence of Au(0) nanoparticles, with a size distribution of 15–80 nm, well dispersed over the Cs2CO3 surface. It is surprising that so large gold particles allow the aerobic oxidation of alcohols under very mild conditions. A second peculiarity has until now no logical explanation: using K2CO3, instead of Cs2CO3, the catalytic system resulted to be almost inactive (<5% conversion of benzyl alcohol) under the same conditions when NaAuCl4–Cs2CO3 allowed >99% conversion.
Similarly to the above reported metal–organic frameworks, porous coordination polymers (PCPs), consisting of metal ions and organic ligands with highly regular nanometre-sized cavities and channels represent a new class of supporting materials.97–100 According to Haruta et al., deposition of gold on PCP, e.g. [Zn4O (bdc)3]n (bdc = benzene-1,4-dicarboxylate) by solid grinding allows a convenient route for preparing 1 nm scale gold clusters.101
This technique is very simple: the volatile organometallic Me2Au (acac) (acac = acetylacetonate) and PCPs are ground in an agate mortar in air for 20 min at room temperature and then reduced with hydrogen.
The resulting catalysts were employed in the liquid phase oxidation of benzylic alcohols in methanol solution. It has been found that the reaction occurs either in the presence or in the absence of alkali and can directed to different products as benzaldehyde and methyl benzoate. Acting on the degree of surface acidity of the supports, the selectivity can be tuned towards methyl benzoate (max. yield 79%) or benzaldehyde (max yield 48%).
A beneficial interaction of bimetallic gold–palladium particles and a polymeric support has been reported by Marx and Baiker for the selective oxidation of benzyl alcohol.102 Colloidal mono- and bimetallic particles (2.4–3.7 nm) were immobilized onto a polymeric organic material, polyaniline, and the resulting catalyst was used in the aerobic oxidation of benzyl alcohol. Compared to pure Au, Au–Pd (1:9 ratio) was shown to be quite active and selective, reaching 98% selectivity at full conversion. The polyaniline supported nanoparticles presented a core–shell structure with an Au-enriched core and a Pd-rich shell. This structure is supposed to be the main reason for the observed changes in the catalytic behavior on adding Pd to Au. A detailed characterization of the catalyst has been presented.
Besides polyaniline, other polymeric organic supports have been utilized (Scheme 8). In particular, poly(o-phenylenediamine) in the form of submicrospheres with an average diameter of 700 nm was used by Han and coworkers for immobilizing nanoparticles of gold.103 In this procedure, metallic gold can be deposited from HAuCl4 without additional reductants. According to the results, the organic submicrospheres containing amino groups and π electrons in benzene rings on their surfaces can significantly stabilize gold nanoparticles against agglomeration. The synthesized submicrosphere-supported gold nanoparticles were used to evaluate their catalytic performances for benzyl alcohol oxidation. The achievements revealed that this catalyst exhibited high yield and selectivity to benzaldehyde under mild conditions (air, in aqueous media). Catalyst size and amount, solvent, temperature, alkali type and reaction time were investigated aiming to optimize the process.
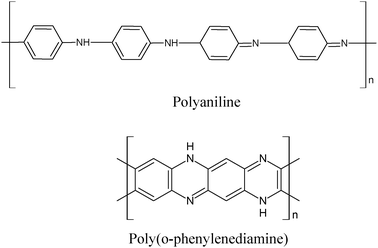 | ||
| Scheme 8 Structures of polyaniline and poly(o-phenylenediamine). | ||
The prolific application of polymeric materials employed as the support for gold catalysis includes the use of a porous polyimide membrane. As reported by Mertens et al.104 gold–palladium clusters, ca. 1.9 nm, stabilized by polyvinylpyrrolidone, were immobilized into the organic membrane without loss of nanodispersion. The resulting catalytic membrane was highly active (TOF 22500 h−1 for benzyl alcohol and 59000 h−1 for 1-phenylethanol) in the solvent-free oxidation of benzylic and allylic alcohols with total selectivity to the carbonyl compounds. Recycling experiments indicated that the catalytic membrane retained its activity (>97% of the original in the fourth reuse). Owing to the high performance and easy use, such kinds of catalytic membrane may represent an useful contribute to clean processes for organic synthesis, worthy of further applications.
The ability of naked colloidal gold particles to catalyze the liquid phase oxidation of organic compounds turned out of scientific interest for separating the role of the metal from the role of the support, when both components participate to the catalyst activity.105,106
It has been reported now that the catalytic system employed by Martens et al. for preparing the catalytic membrane can perform the oxidation of benzyl alcohol also in the absence of any supporting materials.107 Thus, the colloidal bimetallic gold–palladium particles stabilized with polyvinylpyrrolidone when dispersed in N,N-dimethylformamide emerged to be effective for benzyl alcohol oxidation with full selectivity. These catalytic particles can be recycled by nanofiltration with unaffected catalytic performance. As expected, highly active and durable heterogeneous catalysts are formed by immobilization the gold–palladium clusters on high surface area supports, as for example, the basic BaAl2O4.
In order to develop more sustainable processes, the vapor-phase oxidation of benzyl alcohol has been recently investigated by Tsuruya's group108–111 and others,112 because this clean, solvent-free process can provide high-quality benzaldehyde for use in the flavoring and pharmaceutical industries without the need to separate the catalysts. Following previous studies which highlighted the good performance of monometallic gold catalyst in the gas-phase oxidation of a variety of alcohols,113 Della Pina et al. have now investigated improved catalysts based on gold–copper alloys supported on silica.114 Considering various Au:Cu combinations, it has been found that the composition Au/Cu = 4 (w/w) allowed the best result of 99% selectivity at 98% conversion. (Table 2).
Nanoporous gold, fabricated by selective leaching of silver from Ag–Au alloys, is a robust and easy to prepare three-dimensional nano-structured material with continuous pore channels and nanometric scale ligaments.115 In recent years this material has been proposed as an alternative device to conventional supported gold catalysts employed, for example, in the low temperature oxidation of CO.116
Nanoporous gold has been now applied by Han et al.117 in the gas-phase oxidation of benzyl alcohol to benzaldehyde, where it has shown good catalytic activity under moderate reaction conditions. This material could catalyze the oxidation at relatively low temperatures, below 250 °C, where gold on silica nanoparticles are scarcely active.117 However, limited conversions must be achieved in order to obtain a fairly good selectivity which rapidly drops with the conversion: in fact, 98% selectivity could be realized only below 24% conversion.
Owing to the easy handling and good performance, nanoporous gold has attracted new applications as the partial oxidation of methanol. It has been reported by Wittstocks et al. that de-alloyed of AuAg sponge catalyzes the selective oxidative coupling of methanol to methyl formate with selectivity above 97% and high turnover frequencies at temperatures below 80 °C.118
For stoichiometric compositions of methanol and O2 (1:2), the coupling product methyl formate is produced almost exclusively. At room temperature, the selectivity towards methyl formate is ca. 100% at 10% conversion of methanol. No other partial oxidation products, such as formaldehyde or formic acid, were detected. On increasing the temperature up to 80 °C, the selectivity slightly drops down to 97% owing to over-oxidation, while the conversion increases to 60%. This reaction rate corresponds to a turnover frequency of ca. 400 h−1 with respect to all surface atoms. Compared to conventional catalysts, such as supported nanometric gold particles, long-term stability of the catalytic activity was observed: at room temperature, the activity of the catalyst was constant for more than 7 days, while at 60 °C the activity slightly decreased being the selectivity unaltered.
The combustion reactions increase as the partial pressure of O2 is increased. Nevertheless, even at relatively high O2 partial pressures, the selective oxidative coupling is dominant, above 90%, at 20 °C and slowly fades out to 78% at 80 °C.
According to Hereijgers and Weckhuysen, methanol can be converted to CO-free hydrogen by partial oxidation on traditional supported gold catalysts.119 The best results, however, can be gained on doping an alumina-supported catalyst with alkali earth metal oxide. In this case, a promoting effect was observed resulting in a significant improvement of the catalytic performance. For instance, at 85% conversion the selectivity for H2 increased from 15% to 51% on adding BaO as the promoter.
While the gas phase oxidation of alcohol produces mainly carbonyl derivatives, carboxylates can be prepared in the liquid phase oxidation which occurs under mild condition in the presence of alkali.28 However, under more drastic conditions, the liquid phase oxidation can be addressed to the formation of carboxylic acid under appropriate reaction conditions, thus avoiding the expensive transformation of the carboxylate into the free acid.
Related to this scope, Scurrell et al. have studied the liquid-phase oxidation of ethanol to acetic acid using Au catalysts supported on various metal oxides, at 150 °C and 48 bar, using air as the oxidant.120
Catalysts containing 1 wt% Au supported on TiO2, Al2O3, and ZnO were examined for ethanol oxidation which was diluted in aqueous solution (5–40%) for avoiding the formation of the ester. With a slight excess of initially present oxygen, ethanol conversions up to 99.4%, and a selectivity to acetic acid up to 99.8% could be achieved. As a drawback, slight gold leaching has been detected.
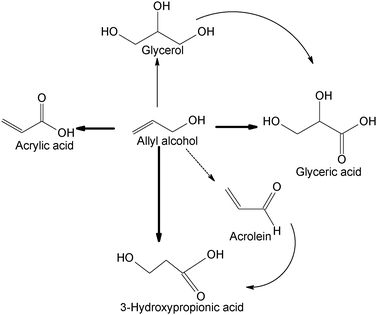
3-Hydroxypropionic acid is considered an important building block for value-added chemicals, in particular polymeric materials, which presently suffers from the absence of a commercially viable production route alternative to biological oxidation, which is carried out using genetically modified bacteria.121 It has been found that, upon reacting allyl alcohol, a common and cheap chemical, with O2 in aqueous alkali solutions a slow oxidation takes place which produces 3-hydroxypropionate besides small amounts of acrylate and glycerate. Nanometric gold particles dispersed on activated carbon turned out to be a preferred catalyst, with respect to Au/TiO2, for the conversion of allyl alcohol, while the selectivity could be controlled by varying the used amount of alkali and the temperature. A high selectivity (79% at full conversion) to the desired 3-hydroxypropionic acid has been obtained by utilizing an excess of NaOH (3:1) and a mild temperature (50 °C).122 The use of so large an amount of alkali is presently a great drawback.
The observation that acrolein is able to produce 3-hydroxypropionic acid under similar reaction conditions has suggested the reaction pathway represented in Scheme 9.
Among different polyols, glycerol oxidation has been subjected to many investigations owing to the commercial interest in transforming bio-glycerol, derived from the bio-diesel industry, into value-added compounds.123
Following a series of investigations, Prati et al.124 have reported new progress in the liquid phase oxidation of glycerol. In a first overview, gold nanoparticles, unsupported, supported on carbon and supported on TiO2, where evaluated in terms of selectivity and activity. The role of the base and the formed hydrogen peroxide has been evaluated. It has been derived that the selectivity towards the desired glycerate is lowered by the C–C bond fission of glycerol, which grows up with the reaction time and partially with the rate of degradation of the by-product H2O2.
In subsequent research, the properties of Au nanoparticles prepared via different techniques and supported on different MgAl2O4 spinels have been discussed.125 After careful characterization of bare and gold-loaded supports, catalytic tests on the selective oxidation of glycerol have been performed in aqueous basic solution. It has emerged that the surface composition, the area of the spinel and, to a lesser extent, the gold particle size plays an important role in determining the catalyst selectivity. When supported on a surface characterized by a similar Al/Mg ratio, derived by XPS technique, the selectivity is not determined by the dimension of gold clusters.
A third study deals with bimetallic Au–Pd nanoparticles supported on activated carbon, which were synthesized according to a two-step procedure: the immobilization of Au sol onto activated carbon followed by immobilization of Pd(0).126 Applied to glycerol oxidation, the bimetallic catalysts showed superior activities compared to monometallic Pd or Au nanoparticles on the same support. A series of catalysts with Au:Pd ratios varying from 9.5:0.5 to 2:8 was prepared. These catalysts were characterized by TEM, HRTEM, EDX, and X-ray mapping techniques to obtain morphological information, particle size distributions, crystalline structure and distribution of the two metals. Correlating with the results from catalytic tests, it has come out that the surface configuration of Pd monomers isolated by Au atoms has a substantial effect on activity and stability. The Au:Pd ratio on the surface of the particles is the paramount parameter and can be finely tuned to achieve an optimal catalytic performance. The segregation or heterogeneity of Pd weakens the synergistic effect of the bimetallic catalyst.
Subsequent research by the same team shows progress in the aqueous-phase oxidation of glycerol by eliminating the need for a base but maintaining high activity and selectivity.127 By using the right alloy of metals, namely Au–Pt, and support material, particularly H-mordenite, it is possible to prepare an active and durable catalyst that is highly selective towards the formation of oxidized C3 molecules from glycerol under acidic conditions. This discovery is significant because previous investigations employing Pd and Pt catalysts at low pH reported that the main products are C1 and C2 molecules produced from C–C bond scission.126,127 Furthermore, it has been formerly reported that by using Pt catalysts it is possible to obtain quite high selectivity to hydroxyacetone or hydroxypyruvic acid, but in this case the selectivity rapidly declines with increasing conversion.128–132
According to the reported experiments, glyceric acid can be synthesized with a 81% selectivity at full conversion. Moreover, it has been pointed out that by alloying Au to Pt the leaching of metals was avoided and the catalyst life improved. In Table 3 the performances of the Au–Pt metals deposited on H-mordenite and on activated carbon are presented.130
| Catalyst | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| 1%AuPd(6:4)/AC |
12 | 65 | - | 24 |
| 1%AuPt(6:4)/AC |
58 | 79 | 1 | 12 |
| 1%AuPt(6:4)/H-mordenite |
70 | 83 | 2 | 10 |
The use of bio-renewable resources, such as glycerol, a by-product from bio-diesel manufacture, can provide a viable route to valuable products using catalytic technology.123 In particular, glycerol can be firstly reduced by copper catalyst to give 1,2-propanediol that can then be selectively oxidized to lactate, a monomer for the synthesis of biodegradable polymers having great commercial potential. As noted in one of the earlier applications of gold catalysis, Au on carbon is an efficient catalyst for the oxidation of 1,2-propanediol to lactate.133 Progress in this application has now been reported by Hutchings et al., who showed that gold–palladium alloy catalysts are superior to the monometallic gold catalyst when applied to the selective oxidation of 1,2-propanediol to lactate.134 In this study, two supports, TiO2 and carbon, and two preparation methods, wet impregnation and sol-immobilization, were compared. The addition of palladium to gold significantly enhanced the activity while retaining the high selectivity to lactate (max. 96% selectivity at 94% conversion, Table 4). The use of the sol-immobilization method led to catalysts with the highest activity for lactate formation. An interesting comparison inside C3 alcohols shows that the reactivity decreases in the order glycerol > 1,2-propanediol > 1,3-propanediol > 1-propanol > 2-propanol.
Ma et al.135 have also investigated the selective oxidation of 1,2-propanediol to lactic acid by using gold nanocrystals supported on a basic material, Mg(OH)2 which were synthesized by sol-immobilization method.
In these experiments, the selectivity resulted to be strongly dependent upon the catalyst amount and the maximum value, 89.3%, was measured at 94.4% conversion by operating at 60 °C and 0.3 MPA of O2.
Considering that the derived catalyst contains gold particles having a large dimension (14–18 nm), it is not surprising to note the lower TOFs, ca. 100 h−1, reported by these researchers with respect to the TOFs exceeding 1000 h−1 previously observed employing smaller (2–4 nm) gold particles.30,135
4. Selective oxidation of sugars and carbonyl compounds
The great interest in the valorization of renewable biological resources has led to extensive investigation on the catalytic oxidation of glucose and other carbohydrates since 2002.136 In this area, efforts have been mainly devoted to the improvement of the catalyst efficiency and process operation.As reported by Haruta et al., solid grinding represents a simple and efficient method for preparing well-dispersed catalysts which have been applied in the aerobic oxidation of alcohols100 and also in glucose oxidation.137
When applied to glucose oxidation, the catalysts prepared according to the solid grinding method gave notable reaction rates. In particular, the highest catalytic activity was observed for Au/ZrO2 catalyst, with a turnover frequency of 45 mol glucose (mol surface Au)−1 s−1 at 50 °C and pH 9.0. This value is superior to any TOF per surface Au atom reported before (e.g. TOF = 42 s−1).138
Kinetic evaluations demonstrated that the catalytic activity of Au/Al2O3 was comparable to that of Au/ZrO2 and slightly higher than that of Au/carbon. The apparent activation energies were calculated to be in the interval 27–96 kJ mol−1 according to the nature of the support and tended to be lower for Au on metal oxides. The apparent activation energy for glucose oxidation over unsupported Au colloids was previously reported to be 47 kJ mol−1.139 Comparing the TOF of various catalysts, the authors conclude that the TOF grows up with the decrease in the diameter of the gold particles.
Former studies reported that the catalytic activity of Au NPs for glucose oxidation was detectable for Au particles smaller than 10 nm and was inversely proportional to the diameter of the gold particles in the range of 4–6 nm. This indicates that the catalytic activity per surface Au atom is almost independent on the diameter of the gold particles in the interval 4–7 nm, while a sharp increment in the specific activity is observed for particles below 4 nm.105 The contribution of geometrical and electronic effects in the nanometric region close to the transition of the metallic/non-metallic properties of the gold particles seems to be responsible of the observed phenomena.73,75
However, a contrasting achievement has been reported by Guczi et al.140 by evaluating two series of gold catalysts supported on TiO2, SiO2, and CeO2, prepared by colloidal gold deposition. The diameter of Au particles stabilized by poly(vinyl alcohol) (PVA) or by tannic acid-citrate was 5–7 nm and 7–13 nm, respectively, after calcination in air. Catalytic activities were compared in glucose oxidation and CO oxidation reactions and an inverse correlation was found between the activity and particle dimension in the two cases.
While the smaller gold particles and the reducible oxide supports favor the activity in CO oxidation, in glucose oxidation the inverse trend has been detected. In particular, with SiO2 support larger sized gold is more active than smaller particles. According to the authors, the effect of the support is more significant than the gold particle size.
A traditional method for preparing an efficient catalyst for glucose oxidation, based on the impregnation of the active component onto the support (0.3% Au/Al2O3), has been optimized by Pruesse et al.141 It has been evaluated how the hydrogen tetrachloroaurate hydrolysis in aqueous solution and the composition of the intermediate complexes depend on the pH value and the chloride concentration. Different impregnation solutions were prepared by dissolving the hydrogen tetrachloroaurate in water and aqueous solutions of hydrochloric acid, nitric acid, sodium chloride, and sodium hydroxide. The resulting gold complexes were characterized by UV/vis spectroscopy, TPR and TEM analyses. Gold catalyst precursors were activated via gas-phase reduction with hydrogen and tested in glucose oxidation. The gold catalyst prepared with a hydrochloric acid solution exhibited the smallest gold particles (mean diameter, 1–2 nm) and, accordingly, the highest activity.
This is a significant finding, because residual chloride in the catalyst precursor has been considered detrimental to the preparation of active catalysts. In addition, the most active catalyst exhibited excellent long-term stability for the conversion of highly concentrated glucose solutions.
As mentioned in section 3, the innovative bulk nanoporous gold material, made by selective leaching of Ag–Au alloy turned out to be a quite effective catalyst for the aerobic oxidation of benzyl alcohol 117 and methanol oxidative coupling.118 A following application by Yin et al. deals with the use in the liquid phase oxidation of glucose to gluconic acid under mild conditions.142 Systematic studies have been carried out including activity dependence as functions of pH value, temperature and porous ligament size. The possible catalytic contribution from the residual Ag atoms trapped in the bulk gold was ruled out, while the correlation of ligament size and activity indicated the low-coordinated surface Au atoms as the active sites for glucose oxidation. Compared to traditional catalysts, however, the catalytic activity of nanoporous gold cannot match that of Au nanoparticles, but it holds additional advantages in terms of easy preparation, recovery and recycling. Owing to structural continuity and excellent electric conductivity, this material is suggested as a component for innovative membrane reactors.
Owing to economical and “green” reasons, air or oxygen is the best reagent for oxidation. Hydrogen peroxide is also attracting interest owing to its biocompatibility and also for mechanistic studies to be compared to the benchmark O2.143 Recently, the selective oxidation of glucose by H2O2 has been re-investigated by Haruta et al.144 Gold nanoparticles were deposited directly from aqueous solution of diethylenediaminegold(III) complex onto commercially available polymer beads, such as poly(methyl methacrylate) (PMMA), polystyrene, and polyaniline (PANI) without any surface modification. The dropwise addition of NaBH4 to reduce Au(III) was found to be very effective to obtain small Au(0) nanoparticles with a narrow size distribution in most cases. The catalytic performance of Au NPs deposited on polymer beads were tested in the competitive reactions of H2O2 decomposition and glucose oxidation: their kinetics appeared to be more significantly affected by the kinds of polymer supports (PMMA > PS > PVC > PANI > MF) rather than by the size of Au particles. The equimolar oxidation of glucose with H2O2 could be operated by avoiding the decomposition rate of H2O2, which occurs better over Au/PMMA.
A second investigation deals with glucose oxidation by H2O2 and using the industrially relevant 0.3% Au/Al2O3 catalyst.145 According to kinetic tests and analytical results carried out under mild conditions (40 °C, pH 9), glucose conversion occurred with high reaction rate (TOF around 8300 mmol min−1 gAu−1) and high selectivity (99% at full conversion). By analyzing the dissolved oxygen it has been demonstrated that the effective oxidizing agent is formed by the decomposition of H2O2 on the gold catalyst. Investigations on the reaction kinetics turned out an apparent activation energy of 48 kJ mol−1 and a reaction order for D-glucose of 0.5, both corresponding to the values reported for D-glucose oxidation with O2. In addition, the oxidation of D-maltose, as a model disaccharide, to sodium D-maltobionate with H2O2 was also investigated.
Despite the wide interest of academia and industry in gold catalysis, the patent literature has not accompanied the scientific progress in the last few years. In this context, glucose oxidation is still attracting commercial interest at Sued Zucker.146 A procedure for the selective carbohydrate oxidation using supported gold catalysts is claimed. The chemoselective oxidation of the aldehyde carbon of a carbohydrate (e.g., glucose) into a carboxyl group (e.g., gluconic acid) has been achieved in the presence of a gold catalyst, comprising gold particles distributed in a nano-dispersed manner on a metal oxide support (e.g., Au/TiO2). Improved yield and selectivity are reported.
Although the D-aldohexose glucose (Fig. 7) has represented the most investigated substrate for aerobic oxidation, gold catalysis has been shown to be of general application for carbohydrate oxidation. According to Kusema et al., the aqueous-phase selective oxidation of the D-aldopentose arabinose by molecular oxygen was studied in an isothermal shaking reactor at moderate conditions of 50–70 C and pH 6–9.147 Different gold catalysts on metal oxide and carbon supports were tested and compared to palladium catalysts. Au supported on metal oxides displayed high catalytic activity and selectivity to arabinonic acid with Au/Al2O3 proving to be the most suitable catalyst. Pd catalysts showed a lower activity and selectivity due to the large amount of by-products. The influence of the kinetic parameters such as pH, temperature and oxygen flow rate was investigated. The optimum conditions for the selective oxidation of arabinose were 60 °C, pH 8. In situ catalyst potential measurements during arabinose oxidation gave information about the extent of the oxygen accumulation on the metal surface and a correlation to activity was obtained. The apparent activation energy was 23.8 kJ mol−1. This value is close to the activation energy barrier found by Haruta et al. in the oxidation of glucose catalyzed by nanometric gold deposited onto oxidic supports.137
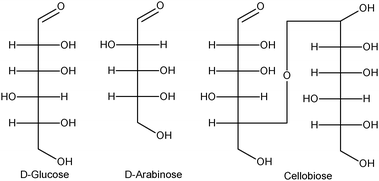 | ||
| Fig. 7 Fischer projections of D-glucose, D-arabinose and cellobiose. | ||
The partial acid hydrolysis of the ubiquitous cellulose gives the disaccharide cellobiose containing two glucose units attached in a 1,4 linkage. This interesting intermediate has been subjected to the selective oxidation by gold nanoparticles.148 A sophisticated catalyst was prepared by loading gold on nitric acid-pretreated carbon nanotubes. The selective oxidation of cellobiose by molecular oxygen produced gluconic acid in aqueous medium without any pH control. An interesting volcano-type curve was obtained by evaluating the gluconic acid yield versus the temperature employed during the reduction step of the catalyst, leading to a maximum around 250 °C. Under optimized conditions, a gluconic acid yield of 80% has been obtained at 145 °C. Although gold nanoparticles deposited on other supports were quite active in converting cellobiose, the selectivity was quite low owing to over-oxidation as in the case of Au/MgO and Au/Al2O3 (Table 5).
5-Hydroxymethyl-2-furfural (HMF) is considered to be a green building block, being derived from renewable resources. Therefore, it is attracting scientific interest for the production of derivatives to be applied in pharmaceuticals, antifungals and polymer precursors.
According to Corma et al.,149 one-pot, base-free oxidative esterification of 5-hydroxymethyl-2-furfural has been performed producing 2,5-dimethylfuroate (DMF), which represents a valuable monomer for the production of biomass-derived polymers. 5-Hydroxymethyl-2-furfural has been selectively converted into DMF (99 mol% yield) under mild conditions (65–130 C, 10 bar O2) in the absence of any base, by using gold on ceria catalyst. The catalyst can be reused several times without any loss of activity or selectivity. A full reaction scheme has been proposed where the rate-limiting step of the reaction is the alcohol oxidation into aldehyde followed by the smoother aldehyde conversion into hemiacetal and further oxidation into the corresponding ester.
5. Oxidation of amines
“Catalytic methods for amine oxidation are almost unknown in organic synthesis”: this incipit of the section dedicated to amines in a previous review nowadays sounds anachronistic. Actually, over the last three years the number of papers on this topic have exponentially flourished.28In basic investigations, it has been pointed out how the doping effect of the amino group on traditional metal catalysts avoids their use in the aerobic oxidation of aminoalcohols, meanwhile emphasizing the pleasant exception of gold: if compared to palladium and platinum under similar conditions, the catalytic activity of nanometric gold represents a strong advancement. The troublesome chemo-selectivity in the catalytic oxidation of amino alcohols by Au catalysts has been outlined, which can lead to the corresponding amino acid as well as the N-oxide derivative, the resulting product being determined by the nature of the nitrogen substituents, experimental conditions and catalytic system.31,150
Angelici and co-workers151,152 discovered the catalytic activity of the large gold particles (around 1000 nm) in the reactions of carbon monoxide, or isocyanides, with primary amines and molecular oxygen under mild conditions to achieve, respectively, ureas or carbodiimides. Furthermore, they showed that bulk gold is also effective in the oxidative dehydrogenation of secondary amines to imines.153 This research team has now reported a comparison between bulk gold powder (∼50 μm particle size) and smaller gold particles (50–150 nm) supported on alumina employed as a catalyst for the oxidative dehydrogenation of secondary and primary amines under moderate reaction conditions (1 atm O2 and 100 °C).154 While both catalytic systems were strongly affected by the nature of the substrate, the smaller Au particles resulted definitely more active.
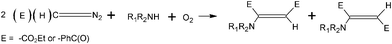
The intensive work of the same research group has led to the discovery of a new type of aerobic reaction catalyzed by bulk gold, which is able to convert diazoalkanes and amines into enamines (Scheme 10).155 This reaction, which occurs in high yields (58–94%), is interesting because no report of other catalytic system is known and the process is quite simple to apply.
 | ||
| Scheme 10 | ||
Also Mullins et al.156 have presented achievements demonstrating that bulk gold is a highly active catalyst for the selective oxidation of propylamine with oxygen. It has been highlighted how the product distribution deeply depends on the coverage of surface oxygen: whereas at low oxygen coverage propylamine initially undergoes N–H bond cleavage, leading to propionitrile and water (Scheme 11), both partial oxidation and combustion channels exist at high oxygen coverage thus producing propionitrile, propionaldehyde, water, CO, CO2, and N2O.
 | ||
| Scheme 11 | ||
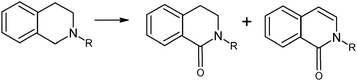
On the other hand, almost two decades of outstanding results in organic synthesis have underpinned the superiority of nanometric gold in several oxidation reactions. It would be, therefore, of considerable interest to compare always bulk gold with nanogold (<5 nm). Regarding this, recent research thoroughly describes the use of graphite-supported gold nanoparticles (AuNPs/C) to catalyze aerobic oxidation of cyclic and acyclic benzylic amines to the corresponding imines with moderate-to-excellent substrate conversions (43–100%) and yields (66–99%).157 The results clearly illustrate the superiority of gold nanoparticles compared to the commercially available bulk gold powder (2–5 μm), as in the case of the aerobic oxidation of dibenzylamine: while bulk gold was almost inert (4% conv.), the large surface area provided by the AuNPs led to complete substrate conversion under similar reaction conditions. As an extension of AuNPs catalysis, the oxidation of N-substituted 1,2,3,4-tetrahydroisoquinolines in the presence of aqueous NaHCO3 solution produced the corresponding amides in good yields (83–93%) and high selectivity (amide/enamide = 93:4) (Scheme 12). It is noteworthy that this latter catalytic oxidation had not been reported in any previous catalytic experiment.
 | ||
| Scheme 12 | ||
Corma's group meanwhile reached notable results applying supported gold nanoparticles for catalyzing the aerobic oxidation of amines.158 While para-substituted benzylamines and heterocyclic methanamines undergo oxidative condensation, 1-phenylethanamine and diphenylmethanamine form imines with much less selectivity than benzylamines. Secondary and tertiary dibenzyl and tribenzylamines give N-benzylidene benzylamines accompanied with aromatic ketones and oximes arising from C–N bond rupture. It has been found that the oxidation rate of amines to benzylidene amines strongly increased as the average particle size was reduced. Corma et al.'s conclusions are: (1) the oxidation of amines to benzylidene amines on gold is a structure sensitive reaction that requires small crystallites; (2) gold supported on carbon selectively catalyzes full oxidative cross-condensation of benzylamines and amines; (3) gold supported on titania is able to selectively catalyze the formation of secondary benzylamines through a one-pot, two-step reaction that involves one oxidative cross condensation step followed by hydrogenation.
Further studies indicate the versatility of the gold-catalyzed oxidation of amines: from anilines, aromatic azo compounds have been produced in high yields, exceeding 98%, using Au/TiO2 as a catalyst under a few bars of oxygen pressure.159 The protocol is displayed with the synthesis of parent azobenzene from aniline. However, Au/TiO2 being an effective hydrogenation catalyst, it is possible to prepare azo compounds directly from nitroaromatics through a two-step (hydrogenation followed by aerobic oxidation), one-pot, one-catalyst reaction. This catalytic process clearly appears to be eclectic, being able to synthesize both symmetric and asymmetric aromatic azo compounds from the mixtures of two anilines substituted with electron-donor and electron-acceptor substituents.
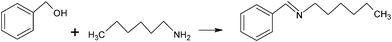
Gold nanoparticles on titania were demonstrated to be an outstanding catalyst in the aerobic oxidation of amines also by Christensen and co-workers. A first study reported on gold ability in promoting the oxidation of n-hexyl amine and 1,6-hexanediamine with high selectivity into the corresponding amides, N-hexyl hexanoic amide and caprolactam, respectively. It was indeed emphasized that pure titania is itself a catalyst working under very mild conditions. Such achievements sound amazing as they open up two new and eco-friendly ways to caprolactam and cyclohexanone oxime, basic precursors for nylon-6.160 In a subsequent paper,161 the formation of imines by aerobic oxidative coupling of mixtures of alcohols and amines was displayed using the same heterogeneous catalyst, Au/TiO2. The reactions were performed at ambient conditions and occurred with excellent selectivity (above 98%) at moderate conversion under optimized conditions. The effect of catalytic amounts of different bases was also investigated, along with reaction temperature and time. As an example, benzyl alcohol and 1-hexylamine produce N-benzylidenehexan-1-amine in 98% selectivity at 51% conversion (Scheme 13). This methodology was then successfully extended to a number of alternative alcohols and amines resulting in a general procedure for imine synthesis.
 | ||
| Scheme 13 | ||
Taking advantage of an unusual supporting material for gold nanoparticles, namely NiO, Haruta et al.162 performed an exciting reaction, the one-pot N-formylation of amines with methanol and molecular oxygen, to produce formamides at a selectivity of 90%. According to experimental evidence, a mechanism where methyl formate is generated in situ has been supposed, followed by reaction of this latter with amines.
As reported by another research team,163N-formylation of amines under aerobic oxidation can be also performed by gold clusters stabilized by poly(N-vinyl-2-pyrrolidone), thus assessing to nanometric gold particles the catalytic role. In the latter experiments, methanol or formalin have been used as a formyl source.
Nanosized particles of gold supported on hydroxyapatite (HAP) gave rise to a versatile multifunctional catalyst for the direct tandem synthesis of imines (Scheme 14) and oximes (Scheme 15) under mild conditions, as well as the one-pot three-component syntheses of α-aminophosphonates in solvent-free conditions (Scheme 16).164 The remarkable yield (90–99%) in the synthesis of a variety of imines and oximes at 60 °C and in the synthesis of the α-aminophosphonates at 100 °C (86% yield) outline the potential of these new catalytic processes.
 | ||
| Scheme 14 | ||
 | ||
| Scheme 15 | ||
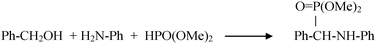 | ||
| Scheme 16 | ||
Last but not least, Baiker and Mallat's research group developed a brand-new generation of gold catalysts for amines oxidation, showing how it is possible to arrive at an efficient catalyst without using any dedicated step for the synthesis of supported gold nanoparticles. In the first paper,165 the authors have prepared the catalyst starting from Au(OAc)3 as the precursor and an oxidic supporting material (CeO2, TiO2, SiO2, Al2O3), which were simply added to the reaction mixture to form in situ supported gold nanoparticles. The aerobic oxidation of the amines produced the corresponding imines in 89–100% yield and the catalyst was proved to be truly heterogeneous and recyclable. The examined substrates were benzylamine, dibenzylamine and indoline, showing that the method could be of general application. Despite the simplicity of the method, the activity of the catalysts was comparable, or even superior, to other known catalysts. In a subsequent manuscript,166 the authors generated the gold nanoparticles in situ in an organic medium prior to the reaction using the simple AuCl3 and HAuCl4·3H2O, as the precursors, and nanoceria as the support obtaining small gold particles, mainly around 5 nm, very active in the dehydrogenation of amines into amines. By comparison, bulk powder gold did not show any detectable catalytic activity under similar conditions. A third application of the in situ formed catalyst is represented by a magnetically separable, recyclable gold catalyst where the metallic nanoparticles were supported on intimately mixed superparamagnetic ceria/iron oxide material. In this case, Au(OAc)3 has been chosen as the gold precursor. Catalytic tests with various amines revealed high selectivity to the corresponding imines (87–100%), easy separation and recycling of the catalyst.167
6. Mechanisms
Many former investigations indicate that the liquid phase oxidation at the alcoholic C–OH and carbonylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds occurs via a dehydrogenation mechanism not involving any incorporation of molecular O2.28 Now accurate studies have been presented, dealing with the mechanism of ethanol oxidation by gold employing temperature-programmed desorption and molecular beam reactive scattering.
O bonds occurs via a dehydrogenation mechanism not involving any incorporation of molecular O2.28 Now accurate studies have been presented, dealing with the mechanism of ethanol oxidation by gold employing temperature-programmed desorption and molecular beam reactive scattering.
According to Gong and Mullins,168 temperature-programmed desorption and molecular beam reactive scattering experiments on clean Au(111), pre-covered with atomic oxygen, indicate that ethanol is smoothly oxidized to acetaldehyde undergoing O–H bond cleavage followed by selective β C–H bond activation. As a consequence, hydroxyl groups are formed via the surface reaction of atomic oxygen and hydroxyl hydrogen of ethanol. Using isotopically labeled18O, it has been demonstrated that only CH3CH16O and H218O evolved, thus indicating the absence of C–O bond cleavage up to 65% ethanol conversion.
Similarly, accurate labeling experiments with 18O2 and H218O demonstrated that oxygen atoms originating from hydroxide ions, instead of molecular oxygen, are incorporated into the alcohols (ethanol, glycerol) during the oxidation reaction which is promoted at high-pH conditions. This property is shared by gold, platinum and palladium supported nanoparticles.169
Moreover, density functional theory calculations indicate that the reaction path involves both solution-mediated and metal-catalyzed elementary steps. Molecular oxygen is proposed to participate in the catalytic cycle not by dissociation to atomic oxygen but by regenerating hydroxide ions formed via the catalytic decomposition of a peroxide intermediate.
Accordingly, activation of O2 occurs at the metal surface through the formation and dissociation of peroxide (OOH) and hydrogen peroxide (HOOH) intermediates (reactions (1)–(3))
| O2 + H2O → OOH + HO | (1) |
| OOH + H2O → H2O2 + HO | (2) |
| HO + e− ↔ HO− | (3) |
An interesting temperature-dependent activity has been detected during the gas-phase selective oxidation of ethanol. As reported by Simakova et al., metallic gold particles, approximately 2 nm in size, supported on TiO2 give rise to a “double-peak” catalytic activity (Fig. 8).170 The temperature at which the first peak of activity occurs, 120 °C, is unusually mild for gas-phase reactions of primary alcohols. In contrast, gold supported on Al2O3 and SiO2 shows more usual behavior, which is similar to the second peak activity of Au/TiO2 at temperatures above 200 °C. To account for these results, it has been proposed the formation of a unidentified active oxygen form at the catalytic sites on the Au/TiO2 surface, as atomic oxygen, adsorbed molecular oxygen, superoxo complexes, or adsorbed OOH species. The amounts of these species can decrease with increasing temperature, as a result of their deactivation or desorption. The XPS spectra of the Au/TiO2 catalysts were unable to interpret the unusual behavior since only one well-defined band, which corresponds to Au(0), was always present, whereas no distinct bands related to positive oxidation states of gold, Au(3+) and Au(+), were ever detected.
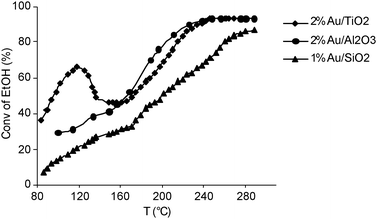
An exhaustive theoretical investigation on adsorption and dissociation of molecular oxygen on different gold surfaces and nanoparticles has been carried out in order to separate the effect of particle size, morphology and support on the mechanism of O2 activation.171
The adsorption and dissociation of molecular O2 on extended gold surfaces, isolated gold nanoparticles and small gold clusters supported on TiO2 have been investigated by means of DFT calculations. The influence of different conditions on the mode of adsorption and activation of O2 have been analyzed separately turning out that O2 dissociation is very sensitive to the arrangement of the gold surface atoms.
Three modes of O2 adsorption have been found: (a) a weakly interacting end-on mode; (b) a top-bridge-top mode; and (c) a bridge-bridge mode. Adsorption energies roughly depend on particle size (Scheme 17).
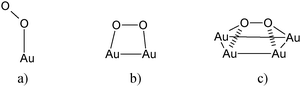 | ||
| Scheme 17 Adsorption modes of O2 on gold. | ||
The highest degree of molecular activation has been found in bridge-bridge conformers and, accordingly, the lowest activation barriers for O2 dissociation are also obtained for these species. Since bridge-bridge conformers are only stable on Au(100) facets, this indicates that particle morphology is a key factor influencing O2 dissociation. However, on TiO2, the most active sites for O2 dissociation are found at the metal-support interface and not on the gold particle.
The selective deactivation of gold nanoparticles has been investigated in order to derive information on the catalytically active sites. In a first study, the selective blocking of the catalytic active sites of gold nanoparticles supported on CeO2 and TiO2 (2.1 and 6.9 nm) was evaluated in the aerobic oxidation of benzyl alcohol, as well as in the hydrogenation of ketopantolactone, by adding various amounts of two chemically distinct thiols with different functionalities (n-octadecanethiol, ODT, and mercaptoacetic acid, MAA).172
ODT poisoned the catalyst in the oxidation reaction much more strongly than MAA. In contrast, MAA was a stronger poisoning agent in the hydrogenation reaction. The characteristics of supported Au particles and the nature of thiol adsorption were investigated by attenuated total reflection infrared spectroscopy, TEM, thermogravimetric desorption and DFT calculations. Based on the adsorption peculiarities of the two thiols, derived from the combined experimental and theoretical study, it is suggested that the hydrogenation of ketopantolactone occurs preferentially at low coordination sites such as corners and edges, which are more dominant in small clusters, while oxidation seems to be favored on extended active sites as prevailing on larger Au clusters.
In a second investigation, the progressive poisoning effect of different molecules on naked and carbon supported gold catalysts has been evaluated during the aerobic oxidation of glucose.75 A geometrical model has been derived for describing the morphological properties of two catalysts made of gold particles, having a known size distribution centered at 3.30 and 7.89 nm respectively. The observed deactivation trend follows the order thiocyanate > cyanide ≈ cysteine > thiourea and it obeys an exponential law. The kinetics of catalyst deactivation has been interpreted by considering the important contribute of electronic factors which overlap the space shielding of active sites, due to long range poison–catalyst interaction influencing the entire metal particle. Considering the nature of the molecules showing a high poisoning effect (soft base) and the promoting effect of OH− (hard base), a molecular model for the electronic interactions on gold nanoparticles during the aerobic oxidation of glucose has been suggested. Accordingly, the step of dioxygen reduction, by hydrated glucose, is differently perturbed by soft and hard-nucleophiles interacting with the gold cluster (Scheme 18). Note that the resulting H2O2 has been experimentally detected as the reduction product of O2 during glucose oxidation.143
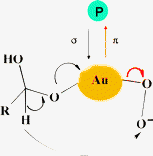 | ||
| Scheme 18 Reaction mechanism and poisoning effect of nucleophilic poisoning compounds (P) during glucose oxidation. | ||
7. Conclusions
Over the last three years, outstanding progress in the selective oxidation of organic compounds has been observed leading to interesting perspectives for gold catalysis. Owing to a continuous optimization of catalyst and experimental conditions, some processes seem to be ready for industrial exploitation. Besides other examples, two emblematic cases, the gas-phase epoxidation of propene and the liquid-phase oxidation of glucose, show how the basic knowledge of molecular mechanisms has favored reaction conditions in the first case, and very high reaction rate in the second case. The explosion of the gold-catalyzed oxidation of amines is a relevant event, which is generating a novel chapter in the organic chemistry of nitrogen compounds. Moreover, the first example of homogeneous oxidative esterification of aldehydes and alcohols with mononuclear gold complexes reported by Hashmi's group173 widens the applications of gold catalysis. In fact, tuning reactivity and stereochemistry of the active centre by suitable ligands opens up unforeseeable perspectives. On the other hand, the report on silane oxidation by gold174 highlights the ubiquitous application of the shining yellow metal. Nowadays, the polyhedric gold active site still represents a place where fantasy meets ability in designing catalytic reactions for clean processes.References
- M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS.
- G. C. Bond and D. T. Thomson, Gold Bull., 2000, 33, 41 CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS.
- S. Carretin, P. Concepción, A. Corma, J. M. López-Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538 CrossRef CAS.
- N. López, T. V. J. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232 CrossRef CAS.
- B. Schumacher, Y. Denkwitz, V. Plzak, M. Kinne and R. J. Behm, J. Catal., 2004, 224, 449 CrossRef CAS.
- G. J. Hutchings, Catal. Today, 2005, 100, 55 CrossRef CAS.
- J. Guzman, S. Carrettin and A. Corma, J. Am. Chem. Soc., 2005, 127, 3286 CrossRef CAS.
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Angew. Chem., Int. Ed., 2005, 44, 1824 CrossRef CAS.
- N. C. Hernández, J. F. Sanz and J. A. Rodriguez, J. Am. Chem. Soc., 2006, 128, 15600 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. M. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbaek, H. Falsig, C. H. Christensen, T. Bligaard and J. K.. Nørskov, Top. Catal., 2007, 44, 15 CrossRef CAS.
- G. J. Hutchings, Chem. Commun., 2008, 1148 RSC.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. Kiely and J. G. Hutchings, Faraday Discuss., 2008, 138, 225 RSC.
- J. K. Edwards, B. Solsona, N. N. Edwin, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- Q. Fu, S. Kudriavtseva, H. Saltsburg and M. Flytzani-Stephanopoulos, Chem. Eng. J., 2003, 93, 41 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS.
- A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejovic, Angew. Chem., Int. Ed., 2004, 43, 6545 CrossRef CAS.
- C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2005, 3451 RSC.
- C. González-Arellano, A. Abad, A. Corma, H. García, M. Iglesias and F. Sánchez, Angew. Chem., Int. Ed., 2007, 46, 1536 CrossRef CAS.
- A. S. K. Hashmi, Pure Appl. Chem., 2010, 82, 657 CrossRef CAS.
- C. Milone, M. L. Tropeano, G. Guline, G. Neri, R. Ingoglia and S. Galvagno, Chem. Commun., 2002, 868 RSC.
- R. Zanella, C. Louis, S. Giorgio and R. Touroude, J. Catal., 2004, 223, 328 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS.
- M. Boronat, P. Concepción, A. Corma, S. González, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230 CrossRef CAS.
- A. Corma, P. Concepción and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- C. Della Pina, E. Falletta, R. Matarrese and M. Rossi, in Metal Nanoclusters in Catalysis and Material Science, ed. B. Corain, N. Toshima and G. Schmid, Elsevier Press, 2008, p. 253 Search PubMed.
- C. Della Pina, E. Falletta and M. Rossi, in Catalysis, ed. J. J. Spivey and K. M. Dooley, RCS Publ., Cambridge, vol. 22, 2010 Search PubMed.
- T. Ishida and M. Haruta, Angew. Chem., Int. Ed., 2007, 46, 7154 CrossRef CAS.
- G. J. Hutchings, Catal. Today, 2008, 138, 9 Search PubMed.
- V. Caps, Act. Chim., 2010, 337, 18 Search PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, N. Glanville, L. Kesavan, C. Hammond, J. K. Edwards, A. F. Carley, C. J. Kiely and G. J. Hutchings, Appl. Catal., A, 2011, 391, 400 Search PubMed.
- L. A. Levchenco, A. P. Sadkov, N. V. Larionsteva, E. M. Koldasheva, A. K. Shilova and A. E. Shilov, Dokl. Biochem. Biophys., 2001, 377, 123 Search PubMed.
- A. Levchenco, N. G. Lobanova, V. M. Martinenko, A. P. Sadkov, A. F. Shestakov, A. K. Shilova and A. E. Shilov, Dokl. Chem., 2010, 430, 50 Search PubMed.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313 CrossRef CAS.
- J. Llorca, A. Casanovas, M. Dominguez, I. Casanova, I. Angurrel, M. Seco and O. Rossell, J. Nanopart. Res., 2007, 10, 537.
- K. Weissermel and H. J. Arpe, in Industrial Organic Chemistry, VCH, Weinheim, 3rd edn, 1997 Search PubMed.
- L. X. Xu, C. H. He, M. Q. Zhu, K. J. Wu and Y. L. Lay, Catal. Lett., 2007, 118, 248 CrossRef CAS.
- L. X. Xu, C. H. He, M. Q. Zhu, K. J. Wu and Y. L. Lai, Catal. Commun., 2008, 9, 816 Search PubMed.
- P. C. Hereijgersa and B. M. Weckhuysen, J. Catal., 2010, 270, 16 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2 Search PubMed.
- R. L. Brutchey, I. J. Drake, A. T. Bell and T. Tilley, Chem. Commun., 2005, 3736 RSC.
- X. Li, J. Xu, L. Zhou, F. Wang, J. Gao, C. Chen, J. Ning and H. Ma, Catal. Lett., 2006, 110, 149 Search PubMed.
- F. Wang, J. Xu, X. Li, J. Gao, L. Zu and L. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987 CrossRef CAS.
- J. Gao, X. Tong, X. Li, H. Miao and J. Xu, J. Chem. Technol. Biotechnol., 2007, 82, 620 Search PubMed.
- A. P. Singh and T. Selvam, J. Mol. Catal. A: Chem., 1996, 113, 489 CrossRef CAS.
- Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS.
- K. Nomiya, K. Hashino, Y. Nemoto and M. Watanabe, J. Mol. Catal. A: Chem., 2001, 176, 79 Search PubMed.
- T. G. Carrell, S. Cohen and G. C. Dismukes, J. Mol. Catal. A: Chem., 2002, 187, 3 CrossRef CAS.
- S. Saravanamurugan, M. Palanichany and V. Murugesan, Appl. Catal., A, 2004, 273, 143 Search PubMed.
- H. V. Borgaonkar, S. R. Raverkar and S. B. Chandalia, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 455 Search PubMed.
- M. L. Kantam, et al. , Catal. Lett., 2002, 81, 223 Search PubMed.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. Izham bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195 Search PubMed.
- K. Bauer, D. Garbe and H. Surburg, Common Fragrance Flavor Materials: Preparation Properties Uses, Wiley, New York, 1997 Search PubMed.
- C. Chapuis and D. Jacoby, Appl. Catal., A, 2001, 221, 93 CrossRef CAS.
- A. Calogirou, B. Larson and D. Kotzias, Atmos. Environ., 1999, 33, 1423 CrossRef CAS.
- P. A. Wende and T. P. Muciaro, J. Am. Chem. Soc., 1992, 114, 5878 CrossRef CAS.
- M. K. Lajunen, J. Mol. Catal. A: Chem., 2001, 169, 33 CrossRef CAS.
- R. Chakrabarty and B. K. Das, J. Mol. Catal. A: Chem., 2004, 223, 39 CrossRef CAS.
- M. de Fatima, T. Gomes and O. A. Antunes, Catal. Lett., 1996, 42, 213 Search PubMed.
- M. Caovilla, A. Caovilla, S. B. Pergher, M. C. Esmelindro, C. Fernandes, C. Dariva, K. B. Gusmao, E. G. Oestreicher and O. A. Antunes, Catal. Today, 2008, 133–135, 695 Search PubMed.
- C. C. Guo, W. J. Yang and Y. L. Mao, J. Mol. Catal. A: Chem., 2005, 226, 279 CrossRef CAS.
- B. A. Allal, L. E. Firdoussi, S. Allaoud, A. Karim, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 2003, 200, 177 CrossRef CAS.
- Y.-W. Suh, N.-K. Kim, W.-S. Ahn and H.-Ku. Rhee, J. Mol. Catal. A: Chem., 2003, 198, 309 CrossRef CAS.
- T. Joseph, D. P. Sawant, C. S. Gopinath and S. B. Halligudi, J. Mol. Catal. A: Chem., 2002, 184, 289 CrossRef.
- S. G. Casuscelli, G. A. Eimer, A. Canepa, A. C. Heredia, C. E. Poncio, M. E. Crivello, C. F. Perez, A. Aguilar and E. R. Herrero, Catal. Today, 2008, 133–135, 678 Search PubMed.
- G. A. Eimer, I. Díaz, E. Sastre, S. G. Casuscelli, M. E. Crivello, E. R. Herrero and J. Pariente, Appl. Catal., A, 2008, 343, 77 Search PubMed.
- S. Ajaikumar, J. Ahlkvist, W. Larsson, A. Shchukarev, A. R. Leino, K. Kordas and J. P. Mikkola, Appl. Catal., A, 2011, 392, 11 Search PubMed.
- G. C. Bond, C. Luis and D. T. Thompson, in Catalysis by Gold, ed. G. C. Hutchings, Imperial College Press, 2006 Search PubMed.
- M. Turner, V. B. Golovko, O. P. Vaughan, P. Abdulkin, A. Berenguer-Marcia, M. S. Tikhov, B. F. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS.
- Y. Zhu, H. Quian and R. Jin, Chem.–Eur. J., 2010, 16, 11455 CrossRef CAS.
- C. Della Pina, E. Falletta, M. Rossi and A. Sacco, J. Catal., 2009, 263, 92 CrossRef CAS.
- Y. Zhu, H. Quian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915 CAS.
- Y. Liu, T. Hironory, A. Tomoki and T. Tsukuda, Chem. Commun., 2010, 46, 550 RSC.
- X. Wang, N. S. Venkataramanan, H. Kawanami and Y. Ikushima, Green Chem., 2007, 9, 1352 RSC.
- Y. Pei, N. Shao, Y. Gao and X. C. Zeng, ACS Nano, 2010, 4, 2009 CrossRef CAS.
- A. Roldan, S. Gonzales, J. M. Ricart and F. Illas, ChemPhysChem, 2009, 9, 1352 Search PubMed.
- O. A. Kholdeeva, O. V. Zalomaeva, A. B. Sorokin, I. D. Ivanchikova, C. Della Pina and M. Rossi, Catal. Today, 2007, 121, 58 CrossRef CAS.
- Y. Chenevierea, V. Caps and A. Tuel, Appl. Catal., A, 2010, 387, 129 Search PubMed.
- S. E. Dapurkar, Z. Yokoyama, Y. Toshirou and H. Kawanami, Catal. Lett., 2009, 130, 42 CrossRef CAS.
- B. Taylor, J. Lauterbach and W. N. Degass, Appl. Catal., A, 2005, 291, 188 CrossRef CAS.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Angew. Chem., Int. Ed., 2009, 48, 1467 CrossRef CAS.
- M. Ojeda and E. Iglesia, Chem. Commun., 2009, 352 RSC.
- M. Haruta, J. Huang, T. Takei and T. Akita, US Patent 20100234623 A1.
- A. Villa, D. Wang, P. Spontoni, R. Arrigo, D. Sub and L. Prati, Catal. Today, 2010, 157, 89 Search PubMed.
- H. Liu, Y. Liu, Y. Li, Z. Tang and H. Jiang, J. Phys. Chem. C, 2010, 114, 13362 CrossRef CAS.
- M. Zahmakiran and S. Ozkar, Mater. Chem. Phys., 2010, 121, 359 Search PubMed.
- C. Y. Ma, B. J. Dou, J. J. Li, J. Cheng, Q. Hu, Z. P. Hao and S. Z. Quiao, Appl. Catal., B, 2009, 92, 202 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Phys. Chem. Lett., 2009, 113, 13457 Search PubMed.
- F. Z. Su, Y. M. Liu, L. C. Wang, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2008, 47, 334 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzig, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- P. J. Miedziak, Z. Tang, T. E. Davies, D. I. Enache, J. K. Bartley, A. F. Carley, A. A. Herzing, C. J. Kiely, S. H. Taylor and G. J. Hutchings, J. Mater. Chem., 2009, 19, 8619 RSC.
- B. Karimi and F. K. Esfahani, Chem. Commun., 2009, 5555 RSC.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., 2004, 116, 2388 CrossRef.
- S. Hermes, M.-K. Schrçter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., 2005, 117, 6394 CrossRef.
- S. Hermes, F. Schrçder, S. Amirjalayer, R. Schmid and R. A. Fischer, J. Mater. Chem., 2006, 16, 2464 RSC.
- F. Schrçder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. Van Tendeloo, B. Walaszek, G. Buntkowsky, H. H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119 CrossRef.
- I. Tamao, M. Nagaoca, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAS.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191 CrossRef CAS.
- J. Han, Y. Liu, l. li and R. Guo, Langmuir, 2009, 25, 11054 CrossRef CAS.
- P. G. N. Mertens, P. Vendezande, X. Ye, H. Poelman, D. E. De Vos and I. F. J. Vankelecom, Adv. Synth. Catal., 2008, 350, 1241 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1 CrossRef CAS.
- P. G. N. Mertens, S. L. F. Corthals, X. Ye, H. Poelman, P. A. Jacobs, B. F. Sels, I. F. J. Vankelecom and D. E. De Vos, J. Mol. Catal. A: Chem., 2009, 313, 14 Search PubMed.
- Y. Li, D. Nakashima, Y. Ichihashi, S. Nishiyama and S. Tsuruya, Ind. Eng. Chem. Res., 2004, 43, 6021 Search PubMed.
- R. Yamamoto, Y. Sawayama, H. Shibahara, Y. Ichihashi, S. Nishiyama and S. Tsuruya, J. Catal., 2005, 234, 308 CrossRef CAS.
- D. Nakashima, Y. Ichihashi, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2006, 259, 108 Search PubMed.
- Y. Sawayama, H. Shibahara, Y. Ichihashi, S. Nishiyama and S. Tsuruya, Ind. Eng. Chem. Res., 2006, 45, 8837 Search PubMed.
- J. Mao, M. Deng, Q. Xue, L. Chen and Y. Lu, Catal. Commun., 2009, 10, 1376 Search PubMed.
- S. Biella and M. Rossi, Chem. Commun., 2003, 378 RSC.
- C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384 CrossRef CAS.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- D. Han, T. Xu, X. Xu and Y. Ding, ChemCatChem, 2010, 2, 383 Search PubMed.
- A. Wittstocks, V. Zielasek, J. Biener, C. M. Friend and M. Baeumer, Science, 2010, 327, 319 CrossRef CAS.
- B. P. C. Hereijgers and M. M. Weckhuysen, ChemSusChem, 2009, 2, 743 Search PubMed.
- S. M. Tembe, G. Patrick and M. S. Scurrell, Gold Bull., 2009, 42, 321 Search PubMed.
- F. Suthers and D. C. Cameron, WO 0116346, 2001.
- C. Della Pina, E. Falletta and M. Rossi, ChemSusChem, 2009, 2, 57 CrossRef.
- M. Pagliaro and M. Rossi, The Future of Glycerol, RCS Publishing, Cambridge, 2nd edn, 2010 Search PubMed.
- L. Prati, P. Spontoni and A. Gaiassi, Top. Catal., 2009, 52, 288 CrossRef CAS.
- A. Villa A. Gaiassi, M. Veith and L. Prati, J. Catal., 2010, 275, 108 CrossRef CAS.
- B. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617 CrossRef CAS.
- A. Villa, A. Gaiassi, M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499 CrossRef CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329 RSC.
- R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Appl. Catal., B, 2008, 82, 157 CrossRef CAS.
- D. Liang, J. Gao, J. Wang, P. Chen and Z. Hou, Catal. Commun., 2009, 10, 1586 CrossRef CAS.
- A. Brandner, K. Lehnert, A. Bienholz, M. Lucas and P. Claus, Top. Catal., 2009, 52, 278 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, S. Meenakshisundaram, J. M. Anthonykutty, G. Brett, A. F. Carley, S. H. Taylor, D. W. Knight and G. J. Hutchings, Green Chem., 2009, 11, 1209 RSC.
- H. Ma, X. Nie, J. Y. Cai, C. Chen, J. Gao, H. Miao and J. Xu, Science China: Chemistry, 2010, 53, 1897 Search PubMed.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2006, 244, 122 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1 CrossRef CAS.
- T. Benkóa, A. Beck, O. Geszti, R. Katona, A. Tungler, K. Frey, L. Guczi and Z. Schay, Appl. Catal., A, 2010, 388, 31 Search PubMed.
- C. Baatz, N. Decker and U. Pruesse, J. Catal., 2008, 258, 165 Search PubMed.
- H. Yin, C. Zhou, C. Xu, P. Liu, X. Xu and Y. Ding, J. Phys. Chem. C, 2008, 112, 9673 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313 CrossRef CAS.
- T. Ishida, K. Kuroda, N. Kinoshita, W. Minagawa and M. Haruta, J. Colloid Interface Sci., 2008, 323, 105 CrossRef CAS.
- R. Salinger, N. Deckera and U. Pruesse, Appl. Catal., B, 2011, 102, 584 Search PubMed.
- H. Berndt, A. Haji, J. Kowalczyk, P. Joerg, I. Pitsch and U. Pruesse, DE 10319917 B4, 2009.
- B. T. Kusema, B. C. Campo, P. Maki-Arvela, T. Sami and D. Yu Murzin, Appl. Catal., A, 2010, 386, 101 Search PubMed.
- X. Tan, W. Deng, M. Liu, Q. Zhang and Y. Wang, Chem. Commun., 2009, 7179 RSC.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Top. Catal., 2007, 44, 325 CrossRef CAS.
- M. Lazar and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 10613 CrossRef CAS.
- B. Zhu and R. J. Angelici, J. Am. Chem. Soc., 2006, 128, 14460 CrossRef CAS.
- B. Zhu and R. J. Angelici, Chem. Commun., 2007, 2157 RSC.
- B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1 CrossRef CAS.
- Y. Zhou, R. J. Angelici and L. K. Woo, Catal. Lett., 2010, 137, 8 CrossRef CAS.
- J. Gong, T. Yan and C. B. Mullins, Chem. Commun., 2009, 761 RSC.
- M.-H. So, Y. Liu, C.-M. Ho and C.-M. Che, Chem.–Asian J., 2009, 4, 1551 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, Nat. Protoc., 2010, 11, 429 Search PubMed.
- S. K. Klitgaard, K. Egeblad, U. V. Mentzel, A. G. Popov, T. Jensen, E. Taarning, I. S. Nielsen and C. H. Christensen, Green Chem., 2008, 10, 419 RSC.
- S. Kegnæs, J. Mielby, U. V. Mentzel, C. H. Christensen and A. Riisager, Green Chem., 2010, 12, 1437 RSC.
- T. Ishida and M. Haruta, ChemSusChem, 2009, 2, 538 CrossRef CAS.
- P. Preedasuriyachai, H. Kitahara, W. Chavasiri and H. Sakurai, Chem. Lett., 2010, 39, 1174 Search PubMed.
- Sun, F.-Z. Su, J. Ni, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2009, 48, 4390 CrossRef CAS.
- L. Aschwanden, T. Mallat, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 309, 57 Search PubMed.
- L. Aschwanden, T. Mallat, M. Maciejewski, F. Krumeich and A. Baiker, ChemCatChem, 2010, 2, 666 Search PubMed.
- L. Aschwanden, B. Panella, P. Rossbach, B. Keller and A. Baiker, ChemCatChem, 2010, 1, 111 Search PubMed.
- J. Gong and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 16458 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74 CrossRef CAS.
- O. A. Simakova, V. I. Sobolev, K. Yu Koltunov, B. Campo, Anne-Riikka Leino, K. Kordμs and D. Yu Murzin, ChemCatChem, 2010, 2, 1535 Search PubMed.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538 RSC.
- P. Haider, A. Urakawa, E. Schmidt and A. Baiker, J. Mol. Catal. A: Chem., 2009, 305, 161 CrossRef CAS.
- A. S. K. Hashmi, C. Lothschütz, M. Ackermann, Renø Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem.–Eur. J., 2010, 16, 8012 CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 5302 RSC.
| This journal is © The Royal Society of Chemistry 2012 |