Mechanochemistry: opportunities for new and cleaner synthesis
Stuart L.
James
*a,
Christopher J.
Adams
b,
Carsten
Bolm
c,
Dario
Braga
d,
Paul
Collier
e,
Tomislav
Friščić
f,
Fabrizia
Grepioni
d,
Kenneth D. M.
Harris
g,
Geoff
Hyett
h,
William
Jones
f,
Anke
Krebs
c,
James
Mack
i,
Lucia
Maini
d,
A. Guy
Orpen
b,
Ivan P.
Parkin
j,
William C.
Shearouse
i,
Jonathan W.
Steed
k and
Daniel C.
Waddell
i
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: s.james@qub.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
cInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52056 Aachen, Germany
dDipartimento di Chimica G.Ciamician, via Selmi 2, 40126 Bologna, Italy
eJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading, RG4 9NH, UK
fUniversity of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK
gSchool of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, Wales, UK
hSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
iDepartment of Chemistry, University of Cincinnati, 301 Clifton Ct, Cincinnati, OH 45221-0172, USA
jMaterials Research Centre, Christopher Ingold Laboratory, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
kDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
First published on 5th September 2011
Abstract
The aim of this critical review is to provide a broad but digestible overview of mechanochemical synthesis, i.e. reactions conducted by grinding solid reactants together with no or minimal solvent. Although mechanochemistry has historically been a sideline approach to synthesis it may soon move into the mainstream because it is increasingly apparent that it can be practical, and even advantageous, and because of the opportunities it provides for developing more sustainable methods. Concentrating on recent advances, this article covers industrial aspects, inorganic materials, organic synthesis, cocrystallisation, pharmaceutical aspects, metal complexes (including metal–organic frameworks), supramolecular aspects and characterization methods. The historical development, mechanistic aspects, limitations and opportunities are also discussed (314 references).
 (From left to right) Stuart James, Carsten Bolm, Dario Braga, Paul Collier, Tomislav Friščić, Fabrizia Grepioni, Kenneth Harris, William Jones, James Mack, Lucia Maini, Guy Orpen, Ivan Parkin, Jonathan Steed |
1. Introduction
1.1 General aspects
‘Mechanochemistry’ refers to reactions, normally of solids, induced by the input of mechanical energy, such as by grinding in ball mills. It is becoming more intensely studied partly because it can promote reactions between solids quickly and quantitatively, with either no added solvent or only nominal amounts. Historically it has been a sideline approach to chemical synthesis, and solution-based methods have been adopted by default. However, mechanochemistry could in future become a more mainstream technique for two reasons. Firstly, it is increasingly clear that is effective, and even advantageous, in ever-widening types of synthesis. Secondly, our current dependence on solvents appears increasingly unsustainable1 since it is wasteful of fossil-derived materials (e.g. 85% of chemicals used in the pharmaceutical industry are solvents and even if recycled typical recovery rates are only 50–80%),1b environmentally problematic, hazardous and energy-demanding with regard to solvent production, purification and recycling.2Here we provide a broad but digestible overview of mechanochemical synthesis (sometimes called mechanosynthesis) including the current state of the art, as well as opportunities and challenges to it becoming a mainstream synthetic technique. The review also covers industrial aspects, inorganic materials, cocrystals, pharmaceutical applications, organic synthesis, discrete metal complexes, extended metal–organic materials (MOFs), supramolecular aspects and characterization methods. It is aimed to be accessible to any chemist or engineer who has no prior knowledge of the subject.
1.2 A brief history
According to Takacs, the earliest documented mechanochemical reaction may have been grinding cinnabar with acetic acid in a copper vessel to give elemental mercury (4th century BC). This may also be the first documented method to obtain an elemental metal from a compound.3 Another early reference point is a statement of Aristotle’s, translated as ‘no reaction proceeds in the absence of solvent’.4 Presented in this way, his statement runs counter to the ease of many solventless mechanochemical reactions. However, an alternative translation, which is less specific and less contentious in this regard, is simply that ‘liquids are the type of bodies most liable to mixing’.5 In the middle ages mechanochemistry was also used in mining and metallurgy, further references to which can be found in ref. 6.Michael Faraday conducted mechanochemical experiments, reducing AgCl to Ag with Zn, Cu, Sn or Fe in a pestle and mortar (1820).7 However, it was work by Carey Lea in the 1890s which showed that mechanochemical reactions could give different products to thermal ones—favouring decomposition of mercury and silver halides to their elements rather than melting or sublimation.8 This work might therefore be seen as the point at which mechanochemistry became a truly distinct sub-topic within chemistry. Wilhelm Ostwald (1853–1932) is credited by some with classifying mechanochemistry as one of four sub-disciplines of chemistry (alongside thermochemistry, electrochemistry and photochemistry) each based on a different type of energy input.6a According to Fernandez Bertran,9 Walther Nernst (1864–1941, one of Ostwald's students), also advocated this classification. An early solvent-free organic mechanochemical reaction, probably a cocrystallization, comes from 1893 by Ling and Baker,10 and during the 1920s research was done into reactions of organic polymers such as cellulose.6a However, where soluble reactants are concerned (generally speaking, molecular synthesis) solution-based reactions have been the default approach throughout the development of synthetic chemistry, and mechanochemistry has been limited largely to insoluble inorganic materials, such as alloys and metal oxides, i.e. perhaps employed only when there was no solvent-based alternative. Molecular mechanochemistry, particularly cocrystallization, developed significantly in the 1980s and 90s (Curtin, Paul,11 Toda,12 Etter,13 Jones,14 Hollingsworth15 and Caira16). These studies showed that mechanochemistry was not only a general way to make cocrystals, but also that it could give products not obtainable by solution-based methods. Regarding covalent organic synthesis, in the 1980s and 90s Toda demonstrated several solvent-free reactions between solids,17 although these often involved grinding followed by heating and may occur via molten phases.18 Reports focusing on organic synthesis in ball mills have been scarce until recently.19 In the areas of organic, metal–organic,20 and supramolecular synthesis (including cocrystals)21 the types of mechanochemical reactions done and the products obtained have broadened greatly in the last ten years. The methodology has also begun to become more sophisticated. This more contemporary work is the focus of Sections 2–9.
1.3 Terminology
The term mechanochemistry is frequently used in a broad sense, covering any chemical reaction induced mechanically (e.g. by grinding etc.).22 This is the sense in which it is used in this review. It has been argued elsewhere that this broad usage is incorrect,19c and that it should only be used when mechanical energy directly ruptures strong bonds (for example in polymers, or indeed in single molecules23). This generates reactive centres (often radicals) which undergo further reactions. This more restrictive use of the term would exclude grinding reactions which may proceed largely due to an increase in the contact surface area between reactants (as the particles become smaller and more intimately mixed). IUPAC defines a mechano-chemical reaction (with hyphen) as a ‘Chemical reaction that is induced by the direct absorption of mechanical energy’ with a note that ‘Shearing, stretching, and grinding are typical methods for the mechano-chemical generation of reactive sites, usually macroradicals, in polymer chains that undergo mechano-chemical reactions’.22 Whilst the note gives guidance for its use in the context of polymers, the basic definition is broad and without restrictions as to the atomic-scale mechanism. Therefore, the general use of the term does appear justified.Grinding is a general term describing mechanical action by hard surfaces on a material, normally to break up the material and reduce its particle size. It may therefore refer to manual methods (mortar and pestle) or non-manual methods such as ball milling, or extrusion etc. Further terminology is associated with grinding solids in the presence of liquids. Very small amounts of added liquid can dramatically accelerate, and even enable, mechanochemical reactions between solids. Often the molar equivalents added are similar to those of the reactants themselves. Such reactions are therefore ‘minimal solvent’ rather than strictly ‘solvent-free’. The original term to describe them, ‘solvent drop grinding’, has been superseded by ‘liquid assisted grinding’, (LAG) so as not to presuppose the role of the liquid (i.e. solvating or non-solvating). LAG is equivalent to the term ‘kneading’, also used in the same context.21,24
There can be confusion over what is meant by ‘solvent-free’. Firstly, ‘mechanochemical’ does necessarily mean ‘solvent-free’ since mechanochemistry can be done in the presence of solvents. Even so, there remains more than one connotation of ‘solvent-free’. It may indicate simply that no solvent was intentionally added to the reaction, e.g. stressing a practical advantage of the approach. However, in interpreting how such reactions proceed mechanistically (particularly how fluidity arises), it may be wrong to think of such a reaction as entirely solvent-free. Solvents can be present in the solid starting materials, such as in hydrated metal salts or in molecular solvates. There may even be (smaller) amounts of moisture in non-formally hydrated materials or in the atmosphere which aid the reaction. Further, species such as water, acetic acid etc. may be generated as condensates. Therefore, whilst use of the term ‘solvent-free’ is often accurate in a practical sense, care must be taken when making mechanistic interpretations.
In the same general context, while a reaction in itself may be described as ‘solvent-free’ (in the practical and/or mechanistic sense), purification may still be needed and this may require a solvent. Therefore a solvent-free reaction does not necessarily correspond to a solvent-free process overall (see also Section 10).
1.4 Mechanistic aspects
Mechanistic studies do not reveal a straightforward, or as yet complete, picture. The situation is complicated by the diversity of reaction types, reaction conditions and reactive materials (from metals and metal oxides to molecular crystals etc.). The inhomogeneous nature of solid–solid reactions, the difficulties of directly observing materials undergoing mechanochemical reactions at microscopic or molecular levels and the lack of studies of some reaction types are further factors. Each mechanistic model developed has a limited area of applicability, whilst more than one may apply to a given reaction. Here, we give an overview of the models developed, organized by the type of material undergoing reaction.Most consideration has been given to extended inorganic materials (metals and metal oxides for example). Several models have been developed as discussed in ref. 6a and 25. Those most widely referred to are hot spot theory and the magma-plasma model.
Hot spot theory originally developed by considering frictional processes between two surfaces sliding against each other. Small protuberances cause plastic deformations associated with dramatic raising of local (within ca. 1 μm2) temperatures to above 1000 °C for short periods (10−3–10−4 s). More brittle (less plastic) materials would tend to crack under strain.25 However, in brittle materials, hot spots can also occur at the tips of propagating cracks where local temperatures are thought to reach several hundreds or thousands of degrees Celsius for very brief periods.6a,25 There is experimental evidence for such high temperatures in the form of gaseous decomposition products from cracking crystals of metal azides as well as organic compounds such as C(CH2NO3)425,26 and glucose.6a
The magma-plasma model arose from considering direct impacts rather than lateral frictional processes. It proposes that local temperatures greater than 104 °C, can be generated at impact points, associated with transient plasmas and the ejection of energetic species including free electrons. This model also was developed largely in the context of extended inorganic materials.6a
It seems unlikely that hot spots and magma-plasma sites are the primary sites of reactivity in molecular organic and metal–organic mechanochemical reactions. If they were, extensive decomposition would be expected. That such decomposition is not seen suggests that these phenomena may be too brief and/or too localized to be the primary reactive sites for molecular organic reactions. It is still possible that they do occur in molecular reactants under mechanochemical conditions and that they contribute to general frictional heating as the localized energy dissipates. Related to such dissipation, but again in the context of inorganic materials, a hierarchical system has been developed delineating several different physical processes, each with an associated timescale, which can occur under mechanochemical conditions following impacts or frictional processes.6a What would seem most relevant to common ball milling reactions of molecular reactants are the temperatures, pressures and processes occurring over larger areas of ca. 1 mm2 as the ball impacts against reactants on the side of the vessel. However, models and measurements over these larger areas have not yet been applied to molecular synthetic reactions to our knowledge.
With regard to cocrystals, mechanochemical mechanisms have recently been reviewed.21 The models developed are different to those described above. On one hand, this distinction can be traced to the inherently different natures of the types of reactants, with molecular crystals being generally softer and more mobile on molecular scales. On the other, this also derives from the fact that the reaction conditions studied are not always actually mechanochemical (and therefore they may not explicitly consider the formation of hot spots) although they are doubtless relevant to mechanochemical conditions. Work from several groups has been summarized under three generic mechanisms,21 specifically (i) molecular transport across surfaces,27 through the vapour phase,27,28 or through the bulk of a crystal,29 (ii) formation of liquid eutectic intermediate phases,.30 and (iii) reaction via an amorphous intermediate phase.31 The first relates to molecules only loosely held in their lattices, with, for example, significant vapour pressures (e.g. naphthalene).27 The second relates to reactants which have low melting points or reaction mixtures which may form low-melting eutectics such as the diphenylamine-benzophenone mixture.30 The third relates to molecules relatively strongly held in their lattice positions (e.g. through substantial hydrogen bonding) but whose reactivity is increased under mechanochemical conditions by forming amorphous phases. An example is grinding carbamazapine with saccharin to form a pharmaceutical cocrystal.31 In the context of these three mechanisms, it is relevant to note the physical effects which grinding can have on molecular crystals. These include (i) breaking down particles to smaller sizes, giving greater surface area and breaking up any product coating layers to expose fresh surfaces, (ii) intimate mixing of reactants, (iii) introducing defects and eventually amorphization of the material, and (iv) frictional heating, both local and bulk. These physical effects can enable or accelerate each of the three mechanisms described above.
Liquid-assisted grinding (LAG) can accelerate cocrystallisation reactions and give products of higher crystallinity compared to neat grinding. Therefore, LAG, as might be expected, seems to provide greater molecular mobility than does neat grinding. Although the term liquid-assisted grinding does not presuppose that the liquid added plays the role of solvent, correlations of reactivity with reactant solubility have been noted in some cases.32 The nature of the added liquid can also determine the product obtained (without being included within it), again suggesting that solvation (and therefore solubility effects) can be significant.33 Regarding molecular scale rearrangements, some crystalline intermediate phases have been observed and structurally characterised.34
Organic reactions in which covalent bonds are formed have been suggested to occur principally, or even exclusively, through bulk liquid eutectic states.18 This mechanism is analogous to the second cocrystallisation mechanism described above. However, in some cases (certain Knoevenagel reactions under temperature-controlled ball milling,35 and organic disulfide metathesis reactions)36 it has been asserted that there was no bulk melt and the reaction was solid state. Further and broader studies of covalent organic reactions would be very valuable in giving a definitive view of the possible transport mechanisms, in particular how generally this type of reaction can proceed via bulk solid phases. However, the possibility that the eutectic mechanism is more dominant in covalent bond-forming reactions than in cocrystal-forming processes would be consistent with some inherent differences between these two reaction classes. In particular, covalent bond forming reactions are more likely to exhibit greater exotherms, to eliminate liquid or low-melting byproducts, and to have additional reactants present (bases, acids etc.). Each of these aspects increases the potential of forming low-melting eutectic intermediate phases. In the solid state Knoevenagel reactions described above the condensate water is believed to be taken up by the crystals of the product.35
Mechanistic studies of metal–organic reactions are relatively sparse. This is also a highly diverse class of reactions. For example, the metal-containing reactant may be an extended covalent metal halide, pseudohalide or oxide, a hydrated/non-hydrated ionic salt or a neutral molecular species. The bond enthalpies involved (and by inference the magnitude of any exotherm) and the labilities of reactants also span very wide ranges. With more labile, molecular examples, the reactions may share mechanistic similarities with cocrystallisations. In fact, coordination polymers can interconvert under liquid-assisted grinding (LAG) via crystalline intermediate phases behavior which is reminiscent of mechanochemical cocrystallisations.37 In studies of LAG reactions of Cu(SCN) complexes, the rate of reactant diffusion in a liquid inter-particle zone has been predicted to be extremely sensitive to the particle size (inversely proportional to the cube of the particle diameter). If the added liquid can dissolve one or both of the reactants, the very high speed of reactant diffusion enabled by small particles may explain the fact that reactions between preground reactants merely placed in contact in the presence of a small amount of the liquid can also proceed at appreciable rates.38 In reactions involving highly hydrated metal salts, or the elimination of condensates such as water or acetic acid, the presence of such ‘internal solvent’ naturally invites comparison with LAG reactions. In fact, the quantities of such internal solvent are similar to the amounts of liquids normally added in LAG. Also, the generally high mechanochemical reactivity of metal acetates with carboxylic acids (favoured by the release of acetic acid as internal solvent) and the accelerating effects of water of crystallization have both been noted.39 Further, at least one reaction (Cu(OAc)2·H2O + NC5H4CO2H) which eliminates acetic acid as a condensate has been noted to be self-sustaining following a brief initiation by grinding.40 This behavior is closely related to the Cu(SCN) system in which reactions between preground reactants merely placed in contact with a small amount of added liquid also proceed without further grinding.38b Whilst some metal–organic complexation reactions can give the product as a paste, which dries to a free-flowing powder after exposure to air (suggesting a eutectic-type mechanism is possible),39 others appear to remain as free-flowing powders throughout the reaction.39,40 With the less labile metal–ligand systems based on stronger bonds, there may be closer mechanistic similarities with covalent organic reactions. However, there are relatively few studies of the more inert systems. Possibly the least labile system studied is the reaction of PtCl2 (a covalent polymer) with PPh3 to give PtCl2(PPh3)2, and the reaction of PtCl2(PPh3)2 with K2CO3 to give Pt(CO3)(PPh3)2.41 The latter reaction is thought to be truly solid state because of the high melting points of the reactants compared to estimated local temperatures generated in the ball mill.
Overall, there is still clearly some way to go to obtain a cohesive and comprehensive picture of the mechanisms of mechanochemical reactions. Further progress is likely to require carefully designed experiments which can provide key insights, as well as a larger body of observations on which to draw. In this regard, as mechanochemical synthesis continues to develop, it will be helpful to report and study reactions which do not proceed under any given mechanochemical conditions as well as those that do.
2. Industrial aspects
2.1 Introduction
The need for sustainability brought about by the Kyoto Treaty and the increasing global demand for products will inevitably lead to an increase in sustainable manufacturing processes which have lower environmental demands.42 Increased sustainability can take the form of lower energy use, reduced waste, less organic solvents and improved selectivity. Big improvements have been made recently in solvent usage in several pharmaceutical processes.43 The manufacture of Viagra (sildenafil) was improved to reduce solvent usage from 1700 to 7 l/kg.44 A new route to metal oxides developed by Süd Chemie replaces dissolution in nitric acid followed by base precipitation with a mild aerobic treatment of the metal in aqueous carboxylic acid, reducing the process water by 95%.45 Mechanochemical methods offer solvent-free (or minimal solvent) routes to industrial materials and are therefore of great interest in devising more sustainable processes.9,46 The potential to access materials not available by other methods is also of great interest.2.2 Overview of patent activity
Fig. 1a shows the growing number of patents filed per year that contain in the full description either of the terms “mechanochemistry” or “mechanochemical”.47 There is little activity up to the mid 1980s at which point there is a dramatic increase. The plot also shows a slowing-off of overall activity since 2005, which merits a more detailed analysis. Patents are classified according to International Patent Classification Codes (IPC) designated by the World Intellectual Property Organization (WIPO),48 which relate to the field of application and the types of inventions etc. and are used here in the four-character form. Fig. 1b–i show the patent activity broken down into eight areas: (1b) A61K (medical and personal care); (1c) H01L (semiconductors and solid state devices); (1d) H01M (mainly batteries); (1e) C01B (compounds of non-metallic elements); (1f) B01J (catalysis); (1g) C01G (metal compounds); (1h) C04B (ceramics) and (1i) G03G (electric charge or magnetic mediated image creation for example in photocopiers). While some areas are static or declining (such as H01L, C01G, C04B and G03G) others are showing steep rises (in particular A61K, H01M, C01B and B01J). Three growth areas of medical/personal care, batteries, and compounds of non metallic elements will be explored in more detail.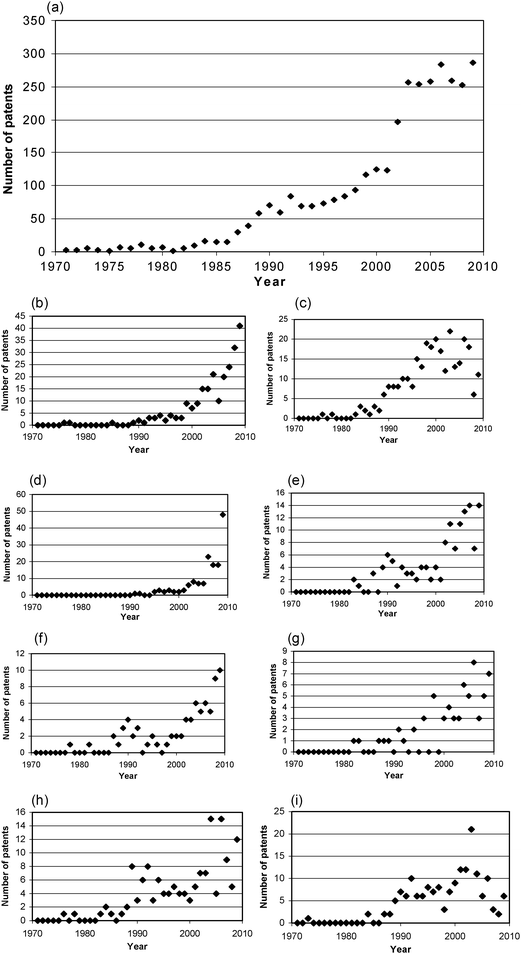 | ||
| Fig. 1 (a) Plot of number of patents vs. year for search terms “mechanochemical” or “mechanochemistry”, and plots showing occurrence of various IPC patent classification codes in patents relating to mechanochemistry; (b) A61K (medical and personal care); (c) H01L (semiconductors and solid state devices); (d) H01M (mainly batteries); (e) C01B (compounds of non metallic elements); (f) B01J (catalysis); (g) C01G (metal compounds); (h) C04B (ceramics) and (i) G03G (electric charge or magnetic mediated image creation for example in photocopiers). See text for full search details. | ||
2.3 Medical/personal care
Iceutica have recently described a mechanochemical route to therapeutically active nanostructured compositions.49 20–30 nm nanoparticles of a pharmaceutical compound are synthesized in a stabilising matrix such as Na2CO3, NH4Cl, (NH4)2CO3etc. Significantly, this process has gone on to commercial scale in a GMP facility.50A further example of production-scale mechanochemistry is by Vectorpharma Spa of Trieste, Italy, who prepared anti-inflammatory drug/carrier composites with β-cyclodextrin by high energy milling.51 β-cyclodextrin finds use as a carrier for pharmaceuticals due to its lipophilic internal cavity and hydrophilic exterior, which allow the formation of water-soluble inclusion complexes.52 The pharmaceutical/β-cyclodextrin complex helps to control the rate of drug delivery. Vectorpharma's mechanochemically-prepared composite shows different properties to those prepared by conventional routes, specifically much higher dissolution rates. The reaction between Nimesulfide and β-cyclodextrin was performed in a high energy vibration mill on pilot (0.5–2 kg) and production (20–50 kg batch) scales with the optimum processing time of a modest 3.5 h. As an indicator of batch-to-batch reproducibility the residual crystallinity of the Nimesulide (an indicator of the amount of free Nimesulide present) was consistently 3–7%.
2.4 Batteries
Li-ion batteries are a major innovation in portable power solutions. They comprise a cathode material such as LiFePO4 or LiCoO2, and an anode material such as graphite.53 Gillette have recently examined the mechanochemical synthesis of the cathode material LiMnO2 from manganese dioxide and lithium hydroxide or lithium carbonate.54 The work was done on the lab scale using a Turbula mixer containing 500 g 1 mm yttria-stabilized zirconia milling media over a 0.5–5 h period. Calcination at 350–420 °C removed residual water. Others have examined such an approach, and High Power Lithium, Lausanne, have also manufactured cathode materials for Li-ion batteries this way.552.5 Compounds of non-metallic elements
Diborane, B2H6, has been prepared mechanochemically without using a solvent for the semiconductor industry.56 Also, silicon nitride is becoming popular for a number of applications such as bearings and high-temperature engine components because it is hard-wearing, lightweight and creep-resistant.57 Commercially, Si3N4 is currently prepared by direct nitridation of silicon powder in the presence of a catalyst at 1200–1400 °C.58 The drawback is that the nitrogen pressure has to be controlled carefully because of the large exotherm. There is also a need for low pressure routes to Si3N4 because high pressure N2 is unattractive regarding safety and capital cost.59 Li et al. with Fujian Sinocera Advanced Materials Co. have used mechanochemical treatment of silicon powder with NH4Cl in steel milling media followed by calcination in the presence of low pressure N2. Pilot work was performed at 400 g scale. The NH4Cl is added principally to improve the texture of the final Si3N4. However, it is possible that during milling it may also improve the characteristics of the silicon. The mechanochemical treatment gives small, highly defected silicon particles which leads to higher reaction rates even at low N2 pressures (10 bar). Mechanochemical treatment reduces the silicon particle size (63 nm after 8 h milling compared to 58 nm after 12 h). The process has been scaled to 3 kg batches and is being transferred to industrial production.2.6 Conclusions
The areas with increasing industrial interest, as indicated by patent activity, are currently medical/personal care, batteries, compounds of non-metallic elements and catalysis. These applications are founded on the types mechanochemistry which have been established for the longest time, i.e. synthesis of inorganic materials and non-covalent organic inclusion complexes/cocrystals (see Sections 3, 4 and 5). Significantly, there are clear examples of processes going into production scale. Of additional interest in future will be patent activity based on more recently-developed uses of mechanochemistry in covalent organic and metal–organic chemistry (Sections 6, 7 and 8).3. Inorganic materials
3.1 Introduction
Inorganic materials represent the most established area of mechanochemical synthesis. As mentioned above, the first recorded example of an entirely solventless mechanochemical reaction can be attributed to Faraday who in 1820 reduced AgCl to Ag using either Zn, Cu, Sn or Fe by grinding in a pestle and mortar.7 Mechanochemistry in the modern era began with mechanical alloying, the process of combining elements or alloys to produce a single homogenous alloy, in high velocity ball mills. The term mechanochemistry is now also widely used to describe this process as well as other chemical reactions to produce alloys and inorganic compounds using ball mills. During such processes there is a significant reduction in crystallite and particle sizes, such that products are often either nanoparticles or amorphous phases,60 which is sometimes desired as providing a top-down route to nano-materials. If more crystalline material is required the product of the mechanochemical process can be sintered.3.2 Alloys
Mechanochemical techniques were first investigated to produce alloys and work in this area is ongoing. General routes are shown in Scheme 1. Recent work using the simple combination of alloys and elements in ball mills has produced Cu–Co,61 Fe–Mo,62 and Mn–Al alloys.63 Elemental combination has also been used for boron-containing alloys in the Ni–Nb–B64 and Ti–Al–B systems.65 The risk of atmospheric oxidation of the metals means that these reactions are carried out under inert gas, typically argon. They generally require relatively long milling times (24 to 300 h).61,63 An alternative to elemental combination is to combine a binary oxide powder and a reducing agent, which can be one of the metals to be alloyed, such as the reaction of TiO2 and Mg to form TiMg,66 or PbO and Te to form PbTe.67 Alternately the reducing agent may not be intended for inclusion, such as carbon, which will be removed as a vapour (CO2)—an example is the synthesis of brass from CuO, ZnO and PbO in the presence of graphite.68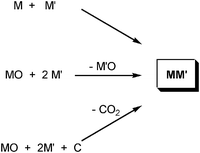 | ||
| Scheme 1 General ball milling routes to alloys. | ||
3.3 Oxides
The mechanochemical synthesis of inorganic oxides can be conducted by several routes (Scheme 2). The simplest is the combination of different binary oxides—similar to the high temperature ceramic synthesis—but relying on the constant fracture and mixing of the grains to produce a homogenous product, as there will often be little thermodynamic driving force in the reaction itself. This method has been used to synthesize numerous materials including CrVO4,69 LaVO4;70 perovskites such as LaCrO3,71 LaMnO3,72 and PbTiO3;73 spinels like MnFe2O4,74 ZnFe2O4,75 and NiFe2O4;76 and Ruddlesden-Popper compounds like Sr3Ti2O7 and Sr2TiO4.77 As for alloys this often produces nano-particulate (<10 nm) products.69,78 However, unlike the alloys, these reactions can be carried out under air, as all the materials are already fully oxidized. Milling times are also generally shorter, typically between 2 and 24 h.69–72,75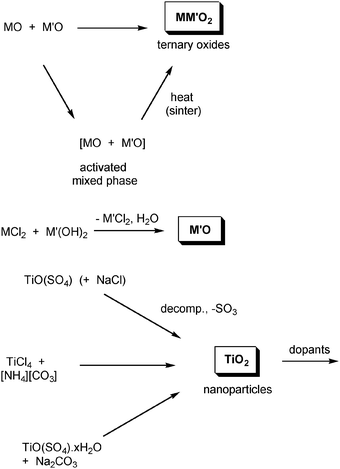 | ||
| Scheme 2 General ball milling routes to metal oxides. | ||
Direct combination of oxide powders to produce a homogenous phase is not always successful, but it can often still be used as an activation step allowing complete reaction at a lower temperature than in traditional ceramic synthesis. For example CaZrO3 synthesized conventionally must be heated to >1100 °C, but only to 800 °C after mechanochemical activation.79 The synthesis temperature of ZrTiO4 could similarly be reduced from 1400 °C to 1100 °C.80 This mechanochemical activation followed by sintering has also been used in the synthesis of MgTa2O6,81 and the aurivillius phases Bi4Srn−3TinO3n+3 (n=4,5).82
An alternative mechanochemical approach involves providing a thermodynamic driving force to the reaction. One method is the presence of a reducing metal, intended for inclusion in the final product and requiring the use of an inert gas, but significantly reducing the necessary milling time—in some cases to 30 min. Examples include the use of titanium in the formation of FeTiO3 and FeTiO4,83 iron in the formation of Fe2GeO4,84 zinc in the formation of ZnFe2O4,85 and aluminium in the formation of FeAl2O4.86 In all these cases the reaction scheme must be devised, as in the case of oxide combination, so that there is a single product. Mechanochemical synthesis can, however, be used for reactions with multiple products as long as the solubilities of the product and by-products are different. An example is the production of silver nanoparticles by reduction of AgCl using either Na or Cu, with the CuCl and NaCl by-products removed by NH4OH leaching and washing respectively.87
Multiple products are also formed in displacement reactions, which represent another method of introducing a driving force to a mechanochemical reaction. An example is the reaction of ZnCl2 and Ca(OH)2, to produce ZnO nanoparticles in a CaCl2 matrix (with loss of water vapour).88 The CaCl2 can be removed and the ZnO nanoparticles isolated by washing with water. Similar displacement reactions with salt products have been used to synthesize ZrO2,89 Cr2O3,90 LaCoO3,91 and Nb2O5.92
A number of researchers have used alkaline and alkaline earth carbonates to introduce Group 1 and 2 metals into compounds, such as CaTiO3,93 Ba1−xSrxTiO3,94 and NaNbO3.95 with loss of CO2, effectively generating MO or M2O in situ for direct oxide synthesis.
The techniques outlined above represent the most common strategies for generating inorganic solids. They have been used to synthesise several potentially important materials for applications. The carbonate method has been used to synthesize Ba2ANb5O15 (A=K, Na, Li),96 while simple combination of oxides allowed the synthesis of Bi4Ti3O12,97 both of which are ferroelectric compounds, with potential use in memory devices. Mechanochemical methods have been used to synthesis solid state electrolytes (silver niobium oxyfluoride and silver molybdenum oxyfluoride),98 fast ion conductors (RbAg4I5 and KAg4I5·)99 and a lithium battery cathode material (Li2Mn2O4).100
Another area in which mechanosynthesis has been used is titania based nanoparticles. TiO2 is a widely used UV semi-conductor photocatalyst with applications in self-cleaning coatings, anti-microbial coatings and photo-activated water splitting.101 Production of TiO2 nanoparticles has been achieved by: mechanochemical decomposition of titanyl sulphate using NaCl diluents;102 reaction of TiCl4 and ammonium carbonate;103 displacement reaction of TiOSO4·xH2O and Na2CO3. After annealing these particles had twice the activity of Degussa P25, the highly active industry standard.104 Attempts to dope TiO2 nanoparticles with carbon, sulphur and nitrogen have also been conducted by mechanochemical reaction of titania with adamantine, sulphur and ammonium carbonate with results indicating that visible-light photoactivity has been induced.105 Fluorine-doped SrTiO3 has been prepared mechanochemically and this has also demonstrated visible light photocatalysis.106
3.4 Halides, sulphides and nitrides
A number of compounds of the form AMF3 have been produced by the mechanochemical combination of AF and MF2, where A is an alkali metal and M is a divalent metal ion (Scheme 3), under inert gas, with milling times of 3–12 h. This has been successful for Na (M=Fe, Mn and Ni)107 and K (M=Mg, Zn, Mn, Ni, Cu, Co and Fe).108 Similarly, a series of chlorides KMCl3 (M=Ti, Cr, Mn, Fe, Co, Ni, Cu and Zn) has been prepared109 and direct combination of CaF2 and LaF3 produced Ca1−xLaxF2+x.110 There has also been interest in mechanochemically synthesized halides as fast ion conductors, including NaSn2F5,111 RbPbF3,112 and Pb1−xSnxF2.113 The range of methods used in the synthesis of LaOF demonstrates a number of possible routes to the mechanochemical introduction fluorine; LaOF has been made by ball milling La2O3 with either PTFE,114 poly(vinyldene fluoride)115 or with LaF3.116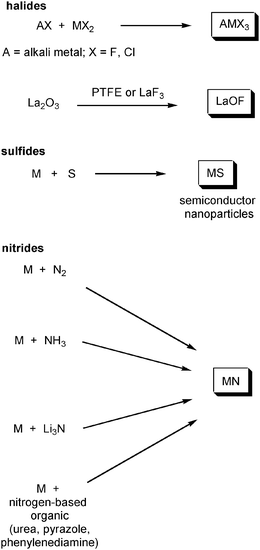 | ||
| Scheme 3 General ball milling routes to halides, sulfides and nitrides. | ||
Mechanochemical synthesis of sulphides has focused principally on semi-conductor nanoparticles, by direct combination of the metal and sulphur. This has been achieved for CdS,117 CdxZn1−xS,118 and FeS.119 Other sulphides of interest that have been investigated include fast ion conductors such as (Ag2S)x(Sb2S3)1−x and the purported anti-cancer agent As4S4 (realgar).120
For several metals nitrides can be synthesized simply by ball milling the metal under a high pressure of nitrogen for greater than 10 h. This method has given TiN,121 ZrN,122 VN,123 NbN124 and CrN.125 Similarly, ball milling the metal under the more reactive ammonia has given Mo2N,126 GaN,127 BN,128 Si3N4.129 Alternative sources of nitrogen include Li3N, which has been used to form GaN,130 ZrN131 and a range of lithium nitridometallates, LiNiN, Li3FeN2, and Li7VN5.132 Its high reactivity can give complete mechanochemical reactions in as little as 7 min.131 Ternary nitrides have been made by alloying a binary nitride (Mo2N) with Fe or Co.133
Solid organic nitrogen compounds may also be used. Examples include reaction of urea with titanium to form TiN;134 pyrazole and iron powder to give Fe3N;135 and phenylene diamine and iron to give Fe2N3.136 This avoids the introduction of a gas, but requires removal with solvent at the end of reaction.
3.5 Composites
For much of the chemistry described above, it has been important to design the reaction such that only a single inorganic product is generated, or if there are by-products, that they can be easily removed as gases or by extraction. However, two or more final products can be deliberately synthesized simultaneously to produce a nano-composite. Most research conducted so far is for Al2O3 nanoparticles embedded in a metal or alloy, to give improved mechanical properties (Scheme 4). Examples include Al2O3 in Zn,137 Nb,138 and Cu.139 This is done by milling aluminium with the metal oxide, driven by the high heat of formation of Al2O3. Use of excess aluminium can give Al2O3 particles in aluminium-based alloys such as TiAl3,140 Al–Zn141 and AlB12.142 | ||
| Scheme 4 Synthesis of a composite material by ball milling. | ||
Mechanochemically-synthesized nano-composites have also been investigated as novel anode materials for Li ion batteries. These include a combination of Sn/C with either TiO2 or Fe,143 or LiH with either Mg or Ti.144a
A related aspect is the mechanochemical dispersion of metal particles on a pre-existing support, which provides a way to generate heterogeneous catalysts. This has been demonstrated for nanoparticulate gold dispersed on coordination polymers, carbon or metal oxides by Haruta using Au(acac)Me2, which is readily vaporised, as the gold source.144b Some of the resulting materials exhibited high catalytic activities.
3.6 Conclusions
The historically most established application of mechanochemical synthesis is for inorganic materials and this continues for current and future technological applications. The formation of nanoparticulate phases and otherwise inaccessible composites are significant aspects. Interestingly it is also possible to react organic compounds with elemental metals this way. Many methods have been devised such that no separation of by-products is needed. This can be seen as an atom-economic approach, even if driven primarily by practical considerations. Analogous reconsideration of routes to organic products will be of interest in applying mechanochemistry to organic and metal–organic synthesis to avoid the need for separations (see Sections 6, 7, 8 and 10).4. Cocrystals
4.1 Introduction
Although mechanochemical synthesis has not been applied as extensively to cocrystals as to inorganic materials, it does have a relatively long history in this context.10 We adopt here the liberal definition of a cocrystal as a “multi-component molecular crystal”.145 This includes solvates and hydrates, and does not discriminate between formally charged systems (salts) versus neutral ones as defined by the extent of proton transfer along a hydrogen bond.145c,d The pharmaceutical applications of cocrystals146 are discussed in Section 5. Mechanical mixing of molecular crystals, manually or by ball milling, is often effective for preparing cocrystals.147 Often the best results are obtained by liquid assisted grinding (LAG, also called kneading) i.e. by grinding with a small amount of a liquid.148 This method is complemented by approaches such as exposure of a solid mixture to solvent vapour149 (vapour digestion) and heating solid mixtures150 including screening by hot stage microscopy.1514.2 Charge transfer cocrystals
Pioneering studies were carried out by Toda et al. in the preparation of crystalline host–guest inclusion compounds152 and charge-transfer systems.153 Formation of charge transfer cocrystals can often be followed by eye due to the change in color. Kuroda et al.28,154 obtained three-component cocrystals based on racemic bis-β-naphthol, benzoquinone and anthracene. Importantly, the resulting cocrystal could not be obtained from solution and so required structure determination from X-ray powder diffraction (see Section 9).155 Sada et al.156 formed brightly colored charge-transfer complexes by mixing a pale-colored electron donor, acting as an analyte, and a pale-coloured electron acceptor, acting as a probe, or vice versa. A series of acceptor molecules was designed as probes to produce a 2D colorimetric indicator array which discriminated between isomers of organic molecules such as di-hydroxynaphthalene, using only the naked eye. All reactions were carried out in molten pastes. In 1 min colour changes were visible but became much brighter after 10–15 min grinding. One probe molecule allowed discrimination between the eight isomers of di-hydroxynaphthalene.4.3 Acid–base cocrystals
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystals of variable chain length dicarboxylic acids HOOC(CH2)nCOOH (n = 1–7) and 1,4-diazabicyclo[2.2.2]octane (dabco) have been prepared mechanochemically and investigated by X-ray diffraction and solid state NMR.157 The melting points of the cocrystals were found to alternate as in the corresponding diacids irrespective of the salt/molecular nature of the cocrystals. It has been recently demonstrated that this behavior is also exhibited by cocrystals of these diacids with dipyridyl molecules 4,4′-bipyridine (bipy), 1,2-bis(4-pyridyl)ethane (bpa), and 1,2-(di-4-pyridyl)ethylene (bpe) which contain an even number of C atoms between the two N atoms; while it is completely reversed in cocrystals with 1,2-bis(4-pyridyl)propane (bpp), which contains an odd number of (CH2) groups.158
:1 cocrystals of variable chain length dicarboxylic acids HOOC(CH2)nCOOH (n = 1–7) and 1,4-diazabicyclo[2.2.2]octane (dabco) have been prepared mechanochemically and investigated by X-ray diffraction and solid state NMR.157 The melting points of the cocrystals were found to alternate as in the corresponding diacids irrespective of the salt/molecular nature of the cocrystals. It has been recently demonstrated that this behavior is also exhibited by cocrystals of these diacids with dipyridyl molecules 4,4′-bipyridine (bipy), 1,2-bis(4-pyridyl)ethane (bpa), and 1,2-(di-4-pyridyl)ethylene (bpe) which contain an even number of C atoms between the two N atoms; while it is completely reversed in cocrystals with 1,2-bis(4-pyridyl)propane (bpp), which contains an odd number of (CH2) groups.158
Competing solid-state exchange between cocrystal components by grinding has also been recently reported on cocrystals of R,R-, S,S-, racemic and R,S-tartaric acid (ta) with pyrazine (py).159 “Supramolecular metathesis” was carried out in methanol slurry by reacting the cocrystal products, (R,R-ta)·(py), (S,S-ta)·(py), (R,S-ta)2·(py) and (R,R/S,S-ta)·(py), with the different forms of tartaric acid showing that coformer exchange could take place according to the sequence of stability (R,S-ta)2·(py) > (R,R/S,S-ta)·(py) > (R,R-ta)·(py) or (S,S-ta)·(py).
Formation and polymorphic transformation by grinding has been studied for 4,4′-bipyridine (bipy)/pimelic acid (H2pma) cocrystals.160 The structures of the three polymorphs (Form I, Form II and Form III) were determined from single crystals grown by seeding of solutions with the microcrystalline mechanochemical product. All polymorphs consisted of chains of alternating bipy and H2pma molecules linked by O–H⋯N hydrogen bonds, but differed in the relative arrangements of the chains. Scheme 5 shows the various interconversions that were possible and illustrates how LAG can be important in enabling some transformations, complementing other more classical methods such as recrystallisation from solvents or from melts.
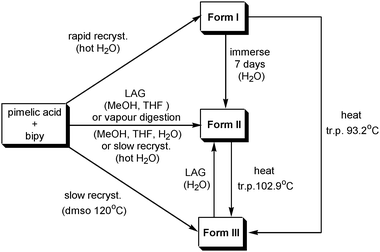 | ||
| Scheme 5 Preparation and transformation conditions of the three crystal forms of (bipy)·(H2pma) (tr.p.: transition point). | ||
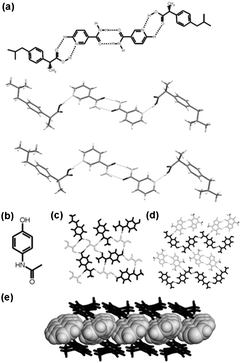
Grinding has also been used to gave a family of organometallic-organic cocrystals of the pyridyl ferrocene derivative Fe(η5-C5H4-C5H4N)2 (Fcpy2) with dicarboxylic acids HOOC(CH2)nCOOH2 of variable chain length (n = 4–7). Compounds of general formula (Fcpy2)·(diacid) were prepared by kneading of solid mixtures with MeOH. Interestingly, all compounds are discrete macrocycles {(Fcpy2)·(diacid)}2 rather than extended networks except for the pimelic acid adduct (n = 5) (Fig. 2).
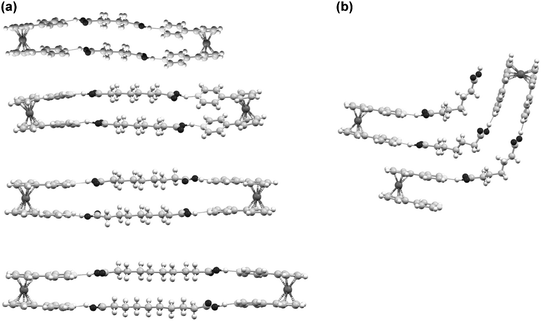 | ||
| Fig. 2 The supramolecular structures of the macrocycles {(Fcpy2)·(HOOC(CH2)nCOOH2)}2 (n=4, 6, 7, 8) (a), and the zig–zag chain found when n = 5(b). | ||
In the search for alternative polymorphs of the pimelic acid adduct vapour digestion of the solid mixture was attempted. The stoichiometry of the products was affected by the protic or aprotic nature of the solvent. The cocrystal with 1:1 stoichiometric ratio as observed in the grinding synthesis, was obtained by exposure to vapours such as CH2Cl2, CHCl3, (CH3CH2)2O, CH3NO2 and ethyl lactate, while a 1:2 cocrystal (Fcpy2)·(HOOC(CH2)5COOH)2 was formed with protic solvents, such as CH3OH, CH3CH2OH, H2O and isopropyl alcohol.149b This indicates that the solvent used in the kneading is not an innocent spectator or lubricant in the diffusion process but takes an active part in the process very likely via slight supersaturation levels over the grain surfaces, i.e. that dissolution, and therefore solubility, in the added liquid is important.
To explore the effect of the preparation method on the nature of the product, the cocrystallisation of Fcpy2 and anthranilic acid, (C6H4)NH2COOH, has been investigated.149a It has been shown that the same product can be obtained, quantitatively, by four different processes, namely kneading with methanol, wet compression (i.e. pressure without mixing in the presence of MeOH), and vapour digestion (i.e. placing a mixture of the solid reactants in an atmosphere of MeOH vapour), and by heating a mixture of the two solid reactants. In contrast, no reaction was observed by dry mixing or dry compression. This demonstrates not only the ability of small amounts of added liquids in LAG to direct the course of a cocrystallization but indeed also to enable cocrystallization. This dramatic influence of the added liquid in LAG is echoed in the mechanosynthesis of metal complexes including metal organic frameworks (Sections 7 and 8).
4.4 Ionic cocrystals
Recently it has also been shown that grinding or kneading of classical ionic crystalline materials (NaBr, KBr, CsI, RbBr etc.) with organic molecules, such as solid barbituric acid (H2ba), gives a new class of “ionic cocrystals” in which BA is present as a neutral component.161 Depending on the metal, hydrated forms were also observed. The structure of (H2ba)·(KBr)·(H2O)2 is shown in Fig. 3. An important aspect is the higher thermal stability and dissolution rates of the cocrystals compared to pure barbituric acid, illustrating that this unusual type of cocrystallization can alter the dissolution behaviour of organic molecules. This ability to modify the properties of organic molecules by creating novel solid forms is of key interest in the pharmaceutical area as expanded upon in Section 5.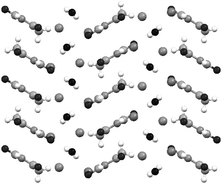 | ||
| Fig. 3 Packing of the ionic cocrystals (H2ba)·(KBr)·(H2O)2. | ||
4.5 Organocatalysis through cocrystallization
A cornerstone organic solid state reaction (not mechanochemical) is the [2+2] photo-induced dimerization of two double bonds in close proximity to one another.162 MacGillivray et al. have used cocrystallization to direct such close arrangements of double bonds using resorcinol derivatives as directing agents.163 Very recently this methodology has been extended to be catalytic by using mechanochemistry (Fig. 4).164 The method involves alternating grinding periods with exposure to UV light (which causes the photochemical cyclisation) in the presence of a substoichiometric amount of a resorcinol directing agent. The catalytic resorcinol directing agent is able to dissociate from the reaction product, allowing it to be redistributed by grinding to complex to further reactant and so enabling catalytic turnover. This can be regarded as a more ‘crystallographic’ example of organocatalytic reactions in general which are discussed in Section 6.4.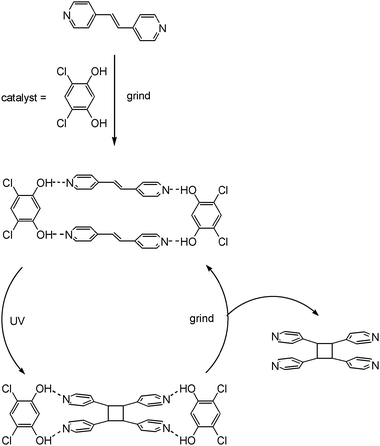 | ||
| Fig. 4 Solid state catalytic system which alternates UV irradiation and grinding-induced cocrystallisation. | ||
4.6 Conclusion
Mechanochemistry is now recognized as one of the most effective ways to generate cocrystals. It is frequently effective regardless of the types of intermolecular interactions which are formed. Importantly, it can provide alternative structures to those obtained by solution crystallization. Whilst there are challenges to determining the structures of such cocrystals advances in structure solution from powder XRD data (see also Section 9) is successful in increasing numbers of cases.5. Pharmaceutical aspects
5.1. Introduction
The discovery of new solid forms of pharmaceuticals (amorphous, crystalline, single and multicomponent) is an important application of mechanochemistry. Although most established for producing amorphous phases,165 attention has recently been given to its use in cocrystallization (see also Section 4).166 Cocrystallization is useful in this context because it provides a way to derivatize active pharmaceutical ingredients (APIs), by modifying their solid-state arrangements rather than their internal molecular structures. Modification of the crystal structure by cocrystallization can improve pharmaceutically relevant properties such as dissolution rate, solubility, thermal and hydration stability or compressibility.167Pharmaceutical cocrystals generally consist of an API and one or more pharmaceutically acceptable molecules, known as the cocrystal formers or “coformers”, assembled into a well-defined crystal lattice.166b,c The coformers are typically compounds “generally regarded as safe” (GRAS compounds). Greater thermodynamic stability168 makes such cocrystals preferred over metastable amorphous forms and alternative polymorphs that may be of higher free energy. Since most pharmaceutical coformers are solids, pharmaceutical cocrystallization is also advantageous compared with solvate formation, since solvates inherently involve the risk of spontaneous desolvation. Finally, cocrystallization is more versatile than salt formation, as it does not require an ionisable centre in the API, and there are considerably more GRAS compounds than pharmaceutically acceptable salt formers. Because of the very large range of potential coformers, the efficiency and convenience of mechanochemical methods makes them particularly advantageous in screening amongst large numbers of potential pharmaceutical cocrystals.
5.2 Pharmaceutical cocrystallization by neat grinding
The simplest mechanochemical method for pharmaceutical cocrystallization is by neat grinding of two or more cocrystal components.169 The first examples were reported independently in 1993 by Caira170 and Etter.171 Caira ground the drug sulfadimidine with a variety of carboxylic acids, including benzoic, anthranilic, salicylic, and acetylsalicylic (aspirin) (Fig. 5a).170 Cocrystals were obtained in all cases, identical to those previously obtained by solution methods. First order kinetic behaviour was observed, interpreted as suggesting a random nucleation reaction mechanism. The exceptional stability of the sulfadimidine-anthranilic acid cocrystal (Fig. 5b) was established through two types of mechanochemical competition experiments either using two different acids or grinding the preformed sulfadimidine cocrystals with an alternative carboxylic acid. The sulfadimidine-anthranilic acid cocrystal was always obtained.170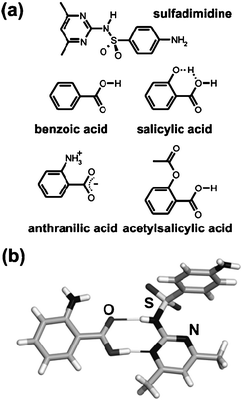 | ||
| Fig. 5 (a) Molecules used in mechanochemical synthesis of pharmaceutical cocrystals by Caira et al.; (b) fragment of the crystal structure of the cocrystal of sulfadimidine with anthranilic acid.170 | ||
Etter described the solid-state cocrystallization of 9-methyladenine and 1-methylthymine,171 driven by the formation of hydrogen-bonded Hoogsteen complexes between the base pairs (Fig. 6).172 Although 9-methyladenine and 1-methylthymine are not API molecules per se, they are derivatives of biologically active molecules.
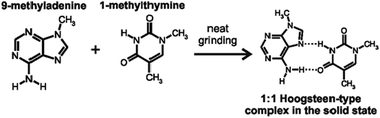 | ||
| Fig. 6 Mechanochemical reaction of 9-methyladenine and 1-methylthymine to form a cocrystal reported by Etter.171,172 | ||
In the later work of Rodríguez-Hornedo, the cocrystal of the API carbamazepine with the coformer saccharin was used to study the mechanism underlying neat grinding cocrystallization (Fig. 7),31,173 which revealed an intermediate amorphous phase. The grinding reaction was faster at temperatures close to the glass transition temperature (Tg) of the ground mixture. Cryogenic grinding, i.e. grinding at low temperatures, allowed observation of the intermediate amorphous phase which crystallized upon warming. Such behaviour is consistent with the mechanochemical behaviour of single-component solids, studied by Descamps et al., i.e. that low temperature grinding leads to amorphization (vitrification), whilst grinding above Tg causes polymorphic transformations.174 Mechanochemical cocrystallization was also accelerated by exposing the intermediate amorphous phase to water (in the atmosphere or as hydrated reactant), effectively lowering the Tg of the reaction mixture.31,173 Higher reactivity of hydrated carbamazepine compared to the anhydrous form was also observed by Rades et al. in mechanosynthesis of the (carbamazepine)·(nicotinamide) cocrystal.175 Similar effects have been seen for caffeine and citric acid and their hydrates.176
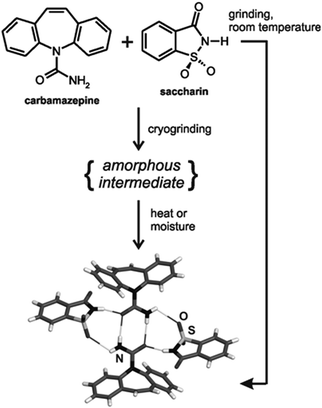 | ||
| Fig. 7 Mechanochemical cocrystallization by neat grinding of carbamazepine and saccharin,31,173 involving an amorphous intermediate. | ||
The ability to scale mechanochemical cocrystal formation is important for industrial applications. This was recently addressed by Medina et al. who described a scalable continuous flow solvent-free process for pharmaceutical cocrystallization using twin screw extrusion.177
5.3 Screening for pharmaceutical cocrystals by liquid-assisted grinding (LAG)
Since the interaction between the components of a pharmaceutical cocrystal is generally based on hydrogen bonding that is likely to be disrupted by interaction with the solvent, cocrystal screening using conventional solution-based methods is not efficient. For example, solution methods are likely to fail in screening for cocrystals of low-solubility APIs, as attempts to form the cocrystal often result in the separation of the solid API with the more soluble coformer retained in solution.178 In such cases, mechanochemical neat grinding179 or liquid-assisted grinding (LAG, also known as kneading or solvent-drop grinding) represent obvious alternatives.180 LAG is preferred to neat grinding in that it is more general, faster (typically 20 min) and gives more highly crystalline products.181 For example, while caffeine and citric acid do not form a cocrystal upon neat grinding, LAG with water or organic solvents gives the pharmaceutical solid (caffeine)·(citric acid).176 The advantage of LAG was also observed in screening for cocrystals of piroxicam.182 The study by Childs, Rodríguez-Hornedo et al. established that LAG was of comparable efficiency to solution-based and thermal methods for cocrystallization of carbamazepine.183 Karki et al. demonstrated that mechanochemistry was more effective than solution- and melt-based methods in screening for cocrystals of nicotinamide.184The application of LAG to form cocrystals of low-solubility APIs was demonstrated using theobromine (Fig. 8). Grinding with trifluoroacetic or malonic acids resulted in cocrystals, while none were obtained by crystallization from solution.185 Additionally, because of the high melting point (>400 °C) of theobromine the two cocrystals could not be obtained from the melt. The failure of solution crystallization also prevented their structural characterization by single crystal X-ray diffraction but characterization was achieved from powder X-ray diffraction (PXRD) data.185 The combined approach of solid-state synthesis and powder structure analysis was also applied to the study of cocrystals of theobromine with acetic acid,186 and of theophylline with chiral and racemic malic acids.187 Among further pharmaceutically interesting molecules that were recently explored through LAG cocrystal screening are the cases of dihydrocarbamazepine,188 indomethacin189 and the drug candidate AMG 517.190
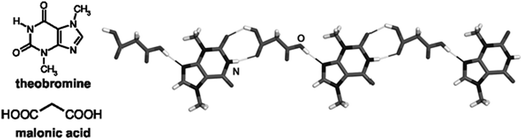 | ||
| Fig. 8 Pharmaceutical cocrystal components and a fragment of crystal structure for theobromine and malonic acid formed by LAG.185 | ||
Zaworotko explored the mechanochemical formation of 25 model cocrystals previously obtained from solution.32 In each case, the cocrystal was successfully obtained using only 4–20 μL of the liquid per 100 mg of the solid. Furthermore, LAG was also shown to be advantageous to cocrystal screening from the melt, as it avoids exposing thermally-sensitive APIs or coformers to high temperatures. This was demonstrated in screening for cocrystals of the model API nicotinamide with dicarboxylic acids: Screening from the melt was not possible with thermally sensitive oxalic acid,184 or the high melting fumaric acid.191
Another potentially interesting role for LAG in the context of pharmaceutical solids is for conducting cocrystal-cocrystal reactions involving chiral and racemic solid forms. In particular, LAG reactions between left- and right-handed pharmaceutical cocrystals of theophylline with tartaric acid were found to give a racemic pharmaceutical cocrystal. In contrast, LAG of left- and right-handed cocrystals of caffeine with tartaric acid decomposed the cocrystal and racemic tartaric acid and solid caffeine separated. This represents a unique example of a solid-state separation of a pharmaceutical cocrystal into the solid active ingredient and the cocrystal former.192
5.4 Control of stoichiometric composition and polymorphism
An attractively versatile aspect of cocrystallization in the synthesis of new API forms is the possibility to form cocrystals containing identical constituents in different stoichiometric ratios.193 Mechanochemistry can often provide such stoichiometric variations by simply grinding different amounts of starting materials. This was first demonstrated for the model API caffeine upon cocrystallization with acetic acid.193 Crystallization of caffeine from liquid acetic acid gives cocrystals of composition (caffeine)·(acetic acid)2. The same product is obtained by grinding the two components in the appropriate ratio. Grinding equimolar amounts of caffeine and acetic acid, however, gave a cocrystal with composition (caffeine)·(acetic acid).193 Stoichiometric variations were also systematically investigated for cocrystals of nicotinamide with dicarboxylic acids and, while readily accomplished mechanochemically, this could not be easily achieved from solution or a melt.100a,184,194The ability to vary the added liquid in LAG allows control over the polymorphic behaviour of mechanochemically obtained cocrystals as noted in Section 4. In the pharmaceutical context this was demonstrated by Trask et al.33,195 Cocrystallization of caffeine with glutaric acid in chloroform solution provided the cocrystal (caffeine)·(glutaric acid) as two concomitant polymorphs. However, LAG with either chloroform or cyclohexane gave each form selectively.196 Such mechanochemical control of polymorphic behaviour has also been observed in other systems.182,195
5.5 Pharmaceutical cocrystals with improved properties
In principle, the formation of pharmaceutical cocrystals can be guided by the concept of supramolecular synthons. However, the physicochemical properties of the resulting material cannot be readily predicted. Furthermore, since the synthon-based approach considers only specific recognition between selected functional groups, its usefulness in fully predicting the three-dimensional structure of the cocrystal is limited. Additionally, the synthon approach is sensitive to competition between different functional groups.196,197 Consequently, pharmaceutical cocrystals with improved properties must be discovered in a trial-and-error process which is strongly assisted by efficient screening methods such as LAG. An example is the use of cocrystals to enhance the hydration stability of a solid API as first demonstrated for model APIs caffeine or theophylline by forming cocrystals with dicarboxylic acids.167c Cocrystallization with oxalic, malonic, maleic and glutaric acids provided cocrystals based on expected R22(7) carboxylic acid-imidazole heterosynthons. In both cases, the cocrystal with oxalic acid demonstrated much greater hydration stability compared to the pure APIs.167cLAG was used to construct cocrystals of nicotinamide with the low melting APIs S-ibuprofen and RS-ibuprofen.179 It was anticipated that cocrystallization would give solid forms with higher melting points, due to extended hydrogen bonding in amide-amide R22(8) homosynthons. Cocrystals with significantly higher melting points compared to the parent APIs were indeed obtained. Single crystals were subsequently grown from solution and structurally characterized, confirming the presence of the expected networks (Fig. 9a).198
 | ||
| Fig. 9 (a) Expected hydrogen-bonded assembly179 (top) and corresponding fragments in observed198 crystal structures of nicotinamide cocrystals with RS- (middle) and S-ibuprofen (bottom); (b) molecular diagram of paracetamol; (c) single layer in the crystal structure of the paracetamol cocrystal with oxalic acid; (d) single layer in the crystal structure of the paracetamol cocrystal with theophylline and (e) stacked layers of paracetamol and naphthalene in the cocrystal. Molecules of paracetamol are shown in black and the molecules of the coformer in grey.167e | ||
Mechanochemical cocrystallization has also been exploited in the synthesis of readily compressible and thermodynamically stable forms of the API paracetamol.167e While tablet formation using the thermodynamically stable paracetamol polymorph is difficult, the metastable orthorhombic polymorph yields tablets much more readily due to its layered crystal structure. Consequently, it was expected that cocrystals having a similar layered structure would also be readily compressible. Screening by LAG revealed four cocrystals of paracetamol with improved ability to compress into tablets. Structural characterization and DFT calculations revealed that enhanced compressibility was indeed related to sheet structures (Fig. 9b–e).167e
5.6 Three-component pharmaceutical solids
Cocrystals of pharmaceutically relevant molecules can contain more than two components. This was first demonstrated with three-component (ternary) inclusion compounds involving a guest molecule within an open hydrogen-bonded host consisting of caffeine and succinic acid (Fig. 10a).180 The formation of ternary solids was attempted with 25 potential guest molecules using solution crystallization, neat grinding and LAG. Solution crystallization provided ternary inclusion compounds in four cases, neat grinding in 15, and LAG in 18 (Fig. 10b).180 Ternary phases of the antibiotic pleuromutilin with succinic acid and methanol or water were studied by Clawson et al.199 From solutions or slurries ternary solids with succinic acid:pleuromutilin ratios between 1:2 and 1.4:1 were obtained, while LAG allowed the construction of materials with a ratio of up to 2:1.199 Solid-state NMR and X-ray crystallography revealed that the material with the 1:2 ratio consisted of a host lattice of protonated pleuromutilin with included succinate anions and solvent. Increasing the relative amount of succinic acid resulted in the progressive replacement of solvent guests with neutral molecules of succinic acid, accompanied by a change in space group.200
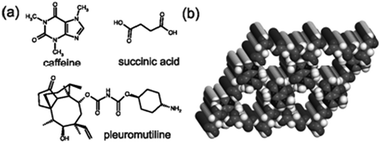 | ||
| Fig. 10 (a) Molecular diagrams of caffeine, succinic acid and pleuromutiline; (b) two-component host of caffeine and succinic acid.180 | ||
A three-component pharmaceutical cocrystal hydrate was obtained while comparing anhydrous and hydrated reactants in mechanochemical cocrystallization of theophylline and citric acid.177 Whereas grinding anhydrous theophylline with anhydrous citric acid gives a binary cocrystal (theophylline)·(citric acid),200 hydrated reactants lead to a ternary solid (theophylline)·(citric acid)·H2O.177
5.7 Pharmaceutical salts
In addition to cocrystallization, LAG is also effective in screening for pharmaceutical salts. In particular, Trask et al. compared LAG and neat grinding in screening for salts of APIs trimethoprim and pyrimethamine.201 LAG was more efficient in forming salts or salt polymorphs. Recently, LAG was used by André et al. to screen for new solvate and salt forms of the antibiotic 4-aminosalicylic acid.2025.8 Conclusion
Mechanochemistry and cocrystallisation are, in tandem, becoming increasingly established as versatile approaches to discovering new solid forms of pharmaceutically active compounds. Mechanochemistry is often preferable to solution or melt-based approaches as a more efficient and general way to screen for potentially new cocrystal forms of APIs.6. Ball milling in organic synthesis: C–C- and C–X-bond formations
6.1 Introduction
Organic synthesis has almost exclusively been restricted to solution-based methods during its development through to the present day. The use of ball mills in solvent-free organic synthesis has recently, however, begun to attract significant attention, and several reviews are now available.19 Concentrating on C–C and C–X bond formations, including catalysed reactions, this section highlights selected examples which show the progress and potential of ball milling in organic synthesis generally.6.2 Stoichiometric organic reactions in ball mills
The Knoevenagel condensation is an important C–C-bond forming reaction, giving access to α,β-unsaturated carbonyl compounds. In 2003, Kaupp introduced a solvent-free version of this reaction carried out in a ball mill (Scheme 6).35 The use of stoichiometric amounts of starting materials led to a quantitative yield of the desired products. Here, as well as in the reported Michael additions, no work-up was required, rendering these waste-free approaches sustainable and eco-friendly.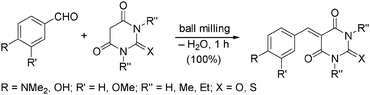 | ||
| Scheme 6 Knoevenagel condensation. | ||
The temperature increase resulting from friction during the milling process was an important factor in the success of these reactions. Various reaction conditions were applied, and a comparison to microwave-accelerated Knoevenagel condensation reactions revealed the superiority of the mechanochemical activation with respect to the energy consumption.
Bräse developed an easy and rapid access to xanthones, a structural motif, which can be found in several natural products.203 A thorough investigation of the reaction between salicylaldehyde and cyclohexenone was performed in the presence of dabco (50 mol%). This report included an optimization of the reaction conditions with respect to time, ratio of starting materials, rotational frequency and number of balls used in the milling process. A fine-tuning of these factors led to a transformation affording tetrahydroxanthenone in 66% yield (Scheme 7).
 | ||
| Scheme 7 Domino oxa-Michael-aldol reaction. | ||
Although the yields of this mechanochemically-induced domino oxa-Michael-aldol reaction were not as high compared to those obtained in solution, the impact of the ball-milling parameters on the yield and chemoselectivity was clearly demonstrated.
Mechanistically related is the Morita–Baylis–Hillman reaction, studied by Mack under ball milling conditions (Scheme 8).204 Using dabco (20 mol%) as the catalyst, a major rate enhancement was observed, producing products in high yields after a short reaction time (30 min).
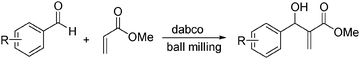 | ||
| Scheme 8 Morita–Baylis–Hillman reaction. | ||
Wittig olefination is important for the formation of alkenes. Balema and Pecharsky showed that various types of phosphorus ylides could be generated mechanochemically in the solid state (Scheme 9).205 Stabilized ylides were isolated in pure form and semi- and non-stabilized ylides directly reacted with solid organic carbonyl compounds. By this approach, phosphoranes were obtained in yields up to 99%, and “one-pot” Wittig reactions afforded olefins in up to 93% yield.
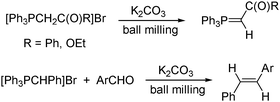 | ||
| Scheme 9 Wittig reaction. | ||
It is noteworthy not only that this was done under solvent-free conditions, but also that K2CO3 was basic enough to deprotonate the phosphonium salt; in solution commonly much stronger bases are required. It can be noted that our common scales of basicity and acidity are normally solvent-specific, and clearly under solventless conditions acidities and basicities may differ from those expected.
Amides play an important role in synthetic and biological chemistry. Traditional methods for the introduction of the amide function often need expensive transition metal catalysts and/or toxic reagents. To overcome these problems Wang developed a solvent-free route for the direct amidation of aryl aldehydes with anilines in a ball mill (Scheme 10).206
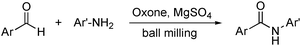 | ||
| Scheme 10 Direct oxidative amidation. | ||
It was found that under those conditions in the presence of MgSO4, Oxone® (potassium peroxymonosulfate, 2KHSO5·KHSO4·K2SO4) promoted the oxidative coupling in moderate to good yields (up to 78%). For comparison, in acetonitrile and toluene the yields were much lower. Also, in the ball mill the chemoselectivity of the oxidant was higher, with only trace amounts of acid (from the oxidation of the corresponding air-sensitive aryl aldehydes with Oxone®) observed.
Although the field of peptide synthesis has made significant progress over the last few decades, major challenges remain. One is to reduce the amount of solvents used. Lamaty studied the opening of urethane-protected α-amino acid N-carboxyanhydrides with α-amino acid derivatives to afford peptidic products under solvent-free conditions in a ball mill (Scheme 11).207
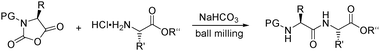 | ||
| Scheme 11 Solvent-free peptide synthesis. | ||
With NaHCO3 as base, a variety of di- and tripeptides were prepared in high yields. It is significant that educts with a wide range of protecting groups (PG) could be applied and that no epimerization was observed.
Thioglycosides are of interest in oligosaccharide synthesis and as enzyme inhibitors. A solvent-free approach towards such compounds under ball milling conditions was reported by Kartha (Scheme 12).208
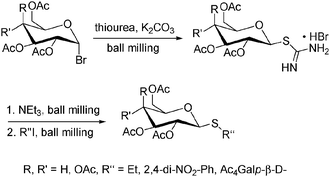 | ||
| Scheme 12 Synthesis of thioglycosides. | ||
Of particular interest is that alkyl, aryl and glycosyl thioglycosides could be accessed without the need for aromatic solvents, toxic thiols or phase transfer catalysts. A key intermediate in this process was glyosyl thiuronium hydrobromide salt, which allowed a “one-pot” preparation of all products in high yields.
Vyle et al. have suggested that ball milling may provide advantages in organic synthesis with biological molecules quite generally.209 This is because the characteristically poor solubilities of nucleosides, nucleobases, sugars etc. have traditionally required the use of polar aprotic solvents such as DMF (dimethylformamide) or pyridine, which are often toxic or carcinogenic. Solventless ball milling has been used effectively for the addition of TBDMS (tbutyldimethylsilyl) protecting groups to phenols as well as a range of nucleosides, avoiding the need for DMF or pyridine altogether (the usual method of purification by chromatography with ethyl acetate and hexane was still required) (Scheme 13).209 Yields were generally at least 95%. Another advantage of using ball milling was that the starting nucleosides did not need to be predried, which further simplifies the overall process. The conditions were also compatible with trityl protecting groups, and one-pot double protections could be done, in particular O-silylation followed by N-benzylation.
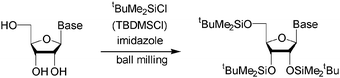 | ||
| Scheme 13 Protection of nucleosides without using DMF or pyridine. | ||
Remarkable results have been achieved in reactions of fullerenes under ball milling conditions.210 Whereas Wudl demonstrated that cyanide added to C60 in toluene/DMF,211 Komatsu found that a C120 dumbbell dimer formed in reactions performed under high-speed vibration milling in the absence of solvent (Scheme 14).212 Other potassium salts such as potassium carbonate and acetate also promoted the reaction, which when optimized gave the dimer and unchanged C60 in a ratio of ca. 3:7.
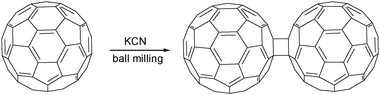 | ||
| Scheme 14 Fullerene dimerization. | ||
6.3 Metal-catalyzed organic reactions in ball mills
Coupling reactions such as the Suzuki,213 Heck,214 Sonogashira215 and many others have become critical to the synthesis of a variety of organic molecules including organic polymers, natural products and organic light emitting compounds. They are especially useful reactions for the synthesis of carbon based building blocks such as graphene and other nanomaterials. Many of these coupling reactions have been shown to be successful under a variety of alternative methods such as in ionic liquids,216 microwave reactors217 or water.218 This section will focus on the reactions of metal-catalyzed reactions that have been performed under solvent-free ball milling conditions.The Sonogashira coupling reaction makes a carbon–carbon bond between the sp2-hybridized carbon of an aryl or alkenyl group with the sp-hybridized carbon of a terminal alkyne. It is typically conducted with an aryl halide (or triflate), a terminal alkyne, and a base in the presence of copper iodide and a palladium catalyst. This reaction was shown to be successful under solvent-free ball milling conditions in high yields with a Spex 8000 M vibratory mixer/mill (Scheme 15).219 Aryl iodides and bromides gave high yields of coupling products with phenyl acetylene and trimethylsilyl acetylene using palladium tetrakis triphenyl phosphine and copper iodide as catalyst and potassium carbonate as base. Following normal reactivity trends in solution, aryl chlorides were unreactive.
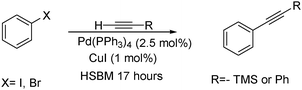 | ||
| Scheme 15 Sonogashira reaction under solvent-free high speed ball milling conditions. | ||
Palladium tetrakistriphenylphosphine and many other palladium(0) catalysts used in coupling reactions are air- and moisture-sensitive. When the Sonogashira reaction is performed in solution, dry solvents and inert atmospheres are needed. However under solvent-free ball milling conditions these reactions can be conducted in an aerobic environment. Most ball milled reactions are carried out in a stainless steel vial with stainless steel balls. When the Sonogashira reaction is ball milled in a stainless steel vial without copper iodide, it proceeds in moderate yield. However if the reaction is conducted in a copper vial it gives the product in high yield (Scheme 16). This demonstrates that the material of the vial and/or ball can be a source of the catalyst.
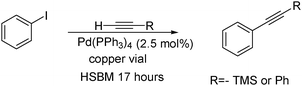
Ondruschka demonstrated that the Sonogashira reaction can be conducted under ball milling conditions using more robust palladium(II) catalysts such as palladium acetate and palladium chloride.220 These reactions were conducted in the absence of additional ligands and copper. The reaction was completed in 20 min using (Scheme 17). This catalytic system was successful with aryl iodides but not with aryl bromides. The rate and yield were highly dependent upon the substrate, grinding media and catalyst used.

The Heck reaction is an important palladium-catalyzed coupling and can be used to synthesize many important compounds such as unsaturated and unnatural amino acids. It forms a carbon-carbon bond between an aryl halide (or triflate) and an olefin. Frejd demonstrated the Heck reaction under solvent-free ball milling conditions (Scheme 18).221 Using various aryl compounds to couple with olefins A and B they were able to synthesize a variety of organic products. Using sodium formate as a reductant for the Pd(II) catalyst improved yields. Similar to solution reactions, aryl iodides were better coupling partners than bromides. Approximately 5 mol% catalyst loading is optimal. The product formed was the Z isomer, showing that diastereoselectivity can still be observed under energetic ball milling conditions. The ball milling conditions gave higher yields than alternative methods. Coupling of iodobenzene and methyl-2-[(tert-butoxycarbonyl)-amino] acrylate gave 77% yield by ball milling, whereas using a hydraulic press to reach pressures of 200 kg cm−2 gave 13% yield. Heating to 80 °C with and without stirring gave 33% and 18% yields respectively. Microwave experiments also generally gave lower yields than in the ball mill. It was concluded that it must be the combination of pressure, heat, grinding, and stirring in the ball mill that accounted for the success of the reaction and not just one of those components individually.
 | ||
| Scheme 18 An example of the ball milled Heck reaction. | ||
Various amino- and hydroxyl-substituted dehydrophenylalanine derivatives could also be made from amido acrylate in modest to good yields under Heck–Jeffery conditions (Scheme 19).222 The system requires a stoichiometric amount of tetraalkylammonium salt but is both solvent- and phosphine-free.
 | ||
| Scheme 19 General scheme for the Heck reaction under ball milling conditions. | ||
The Suzuki reaction couples boronic acids with aryl halide, typically iodides or bromides. Peters et al.223 demonstrated this reaction under solvent-free, ball milling conditions (Scheme 20).
 | ||
| Scheme 20 Suzuki reaction using potassium carbonate and sodium chloride. | ||
Recently, Ondruschka and coworkers224 found that potassium carbonate (base) and sodium chloride (grinding medium) could be replaced with potassium fluoride supported on basic alumina (Scheme 21).
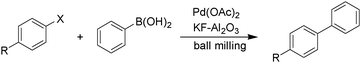 | ||
| Scheme 21 Suzuki reaction in the presence of KF-Al2O3. | ||
Ondruschka also found that under ball milling conditions aryl bromides were more reactive than aryl iodides, which contrasts with Suzuki reactions in solvents. It was also found that the greater water content of the alumina, the greater the reaction.225
6.4 Organocatalytic asymmetric reactions in ball mills
The asymmetric opening of meso-anhydrides has been in the focus of several investigations.226 Using alkaloids or their derivatives as catalysts, high enantioselectivities have been achieved providing synthetically highly useful products. Commonly, the reactions are performed at low temperatures (ambient to −50 °C), and significant amounts of non-polar solvents such as toluene are used.227 Studies of asymmetric anhydride openings with quinidine in a ball mill were performed by Bolm et al. (Scheme 22).228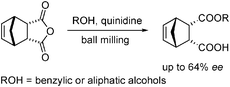 | ||
| Scheme 22 Organocatalytic asymmetric anhydride openings. | ||
Compared to the analogous reactions in toluene, where ee-values of up to 99% had been achieved, the enantioselectivities were lower (up to 64%). The higher temperatures in the ball mill might account for this difference. However, the ball milling proved beneficial in two other aspects: First, no solvent was required as reaction medium and, second, almost equimolar amounts of starting materials could be applied, which contrasted the solution-phase reactions, where a 3-fold excess of the nucleophile was common.
Proline-catalyzed aldol reactions are the most-studied organocatalytic asymmetric C–C-bond forming reactions.229 They proceed via enamine intermediates generated in situ from one of the carbonyl components and the catalyst. Commonly, highly polar solvents such as DMSO, DMF or water are applied, which are difficult to remove after the reaction. Solvent-free reactions of this type under ball milling conditions were first performed by Bolm et al.230 Using 10 mol% of proline and nearly equimolar amounts of starting materials excellent yields (mostly >90%) of the anti-aldol products were obtained and both diastereo- and enantioselectivity were high (up to 99% ee) (Scheme 23).
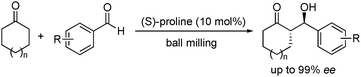 | ||
| Scheme 23 Proline-catalyzed asymmetric aldol reaction. | ||
Various factors affecting yield and stereochemistry were studied,231 and ball milling was superior to conventional stirring. In a similar approach Guillena and Nájera studied direct aldol reactions between ketones and aldehydes under solvent-free conditions.232 A combination of BINAM-prolinamide (5–10 mol%, Fig. 11) and benzoic acid (10–20 mol%) was used. The aldol products had up to 98% ee and were obtained in up to 90% yield.
 | ||
| Fig. 11 Binam-prolinamide organocatalyst applied in asymmetric aldol reactions. | ||
Recently, Bolm et al. studied the phase behaviour of the proline-catalyzed aldol reactions between solid substrates under solvent-free conditions.233 A significant nonlinear relationship between the ee of the catalyst (proline) and that of the aldol product was found, which was suggested to originate from the ternary phase behaviour of scalemic proline. Subsequent studies led to the discovery of an enantioenrichment by iterative retro-aldol/aldol reaction catalyzed by an achiral or racemic base.234
6.5 Synthesis of ligands and hosts
Organic synthesis in balls mills has been applied in a number of cases to ligands and hosts. In the early 2000s Raston and Scott.18,235,236 developed mechanochemical syntheses based on aldol condensation, Baeyer–Villiger oxidation, azomethine synthesis, aromatic bromination, alcohol etherification and benzyl alcohol oligomerization.18 Also reported was a route to Kröhnke type pyridines (Scheme 24).235c The mechanochemical approach involved solventless aldol condensation followed by Michael addition with a second ketone. It gave unsymmetrical compounds in excellent yield, some of which are inaccessible using conventional methods.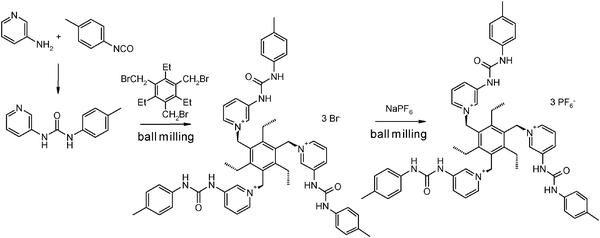
Swinburne and Steed used LAG reactions of pyridine derivatives with benzylbromide derivatives to give tripodal pyridinium anion binding hosts. There are no byproducts and no purification was required. This SN2 substitution, leaving a bromide counter anion is, however, just part of the overall scheme which starts by reaction of 3-aminopyridine with an isocyanate to give a pyridyl urea (Scheme 24). An additional post-reaction step involves metathesis of the bromide counter ion to the less coordinating PF6−. In fact this entire sequence lends itself to mechanochemical synthesis and the combined molecular and supramolecular steps in the sequence were all carried out on the solid product using either neat reagents or LAG in yields generally comparable to the solution based alternatives. While overall the method proved versatile, clean and convenient, some pyridine derivatives did not react at all and in other cases yields were low for reasons that are unclear but which may relate to the sterically hindered nature of the system.237
Mechanochemical methods have also been investigated in the context of macrocyclic hosts, specifically unusual calix[n]arene (n=5 or 7), [4]resorcinarene and cyclotriveratrylene (CTV) derivatives, and, recently, covalent organic cages. A mixture containing p-benzylcalix[5]arene (10–15% when isolated) and p-benzylcalix[7]arene (5–10% when isolated) is produced from ball milling a mixture of the p-benzylcalix[6 or 8]arenes in the presence of KOH and formaldehyde. Dehydration was achieved with sacrificial molecular sieves. Similar conversions were seen in refluxing diphenyl ether solutions. An interesting byproduct was the very unusual p-benzylcalix[10]arene.238Tris-(O-allyl)cyclotriveratrylene is formed after manual grinding of solid benzyl alcohol monomers with a suitable solid acid (the mixture becomes a viscous liquid) and leaving to stand for 10 days. After solvent-base work-up 35% of the product was isolated.239 Related calix[4]resorcinarenes can be obtained in 80–96% yield by manual grinding of resorcinol and benzaldehyde derivatives with p-toluene sulfonic acid as catalyst at ambient temperature.240 The mixture becomes a viscous liquid and then solidifies. Isolation involves washing with water and recrystallization from methanol. Yields are comparable to the solution-mediated routes and the process is convenient. [4]resorcarenes have a tendency to self assemble into giant hydrogen bonded capsules. This tendency is shared by the closely related [4]pyrogallolarenes derived from 1,2,3-trihydroxybenzene (pyrogallol). Reaction of liquid isovaleraldehyde with a fine dispersion of pyrogallol and a catalytic amount of solid p-toluenesulfonic acid with grinding milling using a mortar and pestle results in a condensation reaction to give a brittle white solid within two minutes. This solid is milled to a fine, yellow powder which whose solid state 13C NMR spectrum was consistent with a hexameric hydrogen-bonded capsule.241
Severin et al. found that very large covalent organic cages could be assembled by solvent-free ball milling without solvent. Remarkably, the reaction involves formation of 18 boronate ester and imine linkages between eleven components and was significantly higher-yielding than in solution, although solvent (toluene) was still required for purification.242 Mechanochemistry enabled the synthesis and isolation of the large cage shown in Scheme 25 (71% yield). A smaller version of this cage was obtained in 94% yield by the mechanochemical method, compared to only 24% from solution synthesis.
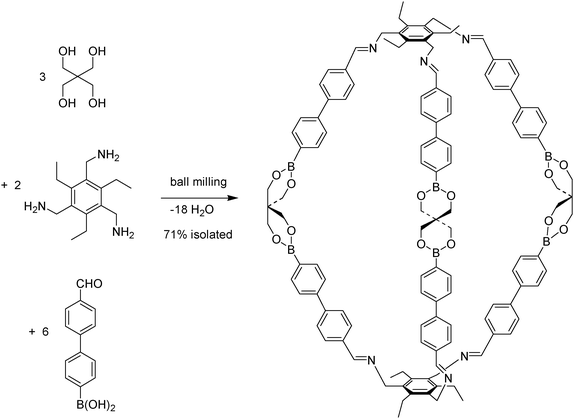 | ||
| Scheme 25 Solventless ball milling synthesis of a large organic cage from ref. 242. | ||
Examples described in this subsection show that macrocyclization and cage formation can be remarkably effective under solventless ball milling conditions, and even more favoured than in solution. This is intriguing given the very high concentrations present and the consequent expectation for larger oligomers or polymers (it seems to be the extreme opposite of traditional high-dilution approaches). Elucidating the underlying reasons for such product speciation would be of great interest.
6.6 Conclusions
Various bond-forming reactions can be accelerated under mechanochemical conditions compared to solution-based methods, and that the use of hazardous or otherwise undesirable solvents can simultaneously be minimized. It is also noteworthy that the energy demands of ball milling have begun to be evaluated in this context and can be low compared to other techniques such as microwave heating.35 Furthermore, previously unknown molecular transformations have been reported, some of which have proved impossible in solution. As with solution-based methods, as the use of ball milling becomes more widely accepted, it would be encouraging to find replacements for palladium catalysts for coupling reactions with less expensive transition metals such as nickel and iron under these conditions.7. Synthesis of discrete metal complexes
7.1 Introduction
The complexation of metal ions is a fundamental and very diverse class of reaction, spanning great ranges of characteristics such as lability, and with applications from small (e.g. radiopharmaceuticals) to very large scales (e.g. metal extraction). As with organic synthesis, it has almost exclusively been developed as solution-state chemistry. However, a growing amount of literature suggests that solvent-free grinding and liquid assisted grinding (LAG) are effective for a wide range of metal complexation reactions. This section deals with discrete metal complexes (coordination polymers are dealt with in Section 8). It is organized by reaction type, specifically ligand additions, ligand additions with elimination, acid–base reactions and main group complexes.7.2 Ligand addition reactions
Many coordination complexes can readily be formed by grinding simple transition metal starting materials with potential ligands. Thus, grinding Ni(NO3)2·6H2O or FeCl2·4H2O with 1,10-phenanthroline gives [Ni(phen)3](NO3)2243 or [Fe(phen)3]Cl2244 respectively, grinding PtCl2 with triphenylphosphine gives PtCl2(PPh3)2,41 and grinding hydrated or unhydrated MCl2 with imidazole gives MCl2(imidazole)2 (M=Co, Cu, Zn).245 Grinding MCl2 (M=Co, Ni, Cu) with ligands L (L=PPh3, OPPh3, OAsPh3246 or toluidine)247 gave the complexes MCl2L2, and in a more risky application of the methodology Ni[ClO4]2 was ground with OPPh3 to give [Ni(OPPh3)2(ClO4)2].246 2-aminopyrimidine (2-Apy) may be ground with either one or one-half equivalents of CuCl2 to form the complexes CuCl2(2-apy) and CuCl2(2-apy)2 respectively,248 and dimethylglyoxime (H2dmg) will react with NiX2 (X=Cl, NO3) to form [Ni(H2dmg)2]X2.249 Grinding thiourea or a derivative with silver salts AgX (X=NO3, SO4, ClO4) gave compounds with various silver:thiourea ratios depending upon the stoichiometry used,250 and whilst grinding PtCl2 with imidazole (Him) gave [PtCl2(Him)2], the analogous reaction with PdCl2 gave the salt [Pd(Him)4]Cl2.251 More complicated ligands may also be used, such as the amino acid gabapentin which can be ground with MCl2 (M=Cu, Zn) to form MCl2(gabapentin)2.252 Simple metal salts may also be made in this way, as grinding two equivalents of imidazolium chloride ([H2im]Cl) with the metal chlorides MCl2 (M=Co, Cu, Zn) gave the imidazolium tetrachlorometallates [H2im]2[MCl4].245
It is also possible to use more complicated metal precursors than simple salts; the iron(III) centre of the protoporphyrin complex hemin will coordinate two imidazole molecules253 or two fluoride ions254 upon co-grinding, and reaction of M(en)(NO3)2 (M=Pd, Pt; en=ethan-1,2-diamine) with 4,4′-bipyridine forms the tetranuclear square [M(bipy)(en)]4(NO3)8 (Scheme 26).255 This last reaction illustrates the benefits that solid-state reactions can have over the equivalent solution reactions—formation of the platinum complex takes 4 weeks at 100 °C in solution, but is complete in 10 min by grinding.
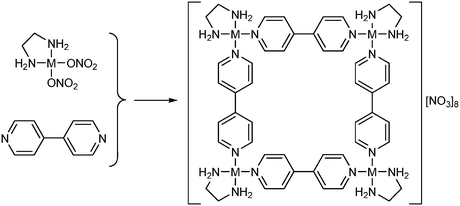
7.3 Ligand addition reactions with elimination of a by-product
New complexes may also be formed by replacing a ligand at a metal centre and eliminating it as a by-product or part of a by-product; for example, reaction of PtCl2(PPh3)2 with K2CO3 displaces the chloride ligands and forms Pt(CO3)(PPh3)2 with elimination of KCl.41 Various tris(pyrazolyl)borate complexes have been made by grinding thallium salts of the ligands with MCl2 starting materials (M=Mn, Co, Ni), eliminating TlCl and forming the desired products with greater efficiency than the corresponding reactions in solution.256 Grinding (in an inert atmosphere) thallium cyclopentadienylide with iron or nickel dichlorides gave ferrocene and nickelocene respectively in good yields; potassium and sodium cyclopentadienylide did not give such good yields, but milling FeCl2 with sodium methylcyclopentadienylide gave 1,1′-dimethylferrocene with 90% conversion.257Mechanochemistry may often prove useful in the conversion of inert and insoluble coordination polymers into more tractable molecules, by reaction with extra ligands that break up the polymeric structure. Thus the inert species [Nb2(E2)2Cl4]∞ (E=S, Se) react with anionic bidentate ligands LL, such as dithiocarbamates, xanthates and oxalate salts, to form [Nb2(E2)2(LL)4] with the elimination of chloride,258 and [M3E7Br4]∞ (M=Mo, W; E=S, Se) can be broken up with oxalate to give [M3E7(ox)3]2− anions259 or with bromide to make [M3E7Br6]2−.260 Grinding polymeric VO(salen) is suggested to give monomers directly without added ligands.261
7.4 Acid–base reactions
A third method of synthesising coordination compounds by grinding involves acid–base reactions of three types. Type 1 is the reaction of a basic metal salt MX and a salt of a protonated ligand [HL]+ to give the complex ML, illustrated by the reaction of imidazolium chloride ([Him]Cl) with cobalt hydroxide or carbonate to form the imidazole complex:245| 2[Him]Cl + CoCO3 → CoCl2(im)2 + CO2 + H2O |
| 2[Him]Cl + Co(OH)2 → CoCl2(im)2 + 2H2O |
| Cu(OAc)2·H2O + 2Hala → Cu(ala)2 + 2HOAc + H2O |
| Al(OiPr)3 + 6NH4F → [NH4]3[AlF6] + 3NH3 + 3iPrOH |
| [Him]2[MCl4] + 2KOH → MCl2(im)2 + 2KCl + H2O |
Type 3 acid–base reactions involve those compounds that release an acid without addition of a base. There are systems that will do this spontaneously, but the gold system reported by Eisenberg (Scheme 27) appears unique in that the acid vapour is only released when the crystals are crushed.270 The change is accompanied by a dramatic change in the luminescence properties of the system, a phenomenon which has been named luminescence tribochromism.271
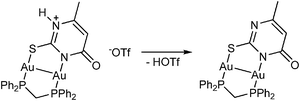 | ||
| Scheme 27 Deprotonation by crushing of a digold(I) compound.270 | ||
7.5 Main-group compounds
Mechanochemistry has also been applied to the synthesis of molecular main-group compounds. A great deal of Russian work in this area was summarized by Volkov,272 who reported syntheses of diborane through the reduction of MBH4 (M=Li, Na, K) by a variety of reducing agents; if the hydrochloride of a nitrogen-donor Lewis-base is used as the reducing agent then the product obtained is the borane adduct. Reactions with larger clusters are also feasible, as the preparation of SnB9C2H11 from CsB9C2H12 demonstrated.272 Another class of reaction summarized in the same work272 is the formation of metal tetrahydridoborates through milling of metal chlorides MClx (M=Zn, Cd, Ti, Zr, Hf, U) with lithium, sodium and potassium borohydrides, which builds on work first reported in 1957.273 In a similar vein, calcium and magnesium tetrahydridoaluminates can be made from appropriate combinations of MAlH4 (M=Li, Na) and M′Cl2 (M′=Ca, Mg).274 Some particularly elegant examples of acid–base reactions were reported by Chandresekar et al., who reacted a range of organotin oxides and hydroxides with protic reagents such as carboxylic, sulphonic or phosphinic acids. This produced in excellent yields a variety of organotin clusters and cages with complicated but well-defined architectures (Scheme 28).275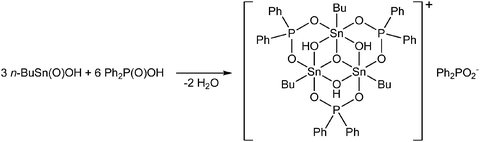 | ||
| Scheme 28 A mechanochemical organotin reaction.275 | ||
7.6 Conclusions
Generally it appears that the formation of metal–ligand bonds in the solid-state can be a powerful and general alternative to solution-based techniques, although it can be noted that most studies so far have concentrated on the more labile metal ions. Mechanochemical methods have also yet to be widely applied to the large range of air-sensitive metal complexes such as those of low-valent platinum-group metals, organometallic compounds of electropositive metals etc., although ball milling is compatible with inert atmosphere techniques. Also with regard to this, some organic reactions catalysed by Pd complexes have been found to be more tolerant to air under ball milling than when done conventionally in solution (see Section 6.3) Therefore, avoidance of solutions might provide advantages in dealing with air-sensitive species.8. Synthesis of coordination polymers (MOFs)
8.1 Introduction
Coordination polymers or metal organic frameworks (MOFs) have become one of the most intensely researched areas of materials chemistry. This section is organized by reaction type in a similar way to the previous section, specifically ligand addition, ligand exchange and acid–base reactions. These three reaction types have been investigated through different mechanochemical methodologies: neat grinding,276 liquid-assisted grinding (LAG) or kneading24 and grinding-annealing.277 There are some analogies with cocrystals (Sections 4 and 5) in that the extended solid state packing is the key point of interest. Because of their growing technological importance, mechanosynthesis of porous MOFs is discussed separately.8.2 Coordination polymers by ligand addition
The addition of neutral ligands to metal-containing building blocks has been extensively used for the construction of coordination polymers. In fact, possibly the first mechanochemical synthesis of a coordination polymer was a reaction of this type.278 In particular, Bourne et al. found that the 1-D zig-zag polymer ZnBr2(pyrazine) could be ground with a further equivalent of pyrazine in a small ‘WIG-L-BUG’ type shaker mill to give the 2-D square grid ZnBr2(pyrazine)2. In a further example, Pichon and James used neat grinding of copper(II) acetylacetonate Cu(acac)2 or hexafluoroacetylacetonate Cu(hfac)2 with 4,4′-bipyridyl (bipy) to give 1-D polymers held together by axial Cu–N bonds.39 The grinding products Cu(acac)2(bipy)n and Cu(hfac)2(bipy)n were obtained quantitatively and identified through powder X-ray diffraction (PXRD) (Fig. 12a).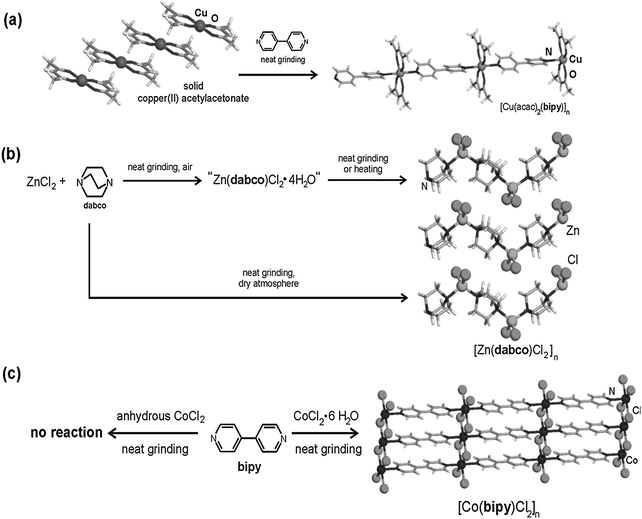 | ||
| Fig. 12 (a) Mechanochemical construction of a 1-D coordination polymer from copper(II) acetylacetonate and bipy;39 (b) formation of the coordination polymer ZnCl2(dabco) by manual grinding in air and grinding in a dry atmosphere;279 and (c) difference in mechanochemical reactivity of bipy towards anhydrous CoCl2 and CoCl2·6H2O.280 | ||
As with mechanochemical cocrystallisation, stepwise mechanisms can occur in such reactions, as noted during neat grinding of anhydrous ZnCl2 with the diamine [2.2.2]-diazabicyclooctane (dabco).279 The first step is the formation of crystalline hydrate ZnCl2(dabco)·4H2O, which upon heating or further grinding dehydrates to the 1-D zigzag polymer ZnCl2(dabco). The formation of an intermediate hydrate was ascribed to the hygroscopic nature of dabco, illustrating how the atmosphere can influence the course of a mechanochemical reaction. If the mechanosynthesis was conducted in dry air with dried reactants the non-hydrated polymer ZnCl2(dabco) formed without observable intermediates.279 With the less hygroscopic 4,4′-bipyridyl (bipy) an analogous zigzag 1-D polymer was formed in a single step.280 In contrast, the construction of a 2-D sheet polymer CoCl2(bipy) from anhydrous CoCl2 and bipy was not possible by neat grinding. The polymer could, however, be obtained by neat grinding of CoCl2·6H2O and bipy, suggesting that the water produced by desolvation of the reagents plays an important role in achieving mechanochemical reactivity (Fig. 12c).280 The construction of polymer CoCl2(bipy) from anhydrous CoCl2 was possible by LAG however.
An increased rate of reaction in LAG over neat grinding was also observed in the synthesis of ethylenethiourea (etu) adducts of silver halides.250a For example, while neat grinding of AgI and etu gave no reaction, LAG with a small amount of water quantitatively gave AgI(etu)2. LAG was also applicable to the construction of coordination polymers based on other silver salts.250b
The addition of a liquid is not only a means to accelerate or enable a mechanochemical reaction, but also an opportunity for molecular inclusion in coordination polymer hosts. This was demonstrated by Braga et al.281 with a versatile 1-D polymer host composed of copper(II) chloride and 1,4-diaminocyclohexane (dace). Although the polymer could not be obtained by neat grinding of CuCl2 and dace, LAG of the two components with a small amount of DMSO gave the host polymer CuCl2(dace) and inclusion of DMSO to form CuCl2(dace)·nDMSO.
Similarly, LAG using water gave inclusion compound CuCl2(dace)·nH2O (dace=1,4-diaminocyclohexane). Both structures are ‘clay-like’ with layers of CuCl2(dace) chains separated by layers of guests.281 Thermal desolvation gave the non-solvated polymer CuCl2(dace) which reversibly included a variety of organic molecules upon kneading and suspension overnight.
The ligand addition reactions of ZnCl2 or CdCl2 with cyanoguanidine (cnge, Fig. 13) illustrate the effect of varying neat grinding conditions on coordination polymer mechanosynthesis.282 The ligand has two different binding sites, enabling the formation of polymers with different metal:ligand ratios and, hence, dimensionality. Grinding ZnCl2 with one or two equivalents of cnge provides the 1-D polymer ZnCl2(cnge) or the discrete complex ZnCl2(cnge)2, respectively. Neat grinding of CdCl2 and cnge in a 1:1 ratio provides the 3-D coordination polymer CdCl2(cnge). The product was always obtained as a pure phase and was structurally characterised using powder XRD data. In contrast to ZnCl2, neat grinding of CdCl2 and cnge in the 1:2 stoichiometric ratio provided only a mixture of CdCl2(cnge) with excess ligand. The 1-D polymer CdCl2(cnge)2 could be obtained only through harsher grinding conditions, i.e. by employing heavier grinding balls (Fig. 13). The difficulty to form 1-D CdCl2(cnge)2 was tentatively related to the higher dimensionality and, hence, kinetic stability of the 3-D CdCl2(cnge). Similar observations were also made for the reaction of CdI2 and cnge.282 The solid-state synthesis and analysis of Cd(cnge)Cl2 and CdCl2(cnge)2 were used as a proof-of-principle of a solvent-free approach to laboratory research.
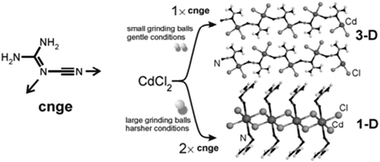 | ||
| Fig. 13 The ligand cnge with different metal binding sites indicated by arrows and its reactions with CdCl2 upon neat grinding under various reaction conditions. | ||
8.3 Coordination polymers by ligand exchange
Manual grinding of copper(II) acetate monohydrate with 1,3-bis(4-pyridyl)propane (pn) replaced the water molecules on the cluster with bridging pn ligands, producing a water inclusion compound of a zigzag 1-D polymer (Fig. 14a).283 The 13C MAS-NMR spectrum was identical to that of the methanol solvate, excluding the resonances of guest methanol.283 Manual grinding of silver acetate and dabco displaces the acetate ligands by dabco and with simultaneous water absorption from the air gives AgOAc(dabco)2·5H2O (Fig. 14b).279 This again illustrates how important the surrounding atmosphere can be in mechanosynthesis.279 Inclusion of moisture was also observed in the mechanochemical reaction of AgOAc with dace.284 Neat grinding gave a coordination polymer tentatively characterized as AgOAc(dace)·nH2O whose crystal structure is not yet known although recrystallization from anhydrous methanol or by passing a stream of dry argon yields two structurally similar products: AgOAc(dace)·3H2O and AgOAc(dace)·H2O0.5·CH3OH, respectively.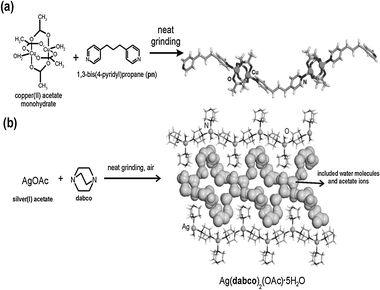 | ||
| Fig. 14 (a) Mechanochemical construction of a 1-D coordination polymer by ligand exchange on the copper(II) acetate paddlewheel complex;283 (b) formation of a hydrated coordination polymer by neat manual grinding of silver(I) acetate and dabco in air. Water and acetate guests are shown using the space-filling model.279 | ||
8.4 Acid–base reactions
In this reaction class the combination of metal acetates with organic acids, accompanied by the release of acetic acid, is particularly noteworthy. For example hydrated nickel(II) acetate and acetylenedicarboxylic acid (H2adc) react to form a hydrated 3-D coordination polymer Ni(adc)(H2O)2.39,285 Analogously zinc(II) acetate dihydrate gave a previously unknown 3-D polymer Zn(adc)(H2O)2, isostructural to the Ni(II) polymer.39 Similarly Stein and Ruschewitz prepared coordination polymers based on alkaline earth metals, by grinding-annealing of magnesium or calcium acetates with H2adc. The resulting materials Ca(adc) and Mg(adc)·2H2O were isostructural to Sr(adc) and Mn(adc)·2H2O.286 The acidic ligand in such reactions does not, however, need to be a carboxylic acid, as demonstrated by Yoshida et al., who conducted neat manual grinding of transition metal acetates with 3-cyanoacetylacetone (HCNacac).268 For Fe(II), Co(II) and Ni(II) non-porous 3-D frameworks were obtained.268 The formation of [Ni(CNacac)2]n in pure form is noteworthy, as solution methods yield a product contaminated with Na[Ni(CNacac)3].268,277,287 Reactions with other acetates gave previously unknown mononuclear complexes Mn(CNacac)2·2H2O, Cu(CNacac)2·H2O and Zn(CNacac)2·H2O. Upon heating to 100 °C, these complexes lose water to form coordination polymers, further illustrating the applicability of grinding followed by annealing in mechanosynthesis. Grinding-annealing is also effective for the thermal dehydrohalogenation of mutually isomorphous 4,4′-bipyridinium salts of FeCl42−, CoCl42− and ZnCl42− anions, prepared by neat grinding of 4,4′-bipyridinium chloride with FeCl2·4H2O, CoCl2 (or CoCl2·6H2O) and ZnCl2, respectively. The heating step eliminates HCl gas to leave 1-D zigzag (with Zn) or 2-D sheet (with Fe and Co) polymers.280,288 The dehydrohalogenation could also be achieved through the addition of an external base, such as KOH, rather than heating, leaving a product containing KCl byproduct.The use of only slightly soluble carbonate or oxide reactants is also possible. Metal oxides are attractive precursors due to low cost, ready availability, and because the only byproduct is water. Mechanochemical reactivity of metal oxides with organic ligands was explored by Fernandez-Bertran,289 who obtained known coordination polymers of Ag(I), Zn, Cd and Hg(II) by neat grinding of respective metal oxides and imidazole, although no reactions occurred with PbO or MgO. Adams et al. obtained the 2-D polymer CoCl2(bipy) by LAG of cobalt(II) carbonate with bipyridinium chloride.288 The product was identical to that obtained by LAG of CoCl2 and bipy with water. LAG of basic zinc carbonate with bipyridinium chloride gave a mixture of two polymorphs of the polymer Zn(bipy)Cl2. This contrasts with the neat grinding reaction of ZnCl2 and bipy which yields only one polymorph (see above).
The reaction of ZnO with fumaric acid (H2fma) was used to rapidly screen for coordination polymers by LAG (Fig. 15).290 Different liquid additives resulted in different products. Anhydrous zinc fumarate and a previously unknown dihydrate were structurally characterized directly from PXRD data. Grinding with three or four equivalents of water gave selectively the zinc fumarate tetrahydrate and the pentahydrate respectively, which form as a mixture from solution.291 Subsequent study revealed that the formation of different products can be correlated with the activity of water in the grinding liquid, and that LAG with pure water proceeds in a stepwise fashion.292 A crystalline hydrate forms first, which depletes the free water in the mixture so as to change the liquid-assisted reaction into a neat grinding process. The latter is speculated to proceed through an amorphous intermediate, deduced by the spontaneous formation of different coordination polymers by ageing of the partially reacted reaction mixture. A stepwise mechanism was also observed in the reaction of CuO with acetic acid.292
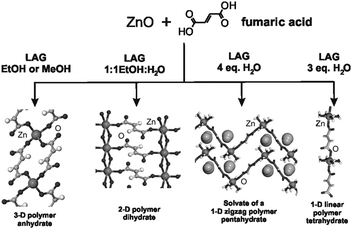
8.5 Porous MOFs by mechanochemistry
Porous metal–organic materials are an intensely researched area. Mechanochemical synthesis of such phases was demonstrated by James et al.,40 by an acid–base reaction between copper acetate and isonicotinic acid (Hina) to give Cu(ina)2. Neat grinding gave the porous framework quantitatively in a few minutes, with the acetic acid and water byproducts partially lost and partially included in the pores (Fig. 16a). The latter could be completely removed by heating. A similar approach gave the industrially relevant open framework Cu3(btc)2 (btc=1,3,5-benzenetricarboxylate) or HKUST-1 (pore diameter ca. 9 Å) by neat grinding of copper(II) acetate with trimesic acid (see below for the properties of the mechanochemically-prepared material).293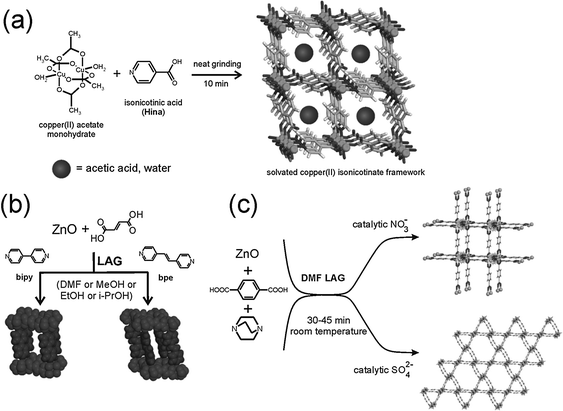 | ||
| Fig. 16 Mechanochemical synthesis of porous MOFs: (a) by neat grinding;40 (b) by liquid-assisted grinding294 and by ion- and liquid-assisted grinding, exploiting the catalytic effect of nitrates and sulfates.295 | ||
Liquid-assisted grinding of ZnO, H2fma and the bridging ligand with DMF, methanol, ethanol or 2-propanol quantitatively yielded porous MOFs pillared by bipy or trans-1,2-bis(4-pyridyl)ethylene (bpe) (Fig. 16b).290 The latter, previously unknown, MOF was structurally characterized from powder diffraction data by Rietveld refinement to the known copper analogue.
Mechanosynthesis of a pillared material with larger pores based on terephthalic acid and dabco proceeded very slowly and in low yield. However, the synthesis could be completed within 45 min by adding catalytic amounts of an alkali metal or ammonium nitrate salt (Fig. 16c).295 This ion- and liquid-assisted grinding (ILAG) gave a pillared framework based on square grid layers with ca. 15 Å pore diameter.296 Replacing nitrate catalysts with sulfates gave the hexagonal isomer of this material framework within 30 min, with pores of ≈18 Å diameter. Although the structural basis of such templating and catalytic effects are not yet known, solid-state NMR studies indicate that salt inclusion within the neutral MOF plays a significant role. In contrast, the analogous pillared framework involving bipy could be readily obtained by LAG without any salt additives.37
Synthesis from metal oxides can also give zeolitic imidazolate frameworks (ZIFs) (Fig. 17a). Whereas neat grinding of ZnO with solid imidazole has limited scope in such synthesis, LAG or ILAG gave rapid and quantitative formation of a series of close-packed and open-frameworks. The grinding liquid and the ionic salt catalyst enhance the reactivity and direct the final product topology. The reactions proceed by stepwise mechanisms in which the most porous structures are formed first and subsequently transform to more close-packed ones. This resembles an Ostwald staging process in which the less stable frameworks of low density (expressed as the ratio of tetrahedral sites (T) and the volume (V) of the unit cell) transform to more stable, denser structures (Fig. 17b).
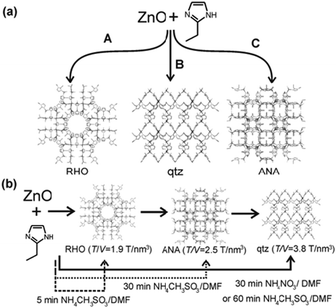 | ||
| Fig. 17 (a) Topologically specific mechanosynthesis of zeolitic imidazolate frameworks (ZIFs) directly from ZnO and 2-ethylimidazole using ILAG. Pathway A represents ILAG with (NH4)2SO4; B is ILAG with NH4NO3 or NH4CH3SO3 in the presence of EtOH and C is ILAG with NH4CH3SO3 and DMF or DEF as the liquid phase; (b) time-dependent ZIF transformations under ILAG conditions, T/V is the number of tetrahedral sites (T) per nm3. | ||
While the synthesis of pillared MOFs using ILAG clearly reveals an anion-directing effect, ZIF synthesis strongly depends on the use of weakly acidic ammonium salts and appears to arise from their influence on the rate of interconversion between different structures, rather than from specific structure-templating effects.
A recently reported aspect of reactivity is the labile nature of MOFs under LAG.37 Three different structural forms of Zn-bdc frameworks (bdc=benzenedicarboxylate) interconverted upon brief grinding with a suitable liquid (Fig. 18). This is probably effectively a grinding-assisted recrystallisation since the product was normally found to be the least soluble form in the liquid used for LAG. Pillared mixed-ligand structures could also be obtained by grinding these Zn-bdc phases with dabco or bipy. Significantly, some of the mixed-ligand products could not be obtained as single step reactions showing that two-step strategies can be useful.37,297
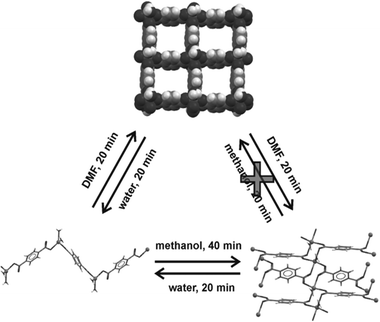
The porosity of mechanochemically prepared MOFs has begun to be investigated. Yuan et al.,297a found that the Brunauer-Emmett-Teller (BET) surface area of Cu3(btc)2 (HKUST-1)298 obtained by neat grinding or LAG of copper(II) acetate monohydrate with benzene-1,3,5-tricarboxylic acid was comparable to that of samples obtained by conventional solution-based routes. Another study by Schlesinger et al.299 compared neat and liquid-assisted mechanosynthesis with a variety of other procedures, involving room-temperature and reflux solution synthesis, solvothermal reactions, microwave-assisted synthesis, sonochemical and electrochemical syntheses. The BET surface area and specific pore volume of HKUST-1 samples could be further increased if the reaction was conducted using LAG with DMF. The resulting surface area and pore volume were comparable to those for samples made solvothermally. Similar observations have been made by Emmerling et al. who also extended the synthesis to MOF-14 Cu3(btb)2 (btb is the larger tricarboxylate 4,4′,4′′-benzenetribenzoate). So far, in order to obtain high surface areas, immersion in bulk solvent has been required during the post-synthetic activation step for mechanochemically-synthesis MOFs.300 It will be interesting to see with other examples, or by adapting the reaction conditions whether this requirement can be avoided.
8.6. Solid solutions
Mechanochemical syntheses are interesting for the formation of solid solutions since they can circumvent troublesome solubility variations between different metal ions or ligands to give homogeneous products more readily than solution methods.301 Formation of solid solutions of coordination polymers has been demonstrated by James et al. LAG reactions of mixtures of different rare earth metal carbonates with 1,3,5-benzenetricarboxylic acid.302,303 Mixed-lanthanide (Sm–Gd, Eu–Gd, Tb–Gd and Dy–Gd) 3-D open frameworks isostructural to those obtained from individual metal carbonate reactants were obtained. Notably, this work also shows the extension of mechanochemical MOF synthesis to trivalent metals. Adams et al.304 explored solid solutions for the synthesis of materials with systematically controllable lattice parameters and physical properties. LAG of anhydrous CoCl2 or CoBr2 with bipy gives the isostructural 2-D sheet coordination polymers CoCl2(bipy) and CoBr2(bipy) respectively. Correspondingly, LAG of mixtures of anhydrous CoCl2 and CoBr2 yields solid solutions of composition CoBr2−xClx(bipy) which were homogeneous at length scales detectable by powder X-ray diffraction. The solid solutions were isostructural to the single phases CoCl2(bipy) and CoBr2(bipy), but with lattice parameters which varied linearly with x over the range 0–2.8.7 Conclusions
Mechanochemical synthesis in the burgeoning field of coordination polymers and MOFs is attractively fast and convenient, does not require additional heating, can sometimes be achieved starting from metal oxides, and avoids bulk solvent in the reaction step (although it may be needed for effective activation). The only waste product of the oxide-based reactions is water. As with cocrystals, there remain challenges in determining the structures of new phases since large single crystals are not obtained directly. However, products can be used to seed the formation of large crystals from solution and advances in structure solution from PXRD are making structure determination from these data more common (see Section 9). Again as with cocrystals, new phases different to those formed from solutions can be obtained, and it will be interesting to establish which generic differences may exist be between solventless mechanochemical and solution-based products.9. Structural characterization of mechanochemically prepared materials
9.1 Introduction
Mechanochemistry is applicable to diverse types of synthesis, and in each case, the appropriate techniques for characterization of the product may differ. In some areas (particularly organic synthesis and with less-labile metal complexes), the normal methods of solution-state NMR or HPLC, etc, remain appropriate for monitoring reactions and for product identification, because of the inert nature of the products and the emphasis on molecular structure rather than crystal structure. However, in other areas (particularly cocrystals and coordination polymers, or MOFs), the crystal packing is the property of key interest. Also, labile molecular products may rearrange in solution. In such cases, it is clearly important to be able to characterize the mechanochemical product directly, without dissolution or solvent-based recrystallization. Such characterization is particularly important for new, previously unknown structures. However, because mechanochemical products are normally microcrystalline powders, single-crystal X-ray diffraction (XRD) usually cannot be applied. Instead, powder XRD (PXRD) is generally the main technique employed, while other techniques (particularly solid-state NMR spectroscopy) can also yield valuable structural insights. Methodology for structure determination of molecular solids from PXRD data305 has advanced significantly in recent years such that this strategy can now be applied relatively routinely to determine crystal structures of moderate complexity, although it remains significantly more challenging than structure determination from single-crystal XRD data. Due to the importance of these techniques for gaining insights into mechanochemical reaction products, methodology and illustrative examples are discussed in this Section.9.2 Assessing whether a mechanochemically prepared material is a new solid phase
To establish whether a material prepared by mechanochemical synthesis represents a new solid phase, its experimental PXRD pattern must be compared with those of known materials (using either experimental data or data simulated from known crystal structures, determined for example from single-crystal XRD data). Such comparison is commonly carried out “by eye” rather than subjecting the PXRD data to rigorous quantitative analysis. Unfortunately, however, this type of visual comparison can leave considerable scope for misinterpretation. Although, in favourable cases, visual comparison may indeed provide unambiguous confirmation of whether two PXRD patterns match or differ, experience shows that deeper scrutiny is frequently required, a discussed below.For example, the PXRD patterns for different sample preparations of the same solid phase may actually look significantly different as a result of instrumental factors, details of the data collection procedure and/or microstructural characteristics of the powder itself (e.g. the size, shape and orientational distribution of the crystallites). Thus, PXRD peak widths and intensities may differ significantly, which can have an important effect on the appearance of the PXRD pattern, particularly in regions with substantial peak overlap. Furthermore, peak positions may also differ due to instrumental factors or differences in the temperature at which data were collected (this issue is particularly relevant when an experimental PXRD pattern recorded at room temperature is compared with simulated PXRD patterns for known crystal structures determined from single-crystal XRD at low temperature).
Conversely, small differences between PXRD patterns, which represent real structural differences between two samples, are often overlooked when comparison is carried out “by eye”. As a consequence, two materials that are genuinely different may be erroneously assigned as having the same crystal structure. Relevant issues here include differences in crystal symmetry, occupancy of framework structures and degrees of disorder, as well as the existence of subtle superstructures. An example, taken in part from ref. 306, is illustrated in Fig. 19. Although the PXRD patterns are very similar, detailed comparison reveals important differences (in this case at 2θ ≈ 29°) which means that the two materials cannot be identical (see Fig. 21 for the actual structural differences).
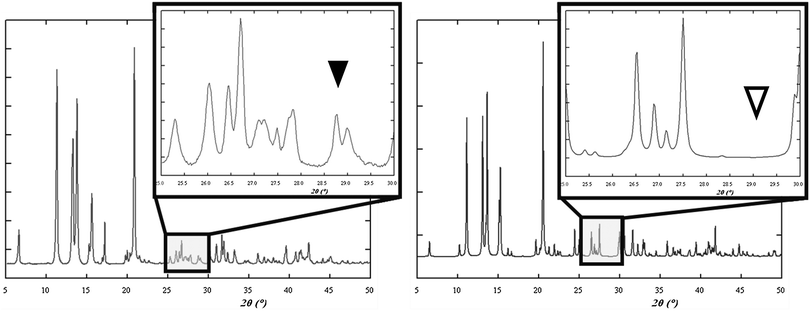 | ||
| Fig. 19 An example of comparison between the experimental powder XRD pattern of a material prepared by mechanochemical synthesis (left) and the simulated powder XRD pattern of a potential candidate of known structure prepared previously by a solvothermal route (right). Adapted from ref. 306. | ||
However, all of the factors discussed above that affect the appearance of a PXRD pattern can be taken into consideration by appropriate quantitative analysis. Thus, in order to establish whether a material prepared by mechanochemical synthesis does or does not correspond to a particular known material, the recommended protocol is to carry out a Rietveld refinement calculation307 using the experimental PXRD data for the material prepared by mechanochemical synthesis and using the crystal structure of the known material as the initial structural model in the refinement.
9.3 Complete structure determination from powder XRD data
The recent upsurge in structure determination of molecular solids from PXRD data has coincided with the development of the “direct-space” strategy for structure solution.305b In this strategy, trial structures are generated independently of the PXRD data, and their quality is assessed against the PXRD data; improvements to the structural models are then made via global-optimization algorithms, leading ultimately to the correct crystal structure. This approach is particularly suited to materials constructed from well-defined modular building units (such as metal–organic-frameworks or molecular solids). However, there have also been several reports of structure solution of molecular solids from PXRD data using traditional approaches analogous to those used for analysis of single-crystal XRD data, or the recently developed charge flipping algorithm.308 These latter approaches may be preferred when peak overlap is not severe and when there is less prior knowledge of the geometries of the molecules present.The use of solid-state NMR in conjunction with PXRD data serves as a particularly powerful combined experimental approach.309,310 For example, solid-state NMR can help to (i) establish the composition, (ii) identify tautomeric forms, (iii) identify specific types of interactions (e.g. hydrogen bonding), (iv) quantify inter-atomic distances, (v) obtain a priori insights on the existence of disorder (including dynamic processes), and (vi) assess whether the molecules occupy general positions or special positions. Such information can be important in setting up the correct structural model for use in structure solution calculations from PXRD data, or for validation of results from Rietveld refinement.
The first155 use of PXRD to determine the structure of a mechanochemically prepared cocrystal was for the three-component material (bis-β-naphthol)(benzoquinone)(anthracene)0.5. Grinding a physical mixture of the three components gives a reddish-purple polycrystalline powder with a different structure to the bluish-black cocrystals obtained from solution. Structure solution was carried out using the direct-space strategy. The final crystal structure obtained following Rietveld refinement is shown in Fig. 20, and may be rationalized on the basis of three different interaction motifs: edge-to-face interactions between benzoquinone (edge) and anthracene (face) molecules, face-to-face interactions between benzoquinone and bis-β-naphthol molecules, and chains of O–H⋯O hydrogen bonds involving bis-β-naphthol and benzoquinone molecules. This structure, with the anthracene molecules lying on a two-fold rotation axis, was supported by the solid-state 13C NMR spectrum.
 | ||
| Fig. 20 Crystal structure of the three-component cocrystal (bis-β-naphthol)(benzoquinone)(anthracene)0.5 determined from PXRD data. Dotted lines indicate π-stacking interactions and hydrogen-bonded chains. Reproduced from ref. 155. | ||
Another example concerns a porous interpenetrated mixed-ligand metal–organic-framework Zn2(fma)2(bipy), prepared mechanochemically from Zn(OAc)2·2H2O, fumaric acid (H2fma) and 4,4′-bipyridine (bipy).306 As shown in Fig. 21, the crystal structure of this material bears some similarity to a previously reported DMF solvate material Zn2(fma)2(bipy)·(DMF)0.5 prepared by a solvothermal route, for which the crystal structure was determined from single-crystal XRD data.294 Nevertheless, there are important structural differences between these materials, primarily concerning the fact that the bipy ligands in the DMF solvate are constrained to be planar (corresponding to the mirror plane in the C2/m space group), whereas there is no such constraint in the structure of the mechanochemically prepared material (for which the space group is P21/a), and the dihedral angle between the two rings of the bipy ligand is 53.2° (see Fig. 19 for a comparison of the PXRD data of the mechanochemical product and the simulated pattern for the DMF solvate). Interestingly, desolvation of the DMF solvate material yields a material identical to that prepared by the mechanochemical synthesis.
 | ||
| Fig. 21 Crystal structure of a metal–organic framework material Zn2(fma)2(bipy) prepared by mechanochemical synthesis, with structure determination carried out directly from powder XRD data,306 viewed (a) along the c-axis and (b) along the b-axis. For comparison, (c) and (d) show the corresponding views of the structure of a DMF solvate material Zn2(fma)2(bipy)·(DMF)0.5 prepared by a solvothermal route.294 Although there is some similarity between these structures, it is nevertheless clear that there are important structural differences. Reproduced from ref. 306. | ||
Other reports of crystal structures of materials prepared under mechanochemical conditions being determined directly from PXRD data include the metal–organic framework Co(dibenzoylmethanate)2(nicotinamide)2. This material was obtained by thermal desolvation of the corresponding acetone solvate, which was prepared by liquid-assisted grinding (LAG).311 The structure comprises “wheel-and-axle” units of composition Co(dibenzoylmethanate)2(nicotinamide)2, which are assembled through hydrogen-bonded amide-amide interactions involving the nicotinamide molecules of neighbouring units, giving rise to anti-parallel chains of amide functionalities in a ladder-type motif. There are channels with approximately hexagonal cross-section running parallel to the hydrogen-bonded amide ladders (Fig. 22).
 | ||
| Fig. 22 Structure of Co(dibenzoylmethanate)2(nicotinamide)2 determined directly from PXRD data. | ||
Further examples of organic materials include a hydrate cocrystal of 5-methyl-2-pyridone and trimesic acid, prepared by grinding a methanol solvate cocrystal of the same components under ambient atmospheric conditions,312 and 1:1 cocrystals of theobromine with trifluoroacetic acid and theobromine with malonic acid, each prepared by LAG.185
9.4 Conclusions
As mechanochemistry becomes more widely used, it is likely that structure determination from PXRD will become applied more extensively in this field. Clearly, structural knowledge elucidated by this approach has the potential to provide important insights towards understanding why the crystal structures of mechanochemical products often differ from those of materials of the same composition prepared by crystallization from solution.10. Obstacles and inherent limitations to the mainstream adoption of mechanochemistry
The previous sections show that mechanochemistry can clearly offer advantages as an alternative to traditional solvent-based synthesis, in that new or improved reactivity can be discovered and less (or even no) solvent may be needed. However, some obstacles and limitations to the technique can be identified as follows.Product purification
As mentioned in Section 1.3, even if a mechanochemical reaction step itself is solvent-free, solvents may still be needed for purification. Although many molecular mechanochemical reactions proceed to completion there will still be cases when non-volatile by-products and/or small traces of starting materials are present at some level, and can only practically be removed by solvent-based extraction or recrystallization. It is clearly unrealistic that mechanochemistry could make all of chemical synthesis completely solvent-free. Therefore it is important to try to identify the types of situations in which mechanochemistry can provide a clear advantage over conventional solvent-based approaches. These include: (i) When, overall, it allows less solvents to be used, or if it avoids the use of particularly undesirable (toxic, carcinogenic etc.) solvents; (ii) when it allows less energy to be used (see also ‘Energy consumption’ below); (iii) when it provides unique or improved reactivity, such as products not accessible through solution chemistry, faster rates, better selectivity etc. A subset of this situation is if it thereby enables fewer steps to be used; (iv) when the product obtained mechanochemically is analytically pure and so requires no purification; (v) when the product contains detectable impurities but which are acceptable for its intended use. A subset of this last situation is if a subsequent synthetic step (potentially solvent-based) removes the impurities.A plausible holistic picture of the future involves use of mechanochemistry in these generic situations, with bulk solvents also being used as required but which are sustainably produced and relatively harmless.313
Scalability
Most of the synthesis described in the above sections has been done on laboratory scales ranging from a few hundred milligrams up to a few grams. Whilst milling equipment for much larger scale work is widely available and used in bulk scale materials processing,6a the issue of scalability in mechanosynthesis has not yet been broadly addressed, and indeed a common perception is that there are difficulties in scaling up such mechanochemistry. Therefore, the clear recent demonstration of production-scale (20–50 kg) synthesis of drug/carrier composites by Vectorpharma Spa described in Section 2, for example,51 is therefore very noteworthy and encouraging. In addition, the recent report of continuous flow mechanochemistry (a cocrystallization) in a twin-screw extruder177 points to interesting new directions for scalable approaches, which do not necessarily have to based on ball milling.Energy consumption
The energy consumption of grinding needs to be weighed up against alternative procedures, including energy-intensive ones such as solvent distillation,2 which it can avoid. Typical laboratory-scale ball mills themselves are, perhaps surprisingly, not energy intensive.297a,314 For example, a laboratory shaker mill such as the Retsch MM400 consumes 100–150 W under typical reaction conditions,297a,314c which is especially attractive when combined with the typically short or modest reaction times needed in mechanosynthesis (often less than one hour, and sometimes only a few minutes). This energy consumption can also compare quite favourably with that of other methods such as microwave heating.314 However, issues of scale will also need to be considered in the move towards larger scale applications.Predictability and mechanistic understanding
It was noted in Section 1.4 that a comprehensive mechanistic understanding of mechanochemical reactions that can underpin a strongly predictive approach to this type of synthesis is still some way off, and consequently much mechanosynthesis is conducted initially on a trial-and-error basis. Despite this there are emerging some general pointers to conducting molecular mechanosynthesis successfully, such as employing, if feasible, lower-melting reactants,19a,39 considering the use of LAG21 and generating internal solvent.39 Also, the basic reactivity principles used for solution-based chemistry can also often be successfully applied to mechanochemistry. A simple but general example is in acid–base reactions; the stronger the acid and the base the more likely the reaction under mechanochemical conditions.39The synthesis ‘mind set’
The usual question when planning synthesis is still ‘which solvent should I use?’ rather than ‘do I need a solvent?’ A change in this regard is needed for the wider acceptance of mechanochemistry, and this aspect may be as important as overcoming technical obstacles.Mills for chemistry
The main intended application of ball mills is not for conducting chemical reactions, but for processing materials, such as in breaking them down to smaller particle sizes. Partly because of this, but also in some cases because of the inherent technical challenge, they are not equipped as standard with capabilities which synthetic chemists take for granted, such as temperature monitoring, temperature control29 (although it should be noted that some such systems have been made and are even commercially available), or in situ monitoring by spectroscopy or diffraction techniques. The development of in situ analysis is likely to help gain mechanistic understanding, as well as to optimize and apply mechanochemical processes.Full life cycle analyses
Above, some general consideration has been given to materials, time and energy usage. ‘Curtate’ life cycle analyses, i.e. limited-scale analyses which neglect upstream or downstream processes, have also begun to be made to compare the overall energy efficiency of ball milling versus other activation methods.314a,b Much further analysis of this type is needed, and full life-cycle analyses particularly are also required for mechanochemical processes.11. General conclusion and outlook
It is clear that solventless (or minimal-solvent) mechanochemistry offers some advantages as an alternative approach to synthesis. These advantages can include greater efficiency with regard to time, materials and energy usage, as well as the discovery of new or improved reactivity and products. Its usage therefore looks likely to continue to grow. We must bear in mind, however, that there are challenges and limitations to it becoming fully adopted as a mainstream technique, which we have attempted to identify in this review. Overall, though, optimism for its future does seem to be justified, since none of the challenges is inherently insurmountable (indeed some, such as scalability are presently being addressed with success), especially given the increasing effort devoted to the topic and the growing requirement to move over to more sustainable synthetic production methods. Mechanochemistry should feature strongly ‘in the mix’ of new and sustainable synthetic chemistry.References
- (a) P. J. Walsh, H. Li and C. A. d. Parrodi, Chem. Rev., 2007, 107, 2503 CrossRef CAS; (b) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC; (c) D. J. C. Constable, C. Jiminez-Gonzales and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- An estimated 6% of total US energy use is due to distillation generally, Emerson Process Management, http://www.emersonprocessxperts.com/archives/2010/04/reducing_distil.html.
- L. Takacs, J. Mineral Met. Mater. Soc., 2000, 52(1), 12 CAS.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS.
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105 CrossRef CAS.
- (a) P. Baláž, Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed; (b) S. Kipp, V. Sepelak and K. D. Becker, Chem. Unserer Zeit, 2005, 39, 384 CrossRef CAS.
- L. Takacs, J. Therm. Anal. Calorim., 2007, 90, 81–84 CrossRef CAS.
- L. Takacs, J. Mater. Sci., 2004, 39, 4987 CrossRef CAS.
- J. F. Fernandez-Bertran, Pure Appl. Chem., 1999, 71, 581 CrossRef CAS.
- R. Ling and J. L. Baker, J. Chem. Soc. Trans., 1893, 63, 1314 RSC.
- A. O. Patil, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1984, 106, 348 CrossRef CAS.
- F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279 RSC.
- (a) M. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 CrossRef CAS; (b) M. Etter and D. Adsmond, J. Chem. Soc., Chem. Commun., 1990, 589 RSC.
- V. Peddiredi, W. Jones, A. Chorlton and R. Docherty, Chem. Commun., 1996, 987 RSC.
- M. Hollingsworth, M. Brown, B. Santarserio, J. Huffman and C. Goss, Chem. Mater., 1996, 6, 1227 CrossRef.
- M. R. Caira, L. R. Nassimbeni and A. F. Wildervanck, J. Chem. Soc., Perkin Trans. 2, 1995, 2213 RSC.
- (a) F. Toda, K. Tanaka and S. Iwata, J. Org. Chem., 1989, 54, 3007 CrossRef CAS; (b) F. Toda, H. Takumi and M. Akehi, J. Chem. Soc., Chem. Commun., 1990, 1270 RSC.
- G. Rothenberg, A. Downie, C. Raston and J. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef CAS.
- (a) A. Bruckman, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131 RSC; (b) G. Kaupp, Top. Curr. Chem., 2005, 254, 95 CAS; (c) G. Kaupp, CrystEngComm, 2009, 11, 388 RSC; (d) B. Rodriguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213 CrossRef CAS; (e) R. S. Varma and V. Polshettiwar, in Eco-friendly synthesis of fine chemicals, ed. R. Ballini, Royal Society of Chemistry, 2009, pp. 275–291 Search PubMed; (f) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC.
- A. Lazuen-Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC.
- T. Friscic and W. Jones, Cryst. Growth Des., 2009, 9, 1621 CAS.
- IUPAC Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. doi:10.1351/goldbook, http://goldbook.iupac.org/MT07141.html.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921 CrossRef CAS.
- D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 1249 RSC.
- P. G. Fox, J. Mater. Sci., 1975, 10, 340 CrossRef CAS.
- P. G. Fox and J. Soria-Ruiz, Proc. R. Soc. London, Ser. A, 1970, 317, 79 CrossRef CAS.
- R. P. Rastogi and N. B. Singh, J. Phys. Chem., 1968, 72, 4446 CrossRef CAS.
- R. Kuroda, K. Higashiguchi, S. Hasebe and Y. Imai, CrystEngComm, 2004, 6, 463 RSC.
- G. Kaupp, CrystEngComm, 2003, 5, 117 RSC.
- K. Chadwick and R. J. Davey, CrystEngComm, 2007, 9, 732 RSC.
- A. Jayasankar, A. Somwangthanaroj, Z. J. Shao and N. Rodríguez-Hornedo, Pharm. Res., 2006, 23, 2381 CrossRef CAS.
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9, 1106 CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890 RSC.
- D. Cincic, T. Friscic and W. Jones, J. Am. Chem. Soc., 2008, 130, 7524 CrossRef CAS.
- G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Tetrahedron, 2003, 59, 3753 CrossRef CAS.
- A. M. Belenguer, T. Friscic, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696 RSC.
- W. Yuan, T. Friscic, D. Apperley and S. L. James, Angew. Chem., Int. Ed., 2010, 49, 3916 CAS.
- (a) G. A. Bowmaker, J. V. Hanna, R. D. Hart, B. W. Skelton and A. H. White, Dalton Trans., 2008, 5290 RSC; (b) G. A. Bowmaker, J. V. Hanna, B. W. Skelton and A. H. White, Chem. Commun., 2009, 2168 RSC.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839 RSC.
- A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211 RSC.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606 RSC.
- M. B. a. M. J. M. Wiktorsson, in Manufacturing Systems and Technologies for the New Frontier: The 41st CIRP Conference on Manufacturing Systems, May 26–28, 2008, Tokyo, Japan, ed. K. U. M. Mitsuishi and F. Kimura, Springer, 2008, pp. 119–122 Search PubMed.
- C. P. F. Marcos, A. P. Martins, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140–4182 CrossRef.
- P. J. G. Dunn, S. Galvin and K. Hettenbach, Green Chem., 2004, 6, 43 RSC.
- J. F. Jenck, F. Agterberg and M. J. Droescher, Green Chem., 2004, 6, 544 RSC.
- A. Dushkin, Chemistry for Sustainable Development, 2004, 12, 251 CAS.
- Search details: US Granted US Applications EP-A EP-B WO JP (bibliographic data only) DE-C,B DE-A DE-T DE-U GB-A FR-A; Full patent spec. Text: mechanochemistry OR mechanochemical. One member per family and duplicates removed. Filing date range 1970–2009: total records 1901.
- “WIPO reformed IPC codes”, http://www.wipo.int/classifications/ipc/ipc8/, World Intellectual Property Organisation.
- F. M. T. Payne, A. Postma, R. Cammarano, F. Caruso, J. Williams, P. McCormick and A. Dodd, Vol. A1, ed. WIPO, Iceutica Pty Ltd, 2006 Search PubMed.
- “iCeutica completes GMP scale-up for its Nano-sized products”, http://www.iceutica.com/pdf/5_aNews_GMPScaleUp_062008.pdf, 2008.
- F. Carli, Proceedings of the International Symposium on Controlled Release of Bioactive Materials, 1999, 26, 873 Search PubMed.
- A. A. R. Challa, J. Ali and R. K. Khar, AAPS PharmSciTech, 2005, 6, E329–E357 CrossRef.
- K. Matzuki and K. Ozawa, in Lithium Ion Rechargeable Batteries: Materials, Technology, and New Applications: Materials, Technology, and Applications, ed. K. Ozawa, Wiley VCH, pp. 1–9 Search PubMed.
- P. A. Christian and O. Mao, US patent 640325781, 2002 Search PubMed.
- “Lithium manganese phosphate/carbon nanocomposites as cathode active materials for secondary lithium batteries”, A. Kay, 2009, WO2009/144600A2.
- N. Anderson, Org. Process Res. Dev., 2001, 5, 613–621 CrossRef CAS.
- J. A. Vaccari, Materials Handbook: An Encyclopedia for Managers, Technical Professionals, Purchasing and Production Managers, Technicians and Supervisors, McGraw-Hill Professional Search PubMed.
- M. Bengisu, Engineering Ceramics, Springer, 2001 Search PubMed.
- J.-T. Li, Y. Yang and H.-B. Jin, Key Eng. Mater., 2007, 336–338, 911–915 CrossRef CAS.
- P. Solsona, S. Doppiu, T. Spassov, S. Surinach and M. D. Baro, J. Alloys Compd., 2004, 381, 66–71 CrossRef CAS.
- L. D. B. Arceo, J. J. Cruz-Rivera, J. G. Cabanas-Moreno, K. Tsuchiya, M. Umemoto and H. Calderson, Mater. Sci. Forum, 2000, 343–346, 641–646 CrossRef CAS.
- M. Karolus, E. Jartych and D. Oleszak, Acta Phys. Pol., A, 2002, 102, 253–258 CAS.
- K. J. Kim, K. Sumiyama and K. Suzuki, J. Magn. Magn. Mater., 1995, 140–144, 49–50 CrossRef CAS.
- L. M. Kubalova, V. I. Fadeeva, I. A. Sviridov and S. A. Fedotov, J. Alloys Compd., 2009, 483, 86–88 CrossRef CAS.
- V. E. Oliker, V. L. Sirovatka, T. Y. Gridasova, I. I. Timofeeva and A. I. Bykov, Powder Metall. Met. Ceram., 2008, 47, 546–556 CrossRef CAS.
- T. Mushove, H. Chikwanda, C. Machio and S. Ndlovu, Mater. Sci. Forum, 2009, 618–619, 517–520 CrossRef CAS.
- H. Rojas-Chavez, S. Diaz-de la Torre, D. Jaramillo-Vigueras and G. Plascencia, J. Alloys Compd., 2009, 483, 275–278 CrossRef CAS.
- I. Farahbakhsh, S. H. Tabaian and J. Vahdati, Adv. Mater. Res. (Zuerich, Switzerland), 2010, 83–86, 36–40 CAS.
- T. Tojo, Q. Zhang and F. Saito, J. Solid State Chem., 2006, 179, 433–437 CrossRef CAS.
- T. Tojo, Q. Zhang and F. Saito, J. Alloys Compd., 2007, 427, 219–222 CrossRef CAS.
- Q. Zhang, J. Lu and F. Saito, Powder Technol., 2002, 122, 145–149 CrossRef CAS.
- C. A. C. Escobedo, F. Sanchez de Jesus, A. M. B. Miro and J. Munoz-Saldana, Phys. Status Solidi C: Curr. Topics Solid State Phys., 2007, 4, 4054–4063 CAS.
- I. Szafraniak-Wiza, B. Hilczer, A. Pietraszko and E. Talik, J. Electroceram., 2008, 20, 21–25 CrossRef CAS.
- V. Berbenni, A. Marini, A. Profumo and L. Cucca, Zeit. Naturforsch., B: Chem. Sci., 2003, 58, 415–422 CAS.
- V. Nachbaur, G. Tauvel, T. Verdier, M. Jean, J. Juraszek and D. Houvet, J. Alloys Compd., 2009, 473, 303–307 CrossRef CAS.
- H. Yang, X. Zhang, W. Ao and G. Qiu, Mater. Res. Bull., 2004, 39, 833–837 CrossRef CAS.
- (a) T. Hungria, I. MacLaren, H. Fuess, J. Galy and A. Castro, Mater. Lett., 2008, 62, 3095–3098 CrossRef CAS; (b) T. Hungria, J. G. Lisoni and A. Castro, Chem. Mater., 2002, 14, 1747–1754 CrossRef CAS.
- (a) T. I. Arbuzova, B. A. Gizhevskii, R. G. Zakharov, S. A. Petrova and N. M. Chebotaev, Phys. Solid State, 2008, 50, 1487–1494 CrossRef CAS; (b) V. Sepelak, I. Bergmann, A. Feldhoff, P. Heitjans, F. Krumeich, D. Menzel, F. J. Litterst, S. J. Campbell and K. D. Becker, J. Phys. Chem. C, 2007, 111, 5026–5033 CrossRef CAS.
- G. Ye and T. Troczynski, J. Am. Ceram. Soc., 2007, 90, 287–290 CrossRef CAS.
- A. Gajovic, I. Djerdj, K. Furic, R. Schlogl and D. S. Su, Cryst. Res. Technol., 2006, 41, 1076–1081 CrossRef CAS.
- A. Mergen, Ceram. Int., 2009, 35, 1151–1157 CrossRef CAS.
- P. Ferrer, J. E. Iglesias, A. P. Ayala, I. Guedes and A. Castro, Solid State Comm., 2005, 136, 621–626 CAS.
- A. A. Cristobal, E. F. Aglietti, M. S. Conconi and J. M. Porto Lopez, Mater. Chem. Phys., 2008, 111, 341–345 CrossRef CAS.
- V. Sepelak, I. Bergmann, A. Diekmann, P. Heitjans and K. D. Becker, Rev. Adv. Mater. Sci., 2008, 18, 349–352 CAS.
- P. M. Botta, E. F. Aglietti and J. M. Porto Lopez, Mater. Res. Bull., 2006, 41, 714–723 CrossRef CAS.
- P. M. Botta, E. F. Aglietti and J. M. Porto Lopez, Mater. Chem. Phys., 2002, 76, 104–109 CrossRef CAS.
- (a) M. T. Le, D. J. Kim, C. G. Kim, J. S. Sohn and J. Lee, J. Exp. Nanosci., 2008, 3, 223–228 CrossRef CAS; (b) J. Keskinen, P. Ruuskanen, M. Karttunen and S. P. Hannula, Appl. Organomet. Chem., 2001, 15, 393–395 CrossRef CAS.
- A. Celikovic, L. Kandic, M. Zdujic and D. Uskokovic, Mater. Sci. Forum, 2007, 555, 279–284 CrossRef CAS.
- A. C. Dodd and P. G. McCormick, J. Aust. Ceram. Soc., 2000, 36, 15–19 CAS.
- T. Tsuzuki and P. G. McCormick, Acta Mater., 2000, 48, 2795–2801 CrossRef CAS.
- T. Ito, Q. Zhang and F. Saito, Powder Tech., 2004, 143–144, 170–173 CAS.
- T. Tsuzuki and P. G. McCormick, Mater. Trans., 2001, 42, 1623–1628 CrossRef CAS.
- S. Palaniandy and N. H. Jamil, J. Alloys Compd., 2009, 476, 894–902 CrossRef CAS.
- R. A. V. Ortiz, J. Munoz-Saldana and F. J. Espinoza-Beltran, Mater. Res. Innovations, 2009, 13, 368–371 CrossRef CAS.
- T. Rojac, M. Kosec, B. Malic and J. Holc, J. Am. Ceram. Soc., 2008, 91, 1559–1565 CrossRef CAS.
- M. Khachane, A. Moure, M. Elaatmani, A. Zegzouti, M. Daoud and A. Castro, J. Alloys Compd., 2006, 424, 231–236 CrossRef CAS.
- K. Han, T. Ko and J. Oh, Scr. Mater., 2008, 59, 143–146 CrossRef CAS.
- (a) W. Tong and G. G. Amatucci, Electrochem. Solid-State Lett., 2009, 12, A219–A224 CrossRef CAS; (b) W. Tong and G. G. Amatucci, ECS Trans., 2008, 11, 19–25 CrossRef CAS.
- H. Peng, N. Machida and T. Shigematsu, Funtai oyobi Funmatsu Yakin, 2002, 49, 69–74 CAS.
- (a) A. J. Cruz-Cabeza, G. M. Day and W. Jones, Chem.–Eur. J., 2008, 14, 8830 CrossRef CAS; (b) H.-J. Choi, K.-M. Lee, G.-H. Kim and J.-G. Lee, J. Am. Ceram. Soc., 2001, 84, 242–244 CrossRef CAS.
- C. W. Dunnill, Z. A. Aiken, A. Kafizas, J. Pratten, M. Wilson, D. J. Morgan and I. P. Parkin, J. Mater. Chem., 2009, 19, 8747–8754 RSC.
- (a) M. Salari, S. M. Mousavi khoie, P. Marashi and M. Rezaee, J. Alloys Compd., 2009, 469, 386–390 CrossRef CAS; (b) M. Salari, M. Rezaee, S. M. Mousavi Koie, P. Marashi and H. Aboutalebi, Int. J. Mod. Phys. B: Cond. Matter Phys., Stat.Phys., Appl. Phys., 2008, 22, 2955–2961 CAS.
- P. Billik and G. Plesch, Scr. Mater., 2007, 56, 979–982 CrossRef CAS.
- P. Billik, G. Plesch, V. Brezova, L. Kuchta, M. Valko and M. Mazur, J. Phys. Chem. Solids, 2007, 68, 1112–1116 CrossRef CAS.
- (a) Q. Zhang, J. Wang, S. Yin, T. Sato and F. Saito, J. Am. Ceram. Soc., 2004, 87, 1161–1163 CrossRef CAS; (b) S. Yin, M. Komatsu, Q. Zhang, F. Saito and T. Sato, J. Mater. Sci., 2007, 42, 2399–2404 CrossRef CAS.
- J. Wang, S. Yin, Q. Zhang, F. Saito and T. Sato, J. Mater. Chem., 2003, 13, 2348–2352 RSC.
- I. D. Gocheva, M. Nishijima, T. Doi, S. Okada, J.-i. Yamaki and T. Nishida, J. Power Sources, 2009, 187, 247–252 CrossRef CAS.
- (a) V. Manivannan, P. Parhi and J. W. Kramer, Bull. Mater. Sci., 2008, 31, 987–993 CrossRef CAS; (b) J. Lee, H. Shin, J. Lee, H. Chung, Q. Zhang and F. Saito, Mater. Trans., 2003, 44, 1457–1460 CrossRef CAS.
- R. H. Pawelke, M. Felderhoff, C. Weidenthaler, B. Bogdanovic and F. Schuth, Z. Anorg. Allg. Chem., 2009, 635, 265–270 CrossRef CAS.
- B. P. Sobolev, I. A. Sviridov, V. I. Fadeeva, S. N. Sul'yanov, N. I. Sorokin, Z. I. Zhmurova, P. Herrero, A. Landa-Canovas and R. M. Rojas, Crystallogr. Rep., 2005, 50, 478–485 CrossRef CAS.
- L. N. Patro and K. Hariharan, Mater. Sci. Eng., B: Adv. Funct. Solid-State Mater., 2009, 162, 173–178 CAS.
- Y. Yamane, K. Yamada and K. Inoue, Solid State Ion., 2008, 179, 605–610 CrossRef CAS.
- M. M. Ahmad, Y. Yamane, K. Yamada and S. Tanaka, J. Phys. D: Appl. Phys., 2007, 40, 6020–6025 CrossRef CAS.
- J. Lee, Q. Zhang and F. Saito, J. Alloys Compd., 2003, 348, 214–219 CrossRef CAS.
- J. Lee, Q. Zhang and F. Saito, Ind. Eng. Chem. Res., 2001, 40, 4785–4788 CrossRef CAS.
- J. Lee, Q. Zhang and F. Saito, J. Am. Ceram. Soc., 2001, 84, 863–865 CrossRef CAS.
- (a) E. Dutkova, P. Balaz and P. Pourghahramani, J. Optoelectr. Adv. Mat., 2009, 11, 2102–2107 CAS; (b) W. Wang, L. Ao, G. He and G. Zhang, Mater. Lett., 2008, 62, 1014–1017 CrossRef CAS.
- E. Dutkova, P. Balaz, P. Pourghahramani, A. V. Nguyen, V. Sepelak, A. Feldhoff, J. Kovac and A. Satka, Solid State Ion., 2008, 179, 1242–1245 CrossRef CAS.
- (a) C.-K. Lin, C.-Y. Chen, P.-Y. Lee and C.-C. Chan, Mater. Sci. Forum, 2007, 561–565, 2099–2102 CrossRef CAS; (b) J. Z. Jiang, R. K. Larsen, R. Lin, S. Morup, I. Chorkendorff, K. Nielsen, K. Hansen and K. West, J. Solid State Chem., 1998, 138, 114–125 CrossRef CAS.
- (a) J. P. Tiwari and K. Shahi, Mater. Sci. Eng., B: Solid-State Mater. Adv. Tech., 2007, 141, 8–15 CAS; (b) J. P. Tiwari and K. Shahi, Solid State Ion., 2005, 176, 1271–1280 CrossRef CAS; (c) P. Balaz, M. Fabian, M. Pastorek, D. Cholujova and J. Sedlak, Mater. Lett., 2009, 63, 1542–1544 CrossRef CAS.
- (a) J. M. Criado, M. D. Alcalá and C. Real, Solid State Ion., 1997, 101–103, 1387–1391 CrossRef CAS; (b) W. Y. Lim, M. Hida, A. Sakakibara, Y. Takemoto and S. Yokomizo, J. Mater. Sci., 1993, 28, 3463–3466 CrossRef CAS.
- A. Calka, Appl. Phys. Lett., 1991, 59, 1568–1569 CrossRef CAS.
- V. Lopez-Flores, M. A. Roldan, C. Real, A. M. Paez and G. R. Castro, J. Appl. Phys., 2008, 104, 023519/023511–023519/023518 CrossRef.
- T. Tsuchida and Y. Azuma, J. Mater. Chem., 1997, 7, 2265–2268 RSC.
- R. Concepción, A. R. Manuel, D. A. María and O. Andrés, J. Am. Ceram. Soc., 2007, 90, 3085–3090 CrossRef.
- G. An and G. J. Liu, Rare Met., 2008, 27, 303–307 CrossRef CAS.
- J. Kano, L. Jenfeng, I. C. Kang, W. Tongamp, E. Kobayashi and F. Saito, Chem. Lett., 2007, 36, 900–901 CrossRef CAS.
- Y. Chen, J. F. Gerald, J. S. Williams and P. Willis, Mater. Sci. Forum, 1999, 312–314, 173–177 CrossRef CAS.
- A. Calka, J. S. Williams and P. Millet, Scr. Metall. Mater., 1992, 27, 1853–1857 CrossRef CAS.
- J. Kano, E. Kobayashi, W. Tongamp and F. Saito, J. Alloys Compd., 2008, 464, 337–339 CrossRef CAS.
- Y. Sun, B. Yao, Q. He, F. Su and H. Z. Wang, J. Alloys Compd., 2009, 479, 599–602 CrossRef CAS.
- S. D. Culligan, H. W. Langmi, V. B. Reddy and G. Sean McGrady, Inorg. Chem. Commun., 13, 540–542 CrossRef CAS.
- C. J. H. Jacobsen, J. J. Zhu, H. Lindelov and J. Z. Jiang, J. Mater. Chem., 2002, 12, 3113–3116 RSC.
- J. F. Sun, M. Z. Wang, Y. C. Zhao, X. P. Li and B. Y. Liang, J. Alloys Compd., 2009, 482, L29–L31 CrossRef CAS.
- G. M. Wang, S. J. Campbell and W. A. Kaczmarek, Mater. Sci. Eng. A, 1997, 226–228, 80–83 CrossRef.
- L. J. Zhuge, W. G. Yao and X. M. Wu, J. Magn. Magn. Mater., 2003, 257, 95–99 CrossRef CAS.
- F. Karimzadeh, M. H. Enayati and M. Tavoosi, Mater. Sci. Eng., A: Struct. Mater.: Prop. Microstruct. Proc., 2008, A486, 45–48 CAS.
- H. Mostaan, M. H. Abbasi and F. Karimzadeh, J. Alloys Compd., 493, 609–612 CrossRef CAS.
- K. Wieczorek-Ciurowa, D. Oleszak and K. Gamrat, Khim. v Interesakh Ustoichivogo Razvitiya, 2007, 15, 255–258 CAS.
- (a) M. M. Verdian and S. Heshmati-Manesh, Int. J. Mod. Phys. B: Cond. Matter Phys., Stat. Phys., Appl. Phys., 2008, 22, 2914–2923 CAS; (b) N. J. Welham, Intermetallics, 1998, 6, 363–368 CrossRef CAS.
- T. G. Durai, K. Das and S. Das, J. Alloys Compd., 2008, 462, 410–415 CrossRef CAS.
- E. Mohammad Sharifi, F. Karimzadeh and M. H. Enayati, J. Alloys Compd., 2009, 482, 110–113 CrossRef CAS.
- (a) C.-M. Park, W.-S. Chang, H. Jung, J.-H. Kim and H.-J. Sohn, Electrochem. Commun., 2009, 11, 2165–2168 CrossRef CAS; (b) S. Yoon, J.-M. Lee, H. Kim, D. Im, S.-G. Doo and H.-J. Sohn, Electrochim. Acta, 2009, 54, 2699–2705 CrossRef CAS.
- (a) Y. Oumellal, A. Rougier, J. M. Tarascon and L. Aymard, J. Power Sources, 2009, 192, 698–702 CrossRef CAS; (b) T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAST. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265 CrossRef CAS; J. Huang, T. Takei, T. Akita, H. Ohashi and M. Haruta, Appl. Catal., B, 2010, 95, 430 CrossRef.
- (a) A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC; (b) S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881–2889 CrossRef CAS; (c) S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS; (d) G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007–1026 CrossRef CAS; (e) C. B. Aakeroy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC; (f) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS.
- S. L. Childs and M. J. Zaworotko, Cryst. Growth. Des., 2009, 9, 4208 CAS.
- (a) D. Braga, L. Maini, M. Polito, L. Mirolo and F. Grepioni, Chem. Commun., 2002, 2960–2961 RSC; (b) D. Braga, L. Maini, M. Polito and F. Grepioni, Chem. Commun., 2002, 2302–2303 RSC; (c) V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 987–988 RSC.
- (a) N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC; (b) D. Braga, L. Maini, S. L. Giaffreda, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2004, 10, 3261–3269 CrossRef CAS; (c) A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890–891 RSC.
- (a) D. Braga, S. L. Giaffreda, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, CrystEngComm, 2007, 9, 39–45 RSC; (b) D. Braga, S. L. Giaffreda, F. Grepioni, M. R. Chierotti, R. Gobetto, G. Palladino and M. Polito, CrystEngComm, 2007, 9, 879–881 RSC; (c) A. Jayasankar, D. J. Good and N. Rodriguez-Hornedo, Mol. Pharmaceutics, 2007, 4, 360–372 CrossRef CAS.
- C. Maheshwari, A. Jayasankar, N. A. Khan, G. E. Amidon and N. Rodriguez-Hornedo, CrystEngComm, 2009, 11, 493–500 RSC.
- (a) L. Kofler and A. Kofler, Thermal Micromethods for the Study of Organic Compounds and Their Mixtures, 1980 Search PubMed; (b) O. Lehmann, Molecularphysik, 1888 Search PubMed; (c) W. C. McCrone, Fusion Methods in Chemical Microscopy, 1957 Search PubMed; (d) D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey, W. Jones, T. Friscic and N. Blagden, Cryst. Growth Des., 2008, 8, 1697–1712 CrossRef CAS; (e) D. Braga, F. Grepioni, L. Maini, P. P. Mazzeo and K. Rubini, Thermochim. Acta, 2010, 507-08, 1 CrossRef.
- (a) F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC; (b) F. Toda, Acc. Chem. Res., 1995, 28, 480–486 CrossRef CAS.
- F. Toda and H. Miyamoto, Chem. Lett., 1995, 24, 861–861 CrossRef.
- (a) Y. Imai, N. Tajima, T. Sato and R. Kuroda, Chirality, 2002, 14, 604 CrossRef CAS; (b) Kuroda, Y. Imai and N. Tajima, Chem. Commun., 2002, 2848 RSC; (c) F. Toda, M. Senzaki and R. Kuroda, Chem. Commun., 2002, 1788 RSC.
- E. Y. Cheung, S. J. Kitchin, K. D. M. Harris, Y. Imai, N. Tajima and R. Kuroda, J. Am. Chem. Soc., 2003, 125, 14658 CrossRef CAS.
- (a) D. R. Trivedi, Y. Fujiki, Y. Goto, N. Fujita, S. Shinkai and K. Sada, Chem. Lett., 2008, 37, 550–551 CrossRef CAS; (b) Darshak R. Trivedi, Y. Fujiki, N. Fujita, S. Shinkai and K. Sada, Chem.–Asian J., 2009, 4, 254–261 CrossRef CAS.
- D. Braga, L. Maini, G. de Sanctis, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2003, 9, 5538–5548 CrossRef CAS.
- D. Braga, E. Dichiarante, G. Palladino, F. Grepioni, M. R. Chierotti, R. Gobetto and L. Pellegrino, CrystEngComm, 2010, 12, 3534 RSC.
- D. Braga, F. Grepioni and G. I. Lampronti, CrystEngComm, 2011, 13, 3122 RSC.
- D. Braga, G. Palladino, M. Polito, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2008, 14, 10149–10159 CrossRef CAS.
- D. Braga, F. Grepioni, L. Maini, S. Prosperi, R. Gobetto and M. R. Chierotti, Chem. Commun., 2010, 46, 7715 RSC.
- M. D. Cohen and G. M. J. Schmidt, Journal of the Chemical Society, 1964, 1996–2000 CAS.
- (a) L. R. Macgillivray, G. S. Papaefstathiou, T. Friscic, T. D. Hamilton, D. K. Bucar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS; (b) I. G. Georgiev and L. R. MacGillivray, Chem. Soc. Rev., 2007, 36, 1239–1248 RSC; (c) X. C. Gao, T. Friscic and L. R. MacGillivray, Angew. Chem., Int. Ed., 2004, 43, 232–236 CrossRef CAS.
- A. N. Sokolov, D.-K. Bučar, J. Baltrusaitis, S. X. Gu and L. R. MacGillivray, Angew. Chem., Int. Ed., 2010, 49, 4273 CrossRef CAS.
- J. F. Willart and M. Descamps, Mol. Pharmaceutics, 2008, 5, 905 CrossRef CAS.
- (a) W. Jones, W. D. S. Motherwell and A. V. T. M. Bulletin, MRS Bull., 2006, 31, 875 CrossRef CAS; (b) N. Shan and M. J. Zaworotko, Drug Discov. Today, 2008, 13, 440 CrossRef CAS; (c) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef CAS.
- (a) D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharm. Res., 2006, 23, 1888 CrossRef CAS; (b) D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 2252 CrossRef CAS; (c) A. V. Trask, W. D. S. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114 CrossRef CAS; (d) A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013 CrossRef CAS; (e) S. Karki, T. Friščić, L. Fábián, P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905 CrossRef CAS.
- R. R. Schartman, Int. J. Pharm., 2009, 365, 77 CrossRef CAS.
- J. Lu and S. Rohani, Org. Process Res. Dev., 2009, 13, 1269 CrossRef CAS.
- M. R. Caira, L. R. Nassimbeni and A. F. Wildervanck, J. Chem. Soc., Perkin. Trans. 2, 1995, 2213 RSC.
- M. C. Etter, S. M. Reutzel and C. G. Choo, J. Am. Chem. Soc., 1993, 115, 4411 CrossRef CAS.
- M. N. Frey, T. F. Koetzle, M. S. Lehmann and W. C. Hamilton, J. Chem. Phys., 1973, 59, 915 CrossRef CAS.
- (a) A. Jayasankar, D. J. Good and N. Rodríguez-Hornedo, Mol. Pharmaceutics, 2007, 4, 360 CrossRef CAS; (b) C. Maheshwari, A. Jayasankar, N. A. Khan, G. E. Amidon and N. Rodríguez-Hornedo, CrystEngComm, 2009, 11, 493 RSC.
- (a) A. D. Gusseme, C. Neves, J. F. Willart, A. Rameau and M. Descamps, J. Pharm. Sci., 2008, 97, 5000 CrossRef; (b) N. Dujardin, J. F. Willart, E. Dudognon, A. Hédoux, Y. Guinet, L. Paccou, B. Chazallon and M. Descamps, Solid State Commun., 2008, 148, 78 CrossRef CAS; (c) For previous work on the amorphisation of pharmaceuticals by cryogrinding, K. J. Crowley and G. Zografi, J. Pharm. Sci., 2002, 91, 492 CrossRef CAS; (d) M. Otsuka, T. Matsumoto and N. Kaneniwa, Chem. Pharm. Bull., 1986, 34, 1784 CAS.
- N. Chieng, M. Hubert, D. Saville, T. Rades and J. Aaltonen, Cryst. Growth Des., 2009, 9, 2377 CAS.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharmaceutics, 2007, 4, 347 CrossRef CAS.
- C. Medina, D. Daurio, K. Nagapudi and F. Alvarez-Nunez, J. Pharm. Sci., 2010, 99, 1693 CAS.
- (a) T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418 RSC; (b) N. Rodríguez-Hornedo, S. J. Nehm, K. F. Seefeldt, Y. Págan-Torres and C. J. Falkiewicz, Mol. Pharmaceutics, 2006, 3, 362 CrossRef.
- T. Friščić and W. Jones, Faraday Discuss., 2007, 136, 167 RSC.
- (a) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546 CrossRef; (b) T. Friščić, A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2008, 8, 1605 CrossRef.
- K. L. Nguyen, T. Friščić, G. M. Day, L. F. Gladden and W. Jones, Nat. Mater., 2007, 6, 206 CrossRef CAS.
- S. L. Childs and K. I. Hardcastle, Cryst. Growth Des., 2007, 7, 1291 CAS.
- S. L. Childs, N. Rodríguez-Hornedo, L. S. Reddy, A. Jayasankar, C. Maheshwari, L. McCausland, R. Shipplett and B. C. Stahly, CrystEngComm, 2008, 10, 856 RSC.
- S. Karki, T. Friščić and W. Jones, CrystEngComm, 2009, 11, 470 RSC.
- S. Karki, L. Fábián, T. Friščić and W. Jones, Org. Lett., 2007, 9, 3133 CrossRef CAS.
- A. J. Cruz-Cabeza, S. Karki, L. Fábián, T. Friščić, G. M. Day and W. Jones, Chem. Commun., 2010, 46, 2224 RSC.
- T. Friščić, L. Fábián, J. C. Burley, D. G. Reid, M. J. Duer and W. Jones, Chem. Commun., 2008, 1644 Search PubMed.
- A. J. Cruz-Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, J. Am. Chem. Soc., 2006, 128, 14466 CrossRef.
- S. Basavoju, D. Boström and S. P. Velaga, Pharm. Res., 2008, 25, 530 CrossRef CAS.
- (a) M. K. Stanton, S. Tufekcic, C. Morgan and A. Bak, Cryst. Growth Des., 2009, 9, 1344 CrossRef CAS; (b) A. Bak, A. Gore, E. Yanez, M. Stanton, S. Tufekcic, R. Syed, A. Akrami, M. Rose, S. Surapaneni, T. Bostick, A. King, S. Neervannan, D. Ostovic and A. K., J. Pharm. Sci., 2008, 97, 3942 CrossRef CAS.
- L. Orola and M. V. Veidis, CrystEngComm, 2009, 11, 415 RSC.
- T. Friščić, L. Fábián, J. C. Burley, W. Jones and W. D. S. Motherwell, Chem. Commun., 2006, 5009 Search PubMed.
- A. V. Trask, J. v. d. Streek, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 2233 CAS.
- C. C. Seaton, A. Parkin, C. C. Wilson and N. Blagden, Cryst. Growth Des., 2009, 9, 47 CAS.
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem. Commun., 2005, 880 RSC.
- (a) C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772 CrossRef; (b) C. B. Aakeröy, N. Schultheiss, J. Desper and C. Moore, Cryst. Growth Des., 2007, 7, 2324 CrossRef; (c) C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef.
- (a) L. S. Reddy, S. K. Chandran, S. George, N. J. Babu and A. Nangia, Cryst. Growth Des., 2007, 7, 2675 CrossRef CAS; (b) N. J. Babu, L. S. Reddy and A. Nangia, Mol. Pharmaceutics, 2007, 4, 417 CrossRef CAS.
- D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey, W. Jones, T. Friščić and N. Blagden, Cryst. Growth Des., 2008, 8, 1697 CAS.
- (a) J. S. Clawson, F. G. Vogt, J. Brum, J. Sisko, D. B. Patience, W. Dai, S. Sharpe, A. D. Jones, T. N. Pham, M. N. Johnson and R. C. P. Copley, Cryst. Growth Des., 2008, 8, 4120 CrossRef CAS; (b) Y. Andemichael, J. Chen, J. S. Clawson, W. Dai, A. Diederich, S. V. Downing, A. J. Freyer, P. Liu, L. M. Oh, D. B. Patience, S. Sharpe, J. Sisko, J. Tsui, F. G. Vogt, J. Wang, L. Wernersbach, E. C. Webb, J. Wertman and L. Zhou, Org. Process Res. Dev., 2009, 13, 729 CrossRef CAS.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, Cryst. Growth Des., 2010, 10, 1435 CAS.
- A. V. Trask, D. A. Haynes, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 51 RSC.
- V. André, D. Braga, F. Grepioni and M. T. Duarte, Cryst. Growth Des., 2009, 9, 5108 Search PubMed.
- E. M. C. Gerard, H. Sahin, A. Encinas and S. Bräse, Synlett, 2008, 2702–2704 CAS.
- J. Mack and M. Shumba, Green Chem., 2007, 9, 328–330 RSC.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, J. Am. Chem. Soc., 2002, 124, 6244–6245 CrossRef CAS.
- J. Gao and G.-W. Wang, J. Org. Chem., 2008, 73, 2955–2958 CrossRef CAS.
- (a) V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318–9321 CrossRef CAS; (b) J. G. Hernandez and E. Juaristi, J. Org. Chem., 2010, 75, 7107 CrossRef CAS.
- P. R. Patil and K. P. R. Kartha, Green Chem., 2009, 11, 953–956 RSC.
- N. Giri, C. Bowen, J. S. Vyle and S. L. James, Green Chem., 2008, 10, 627 RSC.
- K. Komatsu, Topics in Current Chemistry, 2005, 254, 185–206 CAS.
- M. Keshavarz-K, B. Knight, G. Srdanov and F. Wudl, J. Am. Chem. Soc., 1995, 117, 11371–11372 CrossRef CAS.
- G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Nature, 1997, 387, 583–586 CrossRef CAS.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2005, 61, 11771–11835 CrossRef CAS.
- (a) M. S. Viciu and S. P. Nolan, in Modern arylation methods, ed. L. Ackermann, Wiley-VCH, Weinheim, 2009, pp. 183–220 Search PubMed; (b) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS; (c) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441–3461 CrossRef CAS.
- P. Saha, S. Naskar, P. Paira, A. Hazra, K. B. Sahu, R. Paira, S. Banerjee and N. B. Mondal, Green Chem., 2009, 11, 931–934 RSC.
- (a) M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS; (b) V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS.
- D. A. Fulmer, W. C. Shearouse, S. T. Medonza and J. Mack, Green Chem., 2009, 11, 1821–1825 RSC.
- R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985–991 RSC.
- E. Tullberg, D. Peters and T. Frejd, J. Organomet. Chem., 2004, 689, 3778–3781 CrossRef CAS.
- E. Tullberg, F. Schacher, D. Peters and T. Frejd, Synthesis, 2006, 1183–1189 CAS.
- S. F. Nielsen, D. Peters and O. Axelsson, Synth. Commun., 2000, 30, 3501–3509 CrossRef CAS.
- (a) F. Schneider, A. Stolle, B. Ondruschka and H. Hopf, Org. Process Res. Dev., 2009, 13, 44–48 CrossRef CAS; (b) F. Schneider and B. Ondruschka, ChemSusChem, 2008, 1, 622–625 CrossRef CAS.
- F. Bernhardt, R. Trotzki, T. Szuppa, A. Stolle and B. Ondruschka, Beilstein J. Org. Chem., 2010, 6, 1–9 CrossRef.
- (a) Y. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965–2983 CrossRef CAS; (b) S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621–631 CrossRef CAS; (c) I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683–5712 CrossRef CAS.
- C. Bolm, I. Atodiresei and I. Schiffers, Org. Synth., 2005, 82, 120–125 CAS.
- T. Rantanen, I. Schiffers and C. Bolm, Org. Process Res. Dev., 2007, 11, 592–597 CrossRef CAS.
- (a) G. Guillena, C. Najera and D. J. Ramon, Tetrahedron: Asymmetry, 2007, 18, 2249–2293 CrossRef CAS; (b) B. List, Chem. Commun., 2006, 819–824 RSC; (c) B. List, Acc. Chem. Res., 2004, 37, 548–557 CrossRef CAS; (d) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS.
- B. Rodriguez, T. Rantanen and C. Bolm, Angew. Chem., Int. Ed., 2006, 45, 6924–6926 CrossRef CAS.
- B. Rodriguez, A. Bruckmann and C. Bolm, Chem.–Eur. J., 2007, 13, 4710–4722 CrossRef CAS.
- (a) G. Guillena, M. d. C. Hita, C. Najera and S. F. Viozquez, J. Org. Chem., 2008, 73, 5933–5943 CrossRef CAS; (b) G. Guillena, M. d. C. Hita, C. Najera and S. F. Viozquez, Tetrahedron: Asymmetry, 2007, 18, 2300–2304 CrossRef CAS.
- A. Bruckmann, B. Rodriguez and C. Bolm, CrystEngComm, 2009, 11, 404–407 RSC.
- A. M. Flock, C. M. M. Reucher and C. Bolm, Chem.–Eur. J., 2010, 16, 3918–3921 CAS.
- (a) C. L. Raston and J. L. Scott, Green Chem., 2000, 2, 49 RSC; (b) J. L. Scott, D. R. MacFarlane, C. L. Raston and C. M. Teoh, Green Chem., 2000, 2, 123 RSC; (c) G. W. V. Cave, C. L. Raston and J. L. S., Chem. Commun., 2001, 2159 RSC; (d) B. A. Roberts, G. W. V. Cave, C. L. Raston and J. L. Scott, Green Chem., 2001, 3, 280 RSC.
- P. Coppens, S. L. Zheng, M. Gembicky, M. Messerschmidt and P. M. Dominiak, CrystEngComm, 2006, 8, 735–741 RSC.
- A. N. Swinburne and J. W. Steed, CrystEngComm, 2009, 11, 433–438 RSC.
- J. L. Atwood, M. J. Hardie, C. L. Raston and C. A. Sandoval, Org. Lett., 1999, 1, 1523–1526 CrossRef CAS.
- J. L. Scott, D. R. MacFarlane, C. L. Raston and C. M. Teoh, Green Chem., 2000, 2, 123–126 RSC.
- B. A. Roberts, G. W. V. Cave, C. L. Raston and J. L. Scott, Green Chem., 2001, 3, 280–284 RSC.
- J. Antesberger, G. W. V. Cave, M. C. Ferrarelli, M. W. Heaven, C. L. Raston and J. L. Atwood, Chem. Commun., 2005, 892–894 RSC.
- B. Icli, N. Christinat, J. Tonnemann, C. Schuttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154 CrossRef CAS.
- P. J. Nichols, C. L. Raston and J. W. Steed, Chem. Commun., 2001, 1062–1063 RSC.
- T. Ohshita, D. Nakajima, A. Tsukamoto, N. Tsuchiya, T. Isobe, M. Senna, N. Yoshioka and H. Inoue, Ann. Chim. Sci. Mater., 2002, 27, 91–101 CrossRef CAS.
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790 RSC.
- S. K. Thabet, H. A. Tayim and M. U. Karkanawi, Inorg. Nucl. Chem. Lett., 1972, 8, 211–213 CrossRef CAS.
- T. Chen, B. Liang and X. Xin, J. Solid State Chem., 1997, 132, 291–293 CrossRef CAS.
- X. Yao, L. Zheng and X. Xin, J. Solid State Chem., 1995, 117, 333–336 CrossRef CAS.
- G. Hihara, M. Satoh, T. Uchida, F. Ohtsuki and H. Miyamae, Solid State Ion., 2004, 172, 221–223 CrossRef CAS.
- (a) G. A. Bowmaker, N. Chaichit, C. Pakawatchai, B. W. Skelton and A. H. White, Dalton Trans., 2008, 2926 RSC; (b) G. A. Bowmaker, B. W. Skelton and A. H. White, Inorg. Chem., 2009, 48, 3185 CrossRef CAS.
- C. J. Adams, M. F. Haddow, F. Mairi, R. J. I. Hughes, M. A. Kurawa and A. G. Orpen, Dalton Trans., 2010, 39, 3714 RSC.
- D. Braga, F. Grepioni, L. Maini, R. Brescello and L. Cotarca, CrystEngComm, 2008, 10, 469–471 RSC.
- A. Paneque, J. Fernández-Bertrán, E. Reguera and H. Yee-Madeira, Transition Met. Chem., 2001, 26, 76–80 CrossRef CAS.
- A. Paneque, E. Reguera, J. Fernández-Bertrán and H. Yee-Madeira, J. Fluorine Chem., 2002, 113, 1–5 CrossRef CAS.
- A. Orita, L. S. Jiang, T. Nakano, N. C. Ma and J. Otera, Chem. Commun., 2002, 1362–1363 RSC.
- S. V. Kolotilov, A. W. Addison, S. Trofimenko, W. Dougherty and V. V. Pavlishchuk, Inorg. Chem. Commun., 2004, 7, 485–488 CrossRef CAS.
- V. D. Makhaev, A. P. Borisov and L. A. Petrova, J. Organomet. Chem., 1999, 590, 222–226 CrossRef CAS.
- M. N. Sokolov, A. L. Gushchin, S. V. Tkachev, D. Y. Naumov, P. Nuñez, P. Gili, J. G. Platas and V. P. Fedin, Inorg. Chim. Acta, 2005, 358, 2371–2383 CrossRef CAS.
- M. N. Sokolov, A. L. Gushchin, D. Y. Naumov, O. A. Gerasko and V. P. Fedin, Inorg. Chem., 2005, 44, 2431–2436 CrossRef CAS.
- V. P. Fedin, M. N. Sokolov, K. G. Myakishev, O. A. Geras'ko, V. Y. Fedorov and J. Macièek, Polyhedron, 1991, 10, 1311–1317 CrossRef CAS.
- K. Nakajima, M. Kojima, S. Azuma, R. Kasahara, M. Tsuchimoto, Y. Kubozono, H. Maeda, S. Kashino, S. Ohba, Y. Yoshikawa and J. Fujita, Bull. Chem. Soc. Jpn., 1996, 69, 3207–3216 CrossRef CAS.
- H.-K. Liu, T.-H. Tsao, C.-H. Lin and V. Zima, CrystEngComm, 2010, 12, 1044 RSC.
- X.-Q. Xin and L.-M. Zheng, J. Solid State Chem., 1993, 106, 451–460 CrossRef CAS.
- J. Chen, D. He, Y. Di, Y. Kong, W. Yang, W. Dan and Z. Tan, Chin. J. Chem., 2009, 27, 1675–1681 CrossRef CAS.
- Y.-Y. Di, Y.-X. Kong, W.-W. Yang, L. Sun and Z.-C. Tan, J. Chem. Eng. Data, 2008, 53, 2777–2782 CrossRef CAS.
- Y.-Y. Di, Y.-P. Hong, Y.-X. Kong, W.-W. Yang and Z.-C. Tan, J. Chem. Thermodyn., 2009, 41, 80–83 CrossRef CAS.
- Y. Kong, Y. Di, W. Yang, G. Zhao, K. Zhang and Z. Tan, Z. Phys. Chem., 2009, 223, 675–688 CrossRef CAS.
- J. Yoshida, S.-i. Nishikiori and R. Kuroda, Chem.–Eur. J., 2008, 14, 10570 CrossRef CAS.
- G. Scholz and E. Kemnitz, Solid State Sci., 2009, 11, 676–682 CrossRef CAS.
- Y.-A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef CAS.
- Alan L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641–2644 CrossRef CAS.
- V. V. Volkov and K. G. Myakishev, Inorg. Chim. Acta, 1999, 289, 51–57 CrossRef CAS.
- W. E. Reid Jr., J. M. Bish and A. Brenner, J. Electrochem. Soc., 1957, 104, 21–29 CrossRef.
- M. Mamatha, B. Bogdanovic, M. Felderhoff, A. Pommerin, W. Schmidt, F. Schüth and C. Weidenthaler, J. Alloys Compd., 2006, 407, 78–86 CrossRef CAS.
- V. Chandrasekhar, V. Baskar, R. Boomishankar, K. Gopal, S. Zacchini, J. F. Bickley and A. Steiner, Organometallics, 2003, 22, 3710–3716 CrossRef CAS.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002 CrossRef CAS.
- R. Kuroda, J. Yoshida, A. Nakamura and S.-i. Nishikiori, CrystEngComm, 2009, 11, 427 RSC.
- S. A. Bourne, M. Kilkenny and L. R. Nassimbeni, J. Chem. Soc., Dalton Trans., 2001, 1176 RSC.
- D. Braga, S. L. Giaffreda, F. Grepioni and M. Polito, CrystEngComm, 2004, 6, 458 RSC.
- C. J. Adams, H. M. Colquhoun, P. C. Crawford, M. Lusi and A. G. Orpen, Angew. Chem., Int. Ed., 2007, 46, 1124 CrossRef CAS.
- D. Braga, M. Curzi, A. Johansson, M. Polito, K. Rubini and F. Grepioni, Angew. Chem., Int. Ed., 2006, 45, 142 CrossRef CAS.
- V. Štrukil, L. Fábián, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksić and T. Friščić, Chem. Commun., 2010, 46, 9191 RSC.
- W. J. Belcher, C. A. Longstaff, M. R. Neckenig and J. W. Steed, Chem. Commun., 2002, 1602 RSC.
- D. Braga, M. Curzi, F. Grepion and M. Polito, Chem. Commun., 2005, 2915 RSC.
- F. Hohn, H. Billetter, I. Pantenburg and U. Ruschewitz, Z. Naturforsch. B Chem. Sci., 2002, 1375–1381 CAS; F. Hohn, H. Billetter, I. Pantenburg and U. Ruschewitz, Z. Naturforsch. B Chem. Sci., 2002, 57, 1375 Search PubMed.
- I. Stein and U. Ruschewitz, Z. Anorg. Allg. Chem., 2010, 636, 400 CrossRef CAS.
- (a) O. Angelova, J. Macicek, M. Atanasov and G. Petrov, Inorg. Chem., 1991, 30, 1943 CrossRef CAS; (b) O. Angelova, G. Petrov and J. Macicek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 710 CrossRef.
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790 RSC.
- (a) J. F. Fernández-Bertrán, M. P. Hernández, E. Reguera, H. Yee-Madeira, J. Rodriguez, A. Paneque and J. C. Llopiz, J. Phys. Chem. Solids, 2006, 67, 1612 CrossRef; (b) J. Fernández-Bertrán, L. Castellanos-Serra, H. Yee-Madeira and E. Reguera, J. Solid State Chem., 1999, 147, 561 CrossRef.
- T. Friščić and L. Fábián, CrystEngComm, 2009, 11, 743 RSC.
- (a) W. Xu and Y. Q. Zheng, Z. Kristallogr. NCS, 2004, 219, 235 CAS; (b) H. Z. Xie, Y. Q. Zheng and K. Q. Shou, J. Coord. Chem., 2003, 56, 1291 CrossRef CAS.
- F. C. Strobridge, N. Judaš and T. Friščić, CrystEngComm, 2010, 12, 2409 RSC.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839 RSC.
- B.-Q. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912 CrossRef CAS.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712 Search PubMed.
- (a) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS; (b) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS.
- (a) W. Yuan, A. Lazuen-Garay, A. Pichon, R. Clowes, C. D. Wood, A. I. Cooper and S. L. James, CrystEngComm, 2010, 12, 4063 RSC; (b) M. Edgar, R. Mitchell, A. M. Z. Slawin, P. Lightfoot and P. A. Wright, Chem.–Eur. J., 2001, 7, 5168 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- M. Schlesinger, S. Schulze, M. Hietschold and M. Mehring, Microporous Mesoporous Mater., 2010, 132, 121 CrossRef CAS.
- M. Klimakow, P. Klobes, A. F. Thunemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216 CrossRef CAS.
- W. Jones, C. R. Theocharis, J. M. Thomas and G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1983, 1443 RSC.
- (a) W. Yuan, J. O'Connor and S. L. James, CrystEngComm, 2010, 12, 3515 RSC; (b) C. Daiguebonne, O. Guilloa, Y. Gérault, A. Lecerf and K. Boubekeur, Inorg. Chim. Acta, 1999, 284, 139 CrossRef CAS.
- J. Yang, Q. Yue, G.-D. Li, J.-J. Cao, G.-H. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 2857 CrossRef CAS.
- C. J. Adams, M. F. Haddow, M. Lusi and A. G. Orpen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16033 CrossRef CAS.
- (a) P. Lightfoot, M. Tremayne, K. D. M. Harris and P. G. Bruce, J. Chem. Soc., Chem. Commun., 1992, 1012 RSC; (b) K. D. M. Harris, M. Tremayne, P. Lightfoot and P. G. Bruce, J. Am. Chem. Soc., 1994, 116, 3543 CrossRef CAS; (c) B. M. Kariuki, D. M. S. Zin, M. Tremayne and K. D. M. Harris, Chem. Mater., 1996, 8, 565 CrossRef CAS; (d) R. B. Hammond, K. J. Roberts, R. Docherty and M. Edmondson, J. Phys. Chem. B, 1997, 101, 6532 CrossRef CAS; (e) K. D. M. Harris, M. Tremayne and B. M. Kariuki, Angew. Chem., Int. Ed., 2001, 40, 1626 CrossRef CAS; (f) Y. G. Andreev and P. G. Bruce, J. Phys.: Condens. Matter, 2001, 13, 8245 CrossRef CAS; (g) V. V. Chernyshev, Russ. Chem. Bull., 2001, 50, 2273 CrossRef CAS; (h) Structure Determination from Powder Diffraction Data, OUP/IUCr, ed. W. I. F. David, K. Shankland, L. B. McCusker and C. Baerlocher, 2002 Search PubMed; (i) A. Huq and P. W. Stephens, J. Pharm. Sci., 2003, 92, 244 CrossRef CAS; (j) M. Brunelli, J. P. Wright, G. R. M. Vaughan, A. J. Mora and A. N. Fitch, Angew. Chem., Int. Ed., 2003, 2029 CrossRef CAS; (k) K. D. M. Harris, Cryst. Growth Des., 2003, 3, 887 CrossRef CAS; (l) K. D. M. Harris and E. Y. Cheung, Chem. Soc. Rev., 2004, 33, 526 RSC; (m) M. Tremayne, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2691 CrossRef CAS; (n) V. Favre-Nicolin and R. Z. Černý, Z. Kristallogr., 2004, 219, 847 CrossRef CAS; (o) V. Brodski, R. Peschar and H. Schenk, J. Appl. Crystallogr., 2005, 38, 688 CrossRef CAS; (p) H. Tsue, M. Horiguchi, R. Tamura, K. Fujii and H. Uekusa, J. Synth. Org. Chem. Jpn., 2007, 65, 1203 CrossRef CAS; (q) S. Karki, L. Fabian, T. Friscic and W. Jones, Org. Lett., 2007, 9, 3133 CrossRef CAS; (r) W. I. F. David and K. Shankland, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 52 CrossRef CAS; (s) A. Altomare, R. Caliandro, C. Cuocci, C. Giacovazzo, A. G. G. Moliterni, R. Rizzi and C. Platteau, J. Appl. Crystallogr., 2008, 41, 56 CrossRef CAS; (t) K. D. M. Harris, Mater. Manuf. Processes, 2009, 24, 293 CrossRef CAS.
- K. Fujii, A. Lazuen Garay, J. Hill, E. Sbircea, Z. Pan, M. Xu, D. C. Apperley, S. L. James and K. D. M. Harris, Chem. Commun., 2010, 46, 7572 RSC.
- (a) H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS; (b) R. A. Young, The Rietveld Method IUCr: Oxford, 1993 Search PubMed; (c) L. B. McCusker, R. B. V. Dreele, D. E. Cox, D. Louër and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36 CrossRef CAS.
- G. Oszlányi and A. Sütő, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 123 CrossRef.
- K. D. M. Harris and M. Xu, NMR Crystallography, ed. R. K. Harris, R. Wasylishen and M. J. Duer, John Wiley & Sons, Chichester, 2009, p. 275 Search PubMed.
- (a) M. Tremayne, B. M. Kariuki and K. D. M. Harris, Angew. Chem., Int. Ed. Engl., 1997, 36, 770 CrossRef CAS; (b) S. Meejoo, B. M. Kariuki, S. J. Kitchin, E. Y. Cheung, D. Albesa-Jové and K. D. M. Harris, Helv. Chim. Acta, 2003, 86, 1467 CrossRef CAS; (c) R. K. Harris, P. Y. Ghi, R. B. Hammond, C. Y. Ma and K. J. Roberts, Chem. Commun., 2003, 2834 RSC; (d) D. Albesa-Jové, B. M. Kariuki, S. J. Kitchin, L. Grice, E. Y. Cheung and K. D. M. Harris, ChemPhysChem, 2004, 5, 414 CrossRef; (e) D. H. Brouwer, R. J. Darton, R. E. Morris and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 10365 CrossRef CAS; (f) B. Elena, G. Pintacuda, N. Mifsud and L. Emsley, J. Am. Chem. Soc., 2006, 128, 9555 CrossRef CAS.
- T. Friščić, E. Meštrović, D. Škalec-Šamec, B. Kaitner and L. Fábián, Chem.–Eur. J., 2009, 15, 12644 CrossRef.
- K. Fujii, Y. Ashida, H. Uekusa, S. Hirano, S. Toyota, F. Toda, Z. Pan and K. D. M. Harris, Cryst. Growth Des., 2009, 9, 1201 CAS.
- K. Alfonsi, J. Colberg, P. Dunn, T. Fevig, S. Jennings, T. Johnson, H. Kleine, C. Knight, M. Nagy, D. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC.
- (a) R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767–772 RSC; (b) F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894–1899 RSC; (c) R. Thorwirth, F. Bernhardt, A. Stolle, B. Ondruschka and J. Asghari, Chem.–Eur. J., 2010, 16, 13236 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |