Rapid determination of enantiomeric excess: a focus on optical approaches
Diana
Leung
,
Sung Ok
Kang
and
Eric V.
Anslyn
*
Department of Chemistry and Biochemistry, The University of Texas at Austin, 1 University Station A1590 NHB, Austin, Texas 78712, USA. E-mail: anslyn@austin.utexas.edu; Fax: +1 512-471-7791; Tel: +1 512-471-0068
First published on 5th September 2011
Abstract
High-throughput screening (HTS) methods are becoming increasingly essential in discovering chiral catalysts or auxiliaries for asymmetric transformations due to the advent of parallel synthesis and combinatorial chemistry. Both parallel synthesis and combinatorial chemistry can lead to the exploration of a range of structural candidates and reaction conditions as a means to obtain the highest enantiomeric excess (ee) of a desired transformation. One current bottleneck in these approaches to asymmetric reactions is the determination of ee, which has led researchers to explore a wide range of HTS techniques. To be truly high-throughput, it has been proposed that a technique that can analyse a thousand or more samples per day is needed. Many of the current approaches to this goal are based on optical methods because they allow for a rapid determination of ee due to quick data collection and their parallel analysis capabilities. In this critical review these techniques are reviewed with a discussion of their respective advantages and drawbacks, and with a contrast to chromatographic methods (180 references).
 Diana Leung | Diana Leung obtained her BS in chemistry with honours from Binghamton University, while working in Professor Omowunmi A. Sadik's group. She then earned her PhD in Organic Chemistry at the University of Texas at Austin under the supervision of Professor Eric V. Anslyn, working on developing assays for rapid determination of enantiomeric excess using optical signalling techniques. Currently, Diana is a postdoctoral researcher in Professor Christopher N. Bowman's group at the University of Colorado at Boulder. She is working on developing novel monomers for photopolymerizable crosslinked networks to alleviate volumetric shrinkage stress. |
 Sung Ok Kang | Sung Ok Kang was born in Gwangju, South Korea. She studied chemistry at the Chonnam National University where she obtained bachelor, masters and PhD degrees. She conducted MS, and PhD research in supramolecular chemistry with Professor Kye Chun Nam. She has held postdoctoral appointments with Professor Kristin Bowman-James at the University of Kansas (March 2002–September 2009), with Professor Eric V. Anslyn at the University of Texas at Austin (October 2009–January 2011), and currently with Dr Benjamin P. Hay at the Oak Ridge National Laboratory (February 2011–present). |
 Eric V. Anslyn | Eric Anslyn is the Norman Hackerman Professor of Chemistry and Biochemistry, and is a University Distinguished Teaching Professor, at the University of Texas at Austin. He attended the California State University Northridge to receive his BS degree in Chemistry. He performed doctoral research at the California Institute of Technology under the direction of Dr Robert Grubbs, and received his PhD in Organic Chemistry in 1987. From that year to 1989 he was a National Science Foundation post-doctoral Associate at Columbia University working with Dr Ronald Breslow. He started his independent career at the University of Texas at Austin in 1989. |
1. Introduction
The goal of asymmetric synthesis is to obtain one enantiomer preferentially over the other, commonly via the use of chiral catalysts/auxiliaries.1,2 Asymmetric synthesis is cost-effective due to the avoidance of labour-intensive, cumbersome, and often time-consuming separation of enantiomers, and because it does not require the wasteful discarding of the undesired enantiomer.3 With an ever-increasing number of chiral drugs on the market,4 these advantages are critical to drug development. In more general terms, the importance and significance of asymmetric synthesis to the scientific community was highlighted by the 2001 Nobel Prize awarded to W. S. Knowles, R. Noyori, and K. B. Sharpless for their work on asymmetric catalysis.5Enantiomeric excess (ee), defined by eqn (1), is commonly used to quantify the efficiency of an asymmetric transformation. Enantiomeric excess values cover a range from −100% to +100%; however, the absolute value of ee is most frequently reported. To quantify ee, the enantiomers must be separated or distinguished, which requires a chiral environment. The use of a chiral environment generates diastereomers that necessarily are different in energy, which enables enantiomer discrimination and quantification.
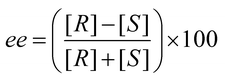 | (1) |
To obtain the highest possible ee, an appropriate chiral catalyst or auxiliary is often used. The discovery of these chiral catalysts/auxiliaries has been traditionally conducted by rational design and iterative trial and error. However, as discussed below, the use of parallel synthesis and/or combinatorial chemistry presents several advantages over traditional rational design. Yet, with these later techniques, the determination of ee values represents a major bottleneck to reaction discovery. The reason is the serial and time-consuming nature of the common chromatographic techniques used to measure ee values. Hence, high-throughput screening (HTS) techniques are becoming increasingly essential if the power of parallel synthesis and combinatorial chemistry are truly to be realized.
There have been a few reviews on determining ee in a high-throughput fashion. These include reviews by Tsukamoto and Kagan on advances in determining ee,6 Reetz on the use of evolutionary mutagenesis for producing large combinatorial libraries and determining ee using kinetics,7 Traverse and Snapper on uses of methods of screening for asymmetric catalysts,4 Finn on emerging methods in determining ee,8 Pu on the use of fluorescent molecules for chiral recognition,9 and a short review by Charbonneau and Ogilvie on methods to screen for asymmetric synthesis.10 In contrast, this review focuses on methods based on optical signalling that have the potential for determining ee in a high-throughput workflow, along with a contrast to chromatographic approaches. Due to the limited space available, only optical techniques are discussed. Nevertheless, we would like to point the readers to results obtained from mass spectrometric techniques that can be used to determine enantiomeric excess.11–15
1.1 Traditional asymmetric synthesis design
As briefly introduced above, rational design is the traditional approach to asymmetric synthesis, whereby a single system (catalyst, auxiliary, other chiral environment) is designed and tested for reactivity and selectivity. After testing, an improved generation is designed, prepared and tested again, and the cycle continues until a system capable of producing the desired ee is obtained (Fig. 1a). The design principles used are typically based on specific structural and electronic properties, which rely heavily on mechanistic data, molecular modelling, and/or blind trial-and-error. Usually a family of compounds is identified as potential chiral catalysts or auxiliaries, since there are only limited methods that identify systems that will give the desired activity and selectivity in advance.16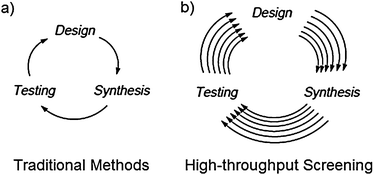 | ||
| Fig. 1 Screening of asymmetric catalysts/auxiliaries by: (a) traditional methods and (b) high-throughput screening. | ||
Most research groups involved in asymmetric reaction discovery use traditional rational design. An example of success comes from Crabtree's search for a chiral catalyst for asymmetric hydrogenation of olefins. He started with a chiral ferrocene ligand which had been used for the industrial asymmetric synthesis of the agrochemical (S)-metolachlor (1), and iridium due to earlier work conducted by Schrock and Osborn with rhodium.17 His group spent two years searching, synthesizing, and characterizing a dozen of different iridium compounds, before achieving satisfactory results.18
In most cases, the activity of a catalyst is affected by several experimental conditions: temperature, solvent, substrate, precursor, co-solvent(s), pressure, pH, reaction time, equivalences, etc., and each requires time for analysis. Hence, although traditional methods are successful, they can be time-consuming due to the iterative creation and testing of many catalysts and conditions. Hence, although iterative rational design is still at the forefront of asymmetric synthesis, the drawbacks have led to the exploration of other approaches, such as parallel synthesis and combinatorial chemistry.7,17,19–23 These methods allow for this same kind of iterative optimization to be performed on larger pools of catalysts/auxiliaries (Fig. 1b). As stated so elegantly by Snapper et al., traditional methods based on a prior mechanistic bias has had less success compared to combinatorial methods within the same time span.19
1.2 Combinatorial chemistry and parallel synthesis for asymmetric catalyst discovery
In the 1990's combinatorial chemistry emerged as a tool for the creation of catalysts due to technological advances that allowed the synthesis of large libraries of compounds. A combinatorial library is usually composed of highly analogous compounds that are rapidly synthesized because similar reaction conditions are used, allowing for the synthesis to be conducted in a parallel fashion. Further, a limited number of reaction vessels can be used in the split-and-pool method.24The screening of a library of catalysts makes a serendipitous discovery more likely than when using traditional rational design due to the large number of catalysts examined. In addition, failures are less disappointing and less disruptive, because of the speed at which the synthesis is conducted. Further, because traditional rational design commonly depends on assumptions about the reaction mechanism, the chemists risk missing unexpected competing mechanisms that may serendipitously arise when a large number of variables are changed in parallel. Hence, combinatorial methods embodied by parallel and split-and-pool techniques allow for more risk, while also lowering the time involved in exploration.
 | ||
| Scheme 1 Model system used to screen for metal ligands for asymmetric epoxidation of olefins. | ||
The split and pool method can quickly lead to thousands of compounds. To reduce the number of analyses, smaller pools of the catalysts/auxiliaries are commonly tested. But such pooling can miss a possible “hit.” A pool may contain two catalysts/auxiliaries producing active hits, but have opposite enantioselectivity thereby producing a racemic mixture. Another disadvantage is an excellent catalyst producing high ee values can be overwhelmed by a majority of low selectivity catalysts. Of course, such drawbacks could be avoided if each nm catalyst was screened individually, but this requires a truly HTS technique for determining ee values. However, such drawbacks have also led many scientists to turn to parallel synthesis, rather than split-and-pool methods, as a second alternative to traditional rational design and testing of chiral catalysts/auxiliaries.
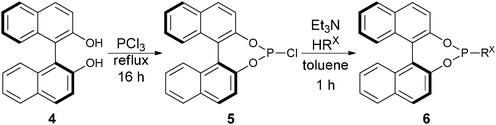 | ||
| Scheme 2 Synthetic scheme for BINOL-based phosphoramidite ligands. | ||
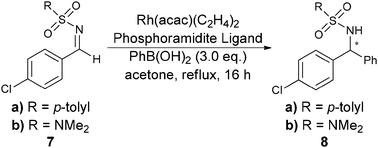 | ||
| Scheme 3 Rhodium-catalyzed asymmetric addition of arylboronic acids. | ||
While parallel synthesis allows for new catalysts to be synthesized and modified simultaneously, it also allows for the optimization of experimental conditions (temperature, solvent, co-solvent(s), pH, reaction time, pressure, equivalences, etc.).27 These factors are otherwise typically unexplored, due to the number of individual reactions that would have to be conducted. The exploration of these parameters has been shown to often be critical due to an interplay that each factor can have on the other, as Minnaard et al.26 and Burgess et al.28 demonstrated recently. However, a week was required to analyse 96 samples. Further, automated parallel synthesis commonly works on small scales, thus reducing costs and waste production.17,19–23,29–33 Despite all these advantages, even parallel synthesis is limited by an inability to determine the ee of samples as quickly as they are created, due to the sheer number of samples/reactions that can be produced.
Hence, both parallel synthesis and combinatorial chemistry approaches are capable of creating more reaction products than can be analysed with the common approaches to ee testing. The methods of ee analysis currently available just don't afford a truly high-throughput workflow. Due to this bottleneck in the workflow, researchers currently restrict the scope of their combinatorial libraries and parallel synthesis routines to reduce the number of samples that ultimately require analysis. Because Reetz has defined a truly high-throughput workflow to be many thousands of samples per day, or more,7 approaches to HTS are targeting this value as a benchmark for success. With this in mind, we review below the current state of the art in the field of optical analysis, starting with a brief overview of the efforts involving the more traditional chromatographic techniques. In addition, the discussion of the convenience of setting up the assay for each specific targeted analyte will be discussed, as well as what type of work-up (if any) of reaction mixtures are required for the implementation of each technique. These are also important factors to consider in addition to the time of analysis.
2. Chromatographic methods
Chromatography has been used for enantiomer separations for decades, and for quantification of ee. Currently, the most common methods used to attempt HTS of ee values are chiral high-performance liquid chromatography (HPLC),34 HPLC coupled with circular dichroism (HPLC-CD), chiral gas-chromatography (GC), or capillary electrophoresis (CE). The main reasons that chromatographic techniques are being explored for HTS are the accuracy they afford, their historical use, and their ability to be automated.There are two chromatographic techniques used to separate enantiomers: direct or indirect. In direct chiral separation, chiral selectors are employed in the mobile phase, and/or stationary phase, that form diastereomeric reversible interactions with the analytes. With indirect chiral separation, chiral derivatizing reagents are used to form diastereomeric complexes with the analytes.35 Direct chiral separation is more commonly used to avoid the extra derivatization step and the additional cost of chiral selectors used by indirect chiral separation. When the chiral selector is loaded on a stationary phase, it is known as a chiral stationary phase (CSP), or more commonly as a chiral column. A number of classes of CSPs exist, such as Pirkle-type (π-complex), polysaccharide derivatives, macrocyclic-type (cyclodextrin, crown ethers, etc.), ligand exchange, proteins, and other polymeric types.35–38 By the very nature of an enantioselective separation, there is no single column that can enantioselectively separate all chiral analytes.37 A CSP must be found and optimized for each specific class of analyte.38 Chiral HPLC columns can often be quite expensive, and they can lose their resolving power over time.39
2.1 HPLC in HTS
A drawback of using chiral HPLC in HTS is the inability to conduct fast parallel analyses, due to it being inherently a serial method. A fairly rapid chiral HPLC analysis requires ten minutes,4 which results in around 144 samples analysed per day, if the HPLC is continuously running. This estimate does not account for any re-equilibration of the solvent system, or the reloading of solvents. Such a time frame does not allow the analysis of thousands of samples that could potentially be generated with combinatorial methods.40 In addition, the injection of crude reaction mixtures is often not possible because some solvents or reagents irreversibly contaminate the column. Hence, sample pre-treatment is commonly necessary.41Recently, chiral HPLC has seen many improvements in the speed of analysis. One is the use of a multiplexing (Fig. 2), which is composed of parallel columns with individual pumps, and detectors. For example, Welch has used eight parallel columns with individual pumps, and individual diode array UV-vis detectors for the quantification of the ee of benzoin. Samples were automatically injected from a microtiter plate, and smaller-bore columns were used to reduce the time and solvent used. 96 samples were analysed in 1.5 to 2 hours, whereas a conventional HPLC would have taken 16 hours.34,41,42
 | ||
| Fig. 2 A multiplex HPLC. | ||
Welch has also been a leader in the use of supercritical fluid chromatography (SFC) for ee analysis. SFC uses a compressible gas, typically carbon dioxide, as the mobile phase in addition to an organic solvent. A chiral selector is still required. The gas is kept at critical temperature and pressure. The main advantage of SFC to HPLC is the increased speed of analysis due to the lower viscosity and higher diffusivity than standard solvents, which allows for higher flow rates with lower column pressures. SFC can reduce a 15 minute HPLC run to 3 minutes, but the resolution can suffer.43
2.2 GC
An alternative chromatographic technique for the rapid determination of ee is the use of chiral gas chromatography (GC). The typical CSPs used with GC rely on inclusion phenomena, coordination, and hydrogen bonding interactions.35,44–46 The advantage of GC over HPLC is the high efficiency, sensitivity, reproducibility, and because the mobile phase is a gas, the speed of analysis is generally much faster. However, GC is limited in the scope of analytes due to the necessity to vaporize analytes for analysis. This method can only be used on compounds that are volatile and thermally stable. GC also has the disadvantage that the CSPs degrade or racemize more quickly due to the temperature at which these experiments are conducted.37 Lastly, it is a serial technique. Some scientists have devised ways to circumvent this limitation. For example, Reetz et al. configured a GC with two injectors, two columns in one oven, and two detectors, such that two samples could be analysed at once.47 To accommodate larger number of samples for analysis, extra columns and detectors will be required.2.3 HPLC-CD
An HPLC coupled with a circular dichroism (HPLC-CD) spectrometer is an alternative chromatographic technique used to quantify ee. Because using CSPs does not always successfully separate enantiomers, CD is used where the chiral environment is provided by circularly polarized light.48–50 To use HPLC-coupled CD a chromophore is required in the analyte, and it must be in intimate electronic contact with the stereocentre(s) to produce a CD signal. Analytes that do not produce a strong enough CD signal or do not contain a chromophore are typically derivatized to amplify or generate a signal.51 Importantly, if CD and absorbance spectra are both taken, the CD response can be linearly related to the ee via the anisotropy factor (g). Anisotropy (g) is defined as the ratio of circular dichroism (Δε) to absorption (ε). The speed of analysis depends on the retention time of the analytes due to the nature of the technique being chromatographic.39,52 Reetz et al. explored the ability of HPLC-CD in a HT analysis that allowed for 700–900 samples a day with a JASCO-CD 1595 instrument. A robotic autosampler on an HPLC was used to purify and determine the ee of alcohol 9 through a correlation of anisotropy (g) to ee.53
2.4 Capillary electrophoresis
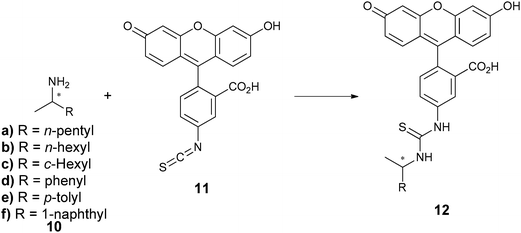
Capillary electrophoresis (CE) is a separation method based on the differential rate of migration of a charged species in a capillary tube. A dc voltage is applied across the capillary tube that leads to a buffer flow and a corresponding migration of analytes dependent on their size and charge. The separation of two enantiomers can be accomplished by the addition of a chiral selector.54–56 There are numerous types of chiral selectors which largely consist of charged cyclodextrins and polysaccharides.57–62The use of capillary array electrophoresis (CAE) with 96 capillaries in parallel has been implemented by Reetz et al. for the analysis of chiral amines (10).63 The amines were derivatized with fluorescein isothiocyanate (11) to create thiourea derivatives (12) for optical detection (Scheme 4), and γ-cyclodextrin was used as the chiral selector. More than 7000 samples per day were analysed with laser-induced fluorescence with excellent baseline resolution (Fig. 3). The authors suggest that the analysis of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 ee samples a day is possible.63
000–30000 ee samples a day is possible.63
 | ||
| Scheme 4 Derivatization of chiral amines for the determination of ee by CAE. | ||
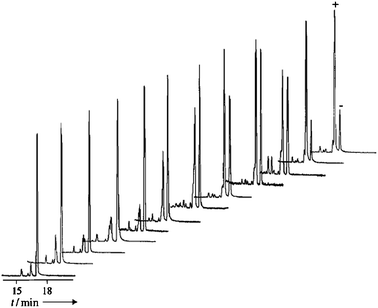 | ||
| Fig. 3 Capillary array electrophoresis separation of derivatized amines (12) at different ee values (12 samples).63 | ||
There are many advantages to using CE for ee analysis. One is the high resolution and minimal sample diffusion. Further, CE uses smaller quantities of chiral selectors and solvents compared to HPLC. Further, CAE is fast and works well with parallel screening in a high-throughput format. A drawback of CE is the requirement that the analyte be ionisable in the analysis buffer. In addition, the analyte must be optically detectable or derivatized to be detected.63
2.5 Summary
By definition, chromatographic techniques separate compounds with associated elution times. They are inherently serial analyses in contrast to the parallel synthesis methods that require their use, thus making them limited to the number of samples that can be analysed. The majority of the advances in the field have been multiplexing, but even when multiplexed, the throughput still depends upon elution time. The fastest methods, such as CE, have limitations on the applicable structures. Hence, there is still an overarching need to develop methods that conduct analyses in parallel.3. Liquid crystals
Nematic and cholesteric liquid crystals64,65 (LCs) have been used for determining ee via visual detection and by spectroscopic measurements. Nematic phases are composed of achiral molecules, where addition of a chiral dopant to a nematic phase generates a chiral nematic phase, more commonly known as a cholesteric phase. In the cases described below, the chiral dopant is the analyte being targeted. The optical properties of the LC are dependent on the ee of the chiral dopant, and the measurement of these properties allows for the determination of ee. Two approaches to the use of liquid crystals for the determination of ee have been explored. In the first, the analyte resembles the achiral mesogenic unit to induce the desirable optical properties, and the second uses ferroelectric liquid crystals.3.1 Derivatization
When a chiral dopant that resembles the mesogenic unit is added to a LC, a helical twist is imparted to the natural layers of the LC (Fig. 4). The helical twist is measured by the pitch (p), which is the distance between the mesogens undergoing a full 360° twist. The pitch is dependent on the concentration and helical twisting power (β) of the dopant, and the ee of the dopant is given by eqn (2). The reflection at a specific wavelength (λ) is dependent on the angle of the incident light (α) relative to the normal of the surface and on the average refractive index of the material (n) (eqn (3)). If one knows the refractive index of the material (n), the ee of the chiral dopant can be measured by determining the wavelength of the reflected light (eqn (4)).| p = (cβee)−1 | (2) |
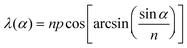 | (3) |
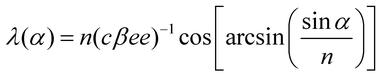 | (4) |
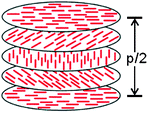 | ||
| Fig. 4 Diagram of liquid crystal film. | ||
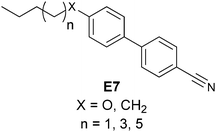
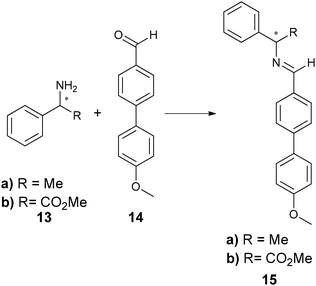
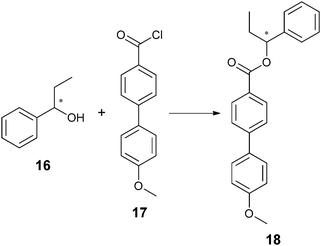
Most analytes of asymmetric reactions do not produce a powerful enough helical twist when used directly as the chiral dopant. To solve this problem, two approaches exist. One is post-reaction derivatization of the analyte to give it mesogenic character. Another is derivatization of the starting material to make it resemble the mesogenic unit prior to an asymmetric reaction. Feringa has exploited both approaches to utilize LCs. He has created systems where the reflected light is within the visible spectrum (360–700 nm) allowing for HTS via “naked eye” detection. He has also modified a microscope to determine λ(α) for the quantification of ee.66–68 For example, when using LC E7, chiral amines (13a) and amino esters (13b) were derivatized by condensation with aldehyde 14 (Scheme 5) to produce imine 15. Chiral alcohol (16) was also targeted by derivatization with acyl chloride 17 to give ester 18 (Scheme 6). As shown, the derivatized analytes 15 and 18 resemble E7, and therefore led to helical twisting that produced pitch angles significant enough to be indicative of differing ee values.
 | ||
| Scheme 5 Derivatization of chiral amines and amino acid esters with mesogenic units. | ||
 | ||
| Scheme 6 Derivatization of chiral alcohol with mesogenic unit. | ||
Upon addition of 10–20% w/w concentrations of 15a to a nematic LC of E7 there was a visible colour change, where addition of 100% ee of 15a produced a violet colour and 50% ee was red (Fig. 5a). Similarly, the colour change upon addition of 18 to E7 ranged from violet to red between 100%–50% ee (Fig. 5b), and calibration curves in this range were generated (Fig. 6a). However, when using 15b as the chiral dopant, a colour change from violet to red was observed on varying the ee from 100% (R) to 100% (S) (Fig. 6b).67
 | ||
| Fig. 5 Colour of the reflected light from (λ(0°)) of doped LC samples with different enantiomeric excesses of: (a) 15a. (b) 18.66 | ||
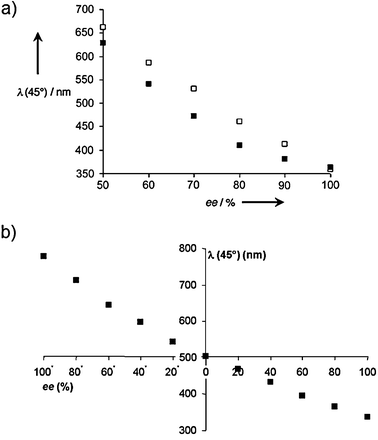 | ||
| Fig. 6 Wavelength of the maximum reflection (λ(31°)) versus the enantiomeric excess for: (a) imine 15a (■) and ester 18 (□), (b) imine 15b.66,67 | ||
Feringa has also explored the derivatization of the starting material.68 This method removes the derivatization step after the parallel asymmetric synthesis, thereby focusing the derivatization effort on only one molecule that can be prepared in bulk, since scientists typically screen only one specific substrate when focusing on a single reaction. Conversely, derivatization of the products of each asymmetric transformation would require individual reactions. He examined a copper-catalyzed asymmetric conjugate addition of diethylzinc to chalcone 19 (Scheme 7) using 21 (Scheme 8). Compound 22, which mimics product 20, induced helical twist films of E7. Upon addition of 22 with varying ee to the LC, blue to deep red colours were produced (Fig. 7). Again, an ee calibration curve could be generated by measuring a reflected wavelength upon addition of 22.
 | ||
| Scheme 7 Copper catalysed asymmetric conjugate addition of diethyl zinc to chalcone (19). | ||
 | ||
| Scheme 8 Asymmetric conjugate addition of diethyl zinc to structure 21, to resemble a mesogenic unit of LC E7 and substrate chalcone (19). | ||
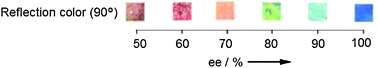 | ||
| Fig. 7 Colour change (90°) observed upon addition of different enantiomeric ratios of 22 to E7 LC.68 | ||
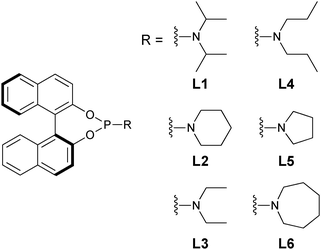
To test the performance of this second approach, six chiral phosphoramidite ligands (L1–L6) were examined in the conjugate addition. The ee of the products could be determined after the reaction mixture was quenched and filtered to remove the zinc and copper salts. The ee was roughly estimated by visual inspection, but then measured by the use of the calibration curve. The values were compared with those obtained by chiral HPLC analysis. The use of visual inspection had a difference of ≤5% compared to chiral HPLC analysis, while the use of calibration curves and reflection wavelength measurements showed excellent correlation.
As demonstrated from these studies, it is possible to quantify ee values using LCs, and the method could be rapid if the proper instrumentation were available. The disadvantage is the extra derivatization steps, which are not optimal for HTS. Even though the second approach requires derivatization of the substrate only, the substrate used in the screening process could be different in reactivity and enantioselectivity relative to the real substrate. For instance, derivatized substrate 21 had solubility differences compared to the original substrate 19, which forced the use of a different solvent in the screening process.
3.2 Non-derivatization
An alternative approach using LCs was demonstrated by Walba et al.69 By leveraging currently established technology in the field of LC displays, Walba et al. used a racemic mixture of a chiral mesogenic unit that is known to give a ferroelectric LC. Upon exposure to an electric field and the addition of a small amount of a chiral dopant (1% w/w), the LC showed electrooptical activity. This means that the LC rotates plane-polarized light, and the degree of rotation depends on the ee of the chiral dopant. Using display technology the LCs can be organised into a matrix of pixels, thereby allowing a very high number of concurrent ee determinations in a truly parallel fashion.In specific, Walba et al. used mesogenic unit 23 for the measurement of the ee of the commercially available drugs naproxen (24) and pseudoephedrine (25). A polarized light microscope was used to measure the rotation of the plane-polarized light at different ee values. Using calibration curves, samples of 24 with unknown enantiomeric purity were analysed, and errors of about ±5% were obtained.
A major advantage of this method is that it does not require derivatization of the analytes or substrates. In addition, the use of a small amount (1% w/w) of dopant/analyte makes this system readily applicable to small-scale HTS of asymmetric catalysts. Finally, the matrix organization is attractive for a truly parallel determination, even though it requires specialized instrumentation to carry out the measurements.
4. Infrared thermography
The use of photovoltaic infrared (IR) cameras with focal-plane array detectors has become popular for monitoring the reactivity of parallel reactions, as pioneered by Maier et al.70 and Willson et al.,71 and explored by others, such as Taylor and Morken.72 The images give a two-dimensional representation of heat emitted during reactions, where the IR radiation is codified by a colour of the image: red is a “hot spot” and blue is a “cold spot.” Hence, to monitor ee values, samples of unknowns undergo reactions that generate heat. A reactive chiral selector (often an asymmetric catalyst) is added to individual enantiomerically pure and racemic mixtures of the analytes as reference values, and the IR images are recorded. The enantiomeric purity of an unknown is then determined by monitoring its reactivity with the same chiral selector.For example, Reetz et al. studied a lipase-catalyzed enantioselective acetylation of a chiral alcohol 9 as a test reaction (Scheme 9).73 A microwell plate was prepared, containing three columns of substrate with differing enantiomeric purity, racemic-9, (S)-9, and (R)-9, with increasing concentrations down the columns (Fig. 8). The lipase Candida antarctica was added to the wells, which selectively acetylates (R)-9 to produce (R)-26 (Scheme 9), leaving (S)-9 unreacted, allowing differentiation between the two enantiomers. A “hot spot” appeared for wells containing (R)-9 after 0.5 min (Fig. 8). Wells containing (S)-9 had no change, and wells containing racemic 9 had a slight rise in temperature.
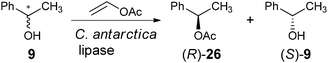 | ||
| Scheme 9 Lipase-catalysed enantioselective acetylation of chiral alcohol. | ||
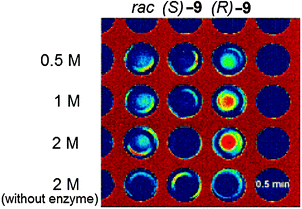 | ||
| Fig. 8 Infrared image of acetylation reaction after 0.5 minutes.73 | ||
Mahmoudian et al. have analysed the same reaction74 in a 96-well plate format (Scheme 9), however, in their study calibration curves were generated. The IR images were recorded before (Fig. 9a) and 60 seconds after the addition of vinyl acetate that starts the reaction (Fig. 9b), and showed elevated heat outputs for racemic-9 and (R)-9, but not for (S)-9. The heat output was measured as a function of time (Fig. 10), and the area under the curve starting at 9 seconds to the maximum temperature rise (117 s) was used to quantify the ee. A five point calibration curve was generated that correlated ee to the area under these curves.
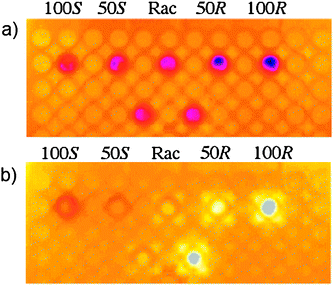 | ||
| Fig. 9 Time-resolved thermal imaging of lipase-catalyzed enantioselective acylation of 1-phenylethanol: (a) before addition of vinyl acetate, (b) 60 seconds after addition of vinyl acetate.74 | ||
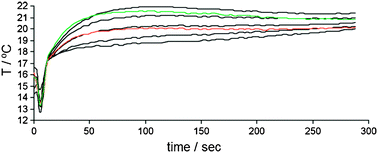 | ||
| Fig. 10 Time-resolved temperature profile for C. antarctica acylation of 1-phenylethanol (9). Black curves are 100% S, 75% S, racemic mixture, 75% R, 100% R. Red and green curves are the racemic 9 and unknown sample of 9, respectively.74 | ||
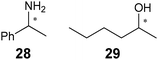
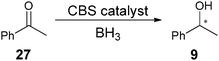
This method was used to determine the ee of the asymmetric reduction of acetophenone (27) using the CBS (Corey–Bakshi–Shibata) oxazaborolidine to produce the unknown sample 9 (Scheme 10). The IR method determined the ee of the sample to be 96%, while chiral HPLC analysis showed 86%. This method was then implemented on other analytes (28 and 29) using the same procedure.
 | ||
| Scheme 10 Reaction for asymmetric reduction of acetophenone (27). | ||
The disadvantage of the IR-thermographic technique is the general inability to quantify ee values, other than Mahmoudian's study.74 Further, some technical issues exist with the method. For example, the uneven dissipation of heat from wells located at the edge vs. at the centre of the plate can give false results.74 Reanalysis of various plates by randomizing the location of wells to achieve reproducibility is necessary. Shaking of the vessel changes the position at which the wells appear in the IR-camera, leading to semicircle-like thermographic emissions near the walls of each well, requiring data to be obtained at the centre only. In addition, the effects of evaporation of solvents or by-products can give skewed results. This later effect was seen in a study conducted by Reetz et al. on ring-closing olefin metathesis, where evolution of ethylene gave false positives.75 These limitations make IR-thermography a difficult technique for reliable rapid determination of ee.
5. Molecularly imprinted polymers
Optical methods that employ the use of molecularly imprinted polymers (MIPs) have also been explored for the determination of ee.76,77 In the creation of MIPs, functional monomers are allowed to self-assemble in solution in the presence of a target molecule to produce a three-dimensional architecture that is then cross-linked to “lock-in” receptor sites with cavities complementary to the target molecules. This makes the method potentially selective for a single enantiomer.Chen and Shimizu78 created MIPs using methacrylic acid (30) and ethylene glycol dimethacrylate (31) in the presence of L-phenylalanine anilide (L-32) (Scheme 11). The L-32 was removed from the MIP by washing with methanol. When a solution containing L-32 was analysed, the MIP retained most of 32 giving a reduction in absorbance of 32, but the opposite was found for samples containing D-32, since it did not bind to the L-MIP (Fig. 11). Different solutions of known ee and concentration were shaken for two hours with the MIP, centrifuged, and the amount of 32 in the supernatant was determined by UV-vis measurements to create a calibration curve (Fig. 12). The accuracy was then tested by determining the ee of unknown samples, resulting in a ±5% standard error compared to chiral HPLC. The advantage of using MIPs is their thermal stability, and tolerance to acidic, basic, and organic treatment due to their highly cross-linked nature. However, the current analytes that have been targeted all contain a chromophore.
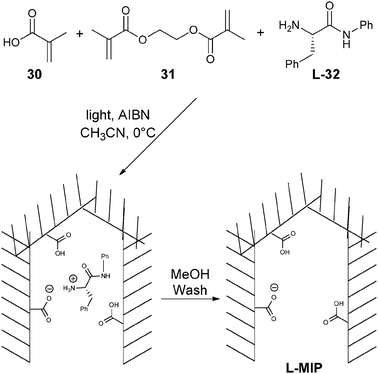 | ||
| Scheme 11 Synthesis of molecular imprinted polymer for enantioselective discrimination of phenylalanine anilide (32). | ||
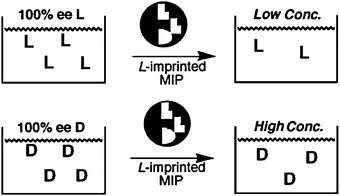 | ||
| Fig. 11 Schematic of using MIP for determining ee of 32.78 | ||
![Correlation of [32] to % ee.78](/image/article/2012/CS/c1cs15135e/c1cs15135e-f12.gif) | ||
| Fig. 12 Correlation of [32] to % ee.78 | ||
6. Enzymatic and antibody methods
The use of enzymes and antibodies is a popular means to determine ee because of their inherent chirality and high selectivity.79–86 Two enzymes or antibodies are commonly used, one being enantioselective thus affording a measurement of the concentration of one enantiomer, while the second binds to both enantiomers giving the total concentration. Also, enzymes or antibodies have been used to target analytes derivatized with a moiety that provides a “handle” for binding.79,82Enzymatic methods for determining enantiomeric excess (EMDee) have been explored by Moberg and Hult et al.87 In their approach a mixture of enantiomers is converted to a mixture of chemically different species with the use of selective enzymatic transformations, thereby making the enantiomers distinguishable. One example involved the dual Lewis acid–Lewis base catalysed addition of acetyl cyanide (33) to benzaldehyde (34) to yield 35 (Scheme 12). Three enzymes were used in this study, and their activities were monitored through UV-vis spectroscopy.
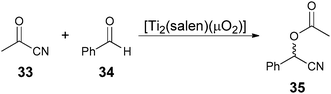 | ||
| Scheme 12 Dual Lewis acid–Lewis base catalysed addition of acetyl cyanide (33) to benzaldehyde (34). | ||
The three enzymes were sequentially added. For example, after the asymmetric reaction, horse liver alcohol dehydrogenase (HLADH) was added, which reduces the unreacted benzaldehyde (34) to phenylmethanol (36) with the co-enzyme NADH (Scheme 13a). Because NADH has an absorbance at 340 nm, while NAD+ does not, the amount of unreacted benzaldehyde could be determined by the decrease in absorbance of NADH. After the reduction of starting material 34, Candida antarctica lipase B (CALB) was added to selectively hydrolyse (S)-acetylated cyanohydrin ((S)-35) to (S)-37, which is in equilibrium with 34. Because HLADH had been previously added, the newly produced 34 will be reduced (Scheme 13b), and decrease [NADH], which can be determined by absorbance measurements at 340 nm, and therefore determines the (S)-35 concentration. The remaining solution contains only (R)-acetylated cyanohydrin ((R)-35), which was then unselectively hydrolysed by the addition of pig liver esterase (PLE). Compound (R)-37 is also in equilibrium with benzaldehyde (34), and the same procedure then determines (R)-35 (Scheme 13c). With the amount of (S)- and (R)-35 determined, the ee can be calculated, and with starting material and product concentration known, percent yield can also be calculated. Equilibration times of 10–50 minutes were required for each addition of enzyme. To demonstrate this system's applicability nine samples with unknown ee values were analysed and compared to a chiral GC analysis, revealing an accuracy of ±10%. This method was then used to analyse a range of O-acylated cyanohydrin samples obtained from addition of ketonitriles to aldehydes. A high-throughput workflow was used, where synthesis and analysis were conducted in a microreactor and microtiter plate, respectively.88 Although the method is a novel advance, the use of three enzymes is problematic due to cost and the associated complicated nature of the technique is not advantageous.
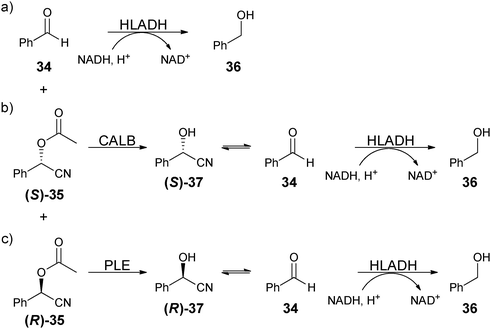 | ||
| Scheme 13 Stepwise reactions used in a EMDee to determine ee of dual Lewis acid–Lewis base catalysed addition of acetyl cyanide to benzaldehyde. | ||
Berkowitz et al. have reported a different approach utilizing enzymes.89,90 In their approach the catalytic reaction under study is coupled in situ with an enzymatic reaction that permits continuous UV-vis spectroscopic monitoring. They called the approach in situ enzymatic screening (ISES). To demonstrate the method they examined [CoIII(salen)] catalysts for the asymmetric hydrolysis of epoxides to create 1,2-diols.
Two enzymes, Thermoanaerobium brockii alcohol dehydrogenase (TBADH) and horse liver alcohol dehydrogenase (HLADH), that have opposite enantioselectivities for the analyte, 1,2-propanediol, were examined in two cuvettes. TBADH reacts with (R)-1,2-propanediol to produce 1-hydroxypropan-2-one, whereas HLADH reacts with (S)-1,2-propanediol to produce (S)-2-hydroxypropanal (Scheme 14). The asymmetric reaction was carried out in a lower organic layer, while the upper aqueous layer contained one of the two enzymes: cuvette 1, TBADH/NADP+; cuvette 2, HLADH/NAD+ (Scheme 14). While the asymmetric reaction in the lower organic layer occurs, the product interacts with the enzymes in the upper aqueous layers, where NADPH and NADH in cuvettes 1 and 2 are created, respectively. Both NADPH and NADH absorb at 340 nm, thereby allowing separate determination of the concentrations of (R)- and (S)-1,2-propanediol, respectively, and thereby also ee values.
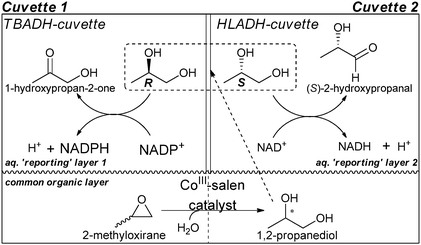 | ||
| Scheme 14 Schematic of a double-cuvette ISES. | ||
Berkowitz et al.89,90 examined CoIII salen ligands that were created from seven chiral diamine scaffolds and seven salicylaldehydes. The ee values were compared to those found from 1H NMR chiral shift experiments. The predictions were within 25% for nine of the ten cases measured. The advantage of ISES is the ability to analyse unknowns without derivatization with a chromophoric unit, while still using an optical signalling technique for rapid analysis. In addition, Berkowitz et al. have developed other ISES methods for diols using Lactobacillus kefir alcohol dehydrogenase (LKADH) and HLADH as enzymes, where both were selective for (S)-alcohol, but to different extents.91 The study was able to analyse alcohols with different chain lengths of alkyl groups.
Competitive enzyme immunoassays (EIA) have also been used for ee analysis. Wagner and Mioskowski et al. used antibodies specific for a product of a particular asymmetric reaction, and immobilized these antibodies in individual wells of a 96-well plate (Fig. 13).92 Subsequently, crude reaction mixtures, enzyme–product conjugates, were added. The products in the crude reaction mixtures compete with the enzyme–product conjugates for binding sites to the antibody. After equilibration, the unbound species are washed off. Then the quantification of enzyme–product conjugates bound to the antibodies is measured by staining and reading the absorbance. The decrease of signal from the stained enzyme–product conjugates when crude was not added compared to when the crude was added will determine the amount of analyte bound to the antibodies. With the use of a specific and non-specific antibody, the ee of the reaction mixture can be determined.
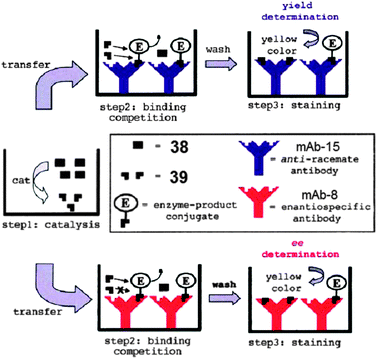
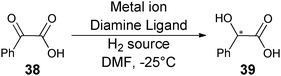
To demonstrate the use of the technique for HTS, 176 catalysts for asymmetric reduction of benzoyl formic acid (38) to mandelic acid (39) (Scheme 15) were examined. Two monoclonal antibodies were used: mAb-15 (anti-racemate antibody) and mAb-8 (enantiospecific antibody). Antibody mAb-15 binds to both (R)- and (S)-39, while mAb-8 binds to (S)-39 preferentially. This allowed for the determination of total concentration of 39 and concentration of (S)-39, respectively. The ee and percent yield of these reactions were determined using calibration curves. Forty-two of the 176 samples were also analysed by HPLC, and the systems accuracy to determine ee was determined to be ±9%. Disadvantages of this method are the long incubation time (12 hours) required with the antibodies, and the necessity to create enzyme–product conjugates.
 | ||
| Scheme 15 Screening different ligands and metals for the symmetric reduction of 38. | ||
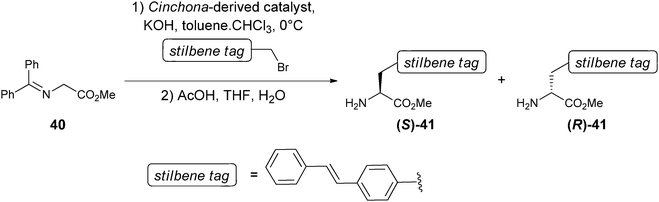
In addition, Matsushita and Janda et al. reported a novel approach to the determination of ee values using antibodies.93,94 In their approach, a fluorophore is conjugated to the product, and/or reactant, of an asymmetric reaction. For example, the monoclonal antibody 19G2 produces a blue fluorescence upon complexation with a trans-stilbene moiety. Because 19G2 is inherently chiral, it can also distinguish enantiomers, such as (R)- and (S)-41, which possess a trans-stilbene tag. Upon binding 19G2 with (S)-41, a blue fluorescence was produced, while binding with (R)-41 showed no fluorescence. An ee calibration curve was generated correlating different ee ratios of 41 with fluorescent measurements using λexc = 327 nm and λem = ∼410 nm.
To demonstrate the system's ability to be used in HTS, 40 Cinchona alkaloid ammonium salts were used as catalysts for the asymmetric synthesis of 41 (Scheme 16). Ten of these reactions were randomly chosen for the determination of their ee values using the calibration curve. The ee values obtained were compared to chiral HPLC analysis, where a variation of ±10% was observed. The sample preparation, plate reading, and catalyst evaluation took less than one hour.
 | ||
| Scheme 16 Chiral ligands asymmetrically synthesized using Cinchona-based phase transfer catalysis. | ||
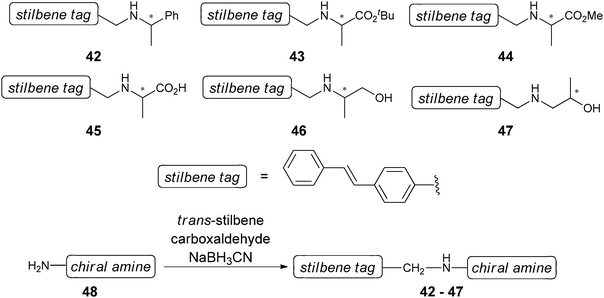
Janda and Matsushita extended the scope of their method to other chiral amines. Introduction of the trans-stilbene moiety by reductive of N-alkylation produces stilbenyzlmethylamines (42–47, Scheme 17). Excellent enantioselectivity by antibody 19G2 for amino alcohols (47) was found, but unfortunately there was low fluorescence for analytes 42–46.
 | ||
| Scheme 17 Synthetic scheme for derivatization of chiral amines with trans-stilbene moiety. | ||
Because there was good discrimination between (R)-47 and (S)-47 by antibody 19G2, the system was used to screen in a 96-well plate a series of Jacobsen's catalysts for the asymmetric synthesis of 47via a catalysed epoxide ring opening (Scheme 18). The ee of 47 was determined using a calibration curve, and showed a variation of <10% when compared to chiral HPLC analysis.
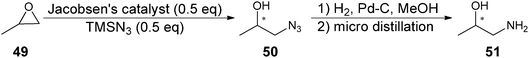 | ||
| Scheme 18 Asymmetric synthesis of 51. | ||
The advantage of their approach is the speed of analysis with the use of fluorescent signals and microwell plate readers. However, this method requires the presence of a trans-stilbene moiety in the analytes. The reactions used to introduce one in the analytes required 10 minutes to 24 hours, with most coming to completion in a couple of hours. In addition to the reaction times, the procedure required extraction and purification steps to obtain the derivatized analytes, but these could ultimately be automated for HTS.
In conclusion, a number of approaches and examples of the use of enzymes and antibodies for the determination of ee values exist. They typically use optical signalling techniques and can be performed in parallel with the use of microwell plates. In addition to the individual disadvantages specified for each approach, the use of enzymes and antibodies as hosts commonly leads the systems to be specific for only one analyte. The techniques therefore require enzymes/antibodies with a reasonable price and effort for each individual analyte.
7. Supramolecular sensors
The methods presented thus far have advanced the field of rapid ee analysis significantly, but each has a specific limitation. The elution times for chromatographic techniques are not favourable, and multiplex methods require specialized instrumentation. The LC methods require derivatization of analytes or substrates. The MIP approach requires polymer synthesis experience and chromogenic analytes. The enzyme and antibody techniques suffer from a lack of generality. Some of these techniques use multi-step protocols or derivatization steps. This is why many research groups have turned to enantioselective molecular sensors using small-molecular artificial hosts, often combined with optical signalling techniques, such that measurements can be conducted rapidly. Below we summarize several approaches, with an examination of specific examples, along with the advantages and disadvantages of each.Optical spectroscopy, such as circular dichroism, fluorescence, and UV-vis absorbance, are well known to be quick. A full spectrum can typically be obtained in less than a minute, whereas optical values at a specific wavelength take seconds or less, potentially allowing for true HTS. Further, these signalling techniques are compatible with microwell plates; the platform of choice for HTS. The ability to use a microwell plate reader is an enormous advantage because they are already present in many parallel and combinatorial synthesis facilities, as are automated liquid dispensers and plate handlers. This provides for a streamlined transition from reaction mixtures to well plates for ee analysis. When using absorbance, the ability for “naked eye” detection is also possible.
Because most of these techniques cannot yet determine ee as accurately as chiral HPLC, their use has been proposed as preliminary screening methods. The technique would identify reactions that produce “good” ee, or trends as a function of experimental conditions. When accuracy is needed, only these samples would be analysed by the more standard chromatography techniques. This would prevent undesirable samples from wasting precious chiral HPLC analysis time. Of course, as the optical methods become more accurate, precise, and user-friendly, these other techniques would become obsolete.
Synthetic receptors have been used as sensors for a variety of different kinds of analytes.95 Typically, the signalling mode is optical, originating from a change in the electronic structure that occurs upon binding between a host and a guest (Scheme 19 and 20). The change in signal allows for the quantification and detection of the analyte (eqn (2), Scheme 20).
 | ||
| Scheme 19 Schematic diagram of a typical molecular sensor with host and guest complexation. | ||
![Equilibrium of molecular sensor. H = host/receptor; G = analyte/guest; [G]t = total guest concentration; KG = binding constant.](/image/article/2012/CS/c1cs15135e/c1cs15135e-s20.gif) | ||
| Scheme 20 Equilibrium of molecular sensor. H = host/receptor; G = analyte/guest; [G]t = total guest concentration; KG = binding constant. | ||
If the synthetic host is chiral, it interacts with chiral analytes to form diastereomers (Scheme 21). Due to the different stabilities of the diastereomers produced, the binding constants (KR and KS) between the chiral host and enantiomeric guests are different (eqn (1) and (2), Scheme 22). This results in a different optical signal for each enantiomer, allowing for the determination of ee (eqn (3), Scheme 22).
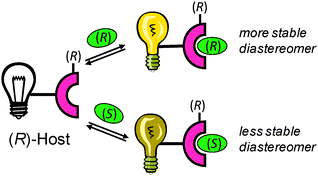 | ||
| Scheme 21 Schematic of discrimination of enantiomers using a chiral host. | ||
![Equilibria of chiral recognition. H* = chiral host/receptor; GR, GS, = analyte/guest; [G]t = total guest concentration; KR, KS = binding constant.](/image/article/2012/CS/c1cs15135e/c1cs15135e-s22.gif) | ||
| Scheme 22 Equilibria of chiral recognition. H* = chiral host/receptor; GR, GS, = analyte/guest; [G]t = total guest concentration; KR, KS = binding constant. | ||
Commonly a chromogenic or fluorogenic unit is incorporated into either the analyte or host in close proximity to the binding site, so that upon binding a change in the spectroscopy is observed. The unit is often synthetically incorporated into the host, not the analytes, so that derivatization after a parallel asymmetric reaction is not required. Supramolecular chemists have explored a number of different host scaffolds, such as crown-ethers, cyclodextrins, calixarenes, 1,1′-bi-2-naphthol (BINOL), and boronic acids. These hosts are readily derivatized to enhance chiral discrimination, and to incorporate a chromogenic unit. Amino acids have been the most common chiral entities used in the hosts to impart chirality due to their availability and variety.
Crown ethers are heterocyclic rings composed of ether groups (52), which have been commonly used for molecular recognition of alkali and alkaline earth cations and amines via interactions with the ether oxygens.96,97 Selectivity can be tuned by the size of the crown ether ring. Cyclodextrins98,99 (53) are highly water soluble and possess a chiral hydrophobic cavity for the inclusion of guest molecules and can be derivatized on the hydroxyl groups at the top or bottom of the macrocycle with a linker/spacer to a fluorophore. Inclusion of the guest into the cyclodextrin cavity leads to an environmental change around an attached fluorophoric unit, thereby signalling the presence of the analyte.100 Calixarenes99,101 are cyclic oligomers of aromatic units, where the para substituents on the phenolic rings define the upper rim, and the lower rim is defined by the phenolic hydroxyl groups (calix[4]arene, 54). Derivatization at the upper and lower rims of calixarenes allows for functionality and incorporation of chirality.102 Calixarenes can be tuned for selectivity by the size of the cavity, and are known to bind ammonium cations through their aromatic cone cavity through cation–π interactions.103,104
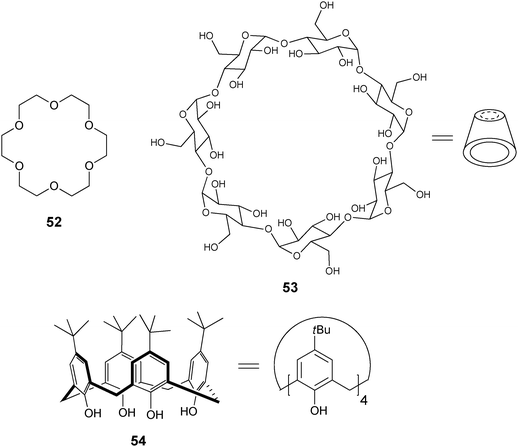
In addition, many groups have used the atropisomers of the easily derivatizable 1,1′-bi-(2-naphthol) (BINOL), for enantioselective discrimination due to the chirogenic unit and the chromophore already present. Kubo has reviewed the use of BINOL as a chromogenic receptor and its use for the colorimetric recognition of amines.105 Boronic acids have also been explored by many groups as a binding moiety due to their ability to reversibly bind saccharides in aqueous media.106 They form reversible covalent bonds with diols, sugars, and α-hydroxycarboxylates, to produce boronate esters.107
The use of synthetic receptors as sensors typically does not require significant equilibration times, work-up for analysis, or specialized equipment. For example, circular dichroism, fluorescence, or UV-vis spectrometers are common. Further, the hosts are typically not very target specific, but are instead primarily functional group selective. This allows for a broader range of analytes to be examined compared to optical techniques based upon antibodies or enzymes.
Of course, there are also disadvantages to the use of synthetic receptors. The first is the effort required in their synthesis. Hence, some researchers in this field, including our group, consciously attempt to minimize the number of steps required to make their receptors. In addition, in some cases the analytes still need to be derivatized to append a group that enhances the binding to the receptor.
Below are several examples of the use of synthetic receptors for the determination of enantiomeric excess, which are divided by the type of signalling paradigm (circular dichroism, fluorescence, and absorbance). The examples are further divided by the host–guest interactions that are being exploited. There are a large number of examples of enantioselective recognition processes studied with synthetic receptors, but due to space constraints, this review is mainly limited to studies that report the measurement of ee. In addition to those, a few systems allowing “naked eye” enantioselective discrimination are also included herein due to their ability to rapidly determine the selectivity of asymmetric reactions. In the fluorescent section, we also limit our coverage to those studies not previously reviewed by Pu.9 Unfortunately, due to space limitations certain fascinating techniques, such as those using fluorescence and colorimetric signalling for identifying activity of enantioselective catalysts, are not covered, since enantiomeric excess or selectivity is not quantified.108–113
7.1 Circular dichroism
When considering optical purity, many chemists think of optical rotation and circular dichroism (CD). The use of optical rotation has the hindrance of requiring concentrated samples, and in HTS of asymmetric reactions the quantity of sample is limited. CD has the advantage of higher sensitivity and does not require the use of a chiral host. In addition, CD can be directly related to ee with the use of anisotropy (g). Exciton coupled circular dichroism (ECCD) is the approach typically used for determining ee.The sign of the Cotton effect is used to determine the relative spatial orientation of the chromophores and thereby the absolute configuration of the analyte. The first Cotton effect (longer wavelength, εr) correlates to the helicity of the system (Fig. 14). A positive Cotton effect (known as P-form, Fig. 14a) reflects the two interacting electronic transitions being oriented in a clockwise orientation, and a negative Cotton effect (known as M-form, Fig. 14b) means they are in a counter clockwise orientation. With the knowledge of the binding mode between the analyte and the host, the CD spectrum can determine the absolute configuration of the analyte and in turn quantify the ee of a sample. For more information on CD refer to the review by Berova et al.50
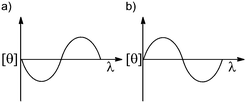 | ||
| Fig. 14 Split Cotton effect: (a) positive Cotton effect, (b) negative Cotton effect. | ||
Most analytes lack a chromophore that can generate a CD signal. This has led chemists to either derivatize the analyte with chromophores, or bind the analyte to chromophores via metal complexation, hydrophobic interactions, electrostatic interactions, or non-covalent interactions. With the advantage of the speedy analysis by a CD spectrophotometer, many groups have created systems exploiting these interactions to produce a signal for the determination of ee.114–123
Metal complexation. Canary exploited ECCD to determine the absolute configuration and ee of amino acids using metal complexation. He derivatized α-amino acids with 2-bromoethyl quinoline, resulting in CuII ligands with a propeller-like twist in their metal complexes 55 (Scheme 23).124 The twist of the structure depends on the configuration at the α-carbon stereocentre. The CuII metal centre binds to the three nitrogens and the carboxylate (Fig. 15) of the ligand, thus locking the spatial orientation of the chromophores. Upon derivatization, amino acids with a L-configuration had a negative Cotton effect, while a D-configuration shows a positive Cotton effect (Fig. 15).
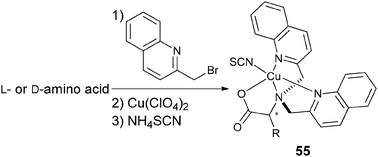 | ||
| Scheme 23 Derivatization scheme of L- or D-α-amino acid. | ||
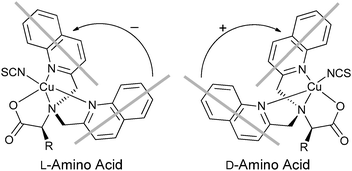 | ||
| Fig. 15 The twist of the propeller-like structure of derivatized L- or D-α-amino acid. | ||
Initially Canary determined the absolute configuration of the amino acids upon derivatization without purification (Scheme 23). However, it was determined that purification of the derivatives was required for the determination of the ee due to an overestimation of the concentration of analyte caused by achiral impurities. Calibration curves for the determination of ee of alanine were generated (Fig. 16), but analysis of unknown samples was not conducted.124 Zahn and Canary conducted similar studies with β-amino alcohols.125 Further, because only the amine functional group is required in the derivatization, the scope of this application can be extended to chiral amines.126
![Using derivatized alanine (Scheme 23) as an analyte, a plot correlating the %ee of [CuII(l/d-55)]ClO4 complex to Δε was generated in methanol at 25 °C at λ = 240 nm.124](/image/article/2012/CS/c1cs15135e/c1cs15135e-f16.gif) | ||
| Fig. 16 Using derivatized alanine (Scheme 23) as an analyte, a plot correlating the %ee of [CuII(L/D-55)]ClO4 complex to Δε was generated in methanol at 25 °C at λ = 240 nm.124 | ||
Hydrophobic interactions. Yashima et al. have shown that ECCD can be used to discriminate the enantiomers of chiral amines using polyacetylene with appended β-cyclodextrin side groups (56, Fig. 17).127 Upon addition of (S)-28 ((1S)-1-phenylethanamine) to host 56 in a H2O
:DMSO mixture (7:3 v/v, buffered at pH 11.7), a colour change from yellow to red resulted (Fig. 18), with a negative first Cotton effect in the CD spectrum. The addition of analyte (R)-28 to host 56 showed no colour change (Fig. 18) and a positive Cotton effect. Other chiral analytes, such as phenylalaninol (57) and 1-phenylethyl alcohol (58), were also tested with this system, but no comparable changes in the CD or absorption spectra were observed.
 | ||
| Fig. 17 Structure of polyacetylene host backbone with β-cyclodextrin as a side group.127 | ||

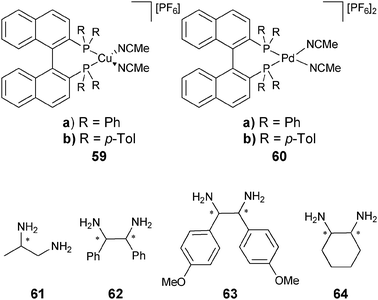
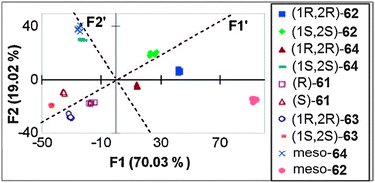
Chemometric techniques, such as linear discriminant analysis (LDA) and artificial neural networks (ANNs), were explored for the interpretation of the experimental results. Using LDA, classification of the diamines (61–64) and their handedness was possible. LDA is a chemometric technique that computes linear functions that maximize the separation between classes and minimize the separation within the classes.130,131 In this case, the identity and handedness of the analytes were determined by their individual clustering (Fig. 19).
An artificial neural network (ANN) was used for the determination of concentration ([G]t) and ee simultaneously. The network correlated the input variables (ellipticities at different wavelengths) to the output variables ([G]t and ee). To demonstrate the system's ability to determine the ee of unknown samples, two samples of 62 were analysed using the ANN and their [G]t and %R were determined, with an average error of 12% and 16% respectively. To explore the systems' ability for HTS of ee, a robotically interfaced 96-well plate CD reader was integrated into the study. Four unknown samples were examined with this assay to determine the [G]t and %R, now with average errors of 8% and 3%, respectively. Using a 96-well plate with 33 wells dedicated to generating the training set for the ANN, the remaining 63 wells could be used for unknown samples. A total of two hours was required to analyse the plate, which correlates to approximately 2 minutes a sample.
In additional studies, racemic mixtures of 59 and 60 were used as hosts, thereby reducing the cost of analysis.128 The racemic mixture was also able to discriminate between the enantiomers of the chiral diamines (61–64), which had equal and opposite CD spectra. The data were analysed by principal component analysis (PCA) to classify chirality, concentration, and identity. PCA is a chemometric technique that classifies and identifies variance in data without prior training.132 The data were then used to train an ANN to determine %R and [G]t, resulting in average errors of 4% and 19%, respectively. This method requires no derivatization of the analytes, and was automated on a 96-well plate CD spectrophotometer, making this system highly compatible with HTS.
In summary, there are a variety of methods for the determination of the absolute configuration and/or ee using circular dichroism with synthetic molecular sensors. As demonstrated by our group, the speed at which analysis can be conducted using a CD spectrophotometer makes the technique applicable to HTS.128,129 Disadvantages are the facts that CD spectrometers are significantly more expensive than UV-vis machines, and commercial 96-well CD plate readers are still extremely rare and even more costly.
7.2 Fluorescence
Fluorescence is another optical technique that has been used to determine ee. There are many advantages, such as its sensitivity, rapid analysis, less expensive instrumentation compared to CD spectrophotometers, and the ability to be used for real-time analysis.9,133,134 Fluorescence has numerous detection modes available, such as intensity, polarization, and anisotropy, all of which can be performed in a steady-state or time-dependent manner. The two most commonly used signalling modes are fluorescent enhancement (a “turn on”) and fluorescence quenching (a “turn off”). The advantage of using a “turn on” signal is its sensitivity because it is carried out against a blank background. In contrast, a “turn off” signal can be too small to detect on top of a high initial signal: i.e. it is easy to spot a single candle being lit in a pitch-dark stadium, while it is almost impossible to see the same candle go off in a stadium full of lit candles.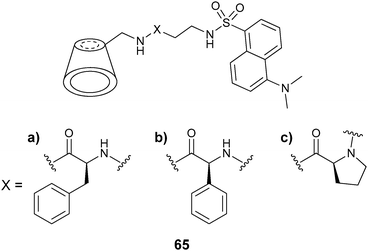
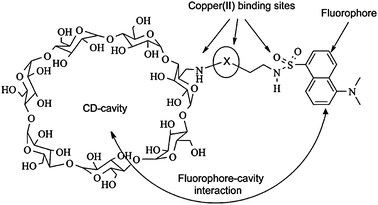 | ||
| Fig. 20 Binding interactions of modified cyclodextrin 65 and CuII(AA).135 | ||
Calibration curves were generated on a microwell plate, where eleven solutions of [CuII(valine)2] were used with varying ee. Six unknown samples (in triplicate) were analysed. When a single host was used to determine the % D-valine in the unknown samples, an accuracy of ±6% was obtained. Upon using three hosts (65a–c) for the analysis of the six samples, the accuracy increased. The analysis of a plate required 2 minutes, which is very favourable for HTS.136
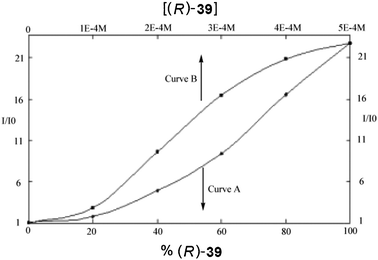 | ||
| Fig. 21 Fluorescent enhancement of host (R)-67 to: curve (A) different enantiomeric compositions of 39, curve (B) varying concentrations of (R)-39.138 | ||
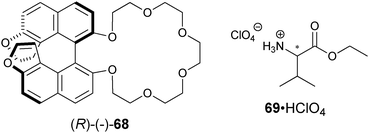
Karnik et al. developed the furan-fused BINOL crown derivative 68 for the enantioselective discrimination of 1-phenylethylammonium perchlorate (28·HClO4) and for the ethyl ester of valine (69·HClO4) in chloroform.141 The observed enantioselectivity of host (R)-68 for 28·HClO4 was KS/KR = 11.3, while for 69·HClO4 it was 7.0. The fluorescent intensity at 378 nm increased more readily with addition of (S)-28·HClO4 and (S)-69·HClO4 compared to their enantiomers. The origin of the enantioselectivity was proposed to be due to a steric clash between the amine substituents and the BINOL moiety (Fig. 22). A linear relationship between I/I0 and the enantiomeric composition of (S)-28·HClO4 allowed for the determination of ee. Similar results were obtained for 69·HClO4.
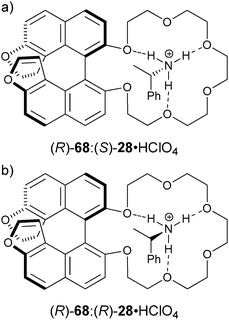 | ||
| Fig. 22 The proposed structures of: (a) (R)-68:(S)-28·HClO4 complex, (b) (R)-68:(R)-28·HClO4 complex.141 | ||
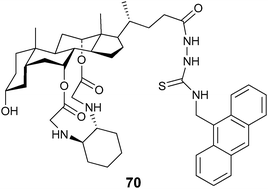
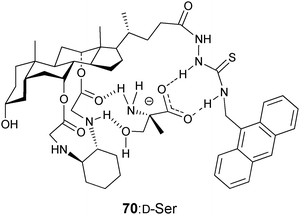
Chan et al. developed cholic acid-based fluorescent probes for the enantioselective recognition of the trifunctional amino acids serine, lysine, threonine, and tyrosine.142 For example, the ditopic probe 70 showed an enantioselectivity for serine of KD/KL = 6.7. In addition, a linear correlation of fluorescence quenching to different compositions of D-serine was obtained. The quenching was attributed to photo-induced electron transfer (PET) from the guest carboxylate when bound to the amidothiourea group of 70 (Fig. 23).
Mei and Wolf have developed a series of 1,8-diacrydylnaphthalene N,N′-dioxide fluorescent sensors for a range of chiral hydrogen bond donor analytes.143,144 For example, ligand 71a is synthesized in six steps, followed by resolution on a chiral HPLC. Chiral carboxylic acids and chiral diamines were examined in acetonitrile,143,144 and fluorescent quenching was observed upon binding. Mei and Wolf used this signal to develop calibration curves for the determination of [G]t and ee using racemic 71a and (+)-71a, respectively. Using calibration curves, six samples of 72 with differing concentration and ee were analysed, giving good agreement between the calculated and actual values.144 Further, six samples of 73 with differing145 concentration and enantiomeric purity were analysed, giving errors of only ±2% and ±3%, respectively. The time for analysis was not reported in these studies, but a typical fluorescent measurement is rapid (1 min), which is optimal for HTS.
In summary, several research groups have demonstrated the ability to perform enantioselective discrimination using fluorescence via the use of a range of binding interactions and scaffolds. In addition, such assays can be readily transitioned to microwell plate readers allowing for fast and parallel analysis, as has been demonstrated by the Corradini group.135
7.3 Absorbance
UV-vis spectroscopy is the most common optical signalling technique used for molecular sensing, and has numerous advantages for rapid determination of ee. The use of absorbance as a signalling mode is inherently quick and requires simple instrumentation. In addition, this technique has the ability to be transition from UV-vis spectrophotometers to a microwell plate reader, giving access to truly parallel analysis. In a 96-well plate, acquiring a full UV-vis spectrum for all wells in the plate takes approximately 30–40 minutes. While measuring absorbance at a single wavelength requires only ∼1 minute. If the absorbance lies in the visible range, “naked eye” detection is also possible, affording a quick approximation of the ee value by visual inspection. These advantages have led numerous research groups to explore the use of absorbance as a mode of detection for the discrimination of enantiomers, and the eventual quantification of ee. Below are selected examples of enantioselective discrimination organized by the type of molecular recognition interaction used.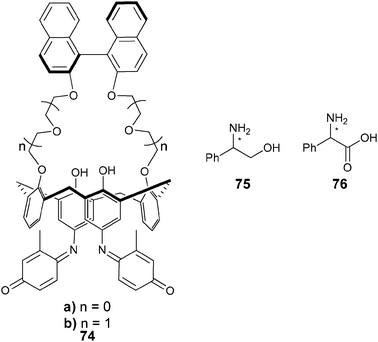
 | ||
| Fig. 24 Solutions containing host 74a, and solutions of host with each enantiomer of 75 demonstrating enantioselective discrimination through “naked eye” detection of guest.146 | ||
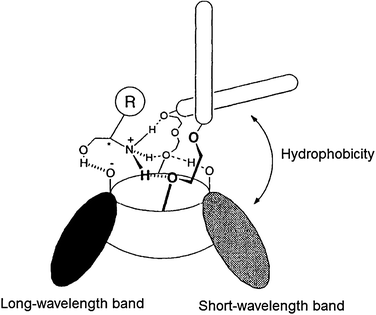
Kubo et al. proposed the binding of 75 to the cavity of 74 through interactions with the ether oxygens of the macrocyclic ring and the phenolate of the indophenol fragment. The colour change of 74a upon addition of (R)-75 was due to ionization of the phenol unit of the indophenol chromophore (a long-wavelength band), along with a binaphthyl-induced hydrophobic effect (a shorter-wavelength band). The binding of the amine alters the position of the binaphthyl unit, which does not occur with (S)-75, leading to the observed enantioselectivity and optical signal.
There was no selectivity between the enantiomers of guest 28 (1-phenylethanamine) by host 74a, demonstrating the importance of the hydroxyl group in the analytes for chiral discrimination. This is due to hydrogen bonding between the phenolate of the indophenol to the guest. Further, the binding of the amine was enhanced by the intramolecular hydrogen bonding between the indophenol hydroxyl group and the adjacent phenolic oxygen in host 74a, which led to the spectral change in the short-wavelength band. Fig. 25 shows the proposed complexation geometry between host 74a and guest 75.
Because host 74a had a larger Ka for (R)-75 than host 74b, it was also used as a colorimetric sensor for the enantiomeric discrimination of other chiral amines (57 (2-amino-3-phenyl-1-propanol) and 76). Enantioselective discrimination and preferential binding of (R)-57 and (R)-76 over (S)-isomers was found.
Fuji and Tsubaki et al. have also used chiral crown ether derivatives for the binding of amines via hydrogen bonding. They conjugated the crown ethers to phenolphthalein (77–81) to discriminate alanine derivatives (84).148 An increase in the absorption at 570 nm was observed with the addition of analytes. Association constants were measured for the five hosts (77–81) for guests (84) in methanol, where hosts 78, 79, and 81 favoured (R)-alanine derivatives, while host 80 had opposite enantioselectivity. The proposed binding between host 79 and guest 84c (Fig. 26) involves a steric repulsion between the methyl group on C-7 of the phenolic 18-crown-6-ring of 79 and the methyl group on the α-position of the guest. Host 79 produced a purple colour upon addition of (R)-84b or (R)-84c, while with the enantiomers a colourless solution was observed (Fig. 27). This allowed for enantiomeric recognition through “naked eye” detection. An ee calibration curve was generated for guest 84c using host 79, but the analysis of unknown samples was not explored.149
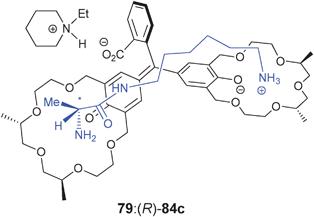 | ||
| Fig. 26 Proposed complexation of host 79 to analyte 84c with addition of N-ethylpiperidine.148 | ||
 | ||
| Fig. 27 Solutions demonstrating “naked eye” detection for enantioselective discrimination of alanine derivatives using host 79.148 | ||
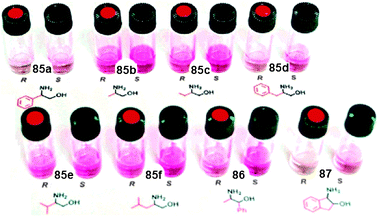
The enantioselective recognition by 79 was extended to β-amino alcohols (85, 86, and (R,S)-87), a simple chiral amine 28 (1-phenylethanamine), and additional alanine derivatives. Negligible differences could be observed between the enantiomeric pairs of guests by “naked eye” detection for 85, 86, and (R,S)-87, while the alanine derivatives showed no increase in enantioselectivity or colour change compared to 84. This led to the development of hosts 82 and 83 containing a phenyl substituent on the crown ether ring.149 Upon interaction of host 82 with β-amino alcohols 85, 86, and (R,S)-87, a colour change was observed (Fig. 28) that was deeper for the (S)-guests.
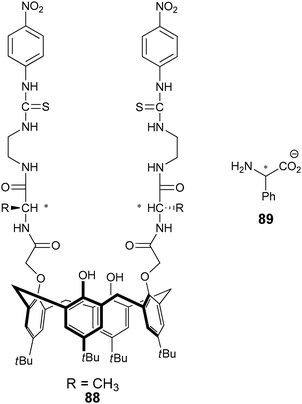
He's research group has developed several enantioselective sensors for the discrimination of a range of chiral analytes. His general design consists of a scaffold with two linkers containing stereocentres for enantioselective discrimination (commonly L-amino acids), and a chromophore for signalling (Fig. 29). He et al. have exploited benzyl,150 BINOL,151 and calixarenes,152 as scaffolds. For example, the calix[4]arene based sensor 88 was used to enantioselectively discriminate α-phenylglycine anion (89).152 The binding event was monitored by a decrease in absorbance at 359 nm, a bathochromic shift, and the formation of a new absorption peak at 481 nm. With D-α-phenylglycine the colour of the solution changed from colourless to saffron, while the addition of L-α-phenylglycine anion produced a yellow solution.
 | ||
| Fig. 29 Schematic of typical fluorescent sensors employed by He's group. | ||
Another example of an enantioselective “naked eye” sensor from He's research group is 90, targeted at mandelate.151 The colour of a solution of host 90b in DMSO changed from yellow to orange upon the addition of D-mandelate anion, while the addition of L-mandelate anion to 90b showed a change from yellow to red (Fig. 30).
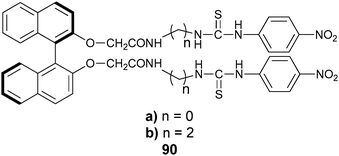
 | ||
| Fig. 30 Colour changes from the addition of mandelate anion to host 90b for enantioselective discrimination.151 | ||
UV-vis titrations revealed that the addition of either D- or L-mandelate to hosts 90a or 90b showed a decrease in absorption at 336 nm, accompanied by an increase at 278 nm, and the formation of a new absorption band at 412 nm. Host 90b showed the highest enantioselectivity for mandelate with Kass(L)/Kass(D) = 13.6. However, host 90a had larger association constants even though it showed lower enantioselectivity. The new absorption peak at 412 nm was attributed to a charge transfer from a thiourea and the electron-deficient p-nitrophenyl moieties of the host induced by coordination of mandelate. The authors suggest that the binding between the hosts and guests was driven primarily by hydrogen bonding interactions, because the addition of methanol or water to the host–guest complexes produced a light yellow solution, rather than red, suggesting that the protic solvents broke up the hydrogen bonding interactions.
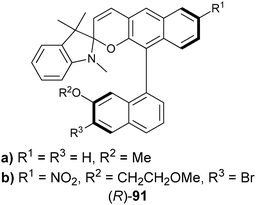
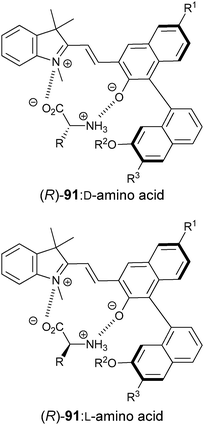 | ||
| Fig. 31 Binding interaction between amino acids and chiral spiropyran derivative receptor ((R)-91). | ||
7.4 Indicator displacement assays
Even though there are a range of chemosensors utilizing optical signalling techniques for enantiomeric discrimination and quantification of ee, there are common limitations. Many of the receptors discussed above require multiple synthetic steps, where the ability to signal the binding event is not known until after the synthesis of the host. This is unfavourable due to the required re-synthesis of the scaffold in cases where the signal or detection is not optimal. The re-synthesis can be time-consuming, especially when multiple synthetic steps and/or purifications, or resolution, are necessary. Even after modification, the new system may not produce the desired signal, thereby needing further refinement. This is very similar to the cycle used in the traditional approach to discovering chiral catalysts and auxiliaries, comprising design, synthesis, and testing. These limitations have led researchers to develop a different class of molecular sensors, generally known as indicator displacement assays (IDAs), also known as a competitive binding assay, and/or the use of ensembles.Indicator displacement assays (IDAs) are competition assays, involving a host/receptor (H), an indicator (I), and a guest/analyte (G). The indicator must reversibly bind to the host (eqn (1), Scheme 24), and the bound indicator must have a different spectroscopic signal than when free in solution. The analyte/guest must bind to the host/receptor (eqn (2), Scheme 24), and upon binding the equilibrium is shifted towards the displacement of the indicator from the host (eqn (3), Scheme 24). The resulting competition between the indicator and the guest for the host can be monitored through the change in the indicator's spectral properties, which are sensitive to the guest total concentration ([G]t) (eqn (4), Scheme 24).154,155 As [G]t increases, more indicator will be displaced, leading to a larger spectroscopic change. The use of IDAs has been popularized by our group,156–160 as well as many other groups.161–169
![Equilibria of an indicator displacement assay and enantioselective indicator displacement assay. H = host/receptor; H* = chiral host/receptor; I = indicator; G, GR, GS, = analyte/guest; [G]t = total guest concentration, KI, KG, KR, KS = binding constant.](/image/article/2012/CS/c1cs15135e/c1cs15135e-s24.gif) | ||
| Scheme 24 Equilibria of an indicator displacement assay and enantioselective indicator displacement assay. H = host/receptor; H* = chiral host/receptor; I = indicator; G, GR, GS, = analyte/guest; [G]t = total guest concentration, KI, KG, KR, KS = binding constant. | ||
Our group has also introduced the use of IDAs for enantioselective discrimination, called enantioselective indicator displacement assays (eIDAs), where a chiral host/receptor (H*) is used.107,133,170–175 The association of a chiral receptor with an enantiomeric mixture of chiral analytes/guests (GR, GS) produces multiple equilibria in solution simultaneously, which leads to the formation of two diastereomeric host–guest complexes (H*:GR, H*:GS, eqn (5) and (6), Scheme 24). The difference in the equilibrium constants (KR and KS) yields differential displacement of indicator (I), and hence a difference in optical properties for the two solutions (eqn (7), Scheme 24). For instance, if the signal is absorbance, mixtures of the two enantiomers will give a colour that is in between the extremes of the two pure enantiomers. An achiral version of the host (H) gives the total concentration of analyte or percent yield of a reaction, while the use of the chiral host (H*) allows for the determination of the ee of the reaction.
Over the last few years eIDAs have been developed for a range of chiral analytes, and the assay's ability to determine the ee of truly unknown samples has been demonstrated numerous times. In general, eIDAs produce ee values of lower accuracy than chromatographic techniques, however the method is much faster. Our group has also demonstrated the power of eIDAs in HTS through the use of microwell plate readers and automated liquid handlers, ultimately allowing for truly rapid screening of asymmetric reactions.
Our group reported two chiral CuII–diamine complexes (92 and 93) for the enantioselective recognition of α-amino acids.170,171 The synthesis of the ligands require one or two steps using commercially available materials, and require no chromatographic purification or resolutions. Chrome azurol S (94) was used as the indicator. Coordination of the indicator to the CuII metal centre produced a blue solution with an absorbance band at 602 nm, while the free indicator 94 produced a yellow solution with an absorbance band at 429 nm. The change in the indicator's optical properties when bound to the host versus free in solution allows for the detection of chiral analytes upon indicator displacement.
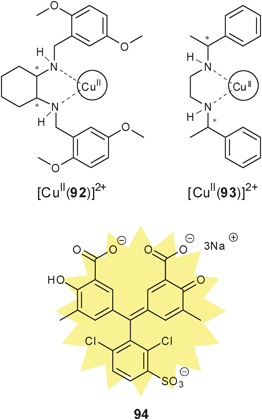
UV-vis titrations were carried out with [CuII((R,R)-92)(94)]2− and amino acids. The absorbance at 602 nm decreased more readily upon addition of L-amino acids than D-amino acids for receptor [CuII((R,R)-92)]2+, indicating its selectivity to L-amino acids (Scheme 25). When using [CuII((S,S)-92)]2+ in the assay, equal but opposite enantioselectivity was observed, confirming the expected enantiomeric cross-reactivity. Enantioselective discrimination for nine of seventeen naturally occurring amino acids was achieved. In the study, three amino acids (His, Asp, Asn) had reverse enantioselectivity, due to the coordination of their side chains to the CuII metal centre instead of the α-carboxylic acid group. The remaining eight amino acids that were not enantioselectively discriminated with receptor [CuII(92)]2+ were analysed with the second receptor [CuII(93)]2+. [CuII((R,R)-93)]2+ was selective towards L-amino acids, and the expected cross-reactivity was indeed observed. Receptor [CuII((R,R)-93)]2+ was able to enantioselectively discriminate four of these eight amino acids.
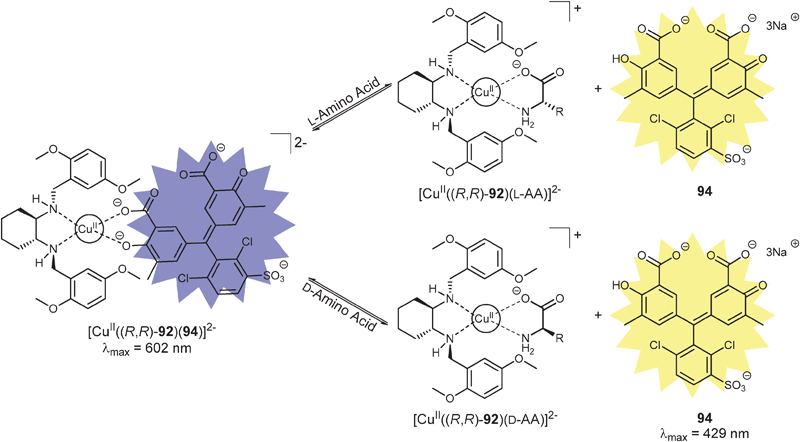 | ||
| Scheme 25 Equilibria in solution for the eIDA of amino acids. | ||
Discrimination through “naked eye” detection was possible for histidine and valine. Solutions containing [CuII((S,S)-92)(94)]2− with L-His produced a yellow solution, while the addition of D-His remained blue (Fig. 32a). Solutions of [CuII((R,R)-93)(94)]2− produced a brown solution upon addition of L-Val, whereas D-Val gave a blue solution (Fig. 32b). This allows for fast visual screening, directly amenable to transfer to a HTS environment.
![Demonstrating enantioselective discrimination through “naked eye” detection of amino acids with solutions containing: (a) [CuII((S,S)-92)(94)]2− with His, (b) [CuII((R,R)-93)(94)]2− with Val.170](/image/article/2012/CS/c1cs15135e/c1cs15135e-f32.gif) | ||
| Fig. 32 Demonstrating enantioselective discrimination through “naked eye” detection of amino acids with solutions containing: (a) [CuII((S,S)-92)(94)]2− with His, (b) [CuII((R,R)-93)(94)]2− with Val.170 | ||
Direct determination of ee was undertaken using calibration curves from standard UV-vis spectra by monitoring the change in absorbance at 602 nm as a function of ee. The ee values of several unknown samples were determined, but with somewhat large errors. Irrespectively, we transitioned the eIDAs from the single-cell cuvettes to microwell plates. Calibration curves for four amino acids were generated, and 24 unknown samples generated independent of the calibration curves were laid out on the 96-well plate, along with [CuII((R,R)-92)(94)]2− (Fig. 33).171 A different plate, also containing four calibration curves and 24 unknown samples was generated, except using receptor [CuII((R,R)-93)]2+. A robotic liquid handler was used to dispense the different components of the eIDA to each well of the microwell plate. Analysis of the unknown samples showed that transition to a microwell plate format from single-cell cuvettes produced little to no loss of accuracy in determining ee, yet all 96 wells were read in less than one minute. This translates to ∼1 s of analysis time per sample, demonstrating the system's ability to be used in a HT format.
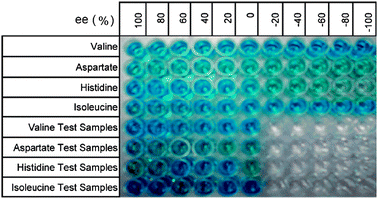 | ||
| Fig. 33 Microwell plate containing four ee calibration curves and 24 unknown samples.171 | ||
To further demonstrate the ability of eIDA's to determine the ee of unknown samples, a reaction to create valine via a Strecker-type asymmetric synthesis was performed.176 The ee of the reaction was determined with receptor [CuII((R,R)-92)]2+ and compared to chiral HPLC and 1H NMR analysis with a samarium(III) chiral shift reagent.177 These two methods showed differences of ±5% and ±12%, respectively, to our method. This study demonstrated the ability of an eIDA to determine the ee of unknown samples from an asymmetric reaction with values comparable to chiral HPLC.
Mei and Wolf have developed a novel system for enantioselective discrimination of amino acids, amines, amino alcohols, and carboxylic acids that is similar to an IDA, but does not use a traditional indicator. Instead, it uses a chromogenic ligand.178 A 2:1 complex between the naphthalene derived ligand 71a and ScIII produces the sensor, which has an absorbance at 410 nm due to charge transfer bands. Ligand 71a was obtained in six steps, requiring resolution on a chiral HPLC. The addition of enantiomerically pure analytes (valine, phenylglycine, methionine, glutamic acid, 85, 95–99) to solutions of [ScIII((+)-71a)2] in acetonitrile:H2O (1:1) displaced one ligand ((+)-71a), producing diastereomers ([ScIII((+)-71a)(analyte)]). This was observed by a decrease in the absorbance at 410 nm. The results indicated that the guest, L-valine, more readily displaced (+)-71a from the [ScIII((+)-71a)2] complex compared to D-valine. The [G]t could be determined using a racemic mixture of the [ScIII(71a)2] complex.
To validate this system's ability to determine ee, calibration curves for determining the [G]t and ee of valine and 97–99 were generated using [ScIII(71a)2] and [ScIII((+)-71a)2] complexes, respectively. Nine samples of unknown [G]t and ee for valine and 97–99 were determined using the corresponding calibration curve, which showed good agreement.
Wolf et al. have extended the scope of their technique by using [ScIII((+)-71b)2] to target α-amino alcohols 96, 100–104.179 The eIDA was similar to the previous work done with [ScIII((+)-71a)2], but a different signalling mode was used. The [ScIII((+)-71b)2] complex has a fluorescence emission at 588 nm upon excitation at 420 nm, and quenching was observed upon addition of the chiral α-amino alcohols. This host was enantioselective towards the (S)-amino alcohols, except for 101 and 104 where (R) and (1S,2R) were preferred, respectively. Once again, [G]t was determined through the use of a racemic mixture of [ScIII(71b)2], while the enantiomeric purity of unknown samples was determined using [ScIII((+)-71b)2]. Nine unknown samples of 100 were generated, and their [G]t and ee were determined using calibration curves, and the calculated ee and [G]t correlated with the true values quite well.
:106 ensemble with different enantiomeric compositions of 107a. Unfortunately, this study did not attempt to determine the ee of unknown samples.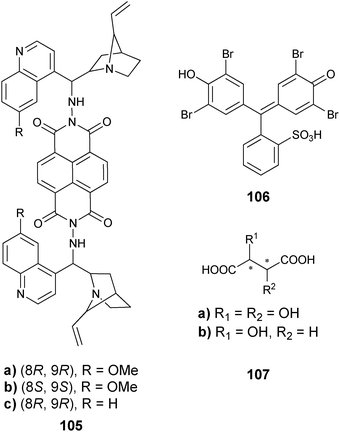
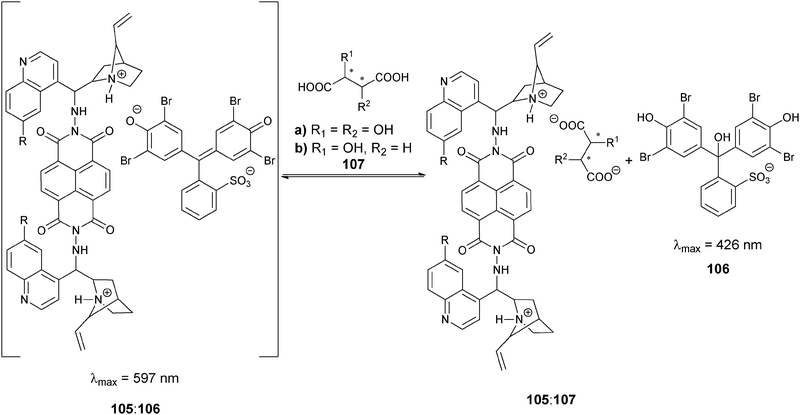 | ||
| Scheme 26 Indicator displacement assay for the recognition of dicarboxylic acids. | ||
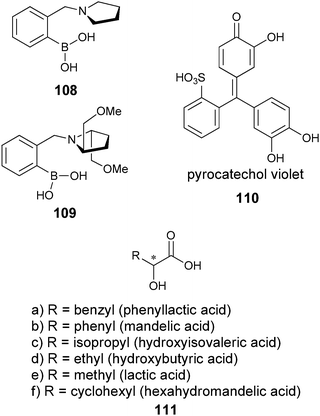
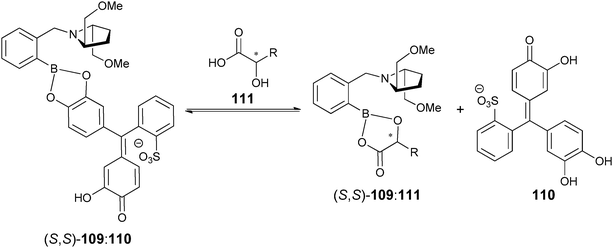 | ||
| Scheme 27 Equilibria in eIDA using boronic acid receptor (S,S)-109 and pyrocatechol violet indicator (110) for discriminating α-hydroxycarboxylic acids. | ||
Instead of using a calibration curve, a mathematical analysis based on mass balance equations and association constants measured from receptor–indicator binding isotherms were used to determine [G]t and ee. This method eliminates the need for an ee calibration curve for each different [G]t. For example, a higher [G]t of a racemic mixture of guest 111a could produce the same change in absorbance upon addition to the (S,S)-109:110 complex as a less concentrated sample of guest 111a enriched in (S)-111a (favoured enantiomer), thereby giving incorrect ee values. If one is using calibration curves, one must consider the [G]t of the curve and sample to quantify ee correctly. The accuracy of the eIDA to determine ee was tested via analysis of three independent samples, and ±20% error was obtained.
As discussed, two measurements are required for the determination of [G]t and ee, in which an achiral host and a chiral host are used, respectively. To eliminate the necessity of two physical measurements, we first employed a dual-chamber cuvette, where one chamber measured [G]t and the other ee.173 The left chamber (Fig. 34) contained achiral host 112 with indicator 113 for determining [G]t, while the right chamber contained (S,S)-109 with indicator 114 for the measurement of ee. Because different indicators are used in the two chambers the signals do not overlap, thereby allowing simultaneous collection of [G]t and ee data. In this case displacement of indicator 113 was monitored at 387 nm, which corresponds to an isosbestic point of the spectral changes for the right chamber. The right chamber determines ee by monitoring the changes at 536 nm, where the absorbance in the left cuvette is silent. This allowed the determination of [G]t and ee with one spectroscopic measurement. Using mathematical protocols previously published, the method gave between 3 and 8% absolute errors in determining the ee values of unknown samples.107,133
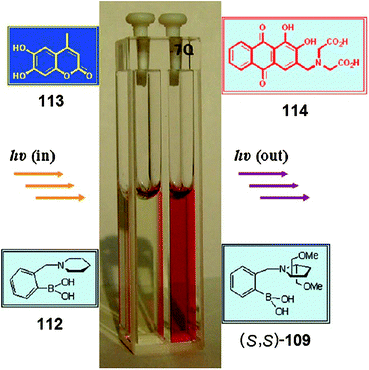
Irrespective of the use of a dual cell cuvette, the study just discussed still uses a mathematical algorithm in which equilibrium constants and molar absorptivities are needed. To avoid having to determine these values in advance when implementing our methods for determining ee and [G]t, we employed the use of artificial neural networks (ANNs). In general, optical data obtained with known [G]t and ee values are used as inputs to generate a network capable of predicting the corresponding [G]t and ee values of unknown samples. Using the receptors and indicators just described, absorbances at 387 nm and 536 nm were used as the input variables. Eleven unknown samples were analysed to determine the validity of the method. Analysis of these samples showed average absolute errors of ±0.45% and ±1.1% for the determination of [G]t and %R, respectively. Because this approach gave improved errors compared to the mathematical analysis methods, we now use ANNs routinely. This reduces the number of measurements required, did away with calibration curves, and thereby resulted in a shorter time for the analysis of data.
As previously stated, one of the limitations of the traditional molecular sensors is the inability to readily optimize a sensor for a specific analyte, or a range of analytes, due to the required synthesis. This is a limitation since not all analytes can be discriminated by only one host. However, because eIDAs have the ability to change or optimize the sensor ensemble either through change of hosts or indicators, as well as experimental conditions, the assay can be more readily optimized for different analytes. To exploit this power of an eIDA, we now use screening plates containing different host and indicator combinations to determine the best host–indicator pairing for a specific targeted analyte.
The entire protocol for using an eIDA to determine ee values in a HT fashion can involve five steps:175 (1) UV-vis titrations and standard binding isotherms to determine the proper concentration of receptor and indicator to be used in the IDAs; (2) using the optimal concentration of receptors and indicators determined in step 1, we screen with a 96-well plate format which host:indicator combination produces the enantioselectivity and dynamic range for ee determination of the specific targeted analyte (this is known as the screening plate); (3) using this best indicator and receptor pairing, a training set for an ANN is generated from optical data for known [G]t and ee values (this is known as the training plate); (4) spectral measurement of unknown samples is conducted to determine [G]t and ee (this is known as the analysis plate); and (5) ANN analysis of the spectral data obtained from step 4, allowing the determination of ee and [G]t. This protocol was tested on hydrobenzoin (125) as the targeted analyte with the use of hosts 108, 109, and 116–121, and indicators 110, 113, 114, and 122–124, in a methanol/water solution buffered to pH 7.4.
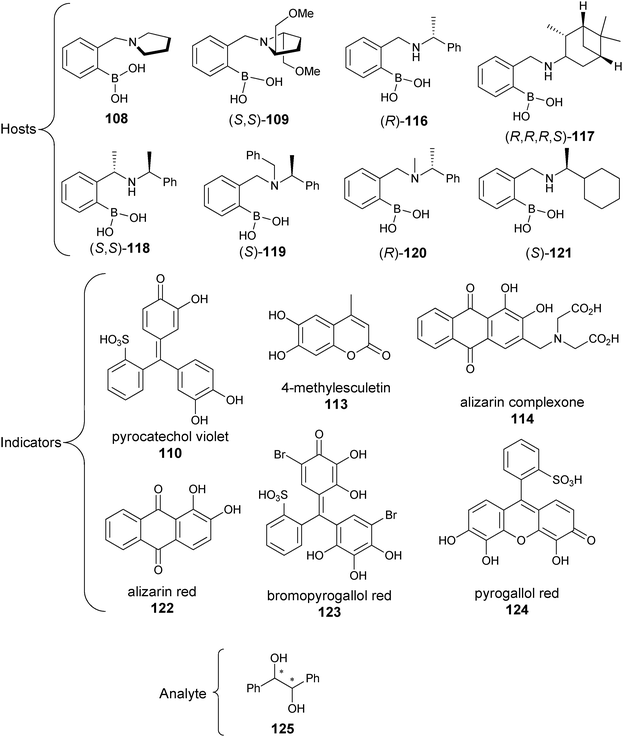
This five-step process was followed for the chiral diol hydrobenzoin (125). The screening plate revealed that (S,S)-109 and indicator 123 was the best combination to produce enantioselective discrimination at two different [G]t concentrations. Three ANN training plates were made using three hosts (108, (S,S)-109, and (R,R)-109) with 123 as the indicator, with varying ee values and four different concentrations of 125. After generating an ANN, achiral host 108 and chiral host 109 with indicator 123 were used to analyse eight different unknowns, with varying [G]t and ee. The spectra of these sixteen samples were recorded on a plate reader and the obtained data analysed with the trained ANN. The errors of the analysis were ±0.17 mM and ±3.5% for [G]t and ee, respectively (steps 4 and 5).
One drawback is that the five-step process includes a labour-intensive first step to determine optimal concentrations of hosts and indicators (step 1). However, because step 1 is not dependent on the analyte, it only needs to be conducted once, and it could be automated. Steps 2 and 3 are analyte specific, but again, they only need to be carried out once per analyte. Microwell plates, liquid dispensers, and robotic stackers make these steps easily automated. After determining the best host and indicator combination, measurement of the actual samples (step 4) and automated ANN analysis of the obtained spectral data (step 5) can be repeated at will until the asymmetric reaction conditions have been optimized.
To demonstrate the system's ability to analyse reactions, two chiral cinchona alkaloid ligands were used for Sharpless asymmetric dihydroxylation of trans-stilbene to give hydrobenzoin. The isolated product's [G]t and ee were determined using the best host–indicator combination (108:123 and 109:123) and an ANN, yielding average absolute errors of ±0.40 mM and ±2.4%, respectively. Although this protocol was applied only to hydrobenzoin, other analytes can be readily approached in the same manner.
8. Summary and outlook
The use of high-throughput screening for chiral catalysts/auxiliaries discovery is increasing in the field of asymmetric synthesis. A remaining challenge is determining the ee of the samples as quickly as they are generated, potentially at a rate of a thousand per day. Current serial analyses are not likely to easily handle this number of samples. Although chromatographic methods still produce the most accurate measurements, these methods are serial in nature and commonly have long elution times. This has led researchers to explore the use of optical signalling techniques, taking advantage of their ability for rapid and parallel analysis. Optical signalling uses simple instrumentation, and more importantly, allows for the transition to microwell plate formats, thereby allowing parallel analysis. The use of the microwell plate format further allows streamlining because it meshes with the facilities already in place in parallel synthesis and HTS laboratories; namely, with automated liquid handlers and plate stackers. Because these methods are not yet as accurate as chromatographic techniques, they are likely to be used as a prior screen for the highest values and trends, allowing for the removal of undesirable samples prior to HPLC analysis.Several groups have demonstrated how optical signalling might meet the challenges involved in HTS. Although advantages exist, there are also some limitations to certain methods (LC, IR-thermography, MIPs, enzymatic/antibodies), such as derivatization steps, narrow scope, specialized equipment, and time-consuming equilibration, or the requirement for pre-treatment of samples before analysis. More importantly, some systems do not actually quantify ee. These limitations have brought a move to the use of synthetic sensors that target functional groups only. Their ability to analyse a class of compounds, instead of targeting one specific analyte, while still using optical signals (CD, fluorescence, absorbance), makes them potentially great vehicles for creating HTS methods. Of course, one disadvantage is the syntheses of the hosts.
Even though synthetic receptors have advantages, current examples show that their ability to quantify ee with high accuracy and precision is limited. In several of the cases reviewed above, the goal was not the determination of ee, but instead demonstration of enantioselective binding. Importantly, most of these systems could be converted to and tested in an analytical method for ee determination. In addition, the transition to microwell plates has not often been implemented but is clearly adoptable with all the optical approaches. Further, the advantage of synthetic receptors to target a functional group has not been explored as extensively as we believe it ultimately will.
The most widespread approach to using synthetic receptors for enantioselective signalling has been through covalent attachment of a chromophore to the host or analyte. This is synthetically challenging, and often does not produce optimal signals for the detection of the analyte, thereby requiring resynthesis of the sensor. In contrast, indicator displacement assays, which do not require the covalent attachment of the chromophore to the host through synthesis, are being explored for the specific purpose of developing assays to quantify ee values. Therefore, eIDAs are widely explored optical techniques as currently described in the literature: they are simple to implement when coupled with either enantiomeric excess calibration curves or pattern recognition protocols; they have been successfully transitioned to the microwell plate format; and they have been applied to unknown samples from real asymmetric reactions. We predict further developments of these methods, eventually leading to their application to real-world HTS scenarios.
Acknowledgements
We gratefully acknowledge the financial support from the National Institutes of Health (GM77437) and Welch Foundation (F-1151). Further, we are indebted to Christopher Welch and others at Merck for first bringing the need for rapid ee analysis to our group's attention.References
- Asymmetric Synthesis: The Essentials, ed. M. Christmann and S. Braese, 2007 Search PubMed.
- G.-Q. Lin, Y.-M. Li and A. S. C. Chan, Principles and Applications of Asymmetric Synthesis, 2001 Search PubMed.
- J. G. de Vries and L. Lefort, Chem.–Eur. J., 2006, 12, 4722–4734 CrossRef CAS.
- J. F. Traverse and M. L. Snapper, Drug Discovery Today, 2002, 7, 1002–1012 CrossRef CAS.
- B. B. Lohray, Curr. Sci., 2001, 81, 1519–1525 CAS.
- M. Tsukamoto and H. B. Kagan, Adv. Synth. Catal., 2002, 344, 453–463 CrossRef CAS.
- M. T. Reetz, Angew. Chem., Int. Ed., 2001, 40, 284–310 CrossRef CAS.
- M. G. Finn, Chirality, 2002, 14, 534–540 CrossRef CAS.
- L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS.
- V. Charbonneau and W. W. Ogilvie, Mini-Rev. Org. Chem., 2005, 2, 313–332 CrossRef CAS.
- J. Guo, J. Wu, G. Siuzdak and M. G. Finn, Angew. Chem., Int. Ed., 1999, 38, 1755–1758 CrossRef CAS.
- S. Yao, J.-C. Meng, G. Siuzdak and M. G. Finn, J. Org. Chem., 2003, 68, 2540–2546 CrossRef CAS.
- A. Teichert and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3360–3362 CrossRef CAS.
- C. A. Mueller and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3363–3366 CrossRef CAS.
- I. Fleischer and A. Pfaltz, Chem.–Eur. J., 2010, 16, 95 CrossRef CAS.
- J. P. Stambuli and J. F. Hartwig, Curr. Opin. Chem. Biol., 2003, 7, 420–426 CrossRef CAS.
- J. A. Loch and R. H. Crabtree, Pure Appl. Chem., 2001, 73, 119–128 CrossRef CAS.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- K. D. Shimizu, M. L. Snapper and A. H. Hoveyda, Chem.–Eur. J., 1998, 4, 1885–1889 CrossRef CAS.
- C. M. Sprout, M. L. Richmond and C. T. Seto, J. Org. Chem., 2005, 70, 7408–7417 CrossRef CAS.
- P. P. Pescarmona, J. C. Van der Waal, I. E. Maxwell and T. Maschmeyer, Catal. Lett., 1999, 63, 1–11 CrossRef CAS.
- K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, Curr. Opin. Chem. Biol., 1999, 3, 313–319 CrossRef CAS.
- M. B. Francis, T. F. Jamison and E. N. Jacobsen, Curr. Opin. Chem. Biol., 1998, 2, 422–428 CrossRef CAS.
- N. K. Terrett, M. Gardner, D. W. Gordon, R. J. Kobylecki and J. Steele, Tetrahedron, 1995, 51, 8135–8173 CrossRef CAS.
- M. B. Francis and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 937–941 CrossRef CAS.
- R. B. C. Jagt, P. Y. Toullec, E. P. Schudde, J. G. De Vries, B. L. Feringa and A. J. Minnaard, J. Comb. Chem., 2007, 9, 407–414 CrossRef CAS.
- N. T. McDougal, S. C. Virgil and B. M. Stoltz, Synlett, 2010, 1712–1716 CAS.
- K. Burgess, H.-J. Lim, A. M. Porte and G. A. Sulikowski, Angew. Chem., Int. Ed. Engl., 1996, 35, 220–222 CrossRef CAS.
- R. H. Crabtree, Chem. Commun., 1999, 1611–1616 RSC.
- J. M. Takacs and J. Han, Org. Lett., 2004, 6, 3099–3102 CrossRef CAS.
- T. Arai, N. Yokoyama and A. Yanagisawa, Chem.–Eur. J., 2008, 14, 2052–2059 CrossRef CAS.
- K. Ding, Pure Appl. Chem., 2005, 77, 1251–1257 CrossRef CAS.
- K. Ding, H. Du, Y. Yuan and J. Long, Chem.–Eur. J., 2004, 10, 2872–2884 CrossRef CAS.
- C. J. Welch, Chirality, 2008, 21, 114–118 CrossRef.
- G. Guebitz and M. G. Schmid, Mol. Biotechnol., 2006, 32, 159–179 CrossRef CAS.
- R. Thompson, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 1215–1231 CrossRef CAS.
- Y. Zhang, D.-R. Wu, D. B. Wang-Iverson and A. A. Tymiak, Drug Discovery Today, 2005, 10, 571–577 CrossRef CAS.
- S. Ahuja, LCGC North Am., 2007, 25, 1112–1128 CAS.
- L. Chen, Y. Zhao, F. Gao and M. Garland, Appl. Spectrosc., 2003, 57, 797–804 CrossRef CAS.
- T. M. Tarasow, S. L. Tarasow and B. E. Eaton, Nature, 1997, 389, 54–57 CrossRef CAS.
- P. Sajonz, W. Schafer, X. Gong, S. Shultz, T. Rosner and C. J. Welch, J. Chromatogr., A, 2007, 1145, 149–154 CrossRef CAS.
- C. J. Welch, P. Sajonz, M. Biba, J. Gouker and J. Fairchild, J. Liq. Chromatogr. Relat. Technol., 2006, 29, 2185–2200 CrossRef CAS.
- Y. Zhao, W. A. Pritts and S. Zhang, J. Chromatogr., A, 2008, 1189, 245–253 CrossRef CAS.
- L. He and T. E. Beesley, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 1075–1114 CrossRef CAS.
- V. Schurig, J. Chromatogr., A, 2001, 906, 275–299 CrossRef CAS.
- V. Schurig, Chirality, 2005, 17, S205–S226 CrossRef CAS.
- M. T. Reetz, K. M. Kuhling, S. Wilensek, H. Husmann, U. W. Hausig and M. Hermes, Catal. Today, 2001, 67, 389–396 CrossRef CAS.
- D. R. Bobbitt and S. W. Linder, TrAC, Trends Anal. Chem. (Pers. Ed.), 2001, 20, 111–123 CrossRef CAS.
- C. Roussel, A. Del Rio, J. Pierrot-Sanders, P. Piras and N. Vanthuyne, J. Chromatogr., A, 2004, 1037, 311–328 CrossRef CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- C. Bertucci, P. Salvadori and L. F. L. Guimaraes, J. Chromatogr., A, 1994, 666, 535–539 CrossRef CAS.
- K. Ding, A. Ishii and K. Mikami, Angew. Chem., Int. Ed., 1999, 38, 497–501 CrossRef CAS.
- M. T. Reetz, K. M. Kuhling, H. Hinrichs and A. Deege, Chirality, 2000, 12, 479–482 CrossRef CAS.
- W. G. Kuhr and C. A. Monnig, Anal. Chem., 1992, 64, 389R–407R CrossRef CAS.
- W. G. Kuhr, Anal. Chem., 1990, 62, 403R–414R CrossRef CAS.
- D. A. Skoog, F. J. Holler and T. A. Nieman, Principles of Instrumental Analysis, Saunders College Pub., Harcourt Brace College Publishers, Philadelphia, Orlando, Fla, 1998 Search PubMed.
- I. Ali, K. Kumerer and H. Y. Aboul-Enein, Chromatographia, 2006, 63, 295–307 CAS.
- G. Gubitz and M. G. Schmid, Electrophoresis, 2000, 21, 4112–4135 CrossRef CAS.
- K. Verleysen and P. Sandra, Electrophoresis, 1998, 19, 2798–2833 CrossRef CAS.
- H. Nishi and S. Terabe, J. Chromatogr., A, 1995, 694, 245–276 CrossRef CAS.
- G. Gubitz and M. G. Schmid, J. Chromatogr., A, 1997, 792, 179–225 CrossRef CAS.
- A. Van Eeckhaut and Y. Michotte, Electrophoresis, 2006, 27, 2880–2895 CrossRef CAS.
- M. T. Reetz, K. M. Kuhling, A. Deege, H. Hinrichs and D. Belder, Angew. Chem., Int. Ed., 2000, 39, 3891–3893 CrossRef CAS.
- D. Demus, J. W. G. Goodby and G. W. Gray, Handbook of Liquid Crystals. Vol. 1, Fundamentals, Wiley-VCH, Weinheim, New York, Chichester, 1998 Search PubMed.
- G. Solladie and R. Zimmermann, Angew. Chem., 1984, 96, 335–349 CrossRef CAS.
- R. A. Van Delden and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 3198–3200 CrossRef CAS.
- R. A. van Delden and B. L. Feringa, Chem. Commun., 2002, 174–175 RSC.
- R. Eelkema, R. A. van Delden and B. L. Feringa, Angew. Chem., Int. Ed., 2004, 43, 5013–5016 CrossRef CAS.
- D. M. Walba, L. Eshdat, E. Korblova, R. Shao and N. A. Clark, Angew. Chem., Int. Ed., 2007, 46, 1473–1475 CrossRef CAS.
- A. Holzwarth, H.-W. Schmidt and W. F. Maier, Angew. Chem., Int. Ed., 1998, 37, 2644–2647 CrossRef CAS.
- F. C. Moates, M. Somani, J. Annamalai, J. T. Richardson, D. Luss and R. C. Willson, Ind. Eng. Chem. Res., 1996, 35, 4801–4803 CrossRef CAS.
- S. J. Taylor and J. P. Morken, Science, 1998, 280, 267–270 CrossRef CAS.
- M. T. Reetz, M. H. Becker, K. M. Kuhling and A. Holzwarth, Angew. Chem., Int. Ed., 1998, 37, 2647–2650 CrossRef CAS.
- N. Millot, P. Borman, M. S. Anson, I. B. Campbell, S. J. F. Macdonald and M. Mahmoudian, Org. Process Res. Dev., 2002, 6, 463–470 CrossRef CAS.
- M. T. Reetz, M. H. Becker, M. Liebl and A. Furstner, Angew. Chem., Int. Ed., 2000, 39, 1236–1239 CrossRef CAS.
- B. Sellergren (editor), Molecularly Imprinted Polymers: Man-Made Mimics of Antibodies and Their Applications in Analytical Chemistry, Tech. Instrum. Anal. Chem., 2001, 23.
- R. A. Bartsch and M. Maeda, Molecular and Ionic Recognition with Imprinted Polymers. (Proceedings of a Symposium held at the 213th National Meeting of the American Chemical Society, 13-17 April 1997, in San Francisco, California.), ACS Symp. Ser., 1998, 703 Search PubMed.
- Y. Chen and K. D. Shimizu, Org. Lett., 2002, 4, 2937–2940 CrossRef CAS.
- F. Moris-Varas, A. Shah, J. Aikens, N. P. Nadkarni, J. D. Rozzell and D. C. Demirjian, Bioorg. Med. Chem., 1999, 7, 2183–2188 CrossRef CAS.
- Z. Li, L. Buetikofer and B. Witholt, Angew. Chem., Int. Ed., 2004, 43, 1698–1702 CrossRef CAS.
- M. Baumann, R. Sturmer and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2001, 40, 4201–4204 CrossRef CAS.
- L. E. Janes, A. C. Lowendahl and R. J. Kazlauskas, Chem.–Eur. J., 1998, 4, 2324–2331 CrossRef CAS.
- G. A. Korbel, G. Lalic and M. D. Shair, J. Am. Chem. Soc., 2001, 123, 361–362 CrossRef CAS.
- F. Badalassi, G. Klein, P. Crotti and J.-L. Reymond, Eur. J. Org. Chem., 2004, 2557–2566 CrossRef CAS.
- N. Bahr, E. Tierney and J.-L. Reymond, Tetrahedron Lett., 1997, 38, 1489–1492 CrossRef CAS.
- F. Badalassi, D. Wahler, G. Klein, P. Crotti and J.-L. Reymond, Angew. Chem., Int. Ed., 2000, 39, 4067–4070 CrossRef CAS.
- A. Hamberg, S. Lundgren, M. Penhoat, C. Moberg and K. Hult, J. Am. Chem. Soc., 2006, 128, 2234–2235 CrossRef CAS.
- A. Hamberg, S. Lundgren, E. Wingstrand, C. Moberg and K. Hult, Chem.–Eur. J., 2007, 13, 4334–4341 CrossRef CAS.
- D. B. Berkowitz, M. Bose and S. Choi, Angew. Chem., Int. Ed., 2002, 41, 1603–1607 CrossRef CAS.
- S. Dey, K. R. Karukurichi, W. Shen and D. B. Berkowitz, J. Am. Chem. Soc., 2005, 127, 8610–8611 CrossRef CAS.
- S. Dey, D. R. Powell, C. Hu and D. B. Berkowitz, Angew. Chem., Int. Ed., 2007, 46, 7010–7014 CrossRef CAS.
- F. Taran, C. Gauchet, B. Mohar, S. Meunier, A. Valleix, P. Y. Renard, C. Creminon, J. Grassi, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2002, 41, 124–127 CrossRef CAS.
- M. Matsushita, K. Yoshida, N. Yamamoto, P. Wirsching, R. A. Lerner and K. D. Janda, Angew. Chem., Int. Ed., 2003, 42, 5984–5987 CrossRef CAS.
- H. Matsushita, N. Yamamoto, M. M. Meijler, P. Wirsching, R. A. Lerner, M. Matsushita and K. D. Janda, Mol. BioSyst., 2005, 1, 303–306 RSC.
- A. K. Yatsimirsky, Encycl. Supramol. Chem., 2004, 809–815 Search PubMed.
- T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS.
- H. Tsukube, Coord. Chem. Rev., 1996, 148, 1–17 CrossRef CAS.
- F. Bellia, D. La Mendola, C. Pedone, E. Rizzarelli, M. Saviano and G. Vecchio, Chem. Soc. Rev., 2009, 38, 2756–2781 RSC.
- S. M. Biros and J. Rebek, Jr., Chem. Soc. Rev., 2007, 36, 93–104 RSC.
- T. Ogoshi and A. Harada, Sensors, 2008, 8, 4961–4982 CrossRef CAS.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS.
- C. Lynam and D. Diamond, J. Mater. Chem., 2005, 15, 307–314 RSC.
- H. N. Demirtas, S. Bozkurt, M. Durmaz, M. Yilmaz and A. Sirit, Tetrahedron, 2009, 65, 3014–3018 CrossRef CAS.
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS.
- Y. Kubo, Synlett, 1999, 161–174 CrossRef CAS.
- T. D. James, M. D. Philips and S. Shinkai, Boronic acids in Saccharide Recognition, The Royal Society of Chemistry, 2006 Search PubMed.
- L. Zhu, Z. Zhong and E. V. Anslyn, J. Am. Chem. Soc., 2005, 127, 4260–4269 CrossRef CAS.
- S. R. Stauffer and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 6977–6985 CrossRef CAS.
- R. Moreira, M. Havranek and D. Sames, J. Am. Chem. Soc., 2001, 123, 3927–3931 CrossRef CAS.
- G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 1999, 121, 4306–4307 CrossRef CAS.
- O. Lavastre and J. P. Morken, Angew. Chem., Int. Ed., 1999, 38, 3163–3165 CrossRef CAS.
- R. F. Harris, A. J. Nation, G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 2000, 122, 11270–11271 CrossRef CAS.
- G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 6496–6502 CrossRef CAS.
- V. V. Borovkov and Y. Inoue, Org. Lett., 2006, 8, 2337–2340 CrossRef CAS.
- H. Imai, H. Munakata, Y. Uemori and N. Sakura, Inorg. Chem., 2004, 43, 1211–1213 CrossRef CAS.
- T. Kurtan, N. Nesnas, Y.-Q. Li, X. Huang, K. Nakanishi and N. Berova, J. Am. Chem. Soc., 2001, 123, 5962–5973 CrossRef CAS.
- X. Huang, B. Borhan, B. H. Rickman, K. Nakanishi and N. Berova, Chem.–Eur. J., 2000, 6, 216–224 CrossRef CAS.
- T. Hayashi, T. Aya, M. Nonoguchi, T. Mizutani, Y. Hisaeda, S. Kitagawa and H. Ogoshi, Tetrahedron, 2002, 58, 2803–2811 CrossRef CAS.
- S. Yagi, H. Kitayama and T. Takagishi, J. Chem. Soc., Trans. Perkin 1, 2000, 925–932 CAS.
- T. Hattori, Y. Minato, S. Yao, M. G. Finn and S. Miyano, Tetrahedron Lett., 2001, 42, 8015–8018 CrossRef CAS.
- G. Cai, N. Bozhkova, J. Odingo, N. Berova and K. Nakanishi, J. Am. Chem. Soc., 1993, 115, 7192–7198 CrossRef CAS.
- W. T. Wiesler and K. Nakanishi, J. Am. Chem. Soc., 1989, 111, 9205–9213 CrossRef CAS.
- J. M. Lintuluoto, K. Nakayama and J.-i. Setsune, Chem. Commun., 2006, 3492–3494 RSC.
- A. E. Holmes, S. Zahn and J. W. Canary, Chirality, 2002, 14, 471–477 CrossRef CAS.
- S. Zahn and J. W. Canary, Org. Lett., 1999, 1, 861–864 CrossRef CAS.
- J. Zhang, A. E. Holmes, A. Sharma, N. R. Brooks, R. S. Rarig, J. Zubieta and J. W. Canary, Chirality, 2003, 15, 180–189 CrossRef CAS.
- K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639–7650 CrossRef CAS.
- S. Nieto, V. M. Lynch, E. V. Anslyn, H. Kim and J. Chin, Org. Lett., 2008, 10, 5167–5170 CrossRef CAS.
- S. Nieto, V. M. Lynch, E. V. Anslyn, H. Kim and J. Chin, J. Am. Chem. Soc., 2008, 130, 9232–9233 CrossRef CAS.
- D. Coomans, D. L. Massart and L. Kaufman, Anal. Chim. Acta, 1979, 112, 97–122 CrossRef CAS.
- Y. Li, J.-H. Jiang, Z.-P. Chen, C.-J. Xu and R.-Q. Yu, J. Chemom., 1999, 13, 3–13 CrossRef CAS.
- M. Ringner, Nat. Biotechnol., 2008, 26, 303–304 CrossRef CAS.
- L. Zhu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 3676–3677 CrossRef CAS.
- X. Mei, R. M. Martin and C. Wolf, J. Org. Chem., 2006, 71, 2854–2861 CrossRef CAS.
- R. Corradini, C. Paganuzzi, R. Marchelli, S. Pagliari, S. Sforza, A. Dossena, G. Galaverna and A. Duchateau, J. Mater. Chem., 2005, 15, 2741–2746 RSC.
- R. Corradini, C. Paganuzzi, R. Marchelli, S. Pagliari, A. Dossena and A. Duchateau, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 625–630 CrossRef CAS.
- G. I. Richard, H. M. Marwani, S. Jiang, S. O. Fakayode, M. Lowry, R. M. Strongin and I. M. Warner, Appl. Spectrosc., 2008, 62, 476–480 CrossRef CAS.
- Z.-B. Li, J. Lin, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2007, 72, 4905–4916 CrossRef CAS.
- Z.-B. Li, J. Lin and L. Pu, Angew. Chem., Int. Ed., 2005, 44, 1690–1693 CrossRef CAS.
- S. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 17698–17700 CrossRef CAS.
- S. P. Upadhyay, R. R. S. Pissurlenkar, E. C. Coutinho and A. V. Karnik, J. Org. Chem., 2007, 72, 5709–5714 CrossRef CAS.
- H. Wang, W.-H. Chan and A. W. M. Lee, Org. Biomol. Chem., 2008, 6, 929–934 CAS.
- X. Mei and C. Wolf, Chem. Commun., 2004, 2078–2079 RSC.
- X. Mei and C. Wolf, Tetrahedron Lett., 2006, 47, 7901–7904 CrossRef CAS.
- C. Wolf, S. Liu and B. C. Reinhardt, Chem. Commun., 2006, 4242–4244 RSC.
- Y. Kubo, S. y. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–524 CrossRef CAS.
- Y. Kubo, N. Hirota, S. n. Maeda and S. Tokita, Anal. Sci., 1998, 14, 183–189 CrossRef CAS.
- K. Tsubaki, M. Nuruzzaman, T. Kusumoto, N. Hayashi, B.-G. Wang and K. Fuji, Org. Lett., 2001, 3, 4071–4073 CrossRef CAS.
- K. Tsubaki, D. Tanima, M. Nuruzzaman, T. Kusumoto, K. Fuji and T. Kawabata, J. Org. Chem., 2005, 70, 4609–4616 CrossRef CAS.
- Z.-H. Chen, Y.-B. He, C.-G. Hu, X.-H. Huang and L. Hu, Aust. J. Chem., 2008, 61, 310–315 CrossRef CAS.
- C. Hu, Y. He, Z. Chen and X. Huang, Tetrahedron: Asymmetry, 2009, 20, 104–110 CrossRef CAS.
- G.-Y. Qing, Y.-B. He, Y. Zhao, C.-G. Hu, S.-Y. Liu and X. Yang, Eur. J. Org. Chem., 2006, 1574–1580 CrossRef CAS.
- K. Tsubaki, K. Mukoyoshi, H. Morikawa, T. Kinoshita and K. Fuji, Chirality, 2002, 14, 713–715 CrossRef CAS.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- S. L. Wiskur, H. Ait-Haddou, J. J. Lavigne and E. V. Anslyn, Acc. Chem. Res., 2001, 34, 963–972 CrossRef CAS.
- T. Zhang and E. V. Anslyn, Org. Lett., 2007, 9, 1627–1629 CrossRef CAS.
- T. Zhang and E. V. Anslyn, Tetrahedron, 2004, 60, 11117–11124 CrossRef CAS.
- B. T. Nguyen, S. L. Wiskur and E. V. Anslyn, Org. Lett., 2004, 6, 2499–2501 CrossRef CAS.
- S. C. McCleskey, P. N. Floriano, S. L. Wiskur, E. V. Anslyn and J. T. McDevitt, Tetrahedron, 2003, 59, 10089–10092 CrossRef CAS.
- A. M. Piatek, Y. J. Bomble, S. L. Wiskur and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 6072–6077 CrossRef CAS.
- M. A. Hortala, L. Fabbrizzi, N. Marcotte, F. Stomeo and A. Taglietti, J. Am. Chem. Soc., 2003, 125, 20–21 CrossRef CAS.
- L. Fabbrizzi, N. Marcotte, F. Stomeo and A. Taglietti, Angew. Chem., Int. Ed., 2002, 41, 3811–3814 CrossRef CAS.
- M. S. Han and D. H. Kim, Angew. Chem., Int. Ed., 2002, 41, 3809–3811 CrossRef CAS.
- M. N. Stojanovic and D. W. Landry, J. Am. Chem. Soc., 2002, 124, 9678–9679 CrossRef CAS.
- L. Fabbrizzi, A. Leone and A. Taglietti, Angew. Chem., Int. Ed., 2001, 40, 3066–3069 CrossRef CAS.
- P. A. Gale, L. J. Twyman, C. I. Handlin and J. L. Sessler, Chem. Commun., 1999, 1851–1852 RSC.
- F. Zaubitzer, A. Buryak and K. Severin, Chem.–Eur. J., 2006, 12, 3928–3934 CrossRef CAS.
- A. Schiller, R. A. Wessling and B. Singarum, Angew. Chem., Int. Ed., 2007, 46, 6457–6459 CrossRef CAS.
- A. Buryak, A. Pozdnoukhov and K. Severin, Chem. Commun., 2007, 2366–2368 RSC.
- D. Leung, J. F. Folmer-Andersen, V. M. Lynch and E. V. Anslyn, J. Am. Chem. Soc., 2008, 130, 12318–12327 CrossRef CAS.
- D. Leung and E. V. Anslyn, J. Am. Chem. Soc., 2008, 130, 12328–12333 CrossRef CAS.
- J. F. Folmer-Andersen, M. Kitamura and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 5652–5653 CrossRef CAS.
- L. Zhu, S. H. Shabbir and E. V. Anslyn, Chem.–Eur. J., 2006, 13, 99–104 CrossRef CAS.
- J. F. Folmer-Andersen, V. M. Lynch and E. V. Anslyn, J. Am. Chem. Soc., 2005, 127, 7986–7987 CrossRef CAS.
- S. H. Shabbir, C. J. Regan and E. V. Anslyn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10487–10492 CrossRef CAS.
- T. K. Chakraborty, K. A. Hussain and G. V. Reddy, Tetrahedron, 1995, 51, 9179–9190 CrossRef CAS.
- A. Inamoto, K. Ogasawara, K. Omata, K. Kabuto and Y. Sasaki, Org. Lett., 2000, 2, 3543–3545 CrossRef CAS.
- X. Mei and C. Wolf, J. Am. Chem. Soc., 2006, 128, 13326–13327 CrossRef CAS.
- S. Liu, J. P. C. Pestano and C. Wolf, J. Org. Chem., 2008, 73, 4267–4270 CrossRef CAS.
- K. Kacprzak, J. Grajewski and J. Gawronski, Tetrahedron: Asymmetry, 2006, 17, 1332–1336 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |