Playing with organic radicals as building blocks for functional molecular materials†‡
Imma
Ratera
and
Jaume
Veciana
*
Institut de Ciència de Materials de Barcelona (ICMAB-CSIC), CIBER-BBN Campus UAB, 08193 Bellaterra (Barcelona), Spain. E-mail: vecianaj@icmab.es; iratera@icmab.es; Fax: +34-935 805 729; Tel: +34-935 801 853
First published on 18th August 2011
Abstract
The literature has shown numerous contributions on the synthesis and physicochemical properties of persistent organic radicals but there are a lesser number of reports about their use as building blocks for obtaining molecular magnetic materials exhibiting an additional and useful physical property or function. These materials show promise for applications in spintronics as well as bistable memory devices and sensing materials. This critical review provides an up-to-date survey to this new generation of multifunctional magnetic materials. For this, a detailed revision of the most common families of persistent organic radicals—nitroxide, triphenylmethyl, verdazyl, phenalenyl, and dithiadiazolyl—so far reported will be presented, classified into three different sections: materials with magnetic, conducting and optical properties. An additional section reporting switchable materials based on these radicals is presented (257 references).
 Imma Ratera | Dr Imma Ratera PhD in Materials Science and Chemistry from Institute of Materials Science of Barcelona (ICMAB-CSIC) with Prof. J. Veciana and Dr D. Ruiz-Molina. Her work was focused on the preparation and characterization of multifunctional molecular materials and switches with optical, magnetic and electronic properties. Postdoc at Lawrence Berkeley National Laboratory (LBNL), Materials and Surface Science Division. Her project involved the nanoscale manipulation of organic molecules, from nano-tribology to electrical properties. Since 2008 she has a research position at ICMAB. Her main interests are on molecular functional materials and molecular nanoscience. |
 Jaume Veciana | Prof. Jaume Veciana (Institut de Ciencia de Materials de Barcelona, ICMAB-CSIC) was appointed as Colaborador Cientifico of the CSIC in 1979 and in 1982/1983 moved to The Johns Hopkins University, MD (USA) as a Postdoctoral fellow working on molecular conductors and organic metals. In 1991 he moved to the ICMAB where he was promoted to Full Professor in 1996. He co-authored more than 370 journal articles and book chapters, 15 patents, and edited two books receiving in 2001 the Solvay Award and in 2004 the Real Sociedad de Quimica Española Award for his research in chemistry. In 2005 he received the DuPont Award for his contributions in Molecular Nanoscience and Nanotechnology. His research interest focuses on molecular functional materials and molecular nanoscience. |
1. Introduction to persistent organic radicals
Organic radicals are open-shell molecules composed of light elements—mostly H, C, N, O and S—with an electronic configuration that makes them very reactive. They are species with an unpaired electron occupying the highest molecular orbital which can easily take part in reactions, such as hydrogen abstraction, dimerization or recombination, leading to the loss of the open-shell character. Nevertheless, it is possible to have persistent organic radicals by screening the paramagnetic center with bulky substituents, hence protecting it from undesired reactions.1,2 The polychlorinated trityl radicals,3 where the central methyl sp2carbon is the spin bearing atom, is one of these radicals being sterically protected. Another way to produce persistent radicals consists in increasing the delocalization of the unpaired electron over a large part of the atomic skeleton. Examples of families of radicals showing persistency are the nitroxides and α-nitronyl nitroxides,4 where the unpaired electrons are delocalized between the nitrogen and the oxygen atoms. Verdazyl radicals5 are another example of radicals with such characteristics where the unpaired electrons are instead delocalized onto the heterocycle containing the nitrogen atoms. These families of persistent radicals find applications in many different fields, such as biochemistry,6 catalysis7 and molecular magnetism.8Since Gomberg's discovery of the triphenylmethyl radical in 19009 physical properties of neutral radicals have been a source of inspiration for research into molecule-based functional materials.10 Magnetism induced by interactions of unpaired electrons is one of the most characteristic properties of organic neutral radicals. The alignment of spins due to the unpaired electrons with adequate interactions through π electrons or one pair of electrons of heteroatoms in the solid state give place to the obtaining of molecule-based magnetic materials. 2,2,6,6,-Tetramethylpiperidin-N-oxyl (TEMPO) and α-nitronyl nitroxide derivatives among others, synthesized a half-century ago as neutral radicals stable enough to handle in air, have importantly contributed to the development of the molecule-based magnetism and spin sciences, and even now play important roles in other various research fields as stable spin sources. For further progress and development of spin science opened up by the stable neutral radicals, the design and syntheses of stable neutral radicals with novel molecular and electronic structures are essential issues. To achieve this target, the use of structural diversity, which is one of the most important virtues of the organic molecules, should be used to the maximum.
The molecular skeleton of stable neutral radicals has two important features: one regarding the good stabilization of the spin and the second regarding the modulation and control of electronic structure which will control the intermolecular interactions responsible for the physico-chemical properties of the materials. For designing novel neutral radicals, it is vital to utilize these two aspects effectively.11 For example, stabilization of neutral radicals by steric protection not only suppresses the dimerization reaction and the reaction with oxygen, but also greatly influences crystal structures and, thus, intermolecular spin–spin interactions. Introduction of heteroatoms is another effective tool for the control of intermolecular electronic interactions through their lone pair electrons. Furthermore, heteroatomic modulation affects the distribution of singly occupied molecular orbitals (SOMOs) and therefore the spin density on the molecular skeleton by the changes in π-topological symmetries,12 and at the same time influences the molecular orbital energy levels modifying their redox ability.
This review forms part of a themed issue recently published about molecule-based magnets.13 Here, we describe the structural characterization and the chemical and physical properties of materials based on persistent organic radicals mainly from the molecular material point of view. Many reviews have been devoted along the years to the various families of spin containing compounds. We therefore do not attempt to present an exhaustive description of the field but only to report on functional molecular materials based on these organic compounds. Thus, the main focus will be on organic materials exhibiting a physical or chemical characteristic, giving different insights to not only the magnetic but also electronic and optical properties of radical-based materials. We will focus mainly on the most extensively used families of persistent free radicals; like triphenylmethyl, phenalenyl, nitroxide, verdazyl and dithiazolyl based radicals.
1.1 Triphenylmethyl radicals
Triphenylmethyl radicals were the first example of an open-shell organic radical prepared and studied.9 From then, several derived diradicals and polyradicals have been obtained.14 However their high reactivity prevented any practical use. The increase of its persistence was achieved by a proper screening of the central carbon atom leading to a wide variety of derivatives that show larger life expectancies.15 Among them we may mention the polychlorotriphenylmethyl (PTM) radicals which are characterized by an extremely high persistence and stability. They are composed of three totally or partially chlorinated phenyl rings connected to a central carbon atom with a sp2 hybridization (Fig. 1).16 The steric strain that exists in a hypothetical planar conformation of this molecule, mainly originated by the bulky chlorine atoms at the ortho position, is released by the rotation of the aromatic rings around the bonds linking the central to the ipso-carbon atoms of the molecule. This forces the molecule to adopt a propeller-like conformation. As a consequence, from the stereochemical point of view, such radicals exist in two different enantiomeric forms that can interconvert at high temperatures with an energy barrier of ca. 23 kcal mol−1.17 Following the Cahn–Ingold–Prelog nomenclature,18 the two atropoisomeric forms are called Plus (P), when the rings present a clockwise torsion, and Minus (M) when the torsion is the opposite one.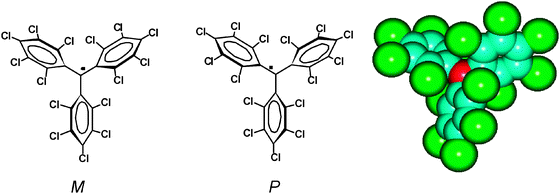 | ||
| Fig. 1 Left: representation of the Plus (P) and Minus (M) atropoisomeric forms of perchlorotriphenylmetyl radical 1˙. Right: radical 1˙ showing the high steric shielding of the central carbon atom (in red) surrounded by the six bulky chlorine atoms at the ortho positions. | ||
Besides the generation of a molecular propeller-like conformation, the existence of the bulky chlorine atoms at the ortho positions and the three phenyl groups where they are allocated also provides the main source of steric protection of the central (or α) carbon atom, which is the atom with the major spin density. In Fig. 1 is shown a molecular model of the perchlorotriphenylmethyl radical (1˙) showing that the central carbon atom is surrounded and protected by three ortho-chlorine atoms at each side of the molecule. Indeed, the protection of the methyl carbon is translated into a large persistence and a very high chemical and thermal stability for this family of radicals.19–23 Thus, unlike in the case of the non-chlorinated analogue which dimerizes very fast and is very reactive to oxygen avoiding its isolation as radical, in solution PTM radicals remain perfectly inert to oxygen and to many other aggressive reagents, decomposing only in the presence of light. Consequently such radicals behave as classical organic compounds and they can be used as intermediates in many reactions and treated with usual purification techniques, such as chromatography, crystallization, sublimation, etc. These radicals in solid state decompose with high melting points,24 usually near 300 °C, and they are stable to light and air practically indefinitely.25
The synthesis of PTM radicals is usually achieved from the corresponding polychlorotriphenylmethanes through the formation of their carbanions with a base, like OH−, followed by the oxidation of the latter with a weak oxidant, such as p-chloranil, I2 or Ag+; as exemplified in Scheme 1 for radical 1˙. The synthesis of the radical precursors generally consists in a sequence of Friedel–Crafts condensations at high temperature and pressure with an excess of the chlorinated benzene derivatives; such as it is depicted in Scheme 1 for the hydrocarbon 1HHH.21
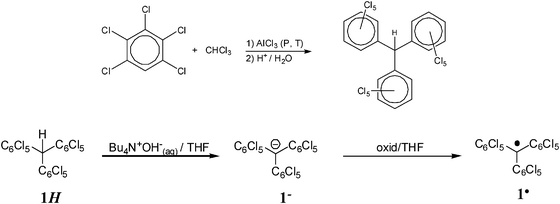 | ||
| Scheme 1 (Top) Synthesis of the PTM radical precursor 1HHH based on a Friedel–Crafts condensation at high temperature and pressure. (Bottom) Synthesis of radical 1˙ from its hydrocarbon precursor 1HHH, through the carbanionic derivative 1−−. | ||
The high persistence of PTM radicals has allowed functionalizing them at the meta and para positions of the aromatic rings. Thanks to the efforts of Ballester and coworkers during the 70s and 80s, these radicals were functionalized using a great variety of reactions,26 which in some cases required extreme conditions. One of the first applications of these radicals was the spin labelling of amino acids and peptides.27PTM functionalization has also allowed the use of such radicals as building blocks of molecular materials with specific properties. Thus, PTM radicals have been used as constitutive units of mixed valence systems that present intramolecular electron transfer phenomena,28 bistable molecular switches,29,30 and new valence tautomeric systems,31 as well as of push–pull compounds with non-linear optical (NLO) properties.32 The particular structural and electronic characteristics of PTM radicals have permitted, not only to obtain organic open-shell dendrimers with high-spin ground states and low-spin excited states, inaccessible even at room temperature,33,34 but also to functionalize them with one or more carboxylic groups. These radicals have been used as molecular synthons to build up open-shell supramolecular structures, either with a purely organic or a metal–organic nature, which show relevant porous and magnetic properties.35 More recently, on the search for new molecular spintronic systems, PTM radicals have also been grafted on surfaces exhibiting interesting electronic transport properties.36
PTM radicals are also interesting because they are electroactive species from which it is possible to generate the corresponding carbanionic and carbocationic species. Indeed, as shown in Fig. 2, the cyclic voltammetry of radical 1˙ exhibits two reversible waves that correspond to the oxidation and reduction of this radical, giving rise to the corresponding carbanion 1−− and carbocation 1++, which are also quite stable species both in solution and in solid state. The reduction of PTM radicals at low potentials turns out to be more feasible than the corresponding oxidation that requires rather high potentials. Such a fact makes this type of radicals excellent electron-acceptor units in push–pull systems with NLO properties and/or to achieve intramolecular electron transfer in molecular nanowires. Electroactivity of PTM radicals can be used for making switchable multifunctional molecular materials since the magnetic, optical and electronic properties of these open-shell molecules can be switched on and off with an external electrochemical stimuli.
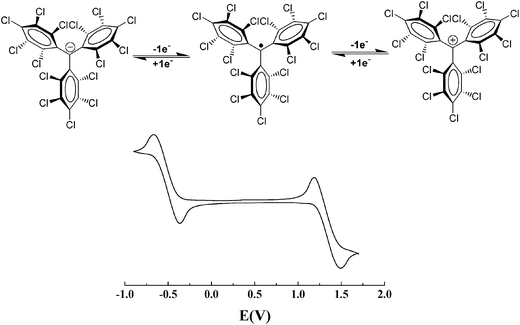 | ||
| Fig. 2 Cyclic voltammogram of radical 1˙ in CH2Cl2, with 0.1 M n-Bu4NPF6 (vs.Ag/AgCl) showing the reversible formation of two different ionic species that correspond to the oxidation and reduction of this radical. | ||
1.2 Nitroxide and verdazyl radicals
Nitroxide, also named as aminoxyl, and verdazyl radicals are two important families of radicals that have been used as building blocks for magnetic materials. In fact, nitroxide and α-nitronyl nitroxide radicals were the first radicals used to prepare molecular materials.37 The second family of radicals used for the same purpose is an emerging one, based on the verdazyl moiety. In both cases, the unpaired electron is located in an antibonding π*-orbital. The delocalisation of the electron on several atoms stabilises the π*-orbital (SOMO) and thereby enhances the stability of the radical. In the field of molecular magnetism, the most studied nitroxide-based radicals are tert-butyl nitroxides (Scheme 2a),38α-nitronyl- and α-imino-nitroxides (Scheme 2b and c). In the last two radicals, the unpaired electron is delocalised on a nitronyl (Scheme 2b) or an imine groups (Scheme 2c) although in both radicals the four methyl groups play a role of hindering the approaching of any reactive species that may destroy its radical character. In the verdazyl family, the radical is stabilised by delocalisation over the four nitrogen atoms of the six membered heterocycle (Scheme 2d).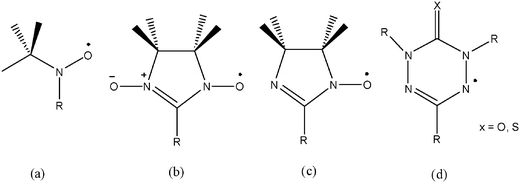 | ||
| Scheme 2 Structure of (a) tert-butylnitroxide, (b) α-nitronyl nitroxide, (c) α-imino nitroxide and (d) verdazyl radicals. | ||
To obtain magnetic materials by using these radicals, one can rely on a wide variety of bond types ranging from strong covalent ones to weak intermolecular ones. Moreover, the heteroatoms of the radicals can be used as ligation sites towards metal ions leading to coordination compounds with organic and inorganic spins bearing strong exchange interactions between the radical and the metal ions. In the case of verdazyl, the coordination ability of the nitrogen atoms of the radical strongly depends on the choice of the R substituent. In α-nitronyl nitroxides, the unpaired electron is equally delocalized on both N–O groups as revealed by EPR spectra. Thus, α-nitronyl nitroxides are characterized by the EPR signal consisting of five lines with an intensity ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:2:1 due to the coupling of the single electron with two equivalent nitrogen atoms (Fig. 3). This kind of electron delocalization makes such radicals ideal building blocks for constructing solids formed by extended chains. The different alternatives for packing such molecules in the solid state brings to a variety of intermolecular interactions between the π-orbitals of the radicals which controls the magnetic properties of the resulting materials. Finally, the functions brought by the R group can introduce complementary bonding pathways that in some cases control also the rich magnetism of this family of extensively used molecules.
:2:3:2:1 due to the coupling of the single electron with two equivalent nitrogen atoms (Fig. 3). This kind of electron delocalization makes such radicals ideal building blocks for constructing solids formed by extended chains. The different alternatives for packing such molecules in the solid state brings to a variety of intermolecular interactions between the π-orbitals of the radicals which controls the magnetic properties of the resulting materials. Finally, the functions brought by the R group can introduce complementary bonding pathways that in some cases control also the rich magnetism of this family of extensively used molecules.
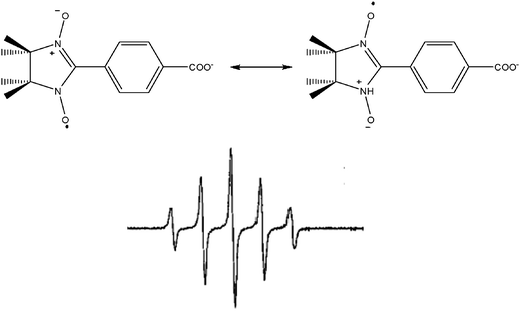 | ||
| Fig. 3 Structure and EPR spectrum of a α-nitronyl nitroxide radical. | ||
The synthesis of these organic radicals follows a multi-step protocol where the key step is the condensation with an aldehyde leading to a ring closure (Scheme 3). This is well documented for both α-nitronyl nitroxides39 and verdazyl40 derivatives. The first steps for both kind of radicals involve the synthesis of bis(hydroxylamino)-2,3-dimethylbutane and the bis(hydrazide)carbonic acid derivatives, respectively. As shown in Scheme 3, the two last steps are common to both types of radicals since they request the condensation with the aldehyde bearing a R substituent followed by the oxidation of the resulting molecule.
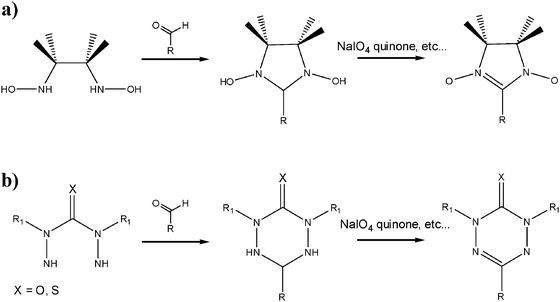 | ||
| Scheme 3 Synthetic pathways for obtaining (a) α-nitronyl nitroxide and (b) verdazyl radicals. | ||
Although many radicals of both families were obtained in the late-1960s, they underwent a huge revival in molecular magnetism after the discovery of the first example of a crystalline purely organic magnet for the p-nitrophenyl α-nitronyl-nitroxide derivative (TC = 0.6 K)41 and of a bisnitroxide biradical with a higher critical temperature (1.48 K).42 Since then hundreds of nitroxides, α-nitronyl-nitroxide and verdazyl derivatives have been synthesised and tested for their solid state properties and suitability as ligands for metal coordination.
1.3 Phenalenyl radicals
Phenalenyl is a hydrocarbon radical with an unpaired electron extensively delocalized on the planar π-conjugated skeleton. The electronic structure can be significantly modulated and perturbed by the choice of the nature and position of their substituents. These features are intrinsic in nature for spin delocalized radical systems, that are difficult to realize in spin localized ones, such as in TEMPO and α-nitronyl nitroxide derivatives. Taking advantage of the extensively delocalized spin on a large π-conjugated molecular skeleton, various novel and intriguing properties, that were previously unknown, have been realized. Phenalenyl is the smallest odd-alternant π-conjugated hydrocarbon neutral radical with a fused-polycyclic planar structure and at the same time in the smallest open-shell graphene-like derivative. As can be interpreted by its resonance structures, most of the spin density of phenalenyl exists in its α positions. This feature is well corroborated by the distribution of the non-bonding molecular orbital. Furthermore, phenalenyl radicals exhibit a high amphoteric redox ability confered by the non-bonding molecular orbital providing to the corresponding anion, neutral radical and cation species huge thermodynamical stabilities (Fig. 4).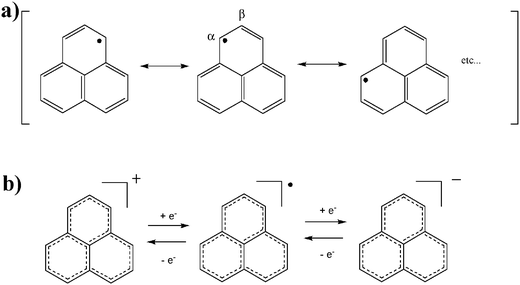 | ||
| Fig. 4 Resonance structures (a) and amphoteric redox processes (b) of phenalenyl radicals. | ||
As the first approach for the stabilization of phenalenyl radicals, the introduction of alkyl groups, such as methyl and trimethylene groups, were carried out. Murata et al. claimed an electronic effect of electron donating and electron accepting groups attached to the phenalenyl skeleton.43 In 1999, these authors synthesized the 2,5,8-tri-tert-butylphenalenyl radical having three tert-butyl groups at the β positions and succeeded for the first time in the isolation of a phenalenyl radical in the solid state in air.44 The decomposition temperature of such a radical under argon atmosphere is 232 °C.
Besides detailed physico-chemical studies, continuous efforts for the stabilization of the phenalenyl derivatives by chemical modifications have enriched its chemistry and have given place to the phenalenyl-based materials. These studies developed exotic magnetic and optical properties by the introduction of substituents into the skeleton and by the extension of its π-conjugated system. Among them one can mention the enhancement of spin multiplicities and redox abilities, the switching of the electronic structures depending on redox states, the creation of single component organic (semi)conductors and their application as electrode active materials in secondary batteries. In this review, recent significant progress in neutral radicals based on the phenalenyl system will be also described.
1.4 Dithiazolyl radicals
Another important family of radicals that has caught a great deal of attention are the dithiazolyl derivatives because of their remarkable electric and magnetic properties in solid state. In more recent years there has been an enormous body of work devoted to the systematic development of these sulfur–nitrogen radicals.45 The thiazyl moiety (–S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) has a three electron π bond with the third electron in a π* orbital. The collective properties of thiazyl moiety are the foundation behind an incredibly diverse array of structure types which have open shell configurations and yet qualify as persistent or stable. Thiazyl radicals have been actively studied since the 1970s and are now well established from both fundamental and applied materials perspectives.
N–) has a three electron π bond with the third electron in a π* orbital. The collective properties of thiazyl moiety are the foundation behind an incredibly diverse array of structure types which have open shell configurations and yet qualify as persistent or stable. Thiazyl radicals have been actively studied since the 1970s and are now well established from both fundamental and applied materials perspectives.
Fundamental exploration of the chemistry of these C/N/S rings by Banister led to the observation and characterisation of many thiazyl-based radicals such as those schematized in Fig. 5, which are derived by exploitation of the isoelectronic analogy between S+, N and R–C.46 Overviews of the considerable synthetic developments made in this area have been presented by Oakley47,48 and Chivers,49 among others, built on an idea originally proposed by Haddon.50
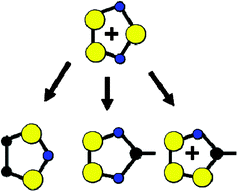 | ||
| Fig. 5 Isoelectronic members of the large family of five-membered heterocyclic thiazyl radicals. | ||
All these thiazyl radicals have been utilised in the design of molecular magnets,51 conductors and switchable materials.52 Regarding the magnetic studies, they have shown that not only a number of thiazyl radicals exhibit bulk magnetic order but also exhibit a bistability in which radicals are able to undergo solid state transformations between diamagnetic and paramagnetic phases. These physical properties make them potential materials in the design of molecular switches. More recent developments include the formation of organic–inorganic composite materials in which thiazyl radicals coordinate to paramagnetic metal complexes with a strong magnetic communication observed between paramagnetic transition metal ions and the coordinated thiazyl radical.
Regarding the magnetic properties of dithiadiazolyl radicals, it is worth mentioning the importance of the dimerization processes that this family of radicals entail. For example, the molecular enthalpy for 1,2,3,5-dithiadiazolyl radicals is ca. 35 kJ mol−1 which is comparable to hydrogen-bonding interactions. Given the prevalence of hydrogen bonding motifs in most compounds containing hydrogen bond donors and acceptors, it is perhaps unsurprising that the majority of 1,2,3,5-dithiadiazolyl radicals form spin-paired π*–π* dimers in the solid state.53 Indeed, the symmetry of the singly occupied molecular orbital permits numerous modes of association and many have been identified in the solid state (Fig. 6).
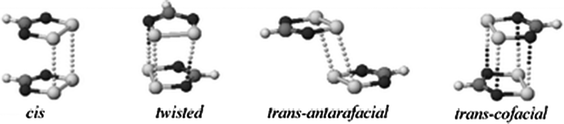 | ||
| Fig. 6 Modes of association of 1,2,3,5-dithiadiazolyl radicals in the solid state; (a) cis-cofacial, (b) twisted; (c) trans-antarafacial; (d) trans-cofacial. | ||
This review focuses predominantly on two families of thiazyl derivatives that have been used as building blocks of functional molecular materials whose physical properties have been studied extensively; the dithiadiazolyl (RCNSSN) and dithiazolyl (RCSNSCR) radicals which are very interesting as molecular building-blocks of magnetic materials.
2. Functional molecular materials based on organic radicals
Much of the current interest in persistent organic radicals arises from the fundamental structure and bonding issues that naturally arise with this class of compounds. There are many areas of chemistry that have taken advantage of the properties afforded by the specific combination of open-shell configuration and chemical stability. Firstly, persistent free radicals have long been used as reporter molecules to obtain structural, dynamic, and reactivity information using electron paramagnetic resonance (EPR) spectroscopy. Techniques such as spin labelling and spin trapping54 and, more recently, EPR imaging55 provide a wealth of information on systems into which persistent radicals have been introduced. Second, there have been widespread efforts nowadays aimed at developing new molecular materials with technologically relevant properties based on stable organic radicals, since they are excellent building blocks simply by the virtue of having unpaired electrons.1c This interest arises from their expectations in future emerging technologies as well as to improve those already existing nowadays.56,57 In this regard, molecular materials based on persistent organic radicals are attracting much attention because they exhibit most of the important physical properties traditionally considered to be only of atom-based inorganic solids—metals and metal oxides—like conductivity and magnetism. But moreover, the properties of such molecular materials can be easily modulated and tuned by small structural modifications of the skeletons of their building blocks using conventional organic synthetic methodologies. This aspect justifies the optimism offered by the molecular approach for the development of novel functional materials. In this regard molecular materials exhibiting two or more properties (magnetic, optical or electrical) can be prepared. For instance, the so-called chiral magnets in which the coupling or synergism between chirality and magnetism may result in the observation of novel physical properties. Switchable magnetic molecular solids, are materials exhibiting bistability at the molecular level and therefore their magnetic properties can be changed by the application of an external stimuli. Due to these switchable properties such materials are potentially useful for technological applications in molecular electronics or information storage.Another class of multifunctional materials are those exhibiting a coexistence or juxtaposition, rather than synergism, of two properties. These materials are generally hybrid materials formed by two networks, where each network furnishes distinct physical properties. Now there is also interest in the preparation of systems where both properties come form the same molecular building block. The latter materials have set the possibility to achieve more relevant goals, including potential applications in spintronics and nanoelectronics. The achievement of such goals will need the development of feasible processing techniques for a convenient organization of the molecules as thin films or size controlled nanostructures. To achieve these goals, the dual-function molecules must be functionalized to favour self-assembling processes, thus generating the derived supramolecular organizations. The evolution of molecular magnetism from materials toward applications in molecular electronics, medicine and other areas, together with the trend towards nanoscale materials/phenomena, expands the multidiscilinarity of the field.
These new type of materials or devices may exhibit different combinations of properties such as magnetism/conductivity, magnetism/optical or conductivity/NLO properties58 and they can have a purely organic or a metal–organic radical nature. The presence of two properties in the same crystal lattice, or in a molecularly functionalized surface or even better in the same molecule, might result, if there is a synergism between them, in new physical phenomena and novel applications that are difficult to achieve in conventional inorganic solids. The design of such multifunctional molecular systems able to change their functions by an external stimuli—like strain, pressure, temperature, light, and electric field—results in more appealing systems because of their potential applications as molecular-scale switches in nanotechnology.59 A molecular-scale switch is a fundamental component of any true molecular magnetic/electronic/photonic device. To achieve a real switching effect, the molecule must be stable in two (or more) states exhibiting very different properties which may be interconverted reversibly in response to an external stimuli, such as redox, temperature, pressure or irradiation. To date, several molecular switches exhibiting changes in one property, such as colour,60 luminescence,61 optical nonlinearity,62 or magnetic properties,63 have been reported. However, only very recently the number of useful properties being simultaneously modulated on a bistable molecule-based system has been extended to three properties (electrical, optical, and magnetic) although in this case the changes in the properties have an intermolecular origin.64
Persistent organic radicals have become nice examples of molecular building blocks for obtaining such multifunctional molecular materials since they permit to combine the intrinsic magnetic characteristics of these molecules with optical, conducting and electrochemical properties.
2.1 Magnetic materials based on organic radicals
The design of molecular materials with relevant magnetic properties based on organic radicals has been one of the major challenges of the last decades, especially of those with a purely organic nature. Due to the light nature of elements that generally compose organic compounds, only the isotropic magnetic exchange interactions determine the magnetic behaviour at temperatures well above 0.1 K. Other kinds of magnetic interactions, such as hyperfine or spin–orbit interactions, as well as sources of magnetic anisotropies can be considered as negligible above this temperature in most open-shell organic materials. Consequently, in practice magnetic organic systems behave magnetically isotropically and they may be described at zero applied magnetic field by the effective spin Hamiltonian approach, that takes the form of eqn (1):| H = −2ΣJijSiSj | (1) |
Most organic magnetic materials exhibit paramagnetic behaviours at high temperatures where the spins of the material behave independently of each other without showing any preferential orientation with respect to the nuclear framework where they are located. However, as the temperature is decreased, the exchange interactions become comparable with the thermal energy of the system and the nearest neighbouring spins tend to align in accordance with the signs of their Jij parameters appearing either as a short- or a long-range ordering of the spins in accordance with the structured dimensionality of the solid. Alignments of spins can be propagated along 1-, 2-, or 3-dimensions of the solid but only if the alignment occurs in 3 spatial dimensions can a real long-range magnetic ordering appear in such organic systems. In this case the solid exhibits a cooperative magnetic property below its critical temperature (TC) behaving as a bulk magnet where all spins are oriented along the same direction dictated by the external magnetic field; a direction which has no relationship with the nuclei of the molecules due to the magnetic isotropic characteristics of organic radicals. Keeping in mind the characteristics previously described, the design of organic molecular materials showing bulk magnetic properties involves three main aspects or steps that must be taken into consideration carefully: (i) the persistence of the spin-containing building blocks, (ii) the coupling routes or magnetic interaction mechanisms between the nearest neighbouring spin-containing building blocks, and (iii) the propagation of the magnetic interactions along the 3 dimensions of the material.
Persistent organic radicals like, triphenylmethyl, α-nitronyl nitroxide, verdazyl or dithazolyl radicals, have been extensively used as the constituent building blocks of molecular magnetic materials. Regarding the coupling routes, a FM interaction between them may result only if an orthogonal arrangement with zero overall overlap integral between the two singly occupied molecular orbitals (SOMOs) of the two interacting subunits is produced. On the other hand, if such an integral is non-zero, the resulting magnetic interaction would be AFM for non-degenerate orbitals. Magnetic interactions can take place either between neighbouring molecules or, in the case that there are two (or more) spin-containing units in the same molecule, intramolecularly. Intermolecular interactions among organic radicals are mainly determined by the isotropic exchange interactions between the unpaired electrons located on the SOMOs of the nearest-neighbouring molecules. The construction of magnetic materials requires that the structural subunits exhibit non-covalent interactions suitable to be controlled in a predictable manner enabling to promote right isotropic exchange interactions between the nearest-neighbouring open-shell molecules. Up to now, the different types of non-covalent intermolecular interactions that have been used for the assembly of such molecular subunits are hydrogen-bonding, transition-metal ligation, π–π stack-type alignment and the ionic bridging of ion radicals by their counterions. In case that the assembly of spin-containing molecules produces the proper magnetic interactions and they propagate along the 3 dimensions, the material shall exhibit a bulk cooperative magnetic property.
Since the pioneering work of Cambi and Szego on spin-crossover magnetic materials in the 1930,65 it has been a holy grail for chemists and physicists to create pure organic magnetic materials. In 1991, Tamura et al. reported the existence of ferromagnetic intermolecular interactions in crystals of p-nitrophenyl α-nitronyl nitroxide, with a critical temperature to a ferromagnetically ordered state below 0.65 K.66 Since then, different strategies have been applied to design molecules or organic free radicals to get ferromagnetic ordering at the highest temperature possible. However, the best result ever achieved was the observation of a long-range magnetic ordering at 35 K in a sulfur–nitrogen radical.67 Up to now, much work has been devoted to the pursuit of organic magnetic materials and many relevant reviews are available.68,69
Intramolecular magnetic interactions play a major role in high-spin organic molecules.55 Thus, when a molecule has two high-lying orthogonal orbitals, close or equal in energy, with two less electrons than necessary for a closed-shell structure, its ground state becomes a triplet, as dictated by Hund's rule. However, the actual nature of the interaction (FM or AFM) depends upon the symmetry and topology of the degenerate or nearly degenerate SOMO's orbitals. One of the general approaches to obtain high-spin organic compounds is based on conjugated polyradicals with topologically polarized α-spins. These compounds are designed using appropriate ferromagnetic coupling units able to align in parallel the spins of a pair (or more) of radical centres connected through such a unit.
In this section we focus our attention in the developments and studies of magnetic materials derived from persistent organic radicals of the families of triphenylmethyl, dithiazolyl, verdazyl and α-nitronyl nitroxide radicals. First, discrete molecules with high spin in their ground state composed of open-shell subunits will be described in detail. Then, molecular solids formed by persistent radicals that have extended structures will be described.
2.1.1.1 Aromatic units as magnetic couplers. m-Phenylene has become the most widely used ferromagnetic organic coupler based on an aromatic unit because it is highly dependable. This unit was used as ferromagnetic organic coupler of triphenylmethyl radical centres in line with the efforts to increase the number of persistent and stable high-spin radicals. Indeed, since the Schlenk's hydrocarbon, first prepared in 1915, high-spin alignments in ground states have been successfully demonstrated for several 1,3-phenylene connected carbenes70 and radicals based on triarylmethyl,71nitrogen-centered72 and aminoxyl units.38b The work done in this direction included the synthesis, characterization and study of physico-chemical properties of molecules with two, three, or more triarylmethyl subunits, that are called bis-, tris-, or poly-triarylic systems or, more generically, extended triarylmethyl systems. A remarkable family of such extended triarylmethyl systems was obtained by Rajca et al. who synthesized the polyradical 2 with the highest spin multiplicity (S = 13) even reported.14PTM polyradicals were also obtained using m-phenylene units as ferromagnetic couplers, the quartet 3 being the species that shows the largest stability ever reported and has a high-spin ground state (S = 3/2) with a low-spin excited state inaccessible even at room temperature.71,73 The synthesis of larger polyradicals like 4 with hyperbranched dendritic nature was also attempted. However, the numerous experimental problems found during the synthesis and purification, due to the presence of abundant magnetic defects and numerous atropisomeric forms, suggested that the high-spin species 4 is the limit of generation for this dendritic radical series.
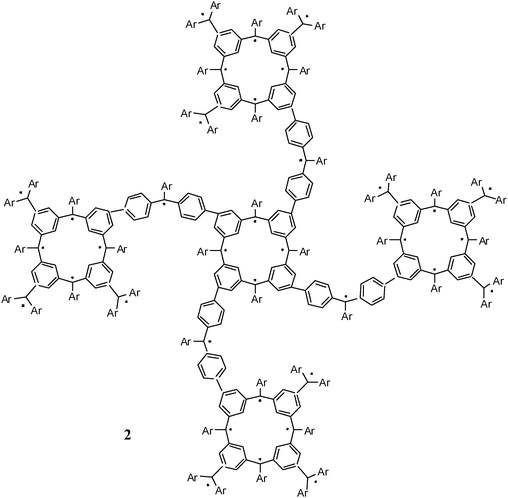
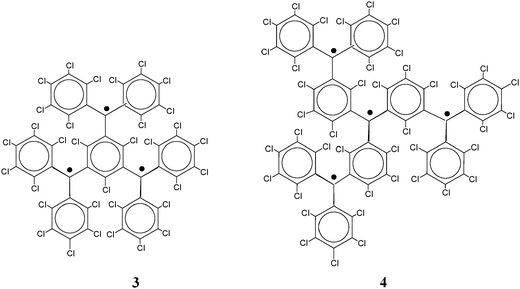
A large number of reported persistent organic diradicals are based on odd electron moieties connected to a central benzene ring. As mentioned before, two radicals linked “meta on a benzene” generally lead to ferromagnetic coupled radicals, while para-linked systems lead to antiferromagnetically coupled systems. With this in mind, several di- and polyradiclas derived from verdazyls have been constructed by the attachment of verdazyl moieties at either one of the nitrogen atoms or the C3 carbon. N linked verdazyl diradicals may be expected to exhibit stronger magnetic coupling because of the substantial spin density on the N atoms.
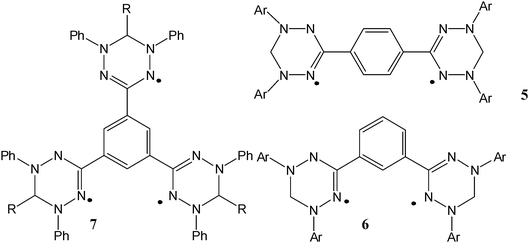
Indeed, for radicals 5–7magnetic susceptibility measurements showed temperature independent moments revealing their spins are weakly or non-interacting. Nevertheless, EPR spectra at low temperatures (77 K) show a typical triplet signal, indicating that a triplet electron state was at least thermally accessible. These results contradicted a previous report claiming that both species possess triplet ground states with exchange energies in excess of 300 K.1f,74
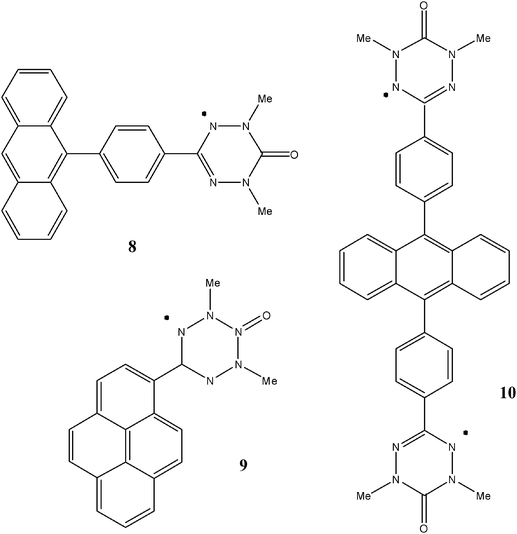
There are other verdazyl-based heterospin systems that represent a somewhat different concept towards magnetic coupling, namely the observation of intermolecular exchange in photoinduced excited states.75 The ground states of compounds 8 and 10 only possess verdazyl-based spins, and consequently have EPR spectra typical of such units. Similarly, Curie analysis of diradical 9 in its ground state suggests a very weak antiferromagnetic exchange (J ≈ −2 cm−1). However, upon continuous irradiation, time-resolved EPR spectroscopy (TREPR) permits the observation of signals readily assigned to spin quartet (S = 3/2) species in the case of 8 or 9, and to a spin quintet (S = 2) for 10. These arise from the photoexcitation of the anthracene or pyrene moieties to produce excited state triplets, which can then interact ferromagnetically with the S = ½ verdazyl radical units. These molecules can be considered as prototypes for organic photo-magnetic materials.
2.1.1.2 Metallocene units as ferromagnetic couplers. Metallocenes have been used very successfully as building blocks of molecular solids promoting intermolecular magnetic interactions.76 Based on their electronic structures and rich chemistry, metallocenes appear to be promising candidates to promote also intramolecular ferromagnetic interactions. With this objective, diradical 11 consisting of two PTM radical sub-units connected by a 1,1′-metallocenylene bridge was constructed. The particular structure and topology of 11 were expected to lead to a non-negligible spin density on the metallocene moiety, making feasible the magnetic coupling between the two organic radical subunits. In addition, the location of both radical units far away from each other avoided any possibility of having intramolecular through-space contacts and, consequently, a significant direct through-space magnetic interaction.
The EPR spectrum of isolated molecules of diradical 11 in a frozen toluene/CH2Cl2 (1:1) mixture shows the characteristic fine structure of a triplet species in which the forbidden Δms = ±2 transition, characteristic of triplet species, is also observed at the half-field region of the spectrum. The intensity of such a signal (Ipp) was measured in the 4–100 K temperature region, and its temperature dependence indicated that the triplet is the ground state with a thermally accessible excited singlet state separated by an energy of +10 K (7 cm−1) (Fig. 7). In conclusion, the metallocene unit in 11 was shown to effectively transmit a ferromagnetic interaction between two PTM radicals.77 This result was justified by ZINDO/1 semiempirical calculations78 where it was observed that the SOMO orbitals are almost degenerate and coextensive, justifying the high-spin ground state experimentally observed.
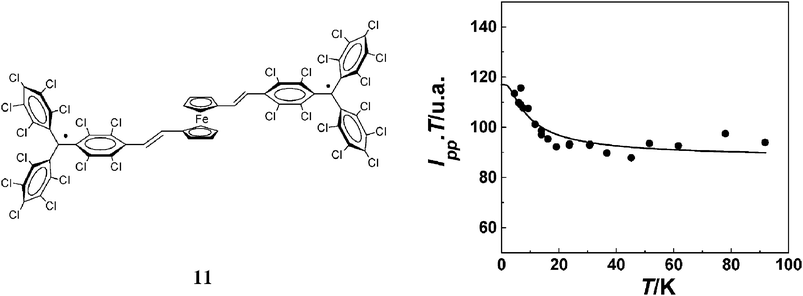 | ||
| Fig. 7 Temperature dependence of Ipp·T of diradical 11. The data represent the experimental data and the continuous lines the fit of experimental data to the Bleaney–Bowers equation. Reproduced from ref. 78 with permission from Elsevier. | ||
Ferrocene was also used to connect two verdazyl radical units promoting their magnetic interactions as observed for diradical 12 in solution where the two spins were magnetically weakly interacting. However such a magnetic interaction was strongly modified in the solid state because of the particular conformation adopted by the molecule in the condensed phase in which the two radical units are π-dimerized inside each molecule producing a complete antiferromagnetic spin pairing that leads to a diamagnetic behaviour for the solid (Fig. 8). Such a π-dimerization is unusual for verdazyl radicals since they do not show trend to dimerize this molecule being the unique example of a verdazyl dimer in which the two rings are eclipsed and have a centroid-to-centroid distance of 3.15 Å which is well within the van der Waals distance and significantly shorter than the cp-to-cp distance of 3.30 Å.79
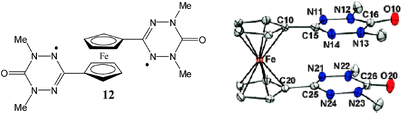 | ||
| Fig. 8 ORTEP drawing (50% probability ellipsoids) of 12 radical. Reproduced from ref. 79 with permission from the American Chemical Society. | ||
2.1.1.3 Hydrogen bonds as magnetic couplers. Investigation of the transmission of magnetic interactions through hydrogen bonds has been carried out with triphenylmethyl and nitroxide radicals functionalized with carboxylic acid groups at the para positions, as in radicals 13 and 14. The general tendency of carboxylic acids to form dimers, both in solution and in solid state, suggested that 13 and 14 were ideal systems to show whether the propagation of the magnetic exchange is efficient at a long distance as well as to determine its FM or AFM nature. An extensive magnetic study, both in solution and in the solid state, with radicals 13 and 14 showed that magnetic exchange interactions are transmitted efficiently through the hydrogen bonds (Fig. 9 and 10).80
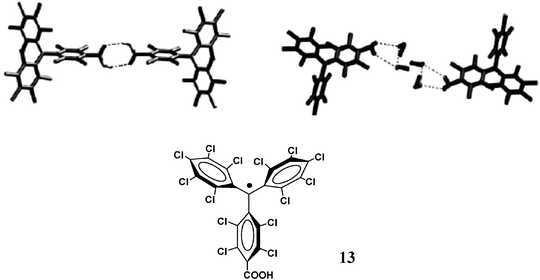 | ||
| Fig. 9 View of hydrogen-bonded dimers present in the α-phase (left) and β-phase (right) of radical 13. Reproduced from ref. 80 with permission from Wiley-VCH. | ||
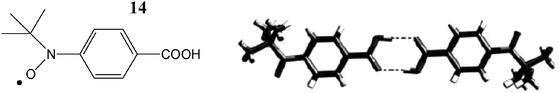 | ||
| Fig. 10 View of the hydrogen-bonded dimer formed by the radical 14. Reproduced from ref. 80 with permission from Wiley-VCH. | ||
In the case of 14, low temperature EPR experiments showed a weak ferromagnetic interaction between the two radicals of the dimer. In contrast low-temperature EPR experiments performed in solution with radical 13 did not lead to any conclusive results, since larger supramolecular aggregates are formed by weak non-covalent interactions, like Cl⋯Cl and π⋯π, distinct from the initially expected hydrogen bonds, which masked the properties of isolated dimers in soltuion. In order to overcome this drawback, studies with radical 13 in solid state were performed. Radical 13 crystallized in two polymorphic phases, the α- and β-phases, depending on the solvent used for the crystallization. As shown in Fig. 9, in an aprotic solvent like CH2Cl2, the α-phase is favoured in which the radicals form nearly isolated R22(8) hydrogen-bonded dimers while in the presence of EtOH the solvent molecules are intercalated by H-bonds among the two radicals of dimeric entities pushing them far away. Moreover, the bulkiness of the two radicals prevents in both phases short contacts between the regions with high spin densities inside each radical subunit of the dimers.
Solid-state magnetic measurements of the two phases of radical 13 provided direct evidence about the nature and strengths of magnetic interactions inside the dimeric entities. Magnetic data of the α-phase clearly show the presence of weak ferromagnetic interactions while such interactions are opposite in the β-phase. The suppression of the ferromagnetic interactions in the β-phase, in which direct H-bonded radical dimers are not present, gave further evidence that the H-bonded dimers are the species uniquely responsible for this interaction. Therefore, such results demonstrated that magnetic exchange through hydrogen-bonded bridges can occur at a long distance even in the cases of well-delocalized radicals. This positive result enables us to use such a coupling tool to prepare materials with extended hydrogen bonding networks showing a long range magnetic order that will be described later on.
2.1.1.4 Metal ions as magnetic couplers. α-Nitronyl nitroxides were the first radical building-blocks used as bridging ligands of metal ions from which the role of metal ions as magnetic coupler was demonstrated. This was done by Rey, Gatteschi and coworkers who pioneered the field using α-nitronyl nitroxide and α-imino nitroxide radicals to bind transition metal ions coining the term metal–radical approach for such a kind of magnetic coupling strategy;81 reporting several examples of such complexes.82
The success of the metal–radical approach is related to the magnetic coupling that takes place in such cases between the metal ions and the spin active organic spacers as well as to the versatility of the chemistry of such radicals. In α-nitronyl nitroxides, independently of the nature of the R substituent, there are already two N–O ligation sites available for metal coordination yielding linear metal–radical chains. When these N–O sites connect the spin center units of the radicals with a metal ion with a large S value it leads to a ferrimagnetic chain due to the antiparallel orientation of such spins and their lack of compensation. However, the nitroxides are rather poor ligands, and this sets large limitations on the nature of the additional ligands that are compatible with them. According to the weak basicity of the N–O groups of this kind of radicals, compared to regular diamagnetic ligands, strong acidic partners were needed for ligation to the radical. A simple way to enhance the acidity of the metal ions is to bind them with additional electron-attracting ligands like fluorinated diketonates and especially the perfluorinated (1,1,1,5,5,5)hexafluoroacetylacetonate (hfac) (Scheme 4).83
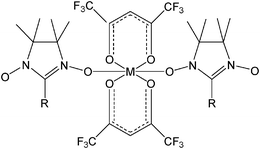 | ||
| Scheme 4 Structure of α-nitroyl nitroxide radicals complexed with M(hfac)2 units commonly used in the metal–radical strategy. | ||
When a nitroxide radical binds through its oxygen atoms to a paramagnetic metal ion, two cases can occur: (i) the spins orient antiparallel to each other or (ii) the spins orient preferentially parallel to each other. The limit of the first case is the formation of a strong covalent bond which totally pairs the spins. Otherwise, if the levels of different total spin multiplicity are thermally populated, an antiferromagnetically coupled system is formed. The second case corresponds to the ferromagnetic coupling. The sign of the coupling of a nitroxide directly bound to a metal ion is easily predicted on the basis of orbital overlap considerations. Thus, if the orbitals containing the unpaired electrons on the metal and the radical have significant overlap, the coupling is antiferromagnetic, while if they are orthogonal to each other, the coupling is ferromagnetic.
In Fig. 11 we show some possible relative orientations of magnetic orbitals leading to ferro- or antiferromagnetic couplings. When the two orbitals are orthogonal to each other the coupling is ferromagnetic as occurs if the radical coordinates in an axial or an equatorial coordination site; a situation that occurs in many copper(II) complexes. Another two cases that may occur in complexes of α-nitronyl nitroxides with nickel(II), cobalt(II), and manganese(II), leading to strong antiferromagnetic coupling are also depicted in Fig. 11.84
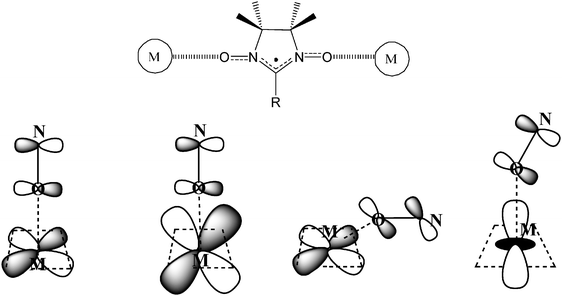 | ||
| Fig. 11 Schemes of possible interactions between the magnetic orbitals of a nitroxide unit and a metal ion. | ||
A great deal of work has also been made with the so-called metal–radical approach combining paramagnetic metal ions and several kinds of radicals, like nitroxides and triphenylmethyl radicals, as ligating sites.85 In particular, functionalized PTM radicals were specially appealing for this purpose. Indeed, carboxylic acid substituted triphenylmethyl radical 13 was an excellent coordinating ligand to obtain new metal complexes with different coordinative geometries following the metal–radical approach. Thus, the reaction of radical 13 with Cu(II) metal ions gave a series of metal–radical complexes like 15, 16, 17 and 18 which were structurally characterized (Scheme 5).86
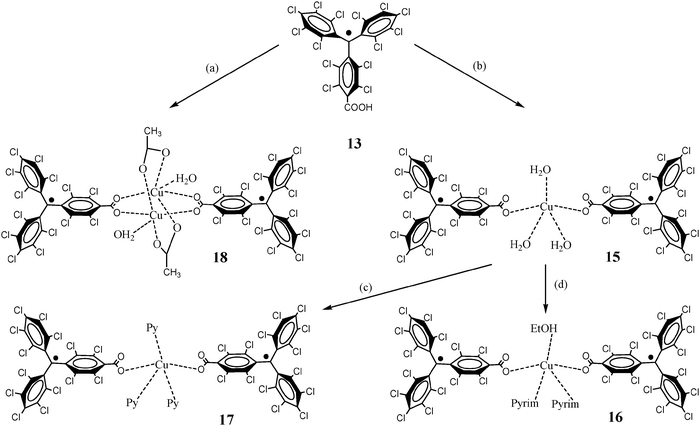 | ||
| Scheme 5 Synthesis of copper complexes based on the PTM radical 13. Reagents and conditions: (a) 13/Cu2(O2CMe)4·H2O (4:1) in EtOH/H2O; (b) 13/Cu2(O2CMe)4·H2O (2:1) in EtOH/H2O; (c) 15/pyridine (excess) in EtOH/n-hexane/THF; (d) 15/pyrimidine (excess) in EtOH/n-hexane/THF. | ||
In complexes 15–17 the copper(II) ions are coordinated to two radical units in a slightly distorted square planar pyramidal geometries, while complex 18 shows a paddle-wheel copper(II) dimeric structure. Each Cu ion in the latter complex is coordinated to one O atom of a H2O molecule and with four O atoms of different carboxylate groups; two of which belong to two radicals and another two to two further acetate ions. The exchange coupling between the copper ions and the radical sub-units is antiferromagnetic for the three complexes 15, 16 and 17, following all of them, the behaviour of a linear three-spin model with a J/kB ≈ −20 K. Magnetic properties of complex 18 resulted more interesting since it is one of the scarce examples of a spin-frustrated discrete system composed of organic radicals and metal ions. In this case, experimental data were fitted to a magnetic model based on a symmetrical butterfly arrangement to give a copper(II)–copper(II) exchange coupling of J/kB = −350 K and a copper(II)–radical exchange coupling of J/kB = −21.3 K, similar to that observed for the copper(II)–radical interactions in complexes 15–17 (Fig. 12).87 Following the same strategy a series of complexes with Zn(II), Ni(II), and Co(II) ions were also synthesized. The magnetic behaviours of the resulting complexes showed the presence of aniferromagnetic interactions of J/kB ≈ −20 K between the radical subunits and these first-row transition ions.87–89 Finally, lanthanide metal ion complexes, such as europium(III), and the carboxylate radical derived from 13 were also reported.90 In the same line, coordination capabilities of the monosulfonate radical using different metals were also explored. In this case the direct coordination of the organic radical through the sulfonate group provides weaker antiferromangetic couplings than with the carboxylated PTM derivatives.91
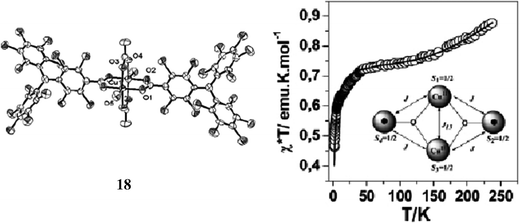 | ||
| Fig. 12 (a) ORTEP view of complex 18. (b) Temperature dependence of the magnetic susceptibilities. The inset shows the schematic arrangements of the metal ions and organic radicals. Reproduced from ref. 87 with permission from the Royal Society of Chemistry. | ||
The above described results demonstrate that carboxylic-substituted PTM radicals turned out to be excellent coordinating paramagnetic ligands able to transmit efficiently moderate metal–radical antiferromagnetic interactions.92 This fact together with the capability of H-bonds to transmit magnetic interactions and link molecules with H-bonding acceptor and donor groups opened the door to the obtaining of magnetic extended systems based on PTM radicals functionalized with several carboxylate/carboxylic groups. These magnetic extended systems resulted in two new families of open-framework materials with either a metal–organic or purely organic nature; named as metal–organic radical open-frameworks (MOROFs) or purely organic radical open-frameworks (POROFs), respectively. A detailed overview of both families is reported below.
The use of bridges or units as magnetic couplers described in the previous section have enabled to prepare radical-based magnetic materials with high structural and magnetic dimensionalities like one-dimensional chains, two-dimensional sheets, and three dimensional networks, using persistent organic radicals as building blocks. This possibility was exploited for more than 20 years to obtain a large variety of systems with 1D, 2D and 3D structures using different kinds of organic free radicals. Remarkable examples of them are detailed below.83
2.1.2.1 Magnetic materials based on nitroxide radicals. Following the metal–radical approach, Rey, Gatteschi and coworkers initiated this field succeeding in the synthesis of one-dimensional (1D) metal–nitroxide compounds with magnet like behaviour.98 These early results demonstrated that coordination of nitroxide radicals with metal ions was an efficient tool for the rational design of molecule based magnetic materials. However, because of the one-dimensional character of the metal–nitroxide network of these compounds, bulk magnetization was observed only at low temperature (<20 K). It soon became evident that any increase of the Curie temperature had to deal with increasing the dimensionality of the metal nitroxide network. In order to do so one way was to add extra coordination sites on the α-nitronyl nitroxide99 or to use polynitroxide radicals.100 Another alternative was to increase the number of radicals around each metal ion. This requires to get rid of the bulky and electron-withdrawing ancillary ligands, such as the hfac. Thus, using the 2-(2-pyridyl)-substituted α-nitronyl nitroxide free radical (NITPy) it was demonstrated with metal chloride complexes that this could be achieved if the N–O group of the α-nitronyl nitroxide was incorporated in a chelate ring in order to force the metal coordination.101 Since then, numerous metal complexes of transition metal and lanthanide ions have been reported with chelating α-nitronyl or α-imino nitroxide radicals grafted on various groups, such as pyridine102 imidazole,103benzimidazole,104bipyridine,105phenanthroline106 or triazole.107 These complexes exhibited different topologies and dimensionalities including 1D and 2D architectures.
Among the large number of compounds prepared up to now with this strategy, the quasi one-dimensional magnetic chains, made with metal ions with a high uniaxial anisotropy, deserve a special comment since they behave in most cases as single chain magnets (SCM).108 These materials present very characteristic magnetic properties among the molecule-based magnetic materials. They show unique magnetic hysteresis and slow dynamics for the relaxation properties109 on the bases of their molecular structures and magnetic anisotropy. Nowadays, these materials are focus of interest because they are good examples where quantum phenomenon can be observed on a macroscale110 and at the same time potential candidates for future high density data storage materials111–113 due to the Ising anisotropy of these metal ions.
In principle slow relaxation of the magnetization can be expected in one-dimensional (1D) materials, as suggested by Glauber in 1963, if two rather stringent conditions are met: (1) the ratio of the magnetic interaction within the chain, J, and that between chains, J′, must be rather high, larger than 100 and (2) the material must behave as a 1D Ising ferro- or ferrimagnet. The first example of a single chain magnet was described by Gatteschi et al. with the [Co(hfac)2-(NITPhOMe)], where NITPhOMe = 4′-methoxyphenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide.114 The spin of the metal ion and that of the radical are strongly interacting and below 20 K ac susceptibility measurements show the appearance of an out-of-phase signal that is strongly dependent on the frequency; as observed in single molecular magnets.115 Furthermore, the compound exhibits a magnetic hysteresis in the absence of three-dimensional magnetic order. The Ising nature of the chain has been attributed to the presence of cobalt(II) as this ion is known to yield large magnetic anisotropy effects when it is hexacoordinated.116
The phenomenology of this type of systems is very rich. In fact, compounds comprising α-nitronyl nitroxide radicals and transition metal or rare earths give rise to a multitude of interesting phenomena, including relatively high temperature transitions to three-dimensional ferromagnetic order,117 single molecule magnets (SMM)118 and SCM119 behaviours and chiral magnetic phases.120–122
The first family of rare-earth-based single chain magnets reported had the general formula [M(hfac)3(NITPhOPh)], where M = Eu, Gd, Tb, Ho, Er, or Yb, and NITPhOPh is the α-nitronyl nitroxide radical: 2,4′-benzoxo-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide. These complexes are isostructural and exhibit isolated chains containing lanthanide centers stabilized by intrachain supramolecular interactions (Fig. 13).117,123 Both static and dynamic magnetic properties of the whole family have been reported exhibiting complex behaviour. By comparing static and dynamic properties, it is evidenced the interplay of anisotropy and SCM behaviour and it has been shown that in some cases, the chains are to be assigned to a dynamical regime dominated by finite size effects.113 Thus, it has been shown that by using rare earths to build SCMs the single ion contribution to the energy barrier can significantly compensate the weak exchange interaction that characterizes 4f ions. Moreover, the wide variability of the pre-exponential τ0 factor, observed in this isostructural family, evidences its key role in the magnetization dynamics of this class of materials.
![(a) View of the crystal structure of [Dy(hfac)3(NITPhOPh)] at 150 K. Fluorine and hydrogen atoms were omitted for clarity. Coordination bonds are represented as segmented. All other chains are found to be isostructural. (b) Temperature dependence of the imaginary χ′′ (top) and real χ′ (bottom) components of the ac susceptibility measured in zero applied field for 10 logarithmically spaced frequencies in the range of 110 Hz (red) to 20 000 Hz (blue). Lines are guides to the eye. Reproduced from ref. 113 with permission from the American Chemical Society.](/image/article/2012/CS/c1cs15165g/c1cs15165g-f13.gif) | ||
| Fig. 13 (a) View of the crystal structure of [Dy(hfac)3(NITPhOPh)] at 150 K. Fluorine and hydrogen atoms were omitted for clarity. Coordination bonds are represented as segmented. All other chains are found to be isostructural. (b) Temperature dependence of the imaginary χ′′ (top) and real χ′ (bottom) components of the ac susceptibility measured in zero applied field for 10 logarithmically spaced frequencies in the range of 110 Hz (red) to 20000 Hz (blue). Lines are guides to the eye. Reproduced from ref. 113 with permission from the American Chemical Society. | ||
The magnetic anisotropy of an exchange-coupled system depends not only on the individual anisotropies of the metal ions but also on the relative orientation of the local axes. The analysis of the magnetic anisotropy and exchange interaction in the above described SCM having a low symmetry environment around the anisotropic lanthanide ions has been done using angle-resolved single crystal magnetometry. In particular, the orientation of the principal axes of the magnetization for the model system [Dy(hfac)3(NITPhOPh)] has been obtained.124,125Fig. 14 shows that the susceptibility is highly dependent on the angle, particularly for the rotation around the crystallographic b′ axis, where magnetization values span the range from 0.74 to 12.9 emu mol−1. Temperature-dependent measurements have also been performed over the temperature range 2–15 K (Fig. 14) showing that χ depends strongly on T, with a 10-fold reduction of the maximum values in going from 2 to 15 K, independent of the rotation axis. On the contrary, the minimum values remain roughly the same, and no shift of the positions of the extrema is observed.
![(a) Angular variation of the molar susceptibility (M/H ratio) of [Dy(hfac)3(NITPhOPh)] at T = 2.5 K along the three axes of the laboratory frame. At θ = 0 and 90°, respectively, H is parallel to b′ and c* for the rotation around a and c* and a for the b′ rotation, and a and b′ for c* rotation. (b) Angular variation of the susceptibility of [Dy(hfac)3(NITPhOPh)] measured at different temperatures, as reported in the color scale, from 2 (blue) to 15 K (red). Rotations were performed around the a axis, and θ = 0 and 90° correspond to measurements along b′ and c*, respectively. Reproduced from ref. 124 with permission from the American Chemical Society.](/image/article/2012/CS/c1cs15165g/c1cs15165g-f14.gif) | ||
| Fig. 14 (a) Angular variation of the molar susceptibility (M/H ratio) of [Dy(hfac)3(NITPhOPh)] at T = 2.5 K along the three axes of the laboratory frame. At θ = 0 and 90°, respectively, H is parallel to b′ and c* for the rotation around a and c* and a for the b′ rotation, and a and b′ for c* rotation. (b) Angular variation of the susceptibility of [Dy(hfac)3(NITPhOPh)] measured at different temperatures, as reported in the color scale, from 2 (blue) to 15 K (red). Rotations were performed around the a axis, and θ = 0 and 90° correspond to measurements along b′ and c*, respectively. Reproduced from ref. 124 with permission from the American Chemical Society. | ||
Other kinds of interesting magnetic phenomena observed in 1D chains made with the metal radical approach are the intrinsic magnetic avalanches and the giant coercivity as observed, respectively, in the [Mn(hfac)2(R)-3MLNN] ferromagnetic chain126 and in the complex [Co(hfac)2·BPNN] where 3MLNN = methyl[3-(4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazolyl-1-oxy-3-oxide) phenoxy]-2-propionate and BPNN = p-butoxyphenyl α-nitronyl nitroxide. Remarkable is the latter127 complex that exhibits a coercive field of HC = 52 kOe (4.1 MA m−1) at 6 K, which is much larger than those previously reported for other molecular materials128 and even of some commercial permanent magnets, like SmCo5 (44 kOe at room temperature) and Nd2Fe14B (19 kOe at room temperature). Up to now, the coordination polymer 19 holds the largest coercive field among hard magnets ever known (Fig. 15).
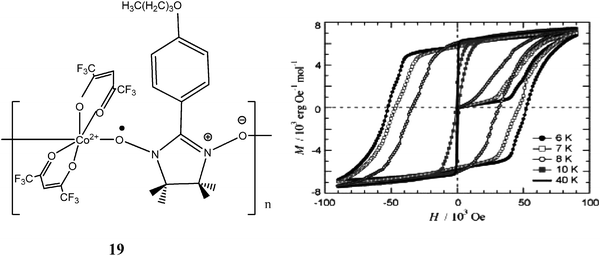 | ||
| Fig. 15 Hysteresis loops of polycrystalline complex 19. Reproduced from ref. 127 with permission from the American Chemical Society. | ||
The metal–radical approach with α-nitroxide radicals has brought also the opportunity to prepare multifunctional magnetic compounds. As an example we can mention the chiral molecule-based magnet, [Co(hfac)2]·BNO* (20); where BNO* is the chiral triplet 1,3-bis(N-tert-butyl-N-oxylamino)-5-{1′-methyl-1′-[2′′-(S)-methylbutoxy]ethyl}benzene.129 In this example, the presence of a pure enantiomeric molecule induces the formation of chiral 1-D chains that are packed parallel to each other in the noncentrosymmetric P1 space group. This compound exhibits in the temperature range of 4–30 K four magnetic states: paramagnetic; antiferromagnetic; forced ferrimagnetic and a metastable field-induced ferrimagnetic. In the paramagnetic state (T > 20 K), it presents a short-range antiferromagnetic interaction between Co ion and nitroxide radical and has a minimum value of χmT at 220 K. The Weiss temperature estimated in the temperature range 220–300 K is found to be 89.9 K. At 20 K, an antiferromagnetic long-range ordering is established. In the temperature range of 4–20 K, the isothermal magnetization curve shows a spin-flip transition to the forced ferrimagnetic state at around 850 Oe (Fig. 16). Below 4 K, this compound enters into a field-induced ferrimagnetic state, which is metastable and is stabilized by the Ising character of the Co ion. In the low-temperature state, the material becomes a very hard magnet with wide hysteresis loop whose coercive field reaches 25 kOe at 2 K.
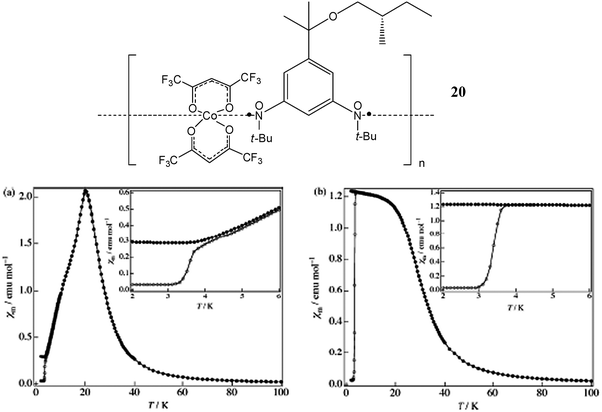 | ||
| Fig. 16 Temperature dependence of molar magnetic susceptibility measured at 500 Oe (a) and 5000 Oe (b) employing the ZFC (open circles) and FC (closed circles) mode. The inset shows a magnified view at the temperature region 2–6 K. Reproduced from ref. 129 with permission from the American Chemical Society. | ||
2.1.2.2 Magnetic materials based on verdazyl radicals. The use of metal–radical approach with verdazyl radicals has also led to magnetic materials with extended structures although it is still in its early stages of development. The first coordination complexes of a verdazyl were reported by Brook et al. using Cu(I) and bisverdazyl diradicals.130 Each diradical simultaneously binds to two Cu(I) ions in a bis-bidentate mode, and the copper ion coordination sphere is completed by bridging halides giving rise to the one-dimensional coordination polymers represented schematically by complexes 21. The magnetic susceptibility of the three complexes is qualitatively very similar, with maxima in the χ versus T plots indicative of the presence of a low-dimensional antiferromagnetic system. The data were modelled based on an alternating one-dimensional chain consisting of two exchange coupling parameters, where one was ascribed to the intramolecular spin–spin interaction within each diradical, while the other is ascribed to intrachain magnetic interactions between radicals and Cu(II) ions.
The current methodology in course to explore this virgin field is closely following the one developed for the α-nitroxide-based radicals over the years.131–134
2.1.2.3 Magnetic materials based on triphenylmethyl radicals. The results presented in the preceding Section 2.1.1.4 demonstrate that carboxylic-substituted triphenylmethyl radicals are excellent coordinating paramagnetic ligands able to transmit efficiently moderate metal–radical antiferromagnetic interactions.92 This fact, together with the ability of H-bonds to transmit magnetic interactions, make possible the obtaining of magnetic extended systems based on polytopic triphenylmethyl radicals functionalized with several carboxylate/carboxylic acid groups. These magnetic extended systems resulted in two families of open-framework materials with extended structures with either a metal–organic or purely organic nature; named as metal–organic radical open-frameworks (MOROFs) or purely organic radical open-frameworks (POROFs), respectively.
By using the di-and tri-carboxylic acid radicals 22 and 23, a series of metal–organic radical open-frameworks (MOROF-n) have been prepared. Interestingly, the crystal structure of Cu3(22)2(py)6(EtOH)2(H2O) (MOROF-1) reveals a 2-D honeycomb (6,3) network composed of assemblies of six metal units and six radicals connected between them. The resulting planar layers are piled up forming very large hexagonal pores which measure 3.1 and 2.8 nm between opposite vertices.135 Moreover, complex MOROF-1 shows additional channels with square and rectangular shapes in the perpendicular directions with estimated sizes of 0.5 × 0.5 and 0.7 × 0.3 nm, respectively. The interconnected pores originate solvent-accessible voids in the crystal structure that amount to 65% of the total unit cell volume. This extremely large void volume is responsible for the exotic properties (vide infra) of this material.
According to the capability of carboxylic substituted PTM radicals to transmit efficiently the magnetic interactions, each radical ligand was able to magnetically bridge three Cu(II) ions and, therefore, to extend the magnetic interactions across the infinite layers, giving rise to ferrimagnetic 2D layers. Indeed, as shown in Fig. 17, the smooth decrease of the χT values below 250 K is a clear signature of the presence of antiferromagnetic couplings between nearest-neighbouring Cu(II) ions and radical ligands within the 2-D layers. The minimum of χT corresponds to a short range order state where the spins of adjacent magnetic centres are antiparallel, provided there is no net compensation due to the 3:2 stoichiometry of the Cu(II) ions and radical units. The increase of χT at lower temperatures indicates an increase of the correlation length of the macroscopic magnetic moment of the 2D as randomising thermal effects are reduced yielding finally to a long range magnetic ordering below 4 K. This bulk magnetic ordering was confirmed by the magnetization curve at 2 K since it exhibits a very rapid increase, as expected for a bulk magnet, although no significant hysteretic behaviour was observed because of the lack of anisotropic sources in the two types of magnetic units. The magnetization value increases much more smoothly at higher fields up to a saturation value of 1.2 μB, which is very close to that expected for a saturated system with an S = ½ magnetic ground state. Thus, this molecular material can be considered as a soft magnet with an overall magnetic ordering below a TC of 4 K.
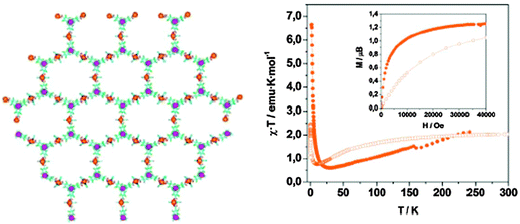 | ||
| Fig. 17 Left: a honeycomb (6,3) layer showing the organization of the copper ions (orange spheres) and the central methyl carbon (magenta spheres) of the 24 radicals. Right: χT as a function of the temperature for as-synthesized (●) and evacuated (○) MOROF-1. Inset: magnetic field dependence of the magnetization at 2 K. Reproduced from ref. 135 with permission from the Nature Publishing Group. | ||
A second remarkable feature of MOROF-1 is the reversible “shrinking–breathing” of its solid state structure upon solvent uptake and release. Indeed, when MOROF-1 is removed from the mother liquor solution in which it was crystallized, and exposed to air at room temperature the crystalline material loses the ethanol and water guest molecules very rapidly becoming an amorphous material with a volume decrease of around 30% (Fig. 18). Even more interesting is the fact that the evacuated sample of MOROF-1 recovers its original crystallinity and up to 90% of its original size after exposure to liquid or vapour EtOH solvent. The quasi-reversible process also occurs with MeOH, but not with other organic solvents, therefore showing a large selectivity for the smallest alcohols. Thus, apparently MOROF-1 behaves as a sponge-like magnet material selective towards MeOH and EtOH. The chemical, structural and morphological quasi-reversibility is also accompanied by changes in the magnetic properties that are macroscopically detected. Magnetic properties of an evacuated amorphous sample of MOROF-1 show similar magnetic behaviour to that shown by the as-synthesized crystals of MOROF-1 with the exception that its critical temperature is one order of magnitude lower, around 0.4 K.
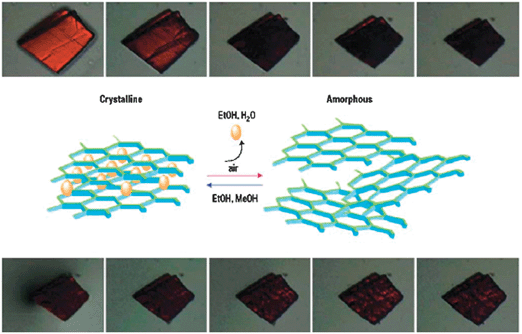 | ||
| Fig. 18 Images of a single crystal of MOROF-1 followed with an optical microscope. The top series show the “shrinking” process, in which a crystal of MOROF-1 exposed to the air experiences a volume decrease of around 30%. In the lower series, the same crystal exposed against ethanol liquid begins to swell. The scheme represents the structural changes of MOROF-1 in contact or not with ethanol or methanol solvent. Reproduced from ref. 135 with permission from the Nature Publishing Group. | ||
Other two supramolecular Co(II)-based coordination polymers, [Co(23)(4,4-bpy)(H2O)3]·6EtOH·2H2O (MOROF-2) and Co6(23)4(py)17(H2O)4-(EtOH) (MOROF-3) have also been reported.89,136 The first coordination polymer shows a paramagnetic non-interpenetrated supramolecular network with an unprecedented (63)·(68.81) topology. Additionally, MOROF-2 exhibits helical nanochannels of dimensions 13.2 × 9.4 Å and a void volume of 55% along with antiferromagnetic interactions. In contrast, MOROF-3 exhibits a (6,3)-helical network with large 17.5 × 6.8 Å 1-D channels and a bulk magnetic ordering below 1.8 K. This was the first reported example of a nanochannel-like structure that in addition to exhibiting an unusual (6,3)-helical coordination network showed mixed ferro- and antiferromagnetic interaction between the Co(II) ions and the radicals.
The hexacarboxylic PTM radical 24 with six carboxylic acid groups at the meta positions of the three phenyl rings have been complexed with Cu(II) ions to obtain a three-dimensional (3-D) coordination polymer. The resulting 3-D structure can be described as two interpenetrating cubic nets. Magnetic properties of this metal radical organic open-framework have been studied showing unexpected metal–radical ferromagnetic interactions.137
Carboxylic acid functionalized triphenylmethyl radicals can also self-assemble into purely organic radical open framework (POROFs) with porous structures and interesting magnetic properties. Thus, radicals 22 and 23 were used to obtain extended H-bonded networks, which in principle would show such a combination of properties. The advantages of these radicals were considerable: (i) their trigonal symmetry provided a typical template for getting channels held together by hydrogen bonds through the carboxylic groups; (ii) the molecular bulkiness and rigidity of PTM radicals was expected to prevent a close packing of molecular units; and (iii) besides their structural control, hydrogen bonds have been shown to favour magnetic coupling between bound radical molecules (Section 2.1.1.3).80 These expectations were confirmed in the H-bonded self-assemblies of 22 and 23 by the formation of robust porous extended networks. Indeed, the dicarboxylic 22 radical crystallizes in a robust porous 2-D extended network (POROF-1) with weak antiferromagnetic interactions.138 The framework shows 1-D tunnels formed by narrowed polar windows (≈5 Å in diameter) and larger hydrophobic cavities, where a sphere of 10 Å in diameter can fit inside them. The combination of supercages and windows gives way to solvent accessible voids in the crystal structure that amount up to 31% of the total volume of the unit cell. Furthermore, it is remarkable that the structural rigidity of the framework permits the evacuation of the guest n-hexane solvent molecules at 373 K without collapsing the self-assembled molecules.
More recently, radical 23 provided another robust porous 2-D H-bonded magnet (POROF-2) where the stacking of the H-bonded radical molecules generates a 3-D structure that exhibits tubular highly hydrophilic channels with a 5.2 Å in diameter with solvent-accessible voids that amount up to 15% of the total unit cell volume. The lack of guest solvent molecules within the channels and its structural rigidity up to 573 K are excellent conditions to explore the porosity properties of this porous material. No less remarkable are the magnetic properties of this material, which orders ferromagnetically at very low temperatures.138
The magnetic properties of structures based on the hexacarboxylic acid-substituted PTM radical 24 have also been reported. The study of its self-assembly in solid state revealed that it is possible, through a tuning of the crystallization conditions, to control the structural dimensionality going from 0- to 2-D solid state structures in which the association of radicals through direct hydrogen bonds to form layers gives rise to the presence of weak ferromagnetic intermolecular interactions between radicals.139
2.1.2.4 Magnetic materials based on dithiazolyl radicals. Finally we want to mention other magnetic extended materials based on radicals where 3-D architectures are glued by weak interactions like S⋯S or π–π interactions, different from hydrogen bonds or metal coordination bonds. This is the case of free radicals based upon heavier p-block elements such as S and Se that self-assemble in solid state exhibiting higher magnetic ordering temperatures.52 In the family of dithiazolyl radicals the complex [BBDTA] [GaCl4], where BBDTA = benzo(bis-1,3,2-dithiazolyl), orders as a ferromagnet below TC = 6.7 K,140 whereas the neutral radical benzo-1,3,2-dithiazolyl (BDTA) exhibits antiferromagnetism below 11 K when rapidly cooled.141 A selenathiazolyl radical has recently been reported to order as a ferromagnet below TC = 12.3 K and its isoelectronic analogues generated by substitution of S for Se have been shown to order at TC = 17 K.142 A series of thia or selenazyl derivatives have also been shown either to order as weak ferromagnets with TC = 18 K and 27 K,143 or to present marked differences in conductivity, which can be enhanced by applying physical pressure. These studies underline the effect that inclusion of heavier heteroatoms enhances not only charge transport but also magnetic exchange interactions.144
Materials based on S/N radicals are of fundamental interest because, in the solid state, some of these radicals behave as magnets with critical temperatures as high as 36 K.145 Nevertheless, as exemplified by the decrease of the TC from 36 K145 to 1.3 K146 upon substitution of the cyano by a nitro group in XC6F4CNSSN (X = CN, NO2) radicals, the magnetic properties of these compounds are highly sensitive to the intermolecular interactions between the radicals and, hence, to the organisation of the radicals in the solid state. It is therefore important to analyse the crystallographic structures of such compounds in order to determine the parameters governing the organisation of the molecules in the solid state.
2.2 Materials with electronic properties based on organic radicals
The interplay between magnetic and conducting properties is becoming a key issue in modern materials science.147 In particular, the investigation of genuine organic materials where conductivity and magnetism coexist is an indispensable ingredient for the development of molecular spin-electronic (or “spintronic”) devices.148Molecular spintronics149 is a new and exciting field that studies the spin transport phenomena 150 in molecular systems which has potential applications such as magnetic recording and memory devices.151–154 In traditional charge-based electronics, equal populations of spin-up and spin-down charge carriers exist and both contribute equally to the associated transport phenomena. In contrast, charge carriers can be induced to undergo a spin preference (i.e. through spin–spin interactions or by the presence of magnetic fields) resulting in spin-polarized current. Electronic devices based on carriers with spin preference have advantages for nonvolatile memory storaging since they can decrease electrical power consumption, increasing the speed of data processing, enabling also to both store and process data on a single chip. Recent concern about spintronics has promoted the operation of a spin valve,155 a spin filter,156detection of a spin-polarized current,157 exploration of a memory device and a quantum computer. 158–160
There are two kinds of such systems enabling to study IET processes which both involve organic radicals as electroactive units (Scheme 6). One consisting of an electron donor (D) and an electron acceptor (A) group as electroactive units linked by a bridge, called D–A dyads, and another composed of the same unit at both sides of the bridge but with different oxidation states, which are named as mixed-valence (M-V) compounds. M-V compounds and D–A dyads belonging to the Class II of Robin Day's classification163 are excellent candidates for IET studies since in them the extra electron (or hole) is localized on one of the extremes but it can move by an external stimuli, like temperature or light. In such species the rate and efficiency of IET can be easily followed and studied from the position, intensity and width of the so-called intervalence transition (IVT) band, also named charge-transfer band that usually appears at the near-infrared (NIR) region. From the position, intensity and width of such a IVT band, the IET can be easily characterized. In most cases, the energy required for the optically induced electron-transfer (Eop) is related with the activation barrier for the thermally induced electron-transfer (ΔG±), the free energy difference between both states (ΔG°) in the case of dyads or asymmetric M-V species, and the total reorganization energy value (λ). The latter energy is composed of two contributions: one inner contribution (λi) corresponding to the reorganization energy inside the molecule and another outer term (λo) that depends on the reorganization of the surrounding media. According to Marcus–Hush theory, the parameters controlling the optical and thermal induced IET processes are closely interrelated by eqn (2) and (3):
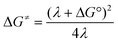 | (2) |
| Eop = λ + ΔG° | (3) |
 | ||
| Scheme 6 Schematic representation of M-V compounds (top) and D–A dyads (bottom). | ||
The effective electronic coupling matrix HAB (expressed in cm−1) between the adiabatic potential energy surface of the two states can be experimentally determined based on eqn (4), where r is the effective separation of the two electroactive sites (in Å), εmax is the maximum extinction coefficient (in M−1·cm−1) of the IVT band and νmax or Eop is the transition energy and Δν1/2 is the full-width at half-height (both in cm−1) of the IVT band.
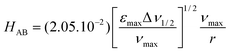 | (4) |
In this section IET phenomena observed in M-V and D–A dyads compounds derived from triphenylmethyl, phenalenyl and thiazyl radicals will be described providing examples of how the IET processes can be modulated by subtle changes of the external environment, such as solvent or temperature. We will also show a few examples of molecular systems based on these open-shell molecules that exhibit bistability phenomena controlled by the temperature and solvent. Efforts for the preparation of organic molecular components to construct molecule-based systems with spin-transportation properties, will be also presented focusing on the concept of a “spin-polarized donor”. Finally the possibility to functionalize surfaces and nanoparticles with radical molecules as spin-containing units towards their use in molecular spintronics will be also described.
2.2.1.1 Electron transfer in mixed valence species. It is worth mentioning that there are very few examples of organic M-V molecules exhibiting IET phenomena showing at the same time high-spin ground-states. Since the nature of the intramolecular magnetic couplings and IET phenomena are not at all independent of each other,164 the study of molecular systems which simultaneously present both phenomena is a task of particular interest.4–6,16 Organic M-V molecules with robust high-spin ground-states were accomplished for the first time using the triradical 3˙˙˙ which has the quartet (S = 3/2) as the ground-state (Scheme 7).13
 | ||
| Scheme 7 Mixed-valence species (3−−˙˙ and 32−2−˙ derived from triradical 3˙˙˙ and its trianionic 33−3− species. | ||
Quartet ranks among the most chemically and thermally stable organic high-spin molecules reported to date (see Section 2.1) and the derived mixed-valence species, like the biradical-anion 3−−˙˙ and the monoradical-dianion , obtained by its partial reduction, also showed a noticeable degree of chemical stability, allowing a reliable study of their electron transfer properties. Spectroelectrochemistry experiments performed with partially reduced solutions of quartet 3˙˙˙ showed two clear isosbestic points together with a progressive decrease of the radical band at 368 nm and a concomitant increase of the band corresponding to the anion chromophore at 508 nm.165 In addition to these changes, the appearance of an intense, and remarkably broad band centred around 2900 nm was observed. Throughout the reduction process the intensity of the latter band increased at the first reduction stages and then started disappearing only after reaching an average reduction state of two electrons per molecule; i.e. after the formation of 32−2−˙ species. This result confirms that the broad band is an IVT band due to the optically induced IET processes occurring in the M-V species 3−−˙˙ and 32−2−˙. 166EPR spectroscopy was used to characterize the magnetic properties of the M-V species 3−−˙˙ and 32−2−˙. For this purpose, CH2Cl2 solutions of partially reduced 3˙˙˙ were diluted with toluene and their spectra of the frozen glassy solutions were recorded. As expected, for the solution containing the M-V radical 32−2−˙, only a single line corresponding to the 32−2−˙ species was observed. In contrast, the solution containing 3−−˙˙ shows a signal with a fine structure characteristic of a non-axial triplet species superimposed to the signal of 3−−˙˙. The 3−−˙˙ species is also responsible for the structureless line observed at the half-field region corresponding to the forbidden Δms = ± 2 transition of a triplet species. In order to identify the ground state of 3−−˙˙, the temperature dependence of such a Δms = ±2 signal was studied revealing a triplet (S = 1) ground state with a thermally inaccessible singlet (S = 0) state that evidenced the robustness of such a high-spin molecule.
In order to study the influence of the bridge topology (or connectivity) on the IET phenomena in M-V wire-like systems diradicals 25 and 26 and their corresponding radical-anions 25−−˙ and 26−−˙ were also studied in detail using magnetic measurements, spectroelectrochemistry and variable temperature EPR.167
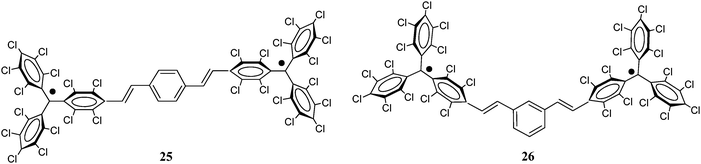
Magnetic susceptibility data of powder samples of diradicals 25 and 26 both follow the Curie–Weiss law with θ = −4 K and −0.4 K, respectively, indicating the presence of very weak intra- or inter-molecular antiferromagnetic interactions. The magnetic moments at room temperature for both diradicals are 2.42 μB indicating the existence of a non-interacting pair of doublets at this temperature. Experimental electronic coupling parameters, HAB, were determined for both systems by means of spectroelectrochemistry experiments. Thus, during the reduction of both diradicals the evolution of the resulting spectra was similar although only for the radical-anion 25−−˙ an IVT band, centred on 1400 nm, was observed. From the position and linewidth of this band it is possible to calculate the effective electronic coupling HAB between the two electroactive sites of 25 which was found to be HAB = 121 cm−1. The absence of any significant IVT band for radical-anion 26−−˙ is an indication that its effective electronic coupling is at least one order of magnitude smaller than in 25−−˙. The thermally activated IET was also studied by variable temperature EPR for radical-anions 25−−˙ and 26−−˙. Thus, the EPR spectrum of 25−−˙ at 200 K displays two symmetrical lines arising from the hyperfine coupling of the unpaired electron with one hydrogen atom of the ethylene moiety and the resulting 1H hyperfine coupling constant is very close to that of related monoradicals. This result clearly demonstrates that at this temperature the unpaired electron of radical-anion 25−−˙ is localized on the EPR timescale on only one-half of the molecule; i.e. on only one electroactive site. When the temperature is increased, a new central line in the EPR spectrum gradually emerges between the two initial ones. This evolution is consistent with the increase of the electron transfer on going from the slow-exchange limit at T < 200 K to the fast-exchange limit at T > 300 K due to a thermally activated IET between the two equivalent sites of this M-V species. At higher temperature, the unpaired electron of 25−−˙ is coupled with two equivalent 1H nuclei. The EPR spectra in the entire temperature range were simulated by using the IET rates, kth, and the resulting kth values were plotted against temperature using a linear Eyring plot [ln(kth/T) vs. 1/T] from which an energy barrier of the thermal electron transfer, ΔG±, of 0.117 eV was obtained.
The EPR spectrum of radical-anion 26−−˙ is unchanged in the temperature range of 150–300 K and appears always as two symmetrical lines arising from the hyperfine coupling of the unpaired electron with one hydrogen atom of the ethylene moiety with a 1H hyperfine coupling constant close to that of the corresponding monoradical-anion at low temperature. This is a clear indication that the extra electron of radical-anion 26−−˙ is always localized on the EPR timescale on one-half of the molecule regardless the temperature. This result can be ascribed to the localization of frontier orbital in the latter radical-anion 26−−˙, because of the meta connectivity of this non-Kekulé molecule.
Very recently we have also studied the influence of the bridge length on the IET phenomena with the series of M-V radical anions 27−−˙ (n = 1–5) derived from diradicals 27 by their partial reduction.168 From a temperature dependence study of the IET rate constants it was revealed the presence of two different regimes at low and high temperatures in which the mechanisms of electron tunnelling via superexchange and thermally activated hopping were competing. Both mechanisms occur to different extents, depending on the sizes of the radical anions, since the lengths of the bridges notable influence the tunnelling efficiency and the activation energy barriers of the hopping processes, the barriers diminishing when the lengths are increased. The nature of solvents also modifies the IET rates by means of the interactions between the bridges and the solvents. Also, in the shortest compounds n = 1 and 2, the IET induced optically through the superexchange mechanism was also observed by the exhibited intervalence bands, whose intensities decrease with the length of the bridge.
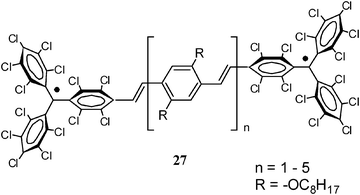
Mixed valence species derived from phenalenyl radicals showing IET have allowed us to prepare bistable systems taking advantage of the multicentre bonding nature of a phenalenyl π-dimer.64a,169,170,183 Haddon and co-workers reported that the ethyl- and butyl-substituted zwitterionic bisphenalenyl boron complexes 28 and 29 show magneto-optical-electronic bistability—that is, remarkable changes in magnetic, optical and electric properties as a function of the temperature (Fig. 19). Thus, M-V compounds form a π-dimer in the crystalline state, similar to that of the tri-t-butyl-phenalenyl in which the interplanar distance within the π-dimer increases from 3.2 Å in the temperature range 20–100 K to 3.3 Å in that between 173 and 293 K. This change is accompanied by a structural phase transition near 120–150 K and by an abrupt change in the magnetic and optical behaviour of the π-dimer. Such compounds exhibit a paramagnetic behaviour at high temperature while antiferromagnetic interactions are present at low temperature.
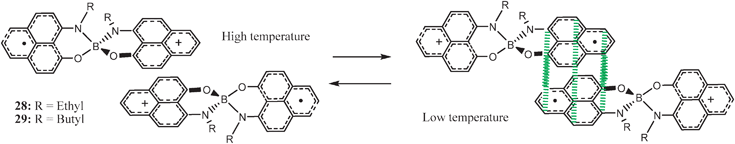 | ||
| Fig. 19 Structural change of intra-π-dimer interaction. Proposed mechanism for the phase transition of zwitterionic bisphenalenyl boron complexes 28 (R = ethyl) and 29 (R = butyl).64a The spins reside on the outer phenalenyl moieties at high temperatures and on the π-dimer moiety at low temperatures. | ||
Moreover, the butyl-substituted zwitterionic bisphenalenyl boron complex 29 is also able to switch between similar states as the ethyl substituted compound but in this case it does so with thermal hysteresis, that is, there is a finite temperature range within which the state of the compound depends on its history. 171
Thiazolyl radicals are other molecules generating M-V species that show bistability based on monomer–dimer transitions,172 where diamagnetic π-dimers are present at low temperature while unassociated paramagnetic radicals are favoured at high temperature. As an example we can mention the polycyclic radical 30 (Fig. 20) prepared by Oakley and coworkers. These authors have demonstrated that radical 30 (R1 = Et, R2 = F) crystallizes in a very unconventional way showing dimers with a four-centre and six-electron σ-type interaction (Fig. 20). This is an extremely rare mode of dimerization for a thiazyl radical, since σ-bond formation is in general the most conventional way for such radicals to dimerize. Even more unusual is that the σ-dimer breaks open at 380 K (TC↑) when increasing the temperature and does not reform until cooling to below 375 K (TC↓). Thus, there is a narrow window of monomer–dimer bistability based on σ-bond making and breaking. The dimer-to-monomer transition can also be induced by application of pressure. The differences in behaviour between 30 and its selenium containing variant which does not show the bistability, can be ascribed to a combination of molecular properties and cooperative effects.
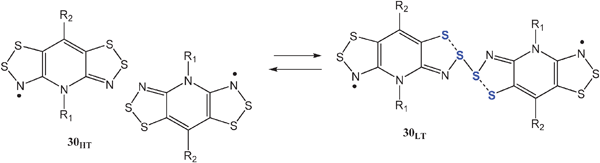 | ||
| Fig. 20 Hysteretic behaviour of dithiazolyl radicals. High-temperature form (30HT) and low-temperature form (30LT) of bis(1,2,3-dithiazolyl) radical 30 (R1 = Et, R2 = F). | ||
2.2.1.2 Electron transfer in donor–acceptor dyads. The excellent electron acceptor abilities of PTM radicals made that they were used as electron acceptor units in D–A dyads. One remarkable example of such D–A dyads is the compound 31 in which a PTM unit is covalently bonded through a vinylene spacer, to a ferrocene moiety that acts as an electron donor. Indeed 31 was shown to exist in solid state in two distinct electronic isomeric forms—one neutral form and another zwitterionic form with separated charges that may be reversibly interconverted.31
Variable temperature Mössbauer experiments (Fig. 21) show that this dyad exists in solid state in a neutral form at low temperature while the zwitterionic form is favoured at higher temperatures, this thermal conversion being completely reversible. Very recently, it has been found that the coexistence in a crystalline sample of both the neutral and zwitterionic forms of 31 is a result of the bistability of crystals induced by the stabilizing electrostatic intermolecular interactions developed in the charge-separated state.173 From this finding it is concluded that novel bistable compounds can be generated in crystals of largely neutral (closed- or open-shell) D–A molecules with attractive electrostatic intermolecular interactions that operate in the charge-separated state. This conclusion has opened a new avenue for the design of valence tautomeric molecules since the variety of valence tautomeric species is up to now mostly limited to transition metal complexes with quinone ligands.174
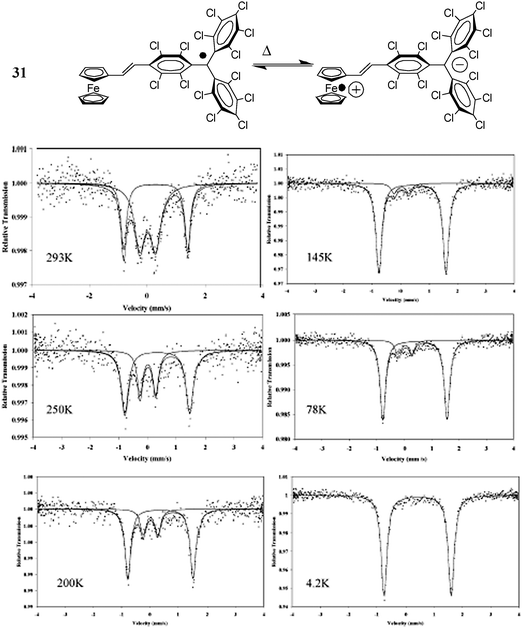 | ||
| Fig. 21 57Fe Mössbauer spectra of D–A dyad 31 in solid state as a function of temperature. Reproduced from ref. 31a with permission from the American Chemical Society. | ||
Solvent dependence of IET phenomena was studied using the D–A dyad 31 and the related species 32 containing a nonamethylferrocene group.175 Both compounds exhibit broad absorptions in the NIR region with band maxima appearing around 1000 and 1500 nm for 31 and 32, respectively. These bands correspond to the excitation of a neutral [DA] ground-state to the charge-separated [D+A−] state, indicative of an optically induced IET process. As shown in Fig. 22, both bands exhibit a large solvatochromic effect due to the different relative stabilization of neutral and charge-separated states. Actually, symmetric M-V compounds exhibit negligible ΔG° values due to their symmetric structures and therefore the IET are governed by the so-called Marcus-normal region energy conditions; where ΔG° is smaller than the λ term (see eqn (3)).176 On the other hand, asymmetric D–A dyads usually exhibit large ΔG° values, which may place them in the Marcus-inverted region that occurs when the ΔG° value is larger than λ.177 In this particular region, the IET process occurs via a different kinetic pathway and unusual features associated with the environment of D–A dyads are quite common.178
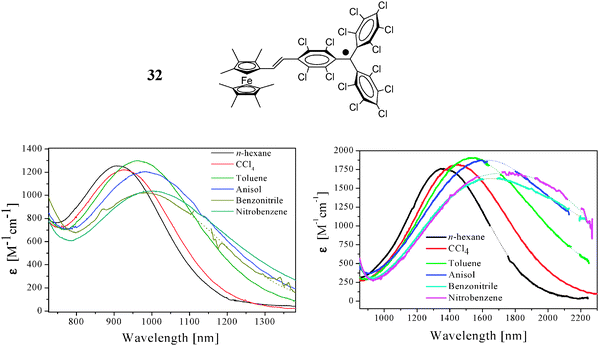 | ||
| Fig. 22 IVT bands of D–A dyads 31 (left) and 32 (right) in some selected solvents. Reproduced from ref. 175 with permission from the American Chemical Society. | ||
D–A dyads 31 and 32 show two reversible one-electron redox processes associated with the oxidation of the ferrocene and the reduction of the PTM subunits (Fig. 23). The solvent dependence of these redox processes was also investigated, allowing the determination of the free energy difference between the neutral and charge-separated states, ΔG°, in different solvents. The ΔG° values, along with the experimental Eopt spectroscopic data in different solvents, allow us to estimate, using the total energy balance Eopt = λ + ΔG° (Eq. 3), the total reorganization energy values, λ, and their solvent polarity dependence. Since ΔG° and λ are of the same order of magnitude but exhibit opposite trends in their solvent polarity dependence, a unique shift from the normal (ΔG° < λ) to the inverted Marcus region (ΔG° > λ) with the change in solvent polarity is found. This fact is of utmost importance since it indicates a crossing of the so-called inverted and normal Marcus regions, where striking differences in the electron transport mechanisms between the donor and acceptor moieties are present. Although it is known that solvent effects can influence strongly the relative values of ΔG° and λ terms in the context of IET in proteins and other biological environment,179 there were no previous examples of molecules that can be shifted from the Marcus-normal to the inverted region simply by changing the polarity of the solvent.
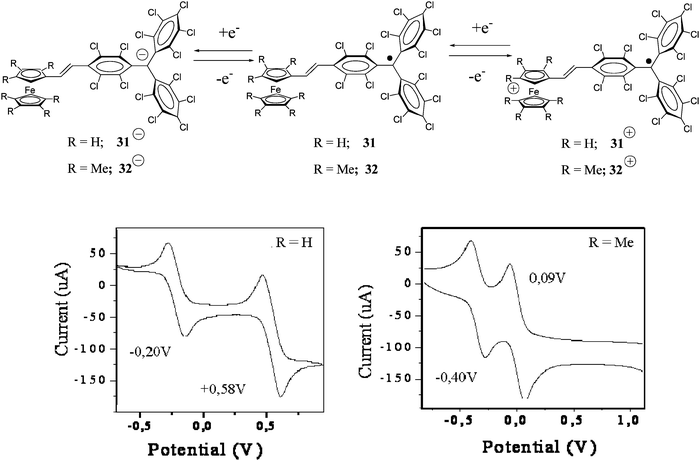 | ||
| Fig. 23 Top: reversible one-electron redox processes associated with the first oxidation and first reduction potentials of dyads 31 and 32. Bottom: cyclic voltammograms of dyads 31 and 32 in CH2Cl2 using (n-Bu4N)PF6 (0.1M) as electrolyte (vs.Ag/AgCl). Reproduced from ref. 175 with permission from the American Chemical Society. | ||
Another interesting family of D–A dyads, studied by Heckmann and Lambert180 consist of PTM radicals linked to different amine donor centres by various spacers. This family of D–A dyads are good model systems for the study of IET due to their uncharged nature in their ground-states and their high solubility in solvents with low polarity. A final example of D–A systems bearing PTM moieties is the C60–(PTM−)2 triad in which two PTM anion units acting in such a compound as electron donors are covalently bonded to a C60 that acts as an electron acceptor. For this triad, photoinduced IET can be confirmed to occur, both in polar and nonpolar solvents, from the quenching of the fluorescence intensities of the excited singlet state of the C60 moiety and of the PTM anion moiety occurring in the regions of 700–750 nm and 560–630 nm, respectively.181
D–A dyads derived from phenalenyl radicals also show interesting bistability phenomena in solution in which IET may take place. In such dyads two electronic isomeric forms can be tuned reversibly by means of an external stimuli such as solvent and temperature changes.182,183 In particular the compound 33 that consists of an 6-oxophenalenoxyl linked with a tetrathiafulvalene (TTF) unit was found to exist in a neutral radical form and a zwitterionic radical form which are in equilibrium (Fig. 24). This equilibrium can be reversibly shifted by a moderate change of the external environment, such as solvent and temperature in a solution, leading to a “spin center transfer”, associated with the IET, which is accompanied with deep colour changes.184
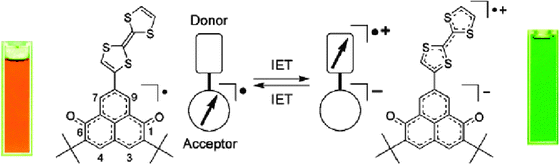 | ||
| Fig. 24 Color changes of D–A dyad 33 due to intramolecular electron transfer produced by a moderate change in the external environment. Reproduced from ref. 182a with permission from Wiley-VCH. | ||
This unconventional dynamic system involves two radical species, a neutral radical and a zwitterionic radical, and is induced by an effective and reversible IET. Thus, in dichloromethane it exists in the neutral radical form while in CF3CH2OH solution the zwitterionic radical form is favoured. The two forms are characterized by ESR spectroscopy (Fig. 25) exhibiting g factors very different depending on the polarity of the solvent used. Thus, 33 shows in CH2Cl2 and toluene a g factor of 2.0047 that is very different to the g factor of 2.0082 shown in CF3CH2OH demonstrating the occurrence of IET from the 6-oxophenalenoxyl moiety to the TTF moiety. This spin switching behaviour is accompanied with a solvatochromism from orange to dark green. Moreover, by controlling the ratio of the two solvents, a temperature dependent spin center transfer was also observed with 100% interconversion within a temperature range of 50 degrees. This thermochromic phenomenon was the first one observed in a purely organic open-shell molecular system, while it was well known for valence tautomeric metal complexes.174,185
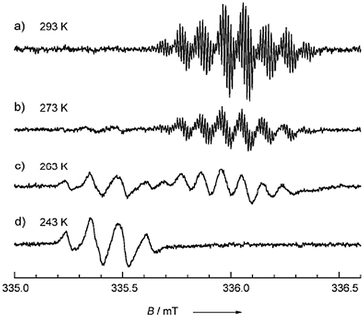 | ||
| Fig. 25 Temperature dependence of the liquid-phase EPR spectra of 33 in a CH2Cl2/CF3CH2OH (199:1) solution (7.7 × 10−5 M) at (a) 293 K, (b) 273 K, (c) 263 K, and (d) 243 K. Reproduced from ref. 182a with permission from Wiley-VCH. | ||
2.2.1.3 Electron transfer mediated by organic radicals. In this section, we focus our attention on the studies of electron transfer phenomena performed with organic radicals that electronically communicate two metallic surfaces. One promising method for accessing to such studies is to use self-assembled monolayer (SAM) of a functionalized radical on a metallic surface, like gold or HOPG. In these cases the tunnelling current between a scanning tunneling microscopy (STM) tip and these types of surfaces is measured. There are several studies about SAM of α-nitronyl nitroxide radicals functionalized with S atoms on a gold substrate.186 Matsushita et al. was the first one to report such kind of studies.187 Since the thiols utilized as ligands to form a SAM on a gold surface acted as radical scavengers destroying the radical character the disulfide derivative 34 was synthesized and successfully used to afford a SAM (Fig. 26) which was studied by STM.
 | ||
| Fig. 26 Formation of a SAM with the thiol-derivatized phenyl α-nitronyl nitroxide (34) on a gold surface. | ||
Another method for studying electron transfer phenomena mediated by organic radicals was the use of nanoparticles functionalized with open-shell molecules. Harada et al. prepared gold nanoparticles with an average diameter of 4.1 nm with many chemisorbed α-nitronyl nitroxide radicals 34-hex on their surfaces (Fig. 27).188 Basically, nanoparticles chemisorbed by 34-hex behave paramagnetically, with an average Curie constant for each of a single nanoparticle as large as 36.4 emu K mol−1 indicating that about 100 radical molecules were chemisorbed on each particle. While the peak-to-peak line width of the ESR spectrum of crystalline 34-hex was only 0.6 mT at 300 K, that of solid chemisorbed nanoparticles was as wide as 30 mT at room temperature which broadened to 180 mT at 11.5 K. This enormous broadening of the linewidth was attributed to the rapid relaxation of the π-localized spins presumably due to the coupling with the conduction electrons of the gold nanoparticle because of the freezing of the librational motion of the ligands when lowering the temperature. This dense assembly of radicals on a metallic nanoparticle is expected to be utilized as building blocks of spintronic systems (vide infra).
 | ||
| Fig. 27 Reaction of 34-hex diradical with stabilized nanoparticles with a cationic surfactant to form 34-hex derivatized gold nanoparticles. Reproduced from ref. 188 with permission from The Chemical Society of Japan. | ||
Studies about electron transfer mediated by PTM radicals using SAMs have also been reported. To investigate in detail the electronic and spin transport properties at the molecular level, PTM radicals were deposited on surfaces of different nature by means of different approaches. Thus, PTM molecules were grafted by physisorption on Au(111) and on graphite (HOPG) and by chemisorption forming SAMs on Au(111) and oxide-based substrates, like SiO2 and ITO (Fig. 28), and the resulting surfaces have been characterised and studied by a variety of techniques.36,189–191,199 The different natures of the substrates employed offer different advantages since gold, graphite and ITO permit to perform transport studies of the functionalised surfaces, while glass substrates are only compatible with the use of optical techniques.
 | ||
| Fig. 28 Approaches employed to anchor PTM radical derivatives on surfaces of different nature by chemisorptions, through covalent or electrostatic interaction bonds. | ||
The PTM radical 35 was chosen as a model compound to address the magnetic behaviour of a metal surface after physisorption. As confirmed by scanning tunnelling microscopy (STM), molecules of these radical were adsorbed on the gold surface forming monolayered aggregates of a few molecules in ordered domains of periodic rows separated by 1.5 ± 0.1 nm which are consistent with the molecular size of 35 (Fig. 29). The paramagnetic character of the surface was confirmed by EPR spectroscopy, showing a sharp line with a weak magnetic anisotropic behaviour demonstrating unambiguously that the radicals preserve their open-shell character on the surface. These samples were also characterized by means of electron spin noise scanning tunnelling microscopy (ESN-STM).192,193 This technique combines the spatial resolution of the STM with the spectral resolution of EPR.194 Analysis of the data collected using this technique leads to the conclusion that it is possible to detect the magnetic character of a decorated surface with 35 radicals.
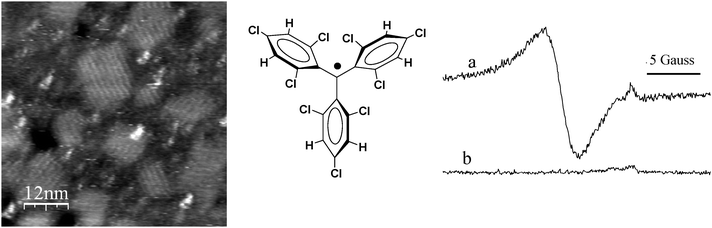 | ||
| Fig. 29 Left: STM image (60 × 60 nm) of PTM radical 35 physisorbed on Au(111). Right: (a) EPR spectrum of a gold surface decorated with physisorbed radicals 35; (b) EPR spectrum of the gold substrate. Reproduced from ref. 193 with permission from Elsevier. | ||
Regarding the chemisorption of PTM radicals on surfaces, different substrates have been investigated by preparing SAMs190,191 For this purpose, two different approaches were followed: (i) direct covalent anchoring of the PTM radical on the surface, and (ii) growth of a prefunctionalized SAM which then reacts with the PTM radical derivative by the formation of either a covalent, a coordination or an electrostatic bond (Fig. 28). The magnetic properties were characterized by EPR in all cases, the electroactive properties by cyclic voltammetry (CV) and the optical properties by UV-Vis and fluorescence spectroscopy. Spectroelectrochemical experiments have also been carried out proving that the PTM radical can be reversibly converted to the corresponding anion and, therefore, the functionalized surfaces act as chemical switches in which the magnetic and optical properties can be used as read-out mechanisms (see Section 3).195 Recently, it has been demonstrated that a gold surface grafted with a PTM radical derivative shows an electron transport rates that are one order of magnitude larger than the surface grafted with the close-shell counterpart of the PTM derivative. In order to gain further insight into the electronic structures of each molecule grafted surfaces density functional calculations were employed. As it can be seen in Fig. 30, the monoradical derivative PTM(αH) has a large energy gap (∼5 eV) between its closed-shell LUMO and HOMO. The PTM(rad) has a SOMO in the α spin, with a similar energy to the PTM(αH) HOMO but a significantly lower energy LUMO in the β spin and significantly close to the Fermi levels of the gold and the conducting tip. This result suggests that the electronic differences between the open and closed-shell PTM derivatives with respect to their electron-accepting orbitals may play an important role in enhancing the conduction in the open-shell PTM(rad) SAMs.196
 | ||
| Fig. 30 Energy levels obtained from DFT calculations for SAMs of PTM(αH) and PTM(rad) derivatives. Reproduced from ref. 196 with permission from the Wiley-VCH. | ||
In the same line, the transport characterization of SAMs based on the closed and open-shell forms of a fully conjugated PTM radical hybridized with a gold substrate reveals that both systems exhibit negative differential resistance (NDR) in their I–V curves at −1.5 V. This result was attributed to similar resonant tunnelling with unoccupied molecular orbitals with analogous energies. When a low bias is applied, the transport mechanisms dominating in both SAMs are different since a non-resonant tunnelling occurs for the PTM(αH) derivatives and a resonant tunnelling is operative for the PTM(rad) SAM. This work demonstrated that distinct transport mechanisms can dominate depending on the bias-voltage applied and also showed that NDR processes are not influenced by the redox character of the molecules.197 Comparative measurements such as the ones reported in these works will clearly pave the way towards the design and development of molecular devices for molecular spintronics.
Several Japanese groups have followed this strategy Sugano and Sugimoto being the first who independently prepared the TTF π-donor 36 connected with a TEMPO radical through an imine linkage and used the π-donor radical for preparing CT complexes with TCNQ.198,199 On the other hand, Nogami et al. prepared a similar π-donor but based on a pyrene derivative carrying a TEMPO radical.200 Later on, Nakatsuji et al. also prepared the TTF derivative 37 carrying a phenoxy radical,201 while Fujiwara and Kobayashi introduced the proxyl radicals into a BEDT-TTF skeleton with a condensed ring structure as in 38.202 Although some of the CT complexes and ion radical salts derived from these π-donor radicals exhibited a coexistence of semiconducting and paramagnetic properties, no direct interaction between conduction electrons and localized spins were detected. This lack of direct interaction may be partly caused by a tiny or null exchange interaction between π-donor units and radical pending groups in the resulting material (Fig. 31).
 | ||
| Fig. 31 Organic TTF donors with pending radicals as building blocks of conducting magnets. | ||
Recently Okada et al. successfully isolated the cation diradical salt of a N,N′-diphenylphenazine-π-donor containing two pending α-nitronyl nitroxide radicals as polarizing units (Fig. 32). Such a substituted cation radical is stable even at room temperature and shows robust ferromagnetic interaction between the three spins on each molecule on the basis of the temperature dependence of the magnetic susceptibility of the salt.203 This result is a direct evidence of a strong exchange magnetic coupling between the localized spin on the radical units and the unpaired spin, delocalized over the entire heterocyclic unit. Similar results were obtained using thianthrene as a π-donor core with α-nitronyl nitroxide pending radical in the 2 position of thianthrenyl, as well as 2,7- and 2,8 positions (Fig. 32) which, respectively, have the triplet and quartet as ground states, when the heterocycles are singly oxidized.204
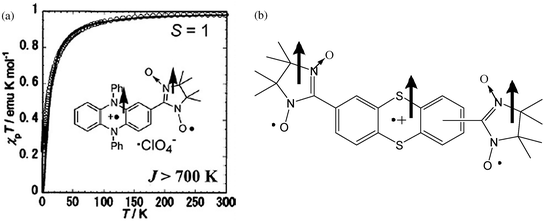 | ||
| Fig. 32 (a) Temperature dependence of the magnetic susceptibility of the ion-diradical salt of a pyrazine-based spin-polarized donor. (b) Ground state cation triradical of thianthrene bis(nitronyl nitroxide). Reproduced from ref. 203 with permission from the American Chemical Society. | ||
The first example of an extended system with polarized conducting electrons was reported by Sugawara and colleagues using the TTF π-donor ESBN with one pending α-nitronyl nitroxide radical such as ESBN.205,206 This molecule upon a one-electron oxidation forms a cation-diradical species which has a triplet ground state, as shown in Fig. 33. This molecule forms by electrocrystallization the 2:1 radical salt (ESBN)2ClO4 where the TTF units are stacked parallel to each other and half of them are oxidized forming a half-filled conduction band where the conducting electrons are magnetically coupled with those localized on the pending radical units (Fig. 33).
 | ||
| Fig. 33 Top: schematic drawing of generation of ground-state triplet cation-diradical species of ESBN upon a one-electron redox process at the donor moiety. Medium: stack of spin-polarized donors in a mixed valence state (A·A+·A·A+…) and spin-dependent intermolecule electron transfer. Bottom: magnetic field dependence of the magnetoresistance of (ESBN)2ClO4 at various temperatures at a bias voltage of 7 V. Reproduced from ref. 154c with permission from the Royal Society of Chemistry. | ||
Due to such characteristics this salt exhibits a giant negative magnetoresistance, as revealed by the magnetic field effect on the resistance of a single crystal of (ESBN)2ClO4 with a constant bias voltage of 7 V (Fig. 33 bottom).206,207 This result serves as direct proof for the presence of interaction between localized spins and conduction electrons being the first genuine organic system ever reported where there is a synergism between conductivity and magnetism.
Even more interesting properties were found for π-donor radical consisting of a dibrominated benzo-TTF core containing a pending α-nitronyl nitroxide radical as the BTBN radical, in which two bromine atoms are introduced into the dithiole ring to increase the intermolecular interactions within its crystal (Fig. 34). It was found that the unicomponent neutral crystals of this compound were conductive at low temperature upon hole injection from carrier injection through electrodes and that the conductance of the crystals can be controlled by the application of an external magnetic field (Fig. 34).208 Then, measuring the resistance of neutral crystals at a bias voltage of 10 V under an external magnetic field between −5 and +5 T, the resistance notably decrease upon application of a magnetic field at temperatures below 30 K, exhibiting therefore a negative magnetoresistance. (Fig. 34)
 | ||
| Fig. 34 Top: plausible mechanism for the increase in the conductance by the application of an external magnetic field. Bottom: structure formula of BTBN and its magnetoresistance at 2 K under an applied magnetic field of 5 T. A negative value of the magnetic field corresponds to the direction antiparallel to the previous magnetic field. Reproduced from ref. 208 with permission from the American Chemical Society. | ||
The appearance of such a magnetoresistive regime below 30 K, determined by the intramolecular ferromagnetic couplings between unpaired and conduction electrons, opens the possibility to increase the temperature range in which magnetoresistance effect is present by an appropriate molecular design leading to the development of molecule-based spintronic devices.
On the basis of the above background of investigations related to spin-carrying molecules grafted on gold surfaces, Sugawara and colleagues cleverly designed an alternative approach based on the spin polarization phenomenon. For that they prepared the bifunctionalized pyrrole-based spin-polarized wire molecule209SPWM. This molecule consists of three functional units: the first unit is a spin-polarizing core, which is made of a α-nitronyl nitroxide radical. This radical is connected to the second unit which is a molecular wire composed of a thiophene–pyrrole–thiophene hybrid trimer. The third unit connects the wire to the surface of gold nanoparticles or gold electrodes by means of a disulfide group. Briefly, in this system the SPWM can act as a spin-polarizer for the tunnelling electrons that pass through it from the two electrodes (Fig. 35). As a reference compound, the spinless wire molecule SLWM, having an acetal group instead of the α-nitronyl nitroxide group was prepared. Both wire molecules were mixed with gold nanoparticles in order to assess their efficiency in polarizing the tunneling electrons circulating between the gold nanoparticles.
 | ||
| Fig. 35 Top: molecular structures of the spinless wire molecule SLWM and the spin containing wire SPWM. Bottom: schematic drawing of spin-dependent tunneling through SPWM and of the electronic structure of SPWM at the junction of electrodes. | ||
For measuring the electron tunnelling phenomena the authors used interdigitated electrodes with a 2.4 μm gap which are bridged by 100 nm gold granules, that in turn, consists of 4 nm nanoparticles linked by the wire molecules—SLWM or SPWM—with an inter-particle gap of about 1.6 nm, the length of the molecular wire, as shown in Fig. 36. The SLWM network showed a sharp transition of the slope in the Arrhenius plot around 30 K. This sharp transition cannot be explained by a variable-range hopping mechanism but is expressed by a T-squared dependence which is well explained by the (inelastic) cotunneling mechanism. Indeed, if the bias voltage is sufficiently lower than the charging energy, the conductance at lower temperatures is roughly proportional to T2. This feature is also derived from the bottle-necked network structure, because the contribution of higher-order cotunneling is greatly suppressed in such a bottlenecked structure. However, the temperature dependence of conductance of the network of SPWM in the low temperature region is much larger than that of SLWM. This temperature dependence is substantially suppressed to lower values by 10−3 than those of the SLWM network at the bottom temperature (4.2 K). This means that the tunneling electron must be scattered by the localized spin on the wire molecule, which exists in the tunneling barrier. In summary, in this hybrid organic–inorganic system, it is found that a single tunnel electron that runs between metallic gold nanoparticles is controlled by the single spins of the wire molecules.209
 | ||
| Fig. 36 (a) SEM image of networked gold nanoparticles with SPWMs wire molecules on interdigitated electrodes with a gap of 2.4 μm (b) Expanded image of a bottle-necked structure at a granule–granule junction (c) Expanded idealized picture of a granule–granule junction consisting of a granule, a wire molecule, a nanoparticle, a wire molecule and a granule, in series. Reproduced from ref. 209a with permission from American Physical Society. | ||
2.3 Materials with optical properties based on organic radicals
In recent years there has also been an increasing attention paid to the creation of multifunctional materials with both magnetic and nonlinear optical (NLO) properties210,211 This is due to the fact, on one hand, that novel and unexpectedly high magnetic components on the NLO processes have been identified.212 On the other hand, it is also due to the potential applications of these compounds since nonlinear properties are commonly used in optical rectification, in modern waveguides, optical communication technology and for optical switching and signal processing.213 The addition of a novel degree of freedom, with the introduction of the magnetic effects, opens the way to the unexplored potential of the subject.Anyway, the synthesis of novel molecules and materials displaying both NLO properties and interesting magnetic behaviour is not an easy task. Some prominent results involve the use of a hybrid approach, in which the inorganic magnetic structure and efficient organic NLO chromophores are associated in the same material but forming segregated substructures.214 This approach has the advantage of allowing relatively easy engineering of both the magnetic and NLO moieties, as these always remain separate and distinct. Although it is possible to obtain a multifunctional material in this way, it is very unlikely that a system can be obtained in which the magnetic and optical behaviours show some form of interplay, as they arise from separate parts of the system. To this end a different approach, which involves the creation of all organic or inorganic–organic magnetic structures directly responsible for the NLO properties is preferred.
Traditionally, materials exhibiting second-order NLO behaviours were inorganic crystals, such as lithium niobate (LiNbO3) and potassium dihydrogenphosphate (KDP). However, molecular materials, such as organic crystals and polymers, have shown to offer better nonlinear optical and physical properties, such as ultrafast response times, lower dielectric constants, better processability and a remarkable resistance to optical damage, as compared to inorganic materials. Up to now, most of the efforts to discover new molecular chromophores having large NLO properties have been focused on closed-shell electronic organic species. However, more recently, some efforts have been devoted to the investigation of materials having open-shell electronic structures. As it has been recently pointed out by Marks et al.,215 species having open-shell electronic states, such as organic radicals216 or paramagnetic transition metal complexes,217 can exhibit very large first-order hyperpolarizabilities (β) in comparison with analogous closed-shell systems, thanks to the presence of accessible low-lying charge transfer electronic states. In spite of this interest, only a very few examples of organic open-shell species showing second-order hyperpolarizabilities have been up to now described; mostly due to the low stability of these species.218 Systems presenting NLO properties can be grouped into two categories: (i) octupolar systems219 and (ii) “push–pull” (donor–acceptor systems linked through a π-backbone) systems.220,221
In this section, the capability of discrete organic radicals to generate optical responses, either as octupolar or as a “push–pull” system, will be presented and the corresponding properties will be provided.222 Finally, an example about how intermolecular interactions can enhance the optical responses in materials with an extended structure constructed with an organic radical will be also presented.
On the contrary, the capability of triphenylmethyl radicals to generate non-linear optical responses is higher because of their larger stability. This fact allowed us to measure the corresponding hyperpolarizabilities, using Hyper-Rayleig Scattering (HRS) technique. Indeed, molecular nonlinear optical coefficients of PTM radicals, 1, 39–45 were measured in CH2Cl2 solution at room temperature with the HRS technique irradiating with a laser light at 1064 nm. Data reported in Fig. 37 show that these radicals exhibit very high NLO responses with β values ranging from 118 × 10−30 to 755 × 10−30 esu. This fact is not surprising since this family of radicals is structurally very similar to crystal violet, which also exhibits a large β value due to its octupolar symmetry.226 In fact, all these compounds belong to the non-planar D3 symmetry groups. They have the general formula Ar1Ar2Ar3Z, where Z is an sp2 hybridized C atom—a C+ in crystal violet and C˙ in PTM radicals—and Ari denotes an aromatic group with polarizable substituents like NEt2 in the crystal violet and Cl atoms in PTM radicals. Moreover, the considerably large β values obtained point out to an enhancement of the second-order NLO activities according to their open-shell electronic states.227 However, it is not possible to extract an irrefutable conclusion about the influence of open-shell character on the NLO responses, since a closer look at radicals 1 and 39–45 reveals that they are composed of three individual subunits—three aryl groups—each one providing also a NLO contribution. More precisely, 1,3,5-trichlorobenzene (TCB) and the pentachlorobenzene (PCB) compounds have also octupolar components and belong, respectively, to D3h—a pure octupolar system—and C2v—with dipolar and octupolar contributions—group of symmetries exhibiting β values of 22 × 10−30 and 8 × 10−30 esu, respectively. For this reason, PTM radicals give access to individual pure octupolar molecules, constituted by individual octupolar subunits, for which the term of “super-octupolar” systems was coined. If an octupolar synergistic effect takes place, this would greatly contribute to enhance the NLO responses of this type of materials.
 | ||
| Fig. 37 Hyperpolarizability β values of several PTM radicals measured with the HRS technique. | ||
Along this line, the comparison between purely octupolar radical compounds 1 and 40 may be very interesting. Both systems have D3 symmetry and are composed of three individual building blocks with NLO contributions based on the PCB and TCB subunits, respectively. HRS measurements show that the β value of 1 is lower than that obtained for 40. Interestingly, such values follow the trend found for the molecular hyperpolarizabilities derived from each individual octupolar building block, suggesting that a synergetic effect between individual building blocks may occur. To give more insight to the relationship between the electronic configuration and the NLO responses of PTM radicals, cyclic voltammetry studies of radicals 1, 39–45 in CH2Cl2, using 0.1 M n-Bu4NPF6 as electrolyte, were performed and analysed. Interestingly, the hyperpolarizability β values show a linear dependence in front of the reduction/oxidation potentials of these radicals. Indeed, as the oxidation potential decreases, β value increases whereas on the contrary, the easier to reduce the radicals the lower results the β values. This confirms a direct correlation between the energy of the electronic transition and the “super octupolar” NLO responses.228
The incorporation of PTM radicals into push–pull (dipolar) systems represents an alternative design strategy for NLO active systems.229 Since the pioneering work of Green et al.,230 who reported the obtaining of a ferrocene derivative with excellent NLO responses, there has been considerable effort in using metallocenes as donor groups in NLO molecular materials along with acceptor groups.231 With this aim, the hyperpolarizability β values of the series of radicals shown in Fig. 38 were measured using also the HRS technique. The introduction of a vinylene bridge connecting the PTM to an electron donor substituent, like a p-bromophenyl group leads to the formation of dipolar “push–pull” compound showing a NLO response of 329 × 10−30 esu. Finally, the replacement of the electron donor group by a stronger one like the ferrocenyl group, notably increases the NLO response up to 803 × 10−30 esu, as expected.
 | ||
| Fig. 38 Hyperpolarizability β values of a few PTM-based “push–pull” systems. | ||
Push–pull systems derived from PTM radicals have been also used to prepare luminescent materials taking advantage of the fluorescence properties of the PTM radicals that emit in the red region when irradiated at the energy of the radical absorption band (380 nm). The large Stokes shift exhibited by such radicals makes them good candidates for different photonic applications enabling the preparation of electroluminescent devices when linked to appropriate molecules, such as the carbazole derivatives.232 Indeed, the carbazole derivatives of triphenylmethyl radicals show relatively intense luminescence and undergo reversible oxidation processes to give stable radical cations, making them good hole carriers. All these properties make them good molecular synthons for organic light-emitting diodes, as hole-transporting and light emitting layers. Moreover, it has been shown that some adducts have properties of amorphous glasses, characteristic that must be used to obtain uniform and transparent amorphous films to be easily integrated in devices.233
 | ||
| Fig. 39 Left: thermal equilibrium between the σ-dimer (left) and the π-dimer (right) of 46 in the crystalline state, and the locations of their highest-occupied molecular orbitals according to quantum chemical calculations. The bonding in the σ-dimer is only through the σ-bond, whereas multiple bonding overlaps are seen in the π-dimer. Right: colour change for specific positions of the single crystal of 46 depending on the temperature. Reproduced from ref. 234 with permission from Nature Publishing Group. | ||
Interestingly, the energy gap between the σ- and π-dimers was experimentally determined to be 2.6 kcal mol−1. This value is considerably smaller than the dissociation enthalpy of the σ-dimer of 46 in solution (12 kcal mol−1), which suggests that the π-dimer structure is multibonded. Furthermore, the ESR spectra of 46 in its crystalline state revealed a thermally accessible triplet state of the π-dimer. Radical 46 is the first system in which such three states, σ- and π-dimer singlet states and an excited π-dimer triplet state, have been recognized in an open-shell organic molecule.
3. Multifunctional switchable materials based on organic radicals
As already shown in the previous sections, persistent organic radicals are advantageous building-blocks of functional materials exhibiting magnetic, electronic and optical properties. The intrinsic multifunctionality of organic radicals together with the increasing interest for developing applications and devices with molecule-based materials made interesting the exploration of optical or redox switching systems.235 A few representative examples of such systems will be discussed in this section.3.1 Photoswitchable molecular systems
Currently, there is a special interest in obtaining photomagnetic materials whose magnetic properties are controlled at will by means of an external irradiation with light.236–238 Such a control over the magnetic properties by an optical stimuli may have applications in magneto-optical devices. Whereas, there are several interesting examples of photoswitchable spin systems based on inorganic compounds and metal complexes,239 those of organic compounds remained still scarcely explored. Here photo-responsive spin systems that do not have any metal-based spins in them will be presented. The systems include photo-responsive π-electron moieties as azobenzene, diarylethene, biindenylidene, terphenoquinone, anthracene, naphthopyran, arylimine, and hexaarylbiimidazoles bearing stable organic radicals or generating organic radicals as spin centres.240 Actually, the systems with photo-responsive magnetic properties investigated in recent years are classified, depending on the kind of structural changes, into intramolecular (Class I) or intermolecular (Class II) systems, as shown in Fig. 40. Class I systems can be further categorized in three types: Type A are organic biradicals associated with molecular structural changes produced by irradiation and dealing with changes in the intramolecular magnetic interactions; Type B are organic biradicals generated from the diamagnetic precursors by an irradiation and Type C are organic high-spin multiplets generated from radicals by an irradiation. Class II systems are those organic radical compounds that self-assemble yielding compounds under irradiation (Fig. 40). Representative examples of each of the above mentioned classes and types of photomagnetic system will be described in the following.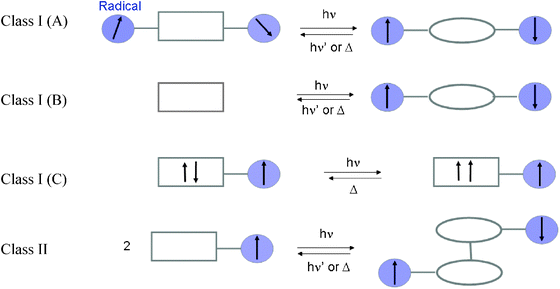 | ||
| Fig. 40 Photoswitchable spin systems based on intra- (Class I) and inter-molecular (class II) structural/electronic changes. | ||
An example of a Class I (A) system is the azobenzene derivative with two α-nitronyl nitroxide radical substituents which is prepared directly as the trans isomer 47a.241 When this trans-isomer is subjected to photoirradiation in a cyclohexane solution using 345–415 nm, the corresponding cis-isomer 47b is formed (Scheme 8), as clearly detected by the UV-vis spectral changes. When the irradiation was halted, the original peaks were found to be recovered by a thermal isomerisation albeit with less intensity. This isomerisation was also followed by EPR at a cryogenic temperature in toluene solution, although the conversion between the isomers is not completely attained (Scheme 8).
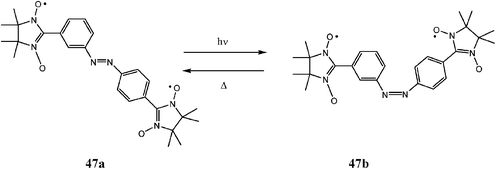 | ||
| Scheme 8 Photoswitchable isomers of the azobenzene biradical 47. | ||
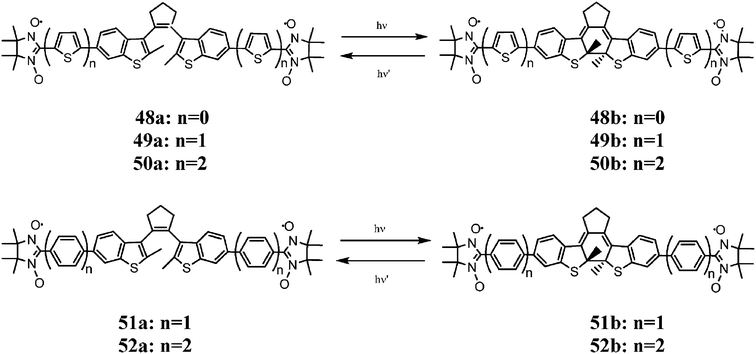 | ||
| Scheme 9 Photoswitchable systems based on diarylethane with two α-nitronyl nitroxide radicals. | ||
Examples of Class I (A) photoswitchable systems that are completely reversible are the series of diarylethane derivatives with two α-nitronyl nitroxide substituents 48a–52a prepared by Matsuda and Irie.242 All of such biradicals show excellent photochemical conversions to the corresponding closed ring isomers 48b–52b (Scheme 9). The backward reactions were also reported to occur by irradiation with light of a suitable wavelength, giving the starting open-ring isomers in each case. The switching behaviour of these diarylethane biradicals has consequences on the intramolecular magnetic interactions in each of the photochromic spin couplers because the conjugation between the two sides is interrupted in the closed ring. Thus, the magnitude of the intramolecular antiferromagnetic interaction between the spins in the open-ring isomer 48a, |2J/kB| = 22 K, increases appreciably to |2J/kB| = 212 K in the closed-ring isomer 48b by the photochemical ring closure such a change being reversible. Interestingly, the photoswitching of intramolecular magnetic interactions can be tuned by expanding the sizes of the spacers of the diarylethane—either using oligo p-phenylene or oligothiophene—as demonstrated with the derivatives 49–52 in which the differences of the exchange interaction of more than 150-fold between open and closed ring form isomers are attained (Scheme 9).243,244
Examples of photochromic materials of Class I (B) were obtained by Toda and Matsura based on closed-shell bisindenylidene derivatives. Single crystals of such compounds gave the corresponding reddish purple biradicals on exposure to sunlight which can be reversed upon stopping the irradiation.245,246
Representative photochromic compounds of Class I (C) are the 9-phenylanthracene derivative with an α-imino nitroxide radical at the para position (53a) and the 9,10-diphenylanthracene derivative 54a with two of such radicals at both para positions (Scheme 10).247 Time-resolved ESR (TRESR) spectra of the first excited states with resolved fine structure splittings were observed for 53a and 54a in frozen solvents upon irradiation with light. These species were unambiguously assigned as an excited quartet (S = 3/2) spin state for 53b and to an excited quintet (S = 2) spin state for 54b. Since a weak antiferromagnetic exchange interaction was observed in the ground state of 54a, the clear detection of the excited quintet state shows that the effective exchange coupling between the two radicals through the diphenylanthracene spin coupler has been changed from antiferromagnetic to ferromagnetic upon excitation and thus a photo-induced spin alignment utilizing the excited triplet was realized in a purely organic p-conjugated spin system.
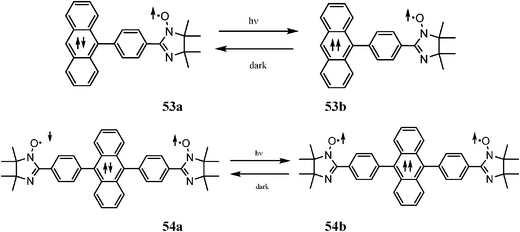 | ||
| Scheme 10 Photochromic derivatives with α-imino nitroxide radicals. | ||
There are very few examples of supramolecular photomagnetic materials classified as Class II since the construction of such systems requires that the structural subunits exhibit non-covalent interactions, suitable to be controlled and changed in a predictable manner by irradiation, and at the same time transmit the magnetic interactions properly. Up to date one of the reported examples is based on an anthracene bearing two aminoxyl radicals. The only example of hydrogen-bonded supramolecular entities with photoswitching properties is the radical 55 which has a PTM subunit connected to a ferrocenyl subunit through a photoisomerizable conjugated imine bridge.248 Specifically, imine derivatives exhibit a trans/cisphotoisomerization in which one of the isomers—the cis one—may be prone to intermolecular hydrogen bonds (Fig. 41). Indeed, the trans isomer of 55 exists in solution as a monomeric species while the cis isomer dimerizes forming a thermodynamically stabilized hydrogen-bonded diradical species. In both species, the donor character of the ferrocene unit and the acceptor character of the radical unit ensure a significant spin delocalization over the imino bridge so that once the supramolecular species is formed, a magnetic exchange coupling between the spins is established. Thus, the trans isomer, that exist as two independent doublets, interconverts by irradiation into the dimeric species of the cis isomer in which relatively strong antiferromagnetic interactions lead to a singlet ground state.
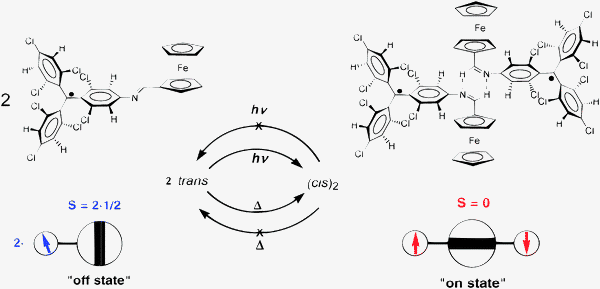 | ||
| Fig. 41 Isomerizations of the PTM Schiff base radical 55. | ||
The above described properties of photoswitchable molecular systems seem very promising in order to prepare optical switching devices at the molecular scale. However, real devices made with these molecules, when are deposited on surfaces or inside polymers, remain unexplored till date.
3.2 Redox switchable molecular systems
The number of redox-switchable molecular systems is also very scarce. Herein we present one of such systems showing simultaneous changes of three different properties—the linear optical, nonlinear optical, and magnetic properties—which is based on the rich electrochemical behaviour of radical 32 (Scheme 11). Thus, radical 32 can be present in three different forms with distinct oxidation states each of them showing distinct physical properties such as magnetism, color and NLO response.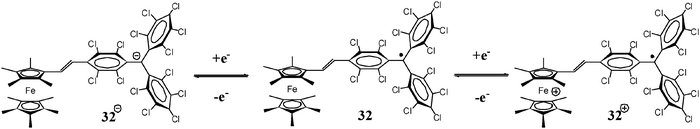 | ||
| Scheme 11 The three redox states of the molecular switch derived from the D–A dyad 32: 32−−, 32 and 32++. | ||
In accordance with the different electronic structures, the three species 32, 32++ and 32−− show strikingly distinct optical properties. Thus, radical 32 shows a red color with the characteristic absorptions of conjugated PTM radicals and a broad IVT band, centered at 1520 nm, associated with an IET phenomenon (see Section 2.2.2). The ferrocenium radical derivative 32++ exhibits the typical band of a ferrocenium derivative with a significant enhancement of the intensity of the radical band at 385 nm, which accounts for the deep yellow colour of this species. On other hand the IVT band is absent in this ferrocenium radical derivative because of the lack of electron-donor character of the organometallic unit when it is oxidized. Carbanion 32−− shows an intense absorption at 534 nm, characteristic of this kind of anions, which is responsible for its intense wine red colour. At the same time, the broad IVT band vanishes in 32−− confirming the absence of the electron-acceptor capability in the reduced PTM unit. Similarly to the striking different optical absorptions of 32, 32++ and 32−−, such species also show distinct NLO responses. The dynamic hyperpolarizabilities of these species, measured by hyper-Rayleigh scattering experiments, gave also remarkable differences. Thus, as suggested by its intense IVT band and its “push–pull” character, the radical 32 gives a large NLO response with a β value of 545 × 10−30 esu. This value is reduced almost ninefold to 66 × 10−30 esu for 32++, and even more for the carbanion 32−−, which has a β value of 30 × 10−30 esu.
Electrochemical interconversions between the three species are completely reversible as shown in Fig. 42 by following the changes of their optical responses of their solutions. This result clearly demonstrates the switching capabilities of this multifunctional molecular system.
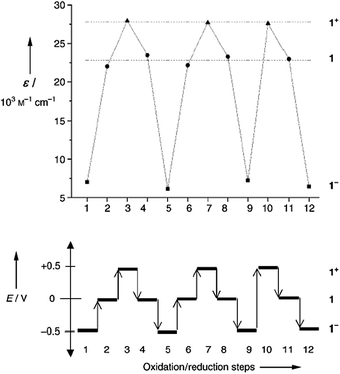 | ||
| Fig. 42 Stepwise oxidations and reductions carried out with 32 in THF with a chronoamperometric technique monitoring the changes in the visible spectrum. Top: changes observed at a wavelength of 385 nm where 32 (●) and 32++ (▲) exhibit the strongest absorption and 32−− (■) shows a very weak absorption. Bottom: fixed potentials E used in the different steps of cyclic redox experiments. Reproduced from ref. 30 with permission from Wiley-VCH. | ||
The magnetic properties of the three studied species are also different. Thus, while the salt [K([18]crown-6)]+32−− in solid state is diamagnetic, the magnetic susceptibility between 4–300 K of crystals of radical 32 and of the salt 32++BF44−− showed quasi-ideal paramagnetic behaviours with effective magnetic moments at 300 K of 1.72 and 2.50 μB, respectively, as expected for systems with S = 1/2 and S = 2 × 1/2 units. In addition, the value of 2.50 μB found for 32++BF44−− indicates that the magnetic interaction between the open-shell ferrocenium moiety and the PTM radical unit is very weak with both spins being apparently uncoupled above 4 K. Analogous differences in the behaviour of the three complexes were found in solution with EPR spectroscopy since 32−− is EPR silent while 32 and 32++ show different complex signals.
In order to move one step forward and use multifunctional switchable systems based on organic radicals for applications like molecular memory devices,249 it is crucial to deposit them on surfaces and demonstrate that they can be reversibly switched between two (or more) stable states when grafted on the surfaces. In addition, it is required that such states exhibit different responses in order to be able to read the status of the switch. A strategy to scale-down to molecular level memory devices is focused on the fabrication of charge storage devices by substrate immobilization and patterning of molecules that can be reversibly oxidized and reduced.250 A simple and versatile technique to address all these molecular building-blocks on surfaces is the preparation of (SAMs),251,252 which allows the functionalization of surfaces with a layer of molecules, two-dimensionally organized, that gives to the substrate new properties governed by the inherent characteristics of the molecules grafted on it.
There are extremely few examples of SAMs based on organic radicals in the literature.187,253 As described in Section 2.2.3, silicon oxide and gold surfaces have been grafted with PTM radicals making use of either covalent or noncovalent interactions.36,190 The magnetic properties of the functionalized surfaces were characterized by EPR in all cases. UV-Vis and fluorescent spectra were used for characterization of their optical properties. Such characterizations corroborated the chemical nature of the monolayer, that exhibits an absorption band at 382 nm and a fluorescent emission band at 690 nm which are characteristic of the radical character of the grafted PTM molecules.254 Electrochemical experiments using cyclic voltammetry were also carried out proving that the grafted PTM radicals can be reversibly converted to the corresponding anion and, therefore, the functionalized surfaces act as chemical switches in which the magnetic and optical properties can be used as read-out mechanisms (Fig. 43).
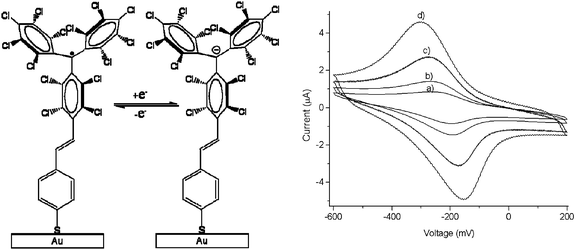 | ||
| Fig. 43 Left: scheme of the redox process on a gold surface with covalently bonded PTM radicals. Right: cyclic voltammograms in CH2Cl2, with 0.1 M n-Bu4NPF6 (vs.Ag/AgCl) at different scan rates: (a) 50, (b) 100, (c) 300 and (d) 400 mV s−1. Reproduced from ref. 36 with permission from the American Chemical Society. | ||
More interesting with respect to potential device applications is the possibility to locally address the PTM radical molecules on the surfaces in order to fabricate multifunctional switchable patterned surfaces. Thus, both the covalently and the electrostatic PTM bonded surfaces have been patterned by microcontact printing and visualized by laser scanning confocal microscopy or fluorescence microscopy due to the fluorescent nature of the PTM molecules.192
Recently, a SAM of a PTM radical on indium tin oxide (ITO) has been investigated for a surface molecular switch in which an electrical input is transduced into optical as well as magnetic outputs. Owing to its electroactive character, the SAM was reversibly interconverted between two redox states: the radical and anion forms. In addition, it has been shown that the inherent magnetic moment that organic radicals exhibit can be exploited. Indeed, the magnetic response can be used as a readout mechanism, as well as the optical response. The extremely high robustness, long-term stability, reversibility and reproducibility of the switch, the fact that it can be patterned and locally addressed, and also that it operates at very low voltages makes this system very promising in the development of non-volatile memory devices based on immobilized molecules (Fig. 44).195
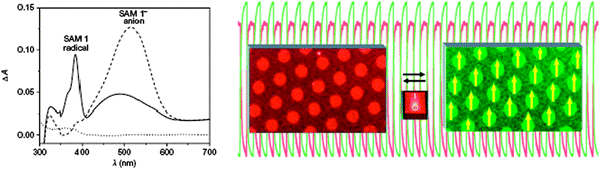 | ||
| Fig. 44 Left: UV-vis absorption spectra of ITO functionalized with PTM radical (solid line) and after in situ electrochemical reduction on applying −0.3 V to the substrate for 30 s (dashed line). The dotted line corresponds to the absorbance of the bare ITO substrate. Right: change of the optical response of the switch versus time during 27 consecutive write–erase cycles (+0.3 V/−0.3 V versus Ag(s)) applied to a SAM of PTM radical monitored at 385 nm (red line) and 515 nm (red line). Reproduced from ref. 195 with permission from Nature Publishing Group. | ||
Reversible redox processes of discrete molecules in a solution can be compared to charge/discharge processes of an electrode active material in a secondary battery. An organic molecule with multistage redox ability can, therefore, accommodate a large number of electrons in the molecular skeleton. Thus, there is a unique possibility to realize a large discharge capacity in the secondary battery systems by utilizing such organic molecules as electrode active material. In 2002, the idea of a secondary battery utilizing phenalenyl radicals like the 6-oxophenalenoxyl derivative was claimed255 and later on a novel secondary battery was developed based on such a derivative as a cathode active material for which the term “molecular crystalline secondary battery” was given.256
The multistage redox ability of the phenalenyl and oxophenalenoxyl systems is attractive for the preparation of active electrode materials in rechargeable batteries with large discharge capacities.11,183,257 Recently, a novel battery of this sort based on 56 in its crystalline form has been developed. This battery has two-step charge (and discharge) behaviour, corresponding to the change from 56 to 56−− and thence to 562−2−. Its charge/discharge capacity (142 A h kg−1) is comparable to that of commercially available lithium ion rechargeable batteries (150–170 A h kg−1) and close to the estimated value (147 A h kg−1) that was calculated from the molecular weight and the number of electrons participating in redox processes. Delocalization greatly contributes to the high stability of anion and radical dianion species in the cathode. This demonstrates that all of the molecules of 56 present in the electrode participate in the charge/discharge processes. Moreover, neither precious nor contaminant transition metals are required in this battery, thus increasing its economic versatility and environmental friendly and energy density. Studies are currently underway to understand the redox processes occurring inside the battery, with a view to designing more efficient electrodes that would make use of the electronic degeneracy of SOMOs. It is noteworthy that the use of the molecular orbital degeneracy contributes to an effective increase in the electric current capacity in the tailor-made molecular batteries. Energy storage devices having multiple functionalities based on redox-driven changes in physical properties of the molecule are anticipated for next generation molecular devices.
4. Summary
In this review the chemical and physical properties of the most common families of persistent organic radicals—nitroxide, triphenylmethyl, verdazyl, phenalenyl, and dithiadiazolyl—are described. In addition, a detailed overview of the use of such organic radicals as building blocks for obtaining molecular magnetic materials exhibiting additional and useful physical properties is also given. Consequently, the main focus of this overview is addressed to the multifunctionality of such magnetic materials providing some representative examples of this kind of novel materials.Regarding the magnetic properties, discrete molecules with high-spin ground states using aromatic units as magnetic couplers have been described. High spin alignments in the ground states of several 1,3-phenylene connected polyradicals (carbenes, triarylmethyl, aminoxyls and verdazyl) have been reviewed, a triarylmethyl polyradical with S = 13 being the one with the largest multiplicity ever reported. Metallocenes have also been used successfully as magnetic couplers of molecular solids promoting intermolecular magnetic interactions between radicals connected through them. We have seen how magnetic exchange through hydrogen-bonded bridges can occur at long-range distances even in the cases of well-delocalized radicals enabling to use such a coupling tool to prepare materials with extended hydrogen bonding networks showing long-range magnetic orderings. The success of the metal–radical approach combining paramagnetic metal ions and different kinds of radicals has been considered. We have described strategies to increase the ligand ability of radicals such as the use of carboxylic functionalized triphenylmethyl radicals as excellent coordinating magnetic ligands. This fact, together with the capability of H-bond to transmit magnetic interactions have opened the door to the obtaining of magnetically extended systems based on triphenylmethyl radicals functionalized with several carboxylic groups. This strategy has resulted in two new families of open-framework materials named as metal organic radical open-frameworks (MOROFs) and purely organic radical open-frameworks (POROFs). Specifically the so-called MOROF-1 materials have been found to behave as a sponge-like magnet material selective towards MeOH and EtOH together with changes in the magnetic properties that are macroscopically detected. Regarding the nitroxide radicals numerous metal complexes with transition metal and lanthanide ions have been reported exhibiting different topologies and dimensionalities including 1D and 2D architectures. Single chain magnets made with such an approach deserved a special mention as they show unique magnetic hysteresis and slow dynamics for the relaxation. Because of the quantum phenomena they exhibit at the macroscale, single chain magnets are potential candidates for future high density data storage materials. Also some examples about the use of metal–radical approach with verdazyl radicals for magnetic materials with extended structures have been described although this field is still in its early stages of development. Finally, regarding the magnetic properties, we have mentioned the case of free radicals based upon heavier p-block elements, such as S and Se, presenting magnetic extended materials with higher temperature magnetic orderings based on weak interactions like S⋯S or π⋯π. A series of thia- or selenazyl derivatives have been shown either to order as weak ferromagnets or to present marked differences in conductivity, which can be enhanced by applying an external pressure. These studies underline the effect that inclusion of heavier heteroatoms enhances not only the charge transport but also magnetic exchange interactions.
Regarding the electrical properties of materials based on organic radicals we have extensively described the intramolecular electron transfer phenomenon which is of prime interest because its understanding could lead to a better design and improvement of molecular-sized electronic devices for spintronics. We have presented some examples of two different approaches based on the use of discrete molecular wires with two identical or distinct electroactive units; known as mixed-valence compounds and donor–acceptor dyads. We have also shown some examples of molecular systems based on open-shell molecules that exhibit bistability controlled by the temperature and solvent. Efforts for the preparation of organic molecular components to construct molecule-based systems with spin-transportation properties, have also been presented focusing on the concept of a “spin-polarized donor”. In this regard, we have reported one example of a material showing a giant negative magnetoresistance which is a direct proof of the presence of an interaction between the localized spins and the conduction electrons. Finally, regarding the electronic properties, the possibility to functionalize surfaces and nanoparticles with radical molecules as spin-containing units towards their use in molecular spintronics has been described.
For the optical properties, we have put a special focus into multifunctional materials with both magnetic and nonlinear optical properties due to their potential applications for optical rectification in modern waveguides and for optical switching and signal processing. Up to now most of the efforts to discover molecules with large NLO properties have been focused on closed-shell organic species. However, it has been seen that open-shell molecules can exhibit very large first-order hyperpolarizabilities thanks to the presence of accessible low-lying charge transfer electronic states which are inherent in such a kind of open-shell molecules. The capability of discrete triphenylmethyl radicals to generate NLO responses, either as octupolar or as push–pull systems, has been described. Finally, an example about how intermolecular interactions can enhance the linear optical responses in materials with an extended structure constructed with a phenalenyl radical derivative has been also presented.
Along the review we have shown that persistent organic radicals are advantageous building blocks of functional materials exhibiting magnetic, electronic and optical properties. This intrinsic multifunctionality of organic radicals together with the increasing interest for developing applications and devices with molecule-based materials made interesting the exploration of optical or redox switching systems. Special emphasis has been put on photomagnetic systems whose magnetic properties can be controlled at will by means of external irradiation with light. Such systems include photo-responsive π-electron moieties, like azobenzene, diarylethene, etc., bearing stable organic radicals. On the other hand, one of the few examples of supramolecular photomagnetic material based on an anthracene bearing two aminoxyl radicals and another example presenting hydrogen bonded interaction with photoswitching properties have also been described. In order to move one step forward and use multifunctional switchable systems based on organic radicals for applications as molecular memories, we have shown that it is crucial to deposit them on surfaces and demonstrate that they can be reversibly switched between two(or more) states. In addition, it is required that such states exhibit different responses in order to be able to read the status of the switch. A strategy to scale-down to the molecule-level memory devices is focused on the fabrication of charge storage devices by substrate immobilization and patterning of molecules that can be reversibly oxidized and reduced. A simple and versatile technique to address all these molecular building-blocks on surfaces is the preparation of self-assembled monolayers, which allows the functionalization of surfaces with a layer of molecules, two-dimensionally organized, that gives rise to substrates with new properties governed by the inherent characteristics of the molecules grafted on it. Here we present one of the few examples using triphenylmethyl radicals grafted on surfaces, which has turned out to be very promising for the development of non-volatile memory devices. Finally, the reversible redox processes of discrete molecules in a solution have also been compared with a charge/discharge process of an electrode of a secondary battery. We have shown the possibility to realize a large discharge capacity in such batteries using phenalenyl radicals as electrode active material developing a novel generation of secondary batteries. Energy storage devices having multiple functionalities based on redox-driven changes of the molecules are anticipated for next generation of molecular devices.
In summary, persistent organic radicals with more than one property coming from the same molecular building-block set the possibility to achieve relevant technological goals, including potential applications in spintronics and nanoelectronics. Specifically, the evolution of molecular magnetism towards applications in molecular electronics, medicine and other related areas, together with the trend towards nanoscale materials, are foreseen for this multidisciplinary field.
References
- (a) A. Caneschi, D. Gatteschi and P. Rey, Prog. Inorg. Chem., 1991, 39, 331; (b) O. Kahn, Magnetism: A Supramolecular Function, Kluwer, Dordrecht, 1996 Search PubMed; (c) P. M. Lahti, Magnetic Properties of Organic Materials, Marcel Dekker, New York, 1999 Search PubMed; (d) K. Itoh and M. Kinoshita, Molecular Magnetism, Kodansha (Gordon and Breach), Tokyo, 2000 Search PubMed; (e) Magnetism: Molecules to Materials, ed. J. S. Miller and M. Drillon, Wiley–VCH,Weinheim, 2001–2005, vol. I–V Search PubMed; (f) B. D. Koivisto and R. G. Hicks, Coord. Chem. Rev., 2005, 249, 2612 CrossRef CAS; (g) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS; (h) K. E. Vostrikova, Coord. Chem. Rev., 2008, 252, 1409 CrossRef CAS.
- (a) P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS; (b) R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321 RSC.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castañer, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608 CrossRef CAS.
- (a) J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90(4), 1078 CrossRef; (b) J. Cirujeda, L. E. Ochando, J. M. Amigo, C. Rovira, J. Rius and J. Veciana, Angew. Chem., Int. Ed. Engl., 1995, 34, 55 CrossRef CAS.
- (a) R. G., Can. J. Chem., 2004, 82, 1119 CrossRef; (b) R. G. Hicks, M. T. Lemaire, L. Öhrström, J. F. Richardson, L. K. Thompson and Z. Xu, J. Am. Chem. Soc., 2001, 123, 7154 CrossRef CAS.
- (a) W. L. Hubbel, A. Gross, R. Langen and M. A. Lietzow, Curr. Opin. Struct. Biol., 1998, 8, 649 CrossRef CAS; (b) G. E. Fanucci and D. S. Cafiso, Curr. Opin. Struct. Biol., 2006, 16, 644 CrossRef CAS; (c) N. Roques, V. Mugnaini and J. Veciana, Top. Curr. Chem., 2010, 207 CAS and references therein.
- (a) J. Stubbe and W. A. Van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS; (b) R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi and L. Greci, J. Org. Chem., 2003, 68, 1747 CrossRef CAS.
- (a) D. Gatteschi, Adv. Mater., 1994, 6, 635 CAS; (b) C. Train, L. Norel and M. Baumgarten, Coord. Chem. Rev., 2009, 253, 2342 CrossRef CAS.
- (a) M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757 CrossRef; (b) M. Gomberg, J. Am. Chem. Soc., 1901, 23, 496 CrossRef; (c) M. Gomberg, Chem. Rev., 1924, 1, 91 CrossRef CAS.
- (a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13 CrossRef CAS; (b) A. R. Forrester and J. M. Hay and R. H. Thomson, Organic Chemistry of Stable Free Radicals, Academic Press, London and New York, 1968 Search PubMed; (c) Metal–Organic and Organic Molecular Magnets, ed. P. Day and A. E. Underhill, The Royal Society of Chemistry, Cambridge, 1999 Search PubMed; (d) Supramolecular Engineering of Synthetic Metallic Materials, ed. J. Veciana, C. Rovira and D. B. Amabilino, Kluwer Academic Publishers, Dordrecht, 1999 Search PubMed; (e) Molecular Magnets: Recent Highlights, ed. W. Linert and M. Verdaguer, Springer-Verlag, Vienna, 2003 Search PubMed; (f) Carbon-Based Magnetism, ed. T. L. Makarova and F. Palacio, Elsevier B. V., Amsterdam, 2006 Search PubMed.
- Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. Hicks, Wiley, New York, 2010 Search PubMed.
- (a) K. Itoh, Chem. Phys. Lett., 1967, 1, 235–238 CrossRef CAS; (b) T. Takui, Y. Teki, K. Sato and K. Itoh, Mol. Cryst. Liq. Cryst., 1995, 272, 1.
- J. Miller and D. Gatteschi, Molecule-based magnets themed issue, Chem. Soc. Rev., 2011, 40(6), 3053–3368 RSC.
- (a) A. Rajca, J. Wongsriratanakul and A. Rajca, Science, 2001, 294, 1503 CrossRef CAS; (b) A. Rajca, K. Lu and S. Rajca, J. Am. Chem. Soc., 1997, 119, 10335 CrossRef CAS; (c) A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 2004, 126, 6608 CrossRef CAS; (d) A. Rajca, Adv. Phys. Org. Chem., 2005, 40, 153 CrossRef CAS; (e) A. Rajca, S. Utamapanya and S. Thayumanavan, J. Am. Chem. Soc., 1992, 144, 1884 CrossRef; (f) A. Rajca, Chem. Rev., 1994, 94, 871 CrossRef CAS.
- W. P. Neumann, A. Penenory, U. Stewen and M. Lehning, J. Am. Chem. Soc., 1967, 89, 2054 CrossRef CAS.
- M. Ballester, Acc. Chem. Res., 1985, 18, 380 CrossRef CAS.
- J. Veciana and M. I. Crespo, Angew. Chem., Int. Ed. Engl., 1991, 30, 74 CrossRef.
- R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl., 1966, 5, 385 CrossRef CAS.
- N. Ventosa, D. Ruiz, C. Rovira and J. Veciana, Mol. Cryst. Liq. Cryst., 1993, 232, 333 CAS.
- M. Ballester, Adv. Phys. Org. Chem., 1989, 25, 267 CAS.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castañer, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608 CrossRef CAS.
- M. Ballester, I. Pascual, J. Riera and J. Castañer, J. Org. Chem., 1991, 56, 217 CrossRef CAS.
- V. M. Domingo and J. Castañer, Chem. Commun., 1995, 895 RSC.
- J. Rius, C. Miravitlles, E. Molins, M. I. Crespo and J. Veciana, Mol. Cryst. Liq. Cryst., 1990, 187, 155.
- M. Ballester, J. Riera, J. Castañer, C. Badía and J. M. Monsó, J. Am. Chem. Soc., 1971, 93, 2215 CrossRef CAS.
- (a) M. Ballester, J. Riera, J. Castañer, C. Rovira and O. Armet, Synthesis, 1986, 64 CrossRef CAS; (b) M. Ballester, J. Veciana, J. Riera, J. Castañer, C. Rovira and O. Armet, J. Org. Chem., 1986, 51, 2472 CrossRef CAS.
- M. Ballester, J. Riera, J. Castañer, C. Rovira, J. Veciana and C. Onrubia, J. Org. Chem., 1983, 48, 3716 CrossRef CAS.
- (a) J. Bonvoisin, J. P. Launay, C. Rovira and J. Veciana, Angew. Chem., Int. Ed. Engl., 1994, 33, 2106 CrossRef; (b) J. Sedó, D. Ruiz, J. Vidal-Gancedo, C. Rovira, J. Bonvoisin, J. P. Launay and J. Veciana, Adv. Mater., 1996, 8, 748 CrossRef CAS.
- I. Ratera, D. Ruiz-Molina, J. Vidal-Gancedo, K. Wurst, N. Daro, J. F. Létard, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2001, 40, 919 CrossRef CAS.
- C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266 CrossRef CAS.
- (a) I. Ratera, D. Ruiz-Molina, F. Renz, J. Ensling, K. Wurst, C. Rovira, P. Gütlich and J. Veciana, J. Am. Chem. Soc., 2003, 125, 1462 CrossRef CAS; (b) G. D'Avino, L. Grisanti, J. Guasch, I. Ratera, J. Veciana and A. Painelli, J. Am. Chem. Soc., 2008, 130, 12064 CrossRef CASG. D'Avino, L. Grisanti, J. Guasch, I. Ratera, J. Veciana and A. Painelli, J. Am. Chem. Soc., 2008, 130, 12072 Search PubMed.
- I. Ratera, S. Marcen, S. Montant, D. Ruiz-Moloina, C. Rovira, J. Veciana, J. F. Létard and E. Freysz, Chem. Phys. Lett., 2002, 363, 245 CrossRef CAS.
- (a) J. Veciana, C. Rovira, M. I. Crespo, O. Armet, V. M. Domingo and F. Palacio, J. Am. Chem. Soc., 1991, 113, 2552 CrossRef CAS; (b) J. Veciana, C. Rovira, N. Ventosa, M. I. Crespo and F. Palacio, J. Am. Chem. Soc., 1993, 115, 57 CrossRef CAS.
- D. Ruiz-Molina, J. Veciana, F. Palacio and C. Rovira, J. Org. Chem., 1997, 62, 9009 CrossRef CAS.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. Tejada, C. Rovira and J. Veciana, C. R. Chim., 2005, 8, 1213 CrossRef CAS.
- N. Crivillers, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2008, 130, 5499 CrossRef CAS.
- (a) T. Ishida, K. Shinozuka, M. Kubota, M. Ohashi and T. Nogami, Chem. Commun., 1995, 1841 RSC; (b) M. D. Pace and A. W. Snow, Macromolecules, 1995, 28, 5300 CrossRef CAS; (c) A. I. Smirnov and T. I. Smirnova, Appl. Magn. Reson., 2001, 21, 453 CrossRef CAS; (d) K. Fukui, T. Ito, M. Tada, M. Aoyama, S. Sato, J. Onodera and H. Ohya, J. Magn. Reson., 2003, 163, 174 CrossRef CAS; (e) M. L. Deschamps, E. S. Pilka, J. R. Potts, I. D. Campbell and J. Boyd, J. Biomol. NMR, 2005, 31, 155 CrossRef CAS; (f) G. Facorro, A. Bianchin, J. Boccio and A. Hager, Appl. Spectrosc., 2006, 60, 1078 CrossRef CAS; (g) D. Marsh, J. Magn. Reson., 2008, 190, 60 CrossRef CAS; (h) M. Tudose, T. Constantinescu, A. T. Balaban and P. Ionita, Appl. Surf. Sci., 2008, 254, 1904 CrossRef CAS; (i) A. Cecchi, L. Ciani, J. Y. Winum, J. L. Montero, A. Scozzafava, S. Ristori and C. T. Supuran, Bioorg. Med. Chem. Lett., 2008, 18, 3475 CrossRef CAS.
- (a) T. Ishida and H. Iwamura, J. Am. Chem. Soc., 1991, 113, 4238 CrossRef CAS; (b) F. Kanno, K. Inoue, N. Koga and H. Iwamura, J. Phys. Chem., 1993, 97, 13267 CrossRef CAS; (c) K. Inoue and H. Iwamura, J. Am. Chem. Soc., 1994, 116, 3173 CrossRef CAS; (d) K. Inoue and H. Iwamura, Adv. Mater., 1996, 8, 73 CAS; (e) K. Inoue, Metal Aminoxyl-Based Molecular Magnets, 2001, p. 61 Search PubMed.
- (a) R. Sayre, J. Am. Chem. Soc., 1955, 77, 6689 CrossRef CAS; (b) M. Lamchen and T. W. Mittag, J. Chem. Soc., 1966, 2300 Search PubMed; (c) J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078 CrossRef; (d) E. F. Ullman, L. Call and J. H. Osiecki, J. Org. Chem., 1970, 35, 3623 CrossRef CAS; (e) E. F. Ullman, J. H. Osiecki, D. G. B. Boocock and R. Darcy, J. Am. Chem. Soc., 1972, 94, 7049 CrossRef CAS; (f) J. F. W. Keana, Chem. Rev., 1978, 78, 37 CrossRef CAS; (g) Imidazoline Nitroxides, ed. L. B. Volodarsky, CRC Press, Boca Raton, FL, 1988, vol. I–II Search PubMed; (h) H. G. Aulich, in Nitrones, Nitronates and Nitroxides, ed. S. Patai and Z. Rappoport, John Wiley & Sons, New York, 1989, p. 313 Search PubMed; (i) M. E. Brik, Heterocycles, 1995, 41, 2827.
- (a) F. A. Neugebauer, Angew. Chem., Int. Ed. Engl., 1973, 12, 455 CrossRef; (b) R. Kuhn and H. Trischmann, Angew. Chem., Int. Ed. Engl., 1963, 2, 155 CrossRef; (c) F. A. Neugebauer, H. Fischer and C. Krieger, J. Chem. Soc., Perkin Trans. 2, 1993, 535 RSC; (d) D. J. R. Brook, V. Lynch, B. Conklin and M. A. Fox, J. Am. Chem. Soc., 1997, 119, 5155 CrossRef CAS; (e) R. G. Hicks, B. D. Koivisto and M. T. Lemaire, Org. Lett., 2004, 6, 1887 CrossRef CAS; (f) T. M. Barclay, R. G. Hicks, M. T. Lemaire and L. K. Thompson, Chem. Commun., 2000, 2141 RSC; (g) T. M. Barclay, R. G. Hicks, A. S. Ichimura and G. W. Patenaude, Can. J. Chem., 2002, 80, 1501 CrossRef CAS; (h) T. M. Barclay, R. G. Hicks, M. T. Lemaire, L. K. Thompson and Z. Q. Xu, Chem. Commun., 2002, 1688 RSC.
- (a) M. Tamura, Y. Nakazawa, D. Shiomi, K. Nozawa, Y. Hosokoshi, M. Ishikawa, M. Takahashi and M. Kinoshita, Chem. Phys. Lett., 1991, 186, 401 CrossRef CAS; (b) A. J. Banister, N. Bricklebank, I. Lavender, J. M. Rawson, C. I. Gregory, B. K. Tannner, W. Clegg, M. R. J. Egglewood and F. Palacio, Angew. Chem., Int. Ed. Engl., 1996, 35, 2533 CrossRef CAS.
- R. Chiarelli, M. A. Novak, A. Rassat and J. L. Tholence, Nature, 1993, 363, 147 CrossRef CAS.
- K. Nakasuji, M. Yamaguchi and I. Murata, et al. , J. Am. Chem. Soc., 1989, 111, 9265 CrossRef CAS.
- K. Goto, T. Kubo and K. Yamamoto, et al. , J. Am. Chem. Soc., 1999, 121, 1619 CrossRef CAS.
- R. T. Oakley, Can. J. Chem., 1993, 71, 1775 CAS.
- A. J. Banister, Nature, 1972, 237, 92 CrossRef CAS.
- R. T. Oakley, Prog. Inorg. Chem., 1988, 36, 299.
- A. W. Cordes, R. C. Haddon and R. T. Oakley, Adv. Mater., 1994, 10, 798 CrossRef CAS.
- T. Chivers, A Guide to Chalcogen–Nitrogen Chemistry, World Scientific, 2005 Search PubMed.
- R. C. Haddon, Nature, 1975, 256, 394 CAS.
- J. M. Rawson and F. Palacio, Struct. Bonding, 2001, 100, 93 CAS.
- J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560 RSC.
- A. J. Banister, I. Lavender and J. M. Rawson, Adv. Heterocycl. Chem., 1995, 62, 137 and references therein.
- (a) M. J. Perkins, Adv. Phys. Org. Chem., 1980, 17, 1 CAS; (b) D. Rehorek, Chem. Soc. Rev., 1991, 20, 341 RSC; (c) A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983 CrossRef CAS; (d) V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992 CrossRef CAS.
- (a) J. L. Zweier and P. Kuppusamy, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5703 CAS.
- M. Kini and H. H. Wang, Proc. NATO Advanced Research Workshop on Molecular Low Dimensional and Nanostructured Materials for Advanced Applications, Poznan, Poland, September 1–5, 2001, ed. A. Graja, B. R. Bulka and F. Kajzar, Kluwer Academic, 2002, p. 139 Search PubMed.
- (a) T. J. Marks, Angew. Chem., Int. Ed. Engl., 1990, 29, 857 CrossRef; (b) J. S. Miller, Adv. Mater., 1990, 2, 98 CrossRef; (c) J. S. Miller, Adv. Mater., 1993, 5, 587 CrossRef; (d) J. S. Miller, Adv. Mater., 1993, 5, 671 CrossRef; (e) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef.
- (a) P. Lacroix, Chem. Mater., 2001, 13, 3495 CrossRef CAS; (b) P. G. Lacroix and K. Nakanti, Adv. Mater., 1997, 9, 1105 CrossRef CAS; (c) R. Andreu, I. Malfant, P. G. Lacroix, H. Gornitzka and K. Nakatani, Chem. Mater., 1999, 11, 840 CrossRef CAS.
- (a) B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) J.-M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed; (c) A. P. de Silva and N. D. McClenaghan, Chem.–Eur. J., 2004, 10, 574 CrossRef; (d) R. L. Carroll and C. B. Gorman, Angew. Chem., Int. Ed., 2002, 41, 4378 CrossRef.
- (a) Photochromism, Molecules and Systems, ed. H. Dekrr and H. Bouas-Laurent, Elsevier, Amsterdam, Revised Edition, 2003 Search PubMed; (b) M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS.
- A. P. de Silva, H. Q. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef.
- (a) B. J. Coe, Chem.–Eur. J., 1999, 5, 2464 CrossRef CAS; (b) M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jeffery, J. A. McCleverty, M. D. Ward, I. Asselberghs, K. Clays and A. Persoons, Chem. Commun., 2001, 49 RSC; (c) F. Paul, K. Costuas, I. Ledoux, S. Deveau, J. Zyss, J.-F. Halet and C. Lapinte, Organometallics, 2002, 21, 5229 CrossRef CAS.
- (a) F. Renz, H. Oshio, V. Ksenofontov, M. Waldeck, H. Spiering and P. Gütlich, Angew. Chem., Int. Ed., 2000, 39, 3699 CrossRef CAS; (b) P. Gütlich, Y. García and T. Woike, Coord. Chem. Rev., 2001, 219, 839 CrossRef; (c) O. Sato, Acc. Chem. Res., 2003, 36, 692 CrossRef CAS.
- (a) M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443 CrossRef CAS; (b) J. S. Miller, Angew. Chem., Int. Ed., 2003, 42, 27 CrossRef CAS.
- L. Cambi and L. Szego, Ber. Dtsch. Chem. Ges. B, 1931, 64, 2591 CrossRef.
- M. Tamura, Y. Nakazawa and D. Hiomi, et al. , Chem. Phys. Lett., 1991, 186, 401 CrossRef CAS.
- A. J. Banister, N. Bricklebank and I. Lavender, et al. , Angew. Chem., Int. Ed. Engl., 1996, 35, 2533 CrossRef CAS.
- (a) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS; (b) C. Bellini and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS.
- J. Veciana and H. Iwamura, MRS Bull., 2000, 36, 41 CrossRef.
- K. Matsuda, N. Nakamura, K. Takahashi, K. Inoue, N. Koga and H. Iwamura, J. Am. Chem. Soc., 1995, 117, 5550 CrossRef CAS.
- (a) N. Ventosa, D. Ruiz, J. Sedo, X. Tomas, C. Rovira, B. Andre and J. Veciana, Chem.–Eur. J., 1999, 12, 3533 CrossRef; (b) J. Sedó, N. Ventosa, D. Ruiz-Molina, M. Mas, E. Molins, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 1998, 37, 330 CrossRef CAS; (c) A. Rajca, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 1999, 121, 6308 CrossRef CAS.
- R. J. Bushby, D. R. McGill, K. M. Ng and N. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1997, 1405 RSC.
- J. Sedó, N. Ventosa, A. Molins, M. Pons, C. Rovira and J. Veciana, J. Org. Chem., 2001, 66, 1567 CrossRef CAS; J. Sedó, N. Ventosa, A. Molins, M. Pons, C. Rovira and J. Veciana, J. Org. Chem., 2001, 66, 1579 CrossRef CAS.
- N. Azuma, K. Ishizu and K. Mukai, J. Chem. Phys., 1974, 61, 2294 CrossRef CAS.
- Y. Teki, M. Kimura, S. Narimatsu, K. Ohara and K. Mukai, Bull. Chem. Soc. Jpn., 2004, 77, 95 CrossRef CAS.
- J. S. Miller and J. A. Epstein, Angew. Chem., Int. Ed. Engl., 1994, 106, 385 CrossRef.
- O. Elsner, D. Ruiz-Molina, J. Vidal-Gancedo, C. Rovira and J. Veciana, Chem. Commun., 1999, 579 RSC.
- O. Elsner, D. Ruiz-Molina, I. Ratera, J. Vidal-Gancedo, C. Rovira and J. Veciana, J. Organomet. Chem., 2001, 637, 251 CrossRef.
- B. D. Koivisto, A. S. Ichimura and R. McDonald, et al. , J. Am. Chem. Soc., 2006, 128, 690–691 CrossRef CAS.
- D. Maspoch, L. Catala, P. Gerbier, D. Ruiz-Molina, J. Vidal-Gancedo, K. Wurst, C. Rovira and J. Veciana, Chem.–Eur. J., 2002, 8(16), 3635 CrossRef CAS.
- (a) A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392 CrossRef CAS; (b) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS.
- (a) A. Caneschi, D. Gatteschi and P. Rey, Prog. Inorg. Chem., 1991, 39, 331; (b) H. Iwamura and K. Inoue, in Magnetism: Molecules to Materials II. Molecule-Based Materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, Germany, 2001, p. 61 Search PubMed; (c) H. Oshio and T. Ito, Coord. Chem. Rev., 2000, 198, 329 CrossRef CAS; (d) R. Ziessel, G. Ulrich, R. C. Lawson and L. Echegoyen, J. Mater. Chem., 1999, 9, 1435 RSC.
- (a) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS; (b) M. Minguet, D. Luneau, E. Lhotel, V. Villar, C. Paulsen, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2002, 41, 586 CrossRef CAS.
- (a) A. Caneschi, D. Gatteschi, J. P. Renard, P. Rey and R. Sessoli, J. Phys., 1988, 49, 859; (b) C. Benelli, A. Caneschi, D. Gatteschi, L. Pardi and P. Rey, J. Phys., 1988, 49, 861; (c) A. Caneschi, D. Gatteschi, R. Sessoli, S. K. Hoffmann, J. Laugier and P. Rey, Inorg. Chem., 1988, 27, 2390 CrossRef; (d) A. Caneschi, D. Gatteschi, J. Laugier, L. Pardi, P. Rey and C. Zanchini, Inorg. Chem., 1988, 27, 2027 CrossRef CAS; (e) A. Caneschi, D. Gatteschi, P. Rey and R. Sessoli, Inorg. Chem., 1988, 27, 1756 CrossRef CAS; (f) A. Caneschi, D. Gatteschi, J. Laugier, P. Rey and R. Sessoli, Inorg. Chem., 1988, 27, 1553 CrossRef CAS; (g) A. Caneschi, D. Gatteschi, J. Laugier, P. Rey, R. Sessoli and C. Zanchini, J. Am. Chem. Soc., 1988, 110, 2795 CrossRef CAS; (h) A. Caneschi, D. Gatteschi, A. Grand, J. Laugier, L. Pardi and P. Rey, Inorg. Chem., 1988, 27, 1031 CrossRef CAS.
- H. Iwamura, K. Inoue and T. Hayamizu, Pure Appl. Chem., 1996, 68, 243 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, J. Vidal-Gancedo, C. Rovira and J. Veciana, Dalton Trans., 2004, 1073 RSC.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, C. Rovira and J. Veciana, Chem. Commun., 2002, 2958 RSC.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, C. Rovira and J. Veciana, Polyhedron, 2003, 22, 1929 CrossRef CAS.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. M. Hernandez, G. Vaughan, C. Rovira, F. Lloret, J. Tejada and J. Veciana, Chem. Commun., 2005, 5035 RSC.
- N. Roques, S. Perruchas, D. Maspoch, A. Datcu, K. Wurst, J.-P. Sutter, C. Rovira and J. Veciana, Inorg. Chim. Acta, 2007, 360, 3861 CrossRef CAS.
- X. Ribas, D. Maspoch, K. Wurst, J. Veciana and C. Rovira, Inorg. Chem., 2006, 45, 14 CrossRef CAS.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. M. Hernández, F. Lloret, J. Tejada, C. Rovira and J. Veciana, Inorg. Chem., 2007, 46, 5 CrossRef.
- S. Nakatsuji and H. Anzai, J. Mater. Chem., 1997, 7, 2161 RSC.
- (a) A. Caneschi, F. Ferraro, D. Gatteschi, A. Le Lirzin, M. A. Novak, E. Rentschler and R. Sessoli, Adv. Mater., 1995, 7, 476–478 CrossRef CAS; (b) A. Caneschi, F. Ferraro, D. Gatteschi, A. Le Lirzin and E. Rentschler, Inorg. Chim. Acta, 1995, 235, 159–164 CrossRef CAS.
- M. P. Allemand, K. C. Khemani, A. Koch, F. Wudl, K. Holczer, S. Donovan, G. Gruner and J. D. Thompson, Science, 1991, 253, 301 CrossRef CAS.
- R. Chiarelli, M. A. Novak, A. Rassat and J. L. Tholence, Nature, 1993, 363, 147 CrossRef CAS.
- M. Mito, H. Nakano, T. Kawae, M. Hitaka, S. Takagi, H. Deguchi, K. Suzuki, K. Mukai and K. Takeda, J. Phys. Soc. Jpn., 1997, 66, 2147 CrossRef CAS.
- (a) A. Caneschi, D. Gatteschi, J. Laugier and P. Rey, J. Am. Chem. Soc., 1987, 109, 2191 CrossRef CAS; (b) A. Caneschi, D. Gatteschi, J. P. Renard, P. Rey and R. Sessoli, Inorg. Chem., 1989, 28, 3314 CrossRef CASA. Caneschi, D. Gatteschi, J. P. Renard, P. Rey and R. Sessoli, Inorg. Chem., 1989, 28, 2940 Search PubMed.
- (a) A. Caneschi, F. Ferraro, D. Gatteschi, P. Rey and R. Sessoli, Inorg. Chem., 1991, 30, 3162 CrossRef CAS; (b) A. Caneschi, P. Chiesi, L. David, F. Ferraro, D. Gatteschi and R. Sessoli, Inorg. Chem., 1993, 32, 1445 CrossRef CAS; (c) A. Caneschi, D. Gatteschi and R. Sessoli, Inorg. Chem., 1993, 32, 4612 CrossRef CAS.
- (a) K. Inoue, T. Hayamizu, H. Iwamura, D. Hashizume and Y. Ohashi, J. Am. Chem. Soc., 1996, 118, 1803 CrossRef CAS; (b) H. Iwamura, K. Inoue and N. Koga, New J. Chem., 1998, 22, 201 RSC; (c) F. Mathevet and D. Luneau, J. Am. Chem. Soc., 2001, 123, 7465 CrossRef CAS; (d) J. Laugier, Inorg. Chem., 1993, 32, 5616 CrossRef CAS.
- D. Luneau, G. Risoan, P. Rey, A. Grand, A. Caneschi, D. Gatteschi and J. Laugier, Inorg. Chem., 1993, 32, 5616 CrossRef CAS.
- (a) D. Luneau, P. Rey, J. Laugier, P. Fries, A. Caneschi, D. Gatteschi and R. Sessoli, J. Am. Chem. Soc., 1991, 113, 1245 CrossRef CAS; (b) P. Rey, D. Luneau and A. Cogne, in NATO ASI Ser., Ser. E, 1991, vol. 198, p. 203 Search PubMed; (c) D. Luneau, P. Rey, J. Laugier, E. Belorizky and A. Cogne, Inorg. Chem., 1992, 31, 3578 CrossRef CAS; (d) C. Benelli, A. Caneschi, D. Gatteschi and L. Pardi, Inorg. Chem., 1992, 31, 741 CrossRef CAS; (e) H. Oshio, T. Watanabe, A. Ohto, T. Ito and U. Nagashima, Angew. Chem., Int. Ed. Engl., 1994, 33, 670 CrossRef; (f) H. Oshio, T. Watanabe, A. Ohto, T. Ito and H. Masuda, Inorg. Chem., 1996, 35, 472 CrossRef CAS; (g) H. Oshio, T. Watanabe, A. Ohto and T. Ito, Inorg. Chem., 1997, 36, 1608 CrossRef CAS; (h) K. Fegy, N. Sanz, D. Luneau, E. Belorizky and P. Rey, Inorg. Chem., 1998, 37, 4518 CrossRef CAS; (i) H. Oshio, M. Yamamoto and T. Ito, J. Chem. Soc., Dalton Trans., 1999, 2641 RSC; (j) C. Lescop, D. Luneau, P. Rey, G. Bussiere and C. Reber, Inorg. Chem., 2002, 41, 5566 CrossRef CAS; (k) A. Marvilliers, Y. Pei, J. Cano Boquera, K. E. Vostrikova, C. Paulsen, E. Riviere, J.-P. Audiere and T. Mallah, Chem. Commun., 1999, 1951 RSC; (l) K. E. Vostrikova, D. Luneau, W. Wernsdorfer, P. Rey and M. Verdaguer, J. Am. Chem. Soc., 2000, 122, 718 CrossRef CAS; (m) G. Francese, F. M. Romero, A. Neels, H. Stoeckli-Evans and S. Decurtins, Inorg. Chem., 2000, 39, 2087 CrossRef CAS.
- (a) K. Fegy, D. Luneau, T. Ohm, C. Paulsen and P. Rey, Angew. Chem., Int. Ed., 1998, 37, 1270 CrossRef CAS; (b) K. Fegy, D. Luneau, E. Belorizky, M. Novac, J.-L. Tholence, C. Paulsen, T. Ohm and P. Rey, Inorg. Chem., 1998, 37, 4524 CrossRef CAS; (c) K. Fegy, C. Lescop, D. Luneau and P. Rey, Mol. Cryst. Liq. Cryst., 1999, 334, 521 CAS; (d) P. Rey and D. Luneau, NATO ASI Ser., Ser. C: Math. Phys. Sci., 1999, 518, 145 Search PubMed.
- (a) C. Lescop, D. Luneau, E. Belorizky, P. Fries, M. Guillot and P. Rey, Inorg. Chem., 1999, 38, 5472 CrossRef CAS; (b) C. Lescop, D. Luneau and P. Rey, MRS Symp. Proc., 2000, 598, BB3.49/1 Search PubMed; (c) C. Lescop, D. Luneau, G. Bussiere, M. Triest and C. Reber, Inorg. Chem., 2000, 39, 3740 CrossRef CAS; (d) C. Lescop, E. Belorizky, D. Luneau and P. Rey, Inorg. Chem., 2002, 41, 3375 CrossRef CAS.
- (a) D. Luneau, F. M. Romero and R. Ziessel, Inorg. Chem., 1998, 37, 5078 CrossRef CAS; (b) F. M. Romero, D. Luneau and R. Ziessel, Chem. Commun., 1998, 551 RSC; (c) H. Oshio, T. Yaginuma and T. Ito, Inorg. Chem., 1999, 38, 2750 CrossRef CAS; (d) C. Stroh, E. Belorizky, P. Turek, H. Bolvin and R. Ziessel, Inorg. Chem., 2003, 42, 2938 CrossRef CAS.
- R. Ziessel, Mol. Cryst. Liq. Cryst., 1995, 273, 101.
- (a) Y. Pei, A. Lang, P. Bergerat, O. Kahn, M. Fettouhi and L. Ouahab, Inorg. Chem., 1996, 35, 193 CrossRef CAS; (b) J. P. Sutter, M. L. Kahn, S. Golhen, L. Ouahab and O. Kahn, Chem.–Eur. J., 1998, 4, 571 CrossRef CAS; (c) J. P. Sutter, M. L. Kahn and O. Kahn, Adv. Mater., 1999, 11, 863 CrossRef CAS; (d) M. L. Kahn, J. P. Sutter, S. Golhen, P. Guionneau, L. Ouahab, O. Kahn and D. Chasseau, J. Am. Chem. Soc., 2000, 122, 3413 CrossRef CAS.
- (a) A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS; (b) H. Miyasaka and R. Clerac, Bull. Chem. Soc. Jpn., 2005, 78, 1725 CrossRef CAS; (c) T. Kajiwara, M. Nakano, Y. Kaneko, S. Takaishi, T. Ito, M. Yamashita, A. Igashira-Kamiyama, H. Nojiri, Y. Ono and N. Kojima, J. Am. Chem. Soc., 2005, 127, 10150 CrossRef CAS; (d) N. Ishii, T. Ishida and T. Nogami, Inorg. Chem., 2006, 45, 3837 CrossRef CAS; (e) K. Bernot, L. Bogani, A. Caneschi, D. Gatteschi and R. Sessoli, J. Am. Chem. Soc., 2006, 128, 7947 CrossRef CAS.
- (a) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66–71 CrossRef CAS; (b) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268 CrossRef CAS.
- B. Barbara, Inorg. Chim. Acta, 2008, 361, 3371 CrossRef CAS.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS.
- (a) H. Miyasaka, M. Julve, M. Yamashita and R. Clerac, Inorg. Chem., 2009, 48, 3420 CrossRef CAS; (b) C. Coulon, H. Miyasaka and R. Clérac, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, Springer, Berlin, 2006, vol. 122, p. 163 Search PubMed.
- K. Bernot, L. Bogani, A. Caneschi, D. Gatteschi and R. Sessoli, J. Am. Chem. Soc., 2006, 128, 7947 CrossRef CAS.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio and R. Sessoli, J. Chem. Soc., Dalton Trans., 2000, 3907 RSC.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., 2001, 113, 1810 CrossRef.
- A. Caneschi, D. Gatteschi, N. Lalioti, R. Sessoli, L. Sorace, V. Tangoulis and A. Vindigni, Chem.–Eur. J., 2002, 8, 286–292 CrossRef CAS.
- (a) C. Benelli, A. Caneschi, D. Gatteschi and R. Sessoli, Inorg. Chem., 1993, 32, 4797 CrossRef CAS; (b) C. Benelli, A. Caneschi, D. Gatteschi and R. Sessoli, Adv. Mater., 1992, 4, 504 CAS.
- (a) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS; (b) A. Caneschi, A. Dei, D. Gatteschi, L. Sorace and K. Vostrikova, Angew. Chem., Int. Ed., 2000, 39, 246 CrossRef; (c) A. Caneschi, A. Dei, D. Gatteschi, C. A. Massa, L. A. Pardi, S. Poussereau and L. Sorace, Chem. Phys. Lett., 2003, 371, 694 CrossRef CAS; (d) G. Poneti, K. Bernot, L. Bogani, A. Caneschi, R. Sessoli, W. Wernsdorfer and D. Gatteschi, Chem. Commun., 2007, 1807 RSC.
- (a) L. Bogani, C. Sangregorio, R. Sessoli and D. Gatteschi, Angew. Chem., Int. Ed., 2005, 44, 5817 CrossRef; (b) K. Bernot, L. Bogani, R. Sessoli and D. Gatteschi, Inorg. Chim. Acta, 2007, 360, 3807 CrossRef CAS.
- (a) F. Bartolome, J. Bartolome, C. Benelli, A. Caneschi, D. Gatteschi, C. Paulsen, M. G. Pini, A. Rettori, R. Sessoli and Y. Volokitin, Phys. Rev. Lett., 1996, 77, 382 CrossRef CAS; (b) F. Cinti, A. Rettori, M. Barucci, E. Olivieri, L. Risegari, G. Ventura, A. Caneschi, D. Gatteschi, D. Rovai, M. G. Pini, M. Affronte, M. Mariani and A. J. Lascialfari, J. Magn. Magn. Mater., 2007, 310, 1460 CrossRef CAS.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS.
- L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750 RSC.
- (a) R. Giraud, W. Wernsdorfer, A. M. Tkachuk, D. Mailly and B. Barbara, Phys. Rev. Lett., 2001, 87, 057203 CrossRef CAS; (b) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. J. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS; (c) T. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650 CrossRef CAS; (d) T. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., 2003, 117, 2991 ( Angew. Chem., Int. Ed. , 2005 , 44 , 2931 ).
- K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gatteschi, J. Am. Chem. Soc., 2009, 131, 5573 CrossRef CAS.
- (a) K. Bernot, J. Luzon, R. Sessoli, A. Vindigni, J. Thion, S. Richeter, D. Leclercq, J. Larionova and A. J. Van Der Lee, J. Am. Chem. Soc., 2008, 130, 1619 CrossRef CAS; (b) A. V. Palii, S. M. Ostrovsky, S. I. Klokishner, O. S. Reu, Z. M. Sun, A. V. Prosvirin, H. H. Zhao, J. G. Mao and K. R. Dunbar, J. Phys. Chem. A, 2006, 110, 14003 CrossRef CAS.
- E. Lhotel, D. B. Amabilino, C. Sporer, D. Luneau, J. Veciana and C. Paulsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 064416 CrossRef.
- N. Ishii, Y. Okamura, S. Chiba, T. Nogami and T. Ishida, J. Am. Chem. Soc., 2008, 130, 24 CrossRef CAS.
- (a) D. K. Rittenberg, K. I. Sugiura, Y. Sakata, S. Mikami, A. J. Epstein and J. S. Miller, Adv. Mater., 2000, 12, 126 CrossRef CAS; (b) A. J. Epstein, MRS Bull., 2000, 25, 33 CrossRef CAS.
- Y. Numata, K. Inoue, N. Baranov, M. Kurmoo and K. Kikuchi, J. Am. Chem. Soc., 2007, 129, 9902 CrossRef CAS.
- D. J. R. Brook, V. Lynch, B. Conklin and M. A. Fox, J. Am. Chem. Soc., 1997, 119, 5155 CrossRef CAS.
- T. M. Barclay, R. G. Hicks, M. T. Lemaire and L. K. Thompson, Inorg. Chem., 2001, 40, 5581 CrossRef CAS.
- L. Norel, F. Pointillart, C. Train, L. M. Chamoreau, K. Boubekeur, Y. Journaux, A. Brieger and D. J. R. Brook, Inorg. Chem., 2008, 47, 2396 CrossRef CAS.
- B. D. Koivisto, A. S. Ichimura, R. McDonald, M. T. Lemaire, L. K. Thompson and R. G. Hicks, J. Am. Chem. Soc., 2006, 128, 690 CrossRef CAS.
- J. Jornet, M. Deumal, J. Ribas-Arino, M. J. Bearpark, M. A. Robb, R. G. Hicks and J. J. Novoa, Chem.–Eur. J., 2006, 12, 3995 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, C. Rovira and J. Veciana, Chem. Commun., 2004, 1164 RSC.
- N. Roques, D. Maspoch, F. Luix, A. Camón, K. Wurst, A. Datcu, D. Ruiz-Molina, C. Rovira and J. Veciana, J. Mater. Chem., 2008, 18, 98 RSC.
- (a) D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. Tejada, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2004, 126, 730 CrossRef CAS; (b) D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, G. Vaughan, J. Tejada, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 1828 CrossRef CAS; (c) N. Roques, D. Maspoch, A. Datcu, K. Wurst, D. Ruiz-Molina, C. Rovira and J. Veciana, Polyhedron, 2007, 26, 1934 CrossRef CAS; (d) D. Maspoch, N. Domingo, N. Roques, K. Wurst, J. Tejada, C. Rovira, D. Ruiz-Molina and J. Veciana, Chem.–Eur. J., 2007, 13, 8153 CrossRef CAS.
- N. Roques, D. Maspoch, K. Wurst, D. Ruiz-Molina, C. Rovira and J. Veciana, Chem.–Eur. J., 2006, 12, 9238 CrossRef CAS.
- W. Fujita and K. Awaga, Chem. Phys. Lett., 2002, 357, 385 CrossRef CAS.
- W. Fujita, K. Awaga, Y. Nakazawa, K. Saito and M. Sorai, Chem. Phys. Lett., 2002, 352, 348 CrossRef CAS.
- (a) C. M. Robertson, D. J. T. Myles, A. A. Leitch, R. W. Reed, B. M. Dooley, N. L. Frank, P. A. Dube, L. K. Thompson and R. T. Oakley, J. Am. Chem. Soc., 2007, 129, 12688 CrossRef CAS; (b) C. M. Robertson, A. A. Leitch, K. Cvrkalj, R. W. Reed, D. J. T. Myles, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2008, 130, 8414 CrossRef CAS.
- (a) A. A. Leitch, J. L. Brusso, K. Cvrkalj, R. W. Reed, C. M. Robertson, P. A. Dube and R. T. Oakley, Chem. Commun., 2007, 3368 RSC; (b) A. A. Leitch, X. Yu, S. M. Winter, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2009, 131, 7112 CrossRef CAS.
- A. A. Leitch, X. Yu, S. M. Winter, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2009, 131, 7112 CrossRef CAS.
- A. J. Banister, N. Bricklebank, I. Lavender, J. M. Rawson, C. I. Gregory, B. K. Tannner, W. Clegg, M. R. J. Egglewood and F. Palacio, Angew. Chem., Int. Ed. Engl., 1996, 35, 2533 CrossRef CAS.
- J. Luzon, J. Campo, F. Palacio, G. J. McIntyre, A. E. Goeta, E. Ressouche, C. M. Pask and J. M. Rawson, Phys. B, 2003, 335, 1 CrossRef CAS.
- (a) G. A. Prinz, Science, 1998, 282, 1660 CrossRef CAS; (b) M. Johnson, J. Phys. Chem. B, 2005, 109, 14278 CrossRef CAS.
- (a) T. Sugawara and M. M. Matsushita, in Carbon-Based Magnetism, ed. T. Makarova and F. Palacio, Elsevier, Amsterdam, 2006, p. 1 Search PubMed; (b) K. Yamaguchi, H. Nishimoto, T. Fueno, T. Nogami and Y. Shirota, Chem. Phys. Lett., 1990, 166, 408 CrossRef CAS; (c) K. Yamaguchi, M. Okumura, T. Fueno and K. Nakasuji, Synth. Met., 1991, 41–43, 3631 CrossRef.
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691 RSC.
- (a) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS; (b) J. F. Gregg, I. Petej, E. Jouguelet and C. Dennis, J. Phys. D: Appl. Phys., 2002, 35, R121 CrossRef CAS.
- (a) H. Ago, Nature, 1999, 401, 572 CrossRef CAS; (b) L. E. Hueso, J. M. Pruneda, V. Ferrari, G. Burnell, J. P. Valdés-Herrera, B. D. Simons, P. B. Littlewood, E. Artacho, A. Fert and N. D. Mathur, Nature, 2007, 445, 410 CrossRef CAS.
- (a) Z. H. Xiong, D. Wu, Z. Valy Vardeny and J. Shi, Nature, 2004, 427, 821 CrossRef CAS; (b) T. X. Wang, H. X. Wei, Z. M. Zeng, X. F. Han, Z. M. Hong and G. Q. Shi, Appl. Phys. Lett., 2006, 88, 242505 CrossRef; (c) F. L. Bloom, W. Wagemans, M. Kemerink and B. Koopmans, Phys. Rev. Lett., 2007, 99, 257201 CrossRef CAS; (d) L. E. Hueso, I. Bergenti, A. Riminucci, Y. Zhan and V. Dediu, Adv. Mater., 2007, 19, 2639 CrossRef CAS.
- (a) T. S. Santos, J. S. Lee, P. Migdal, I. C. Lekshmi, B. Satpati and J. S. Moodera, Phys. Rev. Lett., 2007, 98, 16601 CrossRef CAS; (b) J. R. Petta, S. K. Slater and D. C. Ralph, Phys. Rev. Lett., 2004, 93, 136601 CrossRef CAS; (c) W. Wang and C. Richter, Appl. Phys. Lett., 2006, 89, 153105 CrossRef.
- (a) C. Chappert, A. Fert and F. N. Van Dau, Nat. Mater., 2007, 6, 813 CrossRef CAS; (b) J. Camarero and E. Coronado, J. Mater. Chem., 2009, 19, 1678 RSC; (c) T. Sugawara and M. M. Matsushita, J. Mater. Chem., 2009, 19, 1738 RSC.
- (a) S. Sanvito, J. Mater. Chem., 2007, 17, 4455–4459 RSC; (b) Z. H. Xiong, D. Wu, Z. Valy Vardeny and J. Shi, Nature, 2004, 427, 821 CrossRef CAS; (c) K. Tsukagoshi, B. W. Alphenaar and H. Ago, Nature, 1999, 401, 572 CrossRef CAS.
- P. Parida, A. Kundu and S. K. Pati, Phys. Chem. Chem. Phys., 2010, 12, 6924 RSC.
- (a) R. Fiederling, M. Keim, G. Reuscher, W. Ossau, G. Schmidt, A. Waag and W. Molenkamp, Nature, 1999, 402, 787 CrossRef CAS; (b) Y. Ohno, K. Young, B. Beschoten, F. Matsukura, H. Ohn and D. D. Awschalom, Nature, 1999, 402, 790 CrossRef CAS.
- R. Hanson and D. D. Awschalom, Nature, 2008, 453, 1043 CrossRef CAS.
- (a) R. Metzger, Acc. Chem. Res., 1999, 32, 950 CrossRef CAS; (b) J. M. Tour, Acc. Chem. Res., 2000, 33, 79; (c) R. Lloyd and C. B. Gorman, Angew. Chem., Int. Ed., 2000, 41, 4378; (d) J. M. Tour, M. Kozaki and J. M. Seminario, J. Am. Chem. Soc., 1998, 120, 8486 CrossRef CAS.
- J. M. Seminario and J. M. Tour, Molecular Electronics – Science and Technology, ed. A. Aviram and M. A. Ratner, New York Academy of Science, New York, 1998, p. 69 Search PubMed.
- Mixed Valence Systems: Applications in Chemistry, Physics and Biology, ed. K. Prassides, NATO AS1 Series, Kluwer Academic Publishers, Dordrecht, 1991 Search PubMed.
- (a) J. M. Verhoeven, M. N. Paddon-Row, N. S. Hush, H. Oevering and M. Heppener, Pure Appl. Chem., 1986, 58, 1285 CrossRef CAS; (b) P. Pasman, N. W. Koper, J. W. Verhoeven and J. R. Neth, Chem. Soc., 1983, 102, 55.
- M. B. Robin and P. Day, Mixed valence chemistry, Adv. Inorg. Chem., 1967, 10, 247 CAS.
- (a) P. W. Anderson and H. Hasegawa, Phys. Rev., 1955, 100, 675 CrossRef CAS; (b) L. Noodleman, Inorg. Chem., 1988, 27, 3677 CrossRef CAS; (c) G. Blondin and J.-J. Girerd, Chem. Rev., 1990, 90, 1359 CrossRef CAS.
- J. Veciana, J. Riera, J. Castañer and N. Ferrer, J. Organomet. Chem., 1985, 297, 131 CrossRef CAS.
- J. E. Sutton, P. M. Sutton and H. Taube, Inorg. Chem., 1979, 18, 1017 CrossRef CAS.
- (a) C. Rovira, D. Ruiz-Molina, O. Elsner, J. Vidal-Gancedo, J. Bonvoisin, J. P. Launay and J. Veciana, Chem.–Eur. J., 2001, 7(1), 240 CrossRef CAS; (b) V. Lloveras, J. Vidal-Gancedo, D. Ruiz-Molina, T. M. Figueira-Duarte, J.-F. Nierengarten, J. Vecaiana and C. Rovira, Faraday Discuss., 2006, 131, 291 RSC.
- V. Lloveras, J. Vidal-Gancedo, T. M. Figueira-Duarte, J. F. Nierengarten, J. J. Novoa, F. Mota, N. Ventosa, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2011, 133, 5818 CrossRef CAS.
- (a) X. Chi, et al. , J. Am. Chem. Soc., 2001, 123, 4041–4048 CrossRef CAS; (b) S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281 CrossRef CAS.
- R. G. Hicks, Nat. Chem., 2011, 3, 189 CrossRef CAS.
- (a) R. C. Haddon, A. Sarkar, S. K. Pal, X. Chi, M. E. Itkis and F. Tham, J. Am. Chem. Soc., 2010, 132, 17258 CrossRef; (b) K. Lekin, S. M. Winter, L. E. Downie, X. Bao, J. S. Tse, S. Desgreniers, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2010, 132, 16212 CrossRef CAS.
- (a) T. M. Barclay, et al. , J. Am. Chem. Soc., 1998, 120, 352–360 CrossRef CAS; (b) W. Fujita and K. Awaga, Science, 1999, 286, 261 CrossRef CAS.
- G. D'Avino, L. Grisanti, J. Guasch, I. Ratera, J. Veciana and A. Painelli, J. Am. Chem. Soc., 2008, 130, 12064 CrossRef CAS.
- (a) R. M. Buchanan and C. G. Pierpont, J. Am. Chem. Soc., 1980, 102, 4951 CrossRef CAS; (b) C. Roux, D. M. Adams, J. P. Itie, A. Polina, D. N. Hendrickson and M. Verdaguer, Inorg. Chem., 1996, 35, 2846 CrossRef CAS; (c) D. Ruiz-Molina, J. Veciana, K. Wurst, D. N. Hendrickson and C. Rovira, Inorg. Chem., 2000, 39, 617 CrossRef CAS; (d) D. Ruiz, J. Yoo, I. Guzei, A. Rheingold and D. N. Hendrickson, Chem. Commun., 1998, 2089 RSC; (e) S. H. Bodnar, A. Caneschi, A. Dei, D. A. Shultz and L. Sorace, Chem. Commun., 2001, 20, 2150 Search PubMed.
- I. Ratera, C. Sporer, D. Ruiz-Molina, N. Ventosa, J. Baggerman, A. M. Brouwer, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2007, 129, 6117 CrossRef CAS.
- (a) C. Creutz and H. Taube, J. Am. Chem. Soc., 1969, 91, 3988 CrossRef CAS; (b) C. Creutz, Prog. Inorg. Chem., 1983, 30, 1 CAS; (c) K. D. Demandis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655 CrossRef CAS; (d) S. F. Nelsen, M. A. Tran and H. Q. Nagy, J. Am. Chem. Soc., 1998, 120, 298 CrossRef CAS; (e) J. Bonvoisin, J.-P. Launay, W. Verbouwe, M. Van der Auweraer and F. C. De Schryver, J. Phys. Chem., 1996, 100, 17079 CrossRef CAS; (f) C. Lambert, G. Nöll and J. Schelter, Nat. Mater., 2002, 1, 69 CrossRef CAS; (g) N. Gautier, F. Dumur, V. Lloveras, J. Vidal-Gancedo, J. Veciana, C. Rovira and P. Hudhomme, Angew. Chem., Int. Ed., 2003, 42, 2765 CrossRef CAS.
- (a) S. Utamapanya and A. Rajca, J. Am. Chem. Soc., 1991, 113, 9242 CrossRef CAS; (b) L. Hviid, A. M. Brouwer, M. N. Paddon-Row and J. W. Verhoeven, ChemPhysChem, 2001, 2, 232 CrossRef CAS; (c) A. S. D. Sandanayaka, H. Sasabe, Y. Araki, Y. Furusho, O. Ito and T. J. Takata, J. Phys. Chem. A, 2004, 108, 5145 CrossRef CAS; (d) G. L. Closs and J. R. Miller, Science, 1988, 240, 440 CrossRef CAS.
- (a) R. A. Marcus, J. Chem. Phys., 1956, 24, 966 CrossRef CAS; (b) R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155 CrossRef CAS; (c) R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CAS.
- (a) J. M. Kriegl and G. Ulrich Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 123 CAS; (b) M. Cascella, A. Magistrato, I. Tavernelli, P. Carloni and U. Rothlisberger, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19641 CrossRef CAS; (c) A. Warshel, Z. T. Chu and W. W. Parson, Science, 1989, 246, 112 CrossRef CAS.
- A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2007, 129, 5515 CrossRef CAS.
- S. Chopin, J. Cousseau, E. Levillain, C. Rovira, J. Veciana, A. S. D. Sandanayaka, Y. Araki and O. Ito, J. Mater. Chem., 2006, 16, 112 RSC.
- (a) S. Nishida, et al. , Angew. Chem., Int. Ed., 2005, 44, 7277–7280 CrossRef CAS; (b) Y. Morita, S. Ishida, J. Kawai, K. Fukui, S. Nakazawa, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Org. Lett., 2002, 4, 1985 CrossRef CAS.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS.
- (a) Y. Morita, J. Kawai and N. Handa, Tetrahedron Lett., 2001, 42, 7991 CrossRef CAS; Y. Morita, J. Kawai and N. Handa, Tetrahedron Lett., 2001, 42, 7995; (b) S. Nishida, Y. Morita and K. Fukui, Angew. Chem., Int. Ed., 2005, 44, 7277 CrossRef CAS.
- (a) D. N. Hendrickson and C. G. Pierpont, Top. Curr. Chem., 2004, 234, 63 CAS; (b) E. Evangelio and D. Ruiz-Molina, Eur. J. Inorg. Chem., 2005, 2957 CrossRef CAS; (c) A. Beni, C. Carbonera, A. Dei, J.-F. Letard, R. Righini, C. Sangregorio and L. Sorace, J. Braz. Chem. Soc., 2006, 17, 1522 CAS; (d) O. Sato, A. Cui, R. Matsuda, J. Tao and S. Hayami, Acc. Chem. Res., 2007, 40, 361 CrossRef CAS; (e) E. Evangelio and D. Ruiz-Molina, C. R. Chim., 2008, 11, 1137 CrossRef CAS.
- M. Mannini, L. Sorace, L. Gorini, F. M. Piras, A. Caneschi, A. Magnani, S. Menichetti and D. Gatteschi, Langmuir, 2007, 23, 2389–2397 CrossRef.
- M. M. Matsushita, N. Ozaki, T. Sugawara, F. Nakamura and M. Hara, Chem. Lett., 2002, 596 CrossRef CAS.
- G. Harada, H. Sakurai, M. M. Matsushita, A. Izuoka and T. Sugawara, Chem. Lett., 2002, 1030 CrossRef CAS.
- N. Crivillers, S. Furukawa, A. Minoia, A. Ver Heyen, C. Sporer, M. Mas-Torrent, M. Linares, A. Volodin, C. Van Haesendonck, M. Van der Auweraer, R. Lazzaroni, S. De Feyter, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2009, 131, 6246 CrossRef CAS.
- N. Crivillers, M. Mas-Torrent, S. Perruchas, N. Roques, J. Vidal-Gancedo, J. Veciana, C. Rovira, L. Basabe-Desmonts, B.-J. Ravoo, M. Crego-Calama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2007, 46, 2215 CrossRef CAS.
- O. Shekhah, N. Roques, V. Mugnaini, C. Munuera, C. Ocal, J. Veciana and C. Wöll, Langmuir, 2008, 24, 6640 CrossRef CAS.
- Y. Manassen, R. J. Hamers, J. E. Demuth and A. J. Castellano, Phys. Rev. Lett., 1989, 62, 2531 CrossRef.
- V. Mugnaini, M. Fabrizioli, I. Ratera, M. Mannini, A. Caneschi, D. Gatteschi, Y. Manassen and J. Veciana, Solid State Sci., 2009, 11, 956 CrossRef CAS.
- P. Messina, M. Mannini, A. Caneschi, D. Gatteschi, L. Sorace, P. Sigalotti, C. Sandrin, S. Prato, P. Pittana and Y. Manassen, J. Appl. Phys., 2007, 101, 053916 CrossRef.
- C. Simao, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artes, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359 CrossRef CAS.
- N. Crivillers, C. Munuera, M. Mas-Torrent, C. Simao, S. T. Bromley, C. Ocal, C. Rovira and J. Veciana, Adv. Mater., 2009, 21, 1177 CrossRef CAS.
- N. Crivillers, M. Pardinals, M. Mas-Torrent, S. T. Bromley, C. Ocal, C. Rovira and J. Veciana, Chem. Commun., 2011, 47, 4664 RSC.
- T. Sugano, T. Fukasawa and M. Kinoshita, Synth. Met., 1991, 41, 3281 CrossRef.
- T. Sugimoto, S. Yamaga, M. Nakai, M. Tsuji, H. Nakatsuji and N. Hoshino, Chem. Lett., 1993, 1817 CAS.
- T. Ishida, K. Tomioka, T. Nogami, K. Yamaguchi, W. Mori and Y. Shirota, Mol. Cryst. Liq. Cryst., 1993, 232, 99 CrossRef CAS.
- S. Nakatsuji, S. Satoki, K. Suzuki, Enoki N. Kinoshita and H. Anzai, Synth. Met., 1995, 71, 1819 CrossRef CAS.
- (a) H. Fujiwara, E. Ojima and H. Kobayashi, Synth. Met., 2001, 120, 971 CrossRef CAS; (b) H. Fujiwara, E. Fujiwara and H. Kobayashi, Chem. Lett., 2002, 1048 CrossRef CAS; (c) H. Fujiwara, E. Fujiwara and H. Kobayashi, Synth. Met., 2002, 133, 359; (d) H. Fujiwara, H. J. Lee, H. Kobayashi, E. Fujiwara and A. Kobayashi, Chem. Lett., 2003, 482 CrossRef CAS; (e) H. Fujiwara, E. Fujiwara and H. Kobayashi, Synth. Met., 2003, 135, 533 CrossRef; (f) E. Fujiwara, A. Kobayashi, H. Fujiwara, T. Sugimoto and H. Kobayashi, Chem. Lett., 2004, 964 CrossRef CAS; (g) H. Fujiwara, H. J. Lee, H. B. Cui, H. Kobayashi, E. Fujiwara and A. Kobayashi, Adv. Mater., 2004, 16, 1765 CrossRef CAS; (h) E. Fujiwara, S. Aonuma, H. Fujiwara, T. Sugimoto and Y. Misaki, Chem. Lett., 2008, 84 CrossRef CAS.
- S. Hiraoka, T. Okamoto, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Am. Chem. Soc., 2004, 26, 58 CrossRef.
- A. Izuoka, M. Hiraishi, T. Abe, T. Sugawara, K. Sato and T. Takui, J. Am. Chem. Soc., 2000, 122, 3234 CrossRef CAS.
- (a) R. Kumai, M. M. Matsushita, A. Izuoka and T. Sugawara, J. Am. Chem. Soc., 1994, 116, 4523 CrossRef CAS; (b) J. Nakazaki, M. M. Matsushita, A. Izuoka and T. Sugawara, Tetrahedron Lett., 1999, 40, 5027 CrossRef CAS; (c) Y. Ishikawa, T. Miyamoto, A. Yoshida, Y. Kawada, J. Nakazaki, A. Izuoka and T. Sugawara, Tetrahedron Lett., 1999, 40, 8819 CrossRef CAS; (d) J. Nakazaki, Y. Ishikawa, A. Izuoka, T. Sugawara and Y. Kawada, Chem. Phys. Lett., 2000, 319, 385 CrossRef CAS.
- (a) M. M. Matsushita, H. Kawakami, T. Sugawara and M. Ogata, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195208 CrossRef; (b) M. M. Matsushita, H. Kawakami, Y. Kawada and T. Sugawara, Chem. Lett., 2007, 110 CrossRef CAS.
- (a) E. Goldstein, B. Beno and K. N. Houk, J. Am. Chem. Soc., 1996, 118, 6063; (b) E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem. Soc., 1997, 119, 1297 CrossRef CAS; (c) M. Abe, W. Adam and W. M. Nau, J. Am. Chem. Soc., 1998, 120, 11304 CrossRef CAS; (d) J. H. Rodriguez, D. E. Wheeler and J. K. McCusker, J. Am. Chem. Soc., 1998, 120, 12051 CrossRef.
- H. Komatsu, M. M. Matsushita, S. Yamamura, Y. Sugawara, K. Suzuki and T. Sugawara, J. Am. Chem. Soc., 2010, 132, 4528 CrossRef CAS.
- (a) T. Sugawara, M. Minamoto, M. M. Matsushita, P. Nickels and S. Komiyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235316 CrossRef; (b) P. Nickels, M. M. Matsushita, M. Minamoto, S. Komiyama and T. Sugawara, Small, 2008, 4, 471 CrossRef CAS.
- See, for reviews on the subject: (a) A. Boardman, A. D. Budker and A. Pisarev, J. Opt. Soc. Am. B, 2005, 22 Search PubMed; (b) C. Sanchez, B. Lebeau, F. Chaput and J. P. Boilot, Adv. Mater., 2003, 15, 1969 CrossRef CAS; (c) P. Rabu and M. Drillon, Adv. Eng. Mater., 2003, 5, 189 CrossRef CAS; (d) S. Sioncke, T. Verbiest and A. Persoons, Mater. Sci. Eng., R, 2003, 42, 115 CrossRef.
- Optical Materials for Non-linar Optics. III, ed. G. J. Ashwell G. J. and D. Bloor, Royal Society of Chemistry, London, 1993 and referencees therein Search PubMed.
- (a) R. V. Pisarev, I. Sanger, G. A. Petrakovskii and M. Fiebig, Phys. Rev. Lett., 2004, 93, 037204 CrossRef CAS; (b) M. Fiebig, T. Lottermoser, D. Frohlich and R. V. Pisarev, Nature, 2002, 419, 818 CrossRef CAS; (c) M. Fiebig, D. Frohlich, T. Lottermoser, V. V. Pavlov, R. V. Pisarev and H. J. Weber, Phys. Rev. Lett., 2001, 87, 137202 CrossRef CAS; (d) M. Kauranen, J. J. Maki, T. Verbiest, S. V. Elshocht and A. Persoons, Phys. Rev. B: Condens. Matter, 1997, 55, R1985 CrossRef CAS.
- (a) W. Nie, Adv. Mater., 1993, 5, 520 CrossRef CAS; (b) Molecular Nonlinear Optics, ed. J. Zyss, P. Kelley and P. Liao, Academic Press, Boston, 1994 Search PubMed.
- (a) P. G. Lacroix, A. V. Veret Lemarinier, R. Clement, K. Nakatani and J. A. Delaire, J. Mater. Chem., 1993, 3, 499 RSC; (b) P. G. Lacroix, R. Clement, K. Nakatani, J. A. Delaire, J. Zyss and I. Ledoux, Science, 1994, 263, 658 CrossRef CAS; (c) T. Corradin, R. Clément, P. G. Lacroix and K. Nakatani, Chem. Mater., 1996, 8, 2153 CrossRef.
- A. Di Bella, I. Fragalà, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1996, 118, 12747 CrossRef CAS.
- (a) C. Nicoud, G. Serbutoviez, G. Puccetti, I. Ledoux and J. Zyss, Chem. Phys. Lett., 1990, 175, 257 CrossRef; (b) L. Angeloni, A. Caneschi, L. David, A. Fabretti, F. Ferraro, D. Gatteschi, A. le Lirzin and R. Sessoli, J. Mater. Chem., 1994, 4, 1047 RSC.
- S. Di Bella, I. Fragalà, I. Ledoux and T. J. Marks, J. Am. Chem. Soc., 1995, 117, 9481 CrossRef CAS.
- (a) M. Takahashi, S. Yamada, H. Matsuda, H. Nakanishi, E. Tsuchida and H. Nishide, Chem. Commun., 1997, 1853 RSC; (b) P. M. Lundquist, S. Yitzchaik, T. J. Marks, G. K. Wong, S. Di Bella, R. Cohen and G. Berkovic, Phys. Rev. B: Condens. Matter, 1997, 55, 14055 CrossRef CAS.
- (a) J. Zyss, Nonlinear Opt., 1991, 1, 3 CAS; (b) J. Zyss and I. Ledoux, Chem. Rev., 1994, 94, 77 CrossRef CAS.
- (a) J. L. Oudar, J. Chem. Phys., 1977, 67, 446 CrossRef CAS; (b) J. L. Oudar and D. S. Chemala, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS.
- S. J. Lalama and A. F. Garito, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 20, 1179 CrossRef CAS.
- K. Clays and A. Persoons, Phys. Rev. Lett., 1991, 66, 2980 CrossRef CAS.
- P. M. Lundquist, S. Yizchaik, T. J. Marks, G. K. Wong, S. Di Bella, R. Cohen and G. Berkovic, Phys. Rev. B: Condens. Matter, 1997, 55, 14055 CrossRef CAS.
- A. Caneschi, P. D. Rey and R. Sessoli, Inorg. Chem., 1991, 30, 3936 CrossRef CAS.
- M. Takahashi, S. Yamada, H. Matsuda, H. Nakanishi, E. Tsuchida and D. Nishide, Chem. Commun., 1997, 1853 RSC.
- (a) J. Zyss, T. Chauvan, C. Dhenaut and I. Ledoux, Chem. Phys., 1993, 177, 281 CrossRef CAS; (b) T. Verbiest, K. Clays, C. Samyn, J. Wolff, D. Reinhoudt and A. Persoons, J. Am. Chem. Soc., 1994, 116, 9320 CrossRef CAS; (c) C. Lambert, E. Schmalzlin, K. Meerholz and C. Brauchle, Chem.–Eur. J., 1998, 4, 512 CrossRef CAS; (d) S. Brasselet, F. Cherioux, P. Audebert and J. Zyss, Chem. Mater., 1999, 11, 1915 CrossRef CAS.
- I. Ratera, D. Ruiz-Molina, C. Sporer, S. Marcen, S. Montant, J. F. Letard, E. Freysz, C. Rovira and J. Veciana, Polyhedron, 2003, 22, 1851 CrossRef CAS.
- I. Ratera, S. Marcen, S. Montant, D. Ruiz-Molina, C. Rovira, J. Veciana, J. F. Letard and E. Freysz, Chem. Phys. Lett., 2002, 363, 245 CrossRef CAS.
- I. Ratera, D. Ruiz-Molina, C. Sánchez, R. Alcalà, C. Rovira and J. Veciana, Synth. Met., 2001, 121, 1834 CrossRef CAS.
- M. L. H. Green, S. R. Marder, M. E. Thompson, D. Bandy, D. Bloor, P. V. Kolinsky and R. J. Jones, Nature, 1987, 330, 360 CrossRef CAS.
- (a) S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715 CrossRef CAS; (b) N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21 CrossRef CAS; (c) T. Verbiest, S. Houbrechts, M. Kauranen, K. Clays and A. Persoons, J. Mater. Chem., 1997, 7, 2175 RSC; (d) B. J. Coe, T. A. Hamor, C. J. Jones, J. A. McCleverty, D. Bloor, G. Cross and T. L. Axon, J. Chem. Soc., Dalton Trans., 1995, 673 RSC.
- D. Velasco, S. Castellanos, M. Lopez, F. Lopez-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2007, 72, 7523 CrossRef CAS.
- S. Castellanos, D. Velasco, F. Lopez-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2008, 73, 3759 CrossRef CAS.
- Y. Morita, S. Suzuki, K. Fukui, S. Nakazawa, H. Kitagawa, H. Kishida, H. Okamoto, A. Naito, A. Sekine, Y. Ohashi, M. Shiro, K. Sasaki, D. Shiomi, K. Sato, T. Takui and K. Nakasuji, Nat. Mater., 2008, 7, 48 CrossRef CAS.
- S. Nakatsuji, Adv. Mater., 2001, 13, 1719 CrossRef CAS.
- (a) M. Verdaguer, Science, 1996, 272, 698 CrossRef CAS; (b) S. Schmitt and K. Hafner, Mol. Cryst. Liq. Cryst., 2000, 348, 1.
- (a) P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024 CrossRef and references therein; (b) K. Nagai, T. Iyoda, A. Fujishima and K. Hashimoto, Solid State Commun., 1997, 102, 809 CrossRef CAS.
- S. Nakatsuji, M. Mizumoto, A. Takai, H. Akutsu, J. Yamada, H. Kawamura, S. Schmitt and K. Hafner, Mol. Cryst. Liq. Cryst., 2000, 348, 1.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704 CrossRef CAS.
- S. Nakatsuji, Chem. Soc. Rev., 2004, 33, 348 RSC.
- K. Hamachi, K. Matsuda, T. Itoh and H. Iwamura, Bull. Chem. Soc. Jpn., 1998, 71, 2937 CAS.
- (a) K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 7195 CrossRef CAS; (b) K. Matsuda and M. Irie, Chem.–Eur. J., 2001, 7, 3466 CrossRef CAS; (c) K. Matsuda, M. Matsuo and M. Irie, J. Org. Chem., 2001, 66, 8799 CrossRef CAS.
- K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 8309 CrossRef CAS.
- K. Matsuda, M. Matsuo and M. Irie, J. Org. Chem., 2001, 66, 8799 CrossRef CAS.
- K. Tanaka and F. Toda, J. Chem. Soc., Perkin Trans. 1, 2000, 873 RSC.
- L. Xu, T. Sugiyama, H. Huang, Z. Song, J. Meng and T. Matsura, J. Chem. Soc., Chem. Commun., 2003, 2328 RSC.
- Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2001, 123, 294 CrossRef CAS.
- (a) I. Ratera, D. Ruiz-Molina, J. Vidal-Gancedo, K. Wurst, N. Daro, J. F. Letard, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2001, 40, 919 CrossRef CAS; (b) I. Ratera, D. Ruiz-Molina, J. Vidal-Gancedo, J. J. Novoa, K. Wurst, J. F. Letard, C. Rovira and J. Veciana, Chem.–Eur. J., 2004, 10, 603 CrossRef CAS.
- (a) J. V. Barth, G. Constantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS; (b) B. D. Gates, Q. Xu, M. Stewart, D. Ryan, G. Willson and G. M. Whitesides, Chem. Rev., 2005, 105, 1171 CrossRef CAS.
- A. D. Shukla, A. Das and M. E. Van der Boom, Angew. Chem., Int. Ed., 2005, 44, 3237 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS.
- Y. Kashiwagi, K. Uchyama, F. Kurashima, J. Anazi and T. Osa, Anal. Sci., 1999, 15, 907 CrossRef.
- (a) M. A. Fox, E. Gaillard and Ch. Chen, J. Am. Chem. Soc., 1987, 109, 7088 CrossRef CAS; (b) T. L. Chu and S. I. Weissmann, J. Chem. Phys., 1954, 22, 21 CrossRef CAS.
- Y. Morita, T. Aoki and K. Fukui, et al. , Angew. Chem., Int. Ed., 2002, 41, 1793 CrossRef CAS.
- Y. Morita, S. Nishida, J. Kawai, T. Takui and K. Nakasuji, Pure Appl. Chem., 2008, 47, 507 CrossRef.
- Y. Morita, T. Okafuji and M. Satoh, Japanese patent JP2007227186 A 20070906, 2007 Search PubMed.
Footnotes |
| † Dedicated to Paul Rey in his memory. |
| ‡ Part of the molecule-based magnets themed issue. |
| This journal is © The Royal Society of Chemistry 2012 |