Anion receptor chemistry: highlights from 2010
Marco
Wenzel
,
Jennifer R.
Hiscock
and
Philip A.
Gale
*
Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Fax: +44 (0)23 8059 6805; Tel: +44 (0)23 8059 3332
First published on 14th November 2011
Abstract
This critical review covers advances in anion complexation in the year 2010. The review covers both organic and inorganic systems and also highlights the applications to which anion receptors can be applied such as sensing, anion transport, control of molecular motion and gelation (179 references).
 Marco Wenzel | Marco Wenzel studied Chemical Engineering at the HTW Dresden, Germany. He completed his PhD at the University of Dresden in 2008 under the supervision of Prof Karsten Gloe with research stays at the University of Nottingham, UK (Marie Curie Fellowship), University of Saga, Japan (DAAD) and University of Sydney, Australia (DFG). Before he joined the group of Prof Philip Gale in 2010 he carried out 2 years of Post-Doctoral research with Dr Paul Plieger at Massey University, New Zealand. His main research interests in supramolecular chemistry are membrane transport and liquid–liquid extraction of anionic species and metal salts. |
 Jennifer R. Hiscock | Jennifer Hiscock graduated in biomedicinal chemistry at the University of Exeter 2007. In 2010 she received her PhD from the University of Southampton where she has continued as a Post Doctoral researcher under the supervision of Prof Philip Gale. Her research interests include the supramolecular chemistry of anionic species and the utilisation of organic molecules in catalysis. |
 Philip A. Gale | Philip A. Gale is Professor of Supramolecular Chemistry at the University of Southampton. A major focus of his research concerns the molecular recognition, sensing and lipid bilayer transport of anionic species. |
Introduction
This review is the latest in a series covering recent advances in anion complexation.1–6 It highlights developments in 2010 in the fields of new receptors, modes of binding, transport and extraction of anions, and sensing mechanisms. It is not intended to be a comprehensive overview of the area. The structure of the review is primarily according to the dominant functional group used to complex the anion and by the potential application of these systems. Many receptors use more than one functional group to bind anions or have various applications and the organisation of this review should be regarded as a flexible guide to the classification of these systems.Amide/carbamate containing receptors
Secondary amides have been widely employed in synthetic anion receptor systems. Yatsimirsky and co-workers have investigated a series of known isomeric N,N′-bis(pyridyl)-2,6-pyridine-dicarboxamides7,84–6 and their dicationic pyridinium analogues 1–3 in solution and the solid state.9 The stability constants of the neutral receptors with chloride and acetate were determined by 1H NMR titrations in CD3CN. The NMR titration curve of the m-isomer 4 illustrates the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 (L:A) complexes with both anions (chloride: log K = 1.8, log β21 = 4.23; acetate: log K = 2.6, log β21 = 4.7). For the p-isomer 5 the formation of 1:1 complexes was observed for chloride (log K = 2.85) whereas 1:1 and 1:2 complexes are formed with acetate (log K = 3.48, log β21 = 6.5). No interaction and weak binding were observed during the titration of the o-isomer 6 with chloride and acetate (log K = 0.7) respectively. The binding affinities of the related pyridinium receptors 1–3 towards halides and nitrate are higher. Spectrophotometric titrations for the m- and p-isomers 1 and 2, and competition experiments by deprotonation with pyridine for o-isomer 3 were carried out in CH3CN in order to calculate the stability constant of the 1:1 complexes. A summary of the obtained stability constants is shown in Table 1. The highest affinities were observed with the o-isomer 3 over the m- and p-isomers 1 and 2, respectively. Four crystal structures of chloride and triflate complexes of the three isomers illustrate the binding of the anion in the ligand cleft via multiple hydrogen bonds with the amide groups and via CH-interactions with adjacent methyl-pyridinium rings. The observed NH⋯Cl distances are between 3.194 and 3.307 Å and the CH⋯Cl distances range from 3.342 to 3.614 Å. The triflate anions in 2 and 3 are bound by NH⋯O interactions with distances between 2.878 and 3.127 Å and by CH⋯O interactions between 3.089 to 3.371 Å. The orientation of the methyl substituents towards the centre of the ligand cleft prevents the formation of inclusion complexes with o-isomer 3 for both chloride and triflate in the solid state. The proximity of the positive charge of the pyridinium function and the ligand NH binding side may result in the observed order of stability in solution.
:1 and 1:2 (L:A) complexes with both anions (chloride: log K = 1.8, log β21 = 4.23; acetate: log K = 2.6, log β21 = 4.7). For the p-isomer 5 the formation of 1:1 complexes was observed for chloride (log K = 2.85) whereas 1:1 and 1:2 complexes are formed with acetate (log K = 3.48, log β21 = 6.5). No interaction and weak binding were observed during the titration of the o-isomer 6 with chloride and acetate (log K = 0.7) respectively. The binding affinities of the related pyridinium receptors 1–3 towards halides and nitrate are higher. Spectrophotometric titrations for the m- and p-isomers 1 and 2, and competition experiments by deprotonation with pyridine for o-isomer 3 were carried out in CH3CN in order to calculate the stability constant of the 1:1 complexes. A summary of the obtained stability constants is shown in Table 1. The highest affinities were observed with the o-isomer 3 over the m- and p-isomers 1 and 2, respectively. Four crystal structures of chloride and triflate complexes of the three isomers illustrate the binding of the anion in the ligand cleft via multiple hydrogen bonds with the amide groups and via CH-interactions with adjacent methyl-pyridinium rings. The observed NH⋯Cl distances are between 3.194 and 3.307 Å and the CH⋯Cl distances range from 3.342 to 3.614 Å. The triflate anions in 2 and 3 are bound by NH⋯O interactions with distances between 2.878 and 3.127 Å and by CH⋯O interactions between 3.089 to 3.371 Å. The orientation of the methyl substituents towards the centre of the ligand cleft prevents the formation of inclusion complexes with o-isomer 3 for both chloride and triflate in the solid state. The proximity of the positive charge of the pyridinium function and the ligand NH binding side may result in the observed order of stability in solution.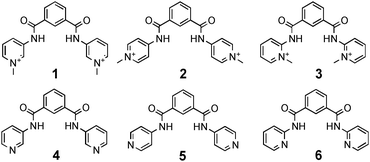
| Anion | 1 | 2 | 3 |
|---|---|---|---|
| a Stability constants for 1 and 2 were obtained by spectrophotometric titrations, for 3 by competition experiments by deprotonation with pyridine. b Values in parentheses are standard deviation in the last significant digit. | |||
| F− | 5.28(8) | 4.36(7) | |
| Cl− | 5.27(9) | 5.65(6) | 5.85(9) |
| Br− | 5.24(7) | 4.29(7) | 5.40(8) |
| I− | 3.80(7) | 3.57(9) | 3.80(8) |
| H2PO4− | 4.20(6) | 5.18(9) | |
| AcO− | 4.40(7) | 5.38(8) | |
| NO3− | 4.11(9) | 4.34(5) | 4.40(6) |
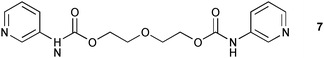
Solid phase investigation of the sulfate, perchlorate and chloride complexes of the protonated aliphatic bis-3-pyridinium carbamate receptor 7 show different modes of interaction with different anionic guests.10 Perchlorate was found to bind via pyridinium NH⋯O interactions between 2.801 and 2.961 Å. Interactions with the carbamate NH hydrogen bond donor occur via a co-crystallised water molecule, but no direct hydrogen bonds were observed between the anion and these groups. The NH⋯Owater distances are between 3.012 and 3.039 Å and the OHwater⋯O–ClO3− distance is 2.858 Å. Direct hydrogen bonds between the carbamate NH and the anion are observed in the sulfate and chloride complexes. The NH⋯Cl distances are between 3.192 and 3.267 Å whereas the pyridinium NH⋯Cl distances are significantly shorter (3.021 to 3.039 Å) presumably due to electrostatic attraction. A similar differentiation was observed for the NH⋯O–SO32− interactions: the pyridinium NH⋯O distances are 2.614 and 2.659 Å whereas the carbamate NH⋯O distance is 2.789 Å.
Simple diamide receptors 8–11 based on anthracene or carbazole linked via amide bonds to pendant pyrrole or phenyl substituents were synthesised and investigated by Sessler and co-workers.11 Proton NMR titrations in DMSO-d6 with chloride, benzoate and dihydrogen phosphate, added as their tetrabutylammonium (n-Bu4N+) salts, show selective complexation of H2PO4− over PhCOO− and Cl− for all receptors. Furthermore, the stability constants show an increase in the anion binding affinity in agreement with the number of N-donor atoms present in the receptor. For example Ka [M−1] for H2PO4− for 8: 2000, for 9: 1400 for 10: 2600 and for 11: 160 (see Table 2). Interestingly, the pyrrolic NH protons do not participate in the coordination of the Cl− ion in the crystal structure of the complex [Cl⊂8](n-Bu4N) (Fig. 1), and the anion is bound by only three of the possible five NH-groups. Instead, the pyrrole protons are involved in the formation of a hydrogen-bonded network with the amide oxygen of another molecule of 8 to form a 1-D hydrogen bonded polymer.
, non-acidic hydrogen atoms and tetrabutylammonium counter cations are omitted for clarity, hydrogen bonds are represented as dotted lines.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f1.gif) | ||
| Fig. 1 Schematic representation of the crystal structure of [Cl⊂8](n-Bu4N), non-acidic hydrogen atoms and tetrabutylammonium counter cations are omitted for clarity, hydrogen bonds are represented as dotted lines. | ||
A series of amide hosts based on a benzene12,13 and tris(2-aminoethyl)amine14,15 (tren) platform have been investigated by Ghosh and co-workers. The bistripodand ligand 12 coordinates multiple anions by dividing the six pendant arms into two compartments with an ababab arrangement or unfavourable aaabbb fashion in the solid state (Fig. 2).12 The latter was found in the 1:2 (L:A) acetate complex with the anion bound by three amide NH⋯O hydrogen bonds (NH⋯O distances in the range 2.677 to 2.792 Å). In the crystal two receptors form a pseudo-cage, composed of six arms that hold two anions separated by 6.101 Å. The authors quote four intermolecular C–F⋯F–C interactions with a F⋯F distance of 2.938 Å as the driving force for pseudo-cage formation. In the nitrate complex of 12 the pendant arms are orientated in an ababab alternate arrangement. A total of four nitrate anions are coordinated by one ligand. Two anions are bound as pair, bridged with one water molecule on either side of the benzene platform (OHwater⋯O–NO2− distance between 2.795 and 3.035 Å). Each of the nitrate anions is coordinated by the ligand via one amide NH⋯O hydrogen bond (NH⋯O distances are 2.893 and 3.135 Å) and an anion–π interaction towards the pentafluoro-phenyl substituent. The anion–centroid distances are 3.201 and 3.511 Å. The anion binding ability of 12 towards the tetrabutylammonium salts of H2PO4−, OAc− and NO3− was studied in acetone-d6 using 1H NMR titrations. Job plot experiments, monitoring the shift of the amide-NH for NO3− and the CH2 resonance for OAc− and H2PO4− show a stoichiometry of 1:4 (L:A) for acetate and dihydrogen phosphate, and a 1:2 host–guest binding for NO3−. The calculated binding constants log Kn for NO3− are 2.83 and 4.91, respectively.
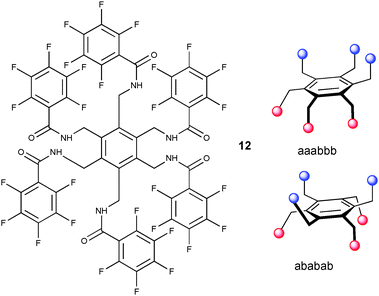 | ||
| Fig. 2 Schematic draw of 12 (left) and aaabbb and ababab orientation of pendant arms in solid phase (right). | ||
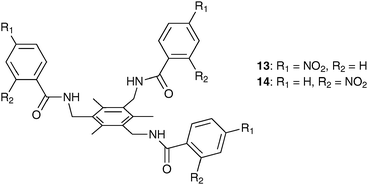
The influence of p- and o-nitrophenyl substituents on anion encapsulation in tripodal amide receptors, based on 1,3,5-trimethylbenzene has been reported by the same research group.13 Comparison of the crystal structures of the nitrate, acetate, fluoride and chloride complexes of p-isomer 13 with known complexes of o-isomer 1416 shows the formation of 2:2 host–guest capsules of 13 with all studied anions. A similar assembly of o-isomer 14 was only observed upon the complexation of [F2(H2O)6]2−. The mutual interdigitation of the six pendant arms of two molecules of 13 in a sandwiched arrangement hosts two NO3− (Fig. 3), [(AcO)2(H2O)4]2−, [F2(H2O)6]2− and [Cl2(H2O)4]2− inside the capsule. The incorporated water molecules in the latter three complexes act as bridges to overcome anion–anion repulsion and are involved in numerous hydrogen bonds with the ligand and the anionic guests. However no water was found in the nitrate structure of 13 ([NO3⊂132]2−). In this case the two anions are arranged in a staggered planar orientation relative to one another with a central N⋯N distance of 3.562 Å. Further, both nitrate anions are in short contact to the respective π-cloud of the aryl platform with N–centroid distances of 3.225 and 3.229 Å. Numerous NH⋯O and CH⋯O hydrogen bonds are formed from the amide groups and the p-nitrophenyl substituents towards the anions. The distances of the observed NH⋯O–NO2− interactions range from 2.984 to 3.297 Å, where the CH⋯O distances are between 3.279 and 3.451 Å. The distance of the NH⋯anion interaction in the acetate complex is 2.893 Å, where a NH⋯F distance of 2.805 Å was obtained in [(F2(H2O)6⊂132]2−. The chloride ions in [(Cl2(H2O)4⊂132]2− are bound by an NH⋯Cl interaction of 3.290 Å and CH⋯Cl contacts of 3.466 and 3.636 Å. The subsequent X-ray analysis of the reaction of 13 with 3 equivalents of the tetrabutylammonium salts of nitrate, acetate, fluoride and chloride from dioxane yields the previously observed complex [(F2(H2O)6⊂132](n-Bu4N)2·2dioxane. Qualitative 1H NMR studies in DMSO-d6 underline the preferred uptake of fluoride over chloride, nitrate and acetate.
![X-Ray structure of [(NO3)2⊂132]2− with selected atom labels and N–H⋯O hydrogen bonds as dotted lines, tetrabutylammonium cations and non-acidic hydrogen atoms are omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f3.gif) | ||
| Fig. 3 X-Ray structure of [(NO3)2⊂132]2− with selected atom labels and N–H⋯O hydrogen bonds as dotted lines, tetrabutylammonium cations and non-acidic hydrogen atoms are omitted for clarity. | ||
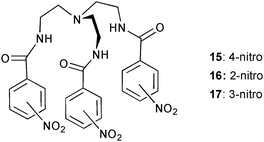
More detailed studies on the influence of positional isomers in solution on anion affinity were performed with the series of tren-based nitrophenyl-amide receptors 15–17.14 Proton NMR titration experiments in DMSO-d6 of the known m-isomer1717 and the two new receptors 15 and 16 with chloride and fluoride show selective binding of fluoride over chloride for the latter two isomers. The calculated stability constants log Ka for a 1:1 binding mode are 4.06 and 2.29 for fluoride and chloride, respectively, with p-isomer 15. A binding constant log Ka of 5.63 was obtained for fluoride with o-isomer 16, whilst no shift in the NH was observed upon addition of tetrabutylammonium chloride. The binding constants are the same order of magnitude for both anions with m-isomer 17 (log Ka = 3.76 and 3.32 for fluoride and chloride, respectively). No uptake of the anion into the pseudo-cage was observed in the solid state. Crystal structures of the iodide, perchlorate and hexafluorosilicate complex of the p-isomer 15 show anion coordination on the ligand periphery only. Presumably strong intermolecular N–H⋯O hydrogen bonds between the amide groups of at least two pendant arms (N⋯O distances between 2.792 and 3.057 Å) prevent the uptake of the anion into the pseudo-cage.
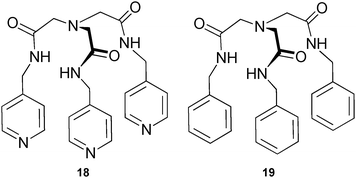
Öztürk and co-workers conducted the solvent free synthesis of two amide-based tripodal receptors.15 The direct treatment of nitrilotriacetic acid with benzylamine and picolylamine under microwave conditions gives receptor 18 and previously reported ligand 1918 in high yield (>80%) and in very short reaction times (<45 min). Binding studies in DMSO-d6 show a pronounced selectivity of both receptors for H2PO4− and PhCOO− over HSO4−, ClO4−, PF6− and Br− (Table 3). Overall higher binding constants were observed for the 4-pyridylmethyl substituted receptor 18 (Ka (M−1) = 810 for H2PO4− and 286 for PhCOO−) than for the benzyl substituted receptor 19 (Ka (M−1) = 241 and 110 for H2PO4− and PhCOO−, respectively).
 :1 complexes of 18 and 19 in DMSO-d6 at 298 K.a Anions added as their tetrabutylammonium salts
:1 complexes of 18 and 19 in DMSO-d6 at 298 K.a Anions added as their tetrabutylammonium salts
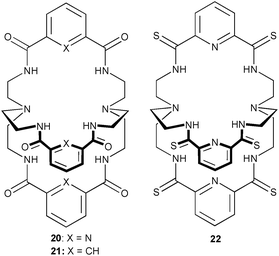
Two known amido-cryptands (20 and 21) and a new thioamido-cryptand 22 have been studied for their fluoride binding ability in solution and the solid state by Bowman-James and co-workers.19 Proton NMR studies in DMSO-d6 reveal binding constants Ka (M−1) of 8.1 × 105, 3.3 × 104 and 3.2 × 104 for 20, 21 and 22, respectively. The significantly lower anion affinity of the thioamide receptor 22 as compared to compound 20 was explained by the authors as being due to the presence of a negative charge on the partly deprotonated thioamide group during ongoing hydrogen/deuterium exchange repelling the incoming fluoride anion. Extensive 19F NMR studies demonstrated the presence of enhanced deuterium-exchange reactions in DMSO for this receptor. The enhanced hydrogen bonding capability of the thioamide group has resulted in higher binding affinities for anions in previous studies of monocyclic thioamides from the same group.20 Two crystal structures of the F− complexes of 20 and 21 show the 1:1 encapsulation of the anion within the pseudo C3-symmetric host. The F− anions are coordinated by N–H⋯F hydrogen bonds with all amide groups in both examples. The observed N⋯F distances range from 2.822 to 2.889 Å in [F⊂20]− and between 2.948 and 3.111 Å in [F⊂21]−, which is longer than most amide N–H⋯F interactions. In the F− complex of 21 additional C–H⋯F interactions are formed between the three isophthaloyl hydrogen atoms and the central anion. The C⋯F distances range from 3.032 to 3.077 Å.
The same group has also reported tricyclic 2,6-pyridinedicarboxamide host 23.21 This larger receptor forms more stable complexes with linear anions, such as FHF− and N3− over anions such as Cl−, Br− and I−. Crystal structures of both bifluoride and azide complexes show encapsulation of the guest inside the receptor cavity. Internal multi-atom hydrogen bonding networks involving the pyridine nitrogen, amido hydrogen and amine nitrogen atom (Npy⋯Hamide⋯Namine) provide a preorganised hydrogen bonding pocket for the coordination of the guest anions (Fig. 4). The observed Namide⋯Namine distances range from 2.839 to 2.944 Å where Namide⋯Npy interactions between 2.734 and 2.772 Å have been observed. In both complexes the anions are coordinated by multiple hydrogen bonds from the remaining amide groups. The N⋯F interactions range from 2.724 to 2.754 Å where the fluorine atoms are separated by 2.475 Å with a FHF− angle of 168°. The azide ion is bound via Namide⋯Nazide interactions between 2.854 and 2.906 Å. The N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN separation is 2.355 Å with nearly equal N–N distances of 1.162 and 1.193 Å in the linear ion (N3− angle 179.3°). In contrast, the receptor in the sulfate complex is protonated with the two sulfate anions coordinated by 23 neutralised by the charge on the four protonated bridgehead amine groups. The anions are located outside the receptor cavity and are bound by multiple hydrogen bonds to the amide and ammonium groups. The Namine⋯O interactions range from 2.638 to 2.704 Å with Namide⋯O distances between 2.874 and 2.891 Å. In addition two co-crystallised water molecules are present, which act as hydrogen bonding bridges between the sulfate ions and the ligand (S–O⋯Owater interaction of 2.672 and 2.836 Å, NH⋯Owater distances 2.854 and 2.937 Å). Proton NMR titration experiments in DMSO-d6 show preferential binding of FHF− over N3−, H2PO4− and CH3COO− by 23. A shift of the HF2− proton resonance from 15.4 to 17.5 ppm was observed upon complexation. The calculated stability constants employing a 1:1 binding mode results in a log Ka of 3.74 for FHF−, 2.87 for H2PO4−, 2.53 for N3− and 2.00 for AcO−, and negligible binding for HSO4−, Cl−, Br−, I−, NO3−, SCN− and ClO4−.
NN separation is 2.355 Å with nearly equal N–N distances of 1.162 and 1.193 Å in the linear ion (N3− angle 179.3°). In contrast, the receptor in the sulfate complex is protonated with the two sulfate anions coordinated by 23 neutralised by the charge on the four protonated bridgehead amine groups. The anions are located outside the receptor cavity and are bound by multiple hydrogen bonds to the amide and ammonium groups. The Namine⋯O interactions range from 2.638 to 2.704 Å with Namide⋯O distances between 2.874 and 2.891 Å. In addition two co-crystallised water molecules are present, which act as hydrogen bonding bridges between the sulfate ions and the ligand (S–O⋯Owater interaction of 2.672 and 2.836 Å, NH⋯Owater distances 2.854 and 2.937 Å). Proton NMR titration experiments in DMSO-d6 show preferential binding of FHF− over N3−, H2PO4− and CH3COO− by 23. A shift of the HF2− proton resonance from 15.4 to 17.5 ppm was observed upon complexation. The calculated stability constants employing a 1:1 binding mode results in a log Ka of 3.74 for FHF−, 2.87 for H2PO4−, 2.53 for N3− and 2.00 for AcO−, and negligible binding for HSO4−, Cl−, Br−, I−, NO3−, SCN− and ClO4−.
·3H2O with selected atom labels and hydrogen bonds as dotted lines, tetrabutylammonium counter cations, co-crystallised H2O molecules and non-acidic hydrogen atoms are omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f4.gif) | ||
| Fig. 4 X-Ray structure of [(FHF)⊂23](n-Bu4N)·3H2O with selected atom labels and hydrogen bonds as dotted lines, tetrabutylammonium counter cations, co-crystallised H2O molecules and non-acidic hydrogen atoms are omitted for clarity. | ||
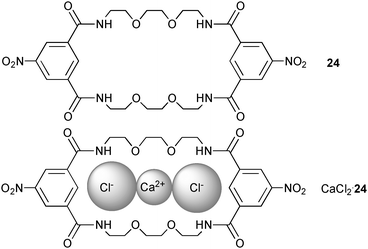
Another extended amide based macrocycle has been employed by Lünig and co-workers to bind chloride as its calcium salt in a cascade-like complex.22 The 2+2 condensation of 5-nitroisophthaloyl dichloride and 1,8-diamino-3,6-dioxaoctane in THF results in macrocycle 24. Both isophthalamide residues provide the binding pocket for anions where the four oxygen atoms of the diethylene glycol linker act as donor atoms for the coordination of the metal centre. Electrospray ionisation mass spec (ESI-MS) investigations give evidence for the coordination of chloride (molecular weight (Cl·24)− = 681.2, m/z observed = 681.2 (negative mode)) and the calcium salt (CaCl·24)+ (m/z = 721.14, calc mass: 721.2). Quantitative 1H NMR experiments of 24 in CDCl3 with 5% DMSO-d6 over an excess of n-Bu4NCl, CaCl2, MgCl2 and BaCl2, present as solids, show a pronounced shift of the amide protons in the case of n-Bu4NCl or CaCl2, only, illustrating the extraction and uptake of CaCl2 over MgCl2 and BaCl2 into 24.
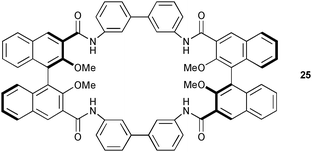
The chiral amide based macrocycle 25 has been synthesised and studied by the group of Pasini.23 The receptor shows no particular interaction towards halide ions (Cl− and Br−) or towards numerous mono- and di-carboxylates, with exception of glutarate (Ka = 30 M−1, 1H NMR titration in CDCl3). The authors explain this modest anion affinity with the rigid receptor structure and strong intermolecular NH⋯O hydrogen bonds.
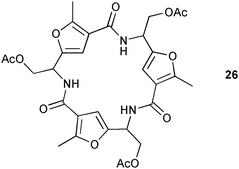
Robina and co-workers have reported the synthesis and anion binding ability of furyl-cyclopeptide receptor 26.24 ESI-MS and 1H NMR in CD3CN studies demonstrate the formation of 1:1 complexes with chloride, acetate and cyanide. The calculated stability constants Ka (M−1) show selectivity for chloride (Ka = 16000) over cyanide (Ka = 1600) and acetate (Ka = 1000) by the receptor.
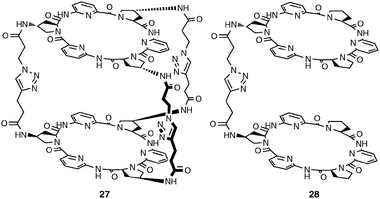
Kubik and co-workers conducted binding studies with sulfate for cyclopeptide based cages 27 and 28 in highly competitive solvents, such as H2O/CH3OH.25 The results of isothermal titration calorimetry (ITC) experiments in H2O/CH3OH mixture with water content between 35 and 65% are summarized in Table 4. A stability constant (log Ka) of 5.70 for 27 and 4.96 for 28 in H2O/CH3OH (1:1 v/v) reveals slightly enhanced binding upon increasing the number of linkers and subsequent higher preorganisation of 27. After detailed analysis of the thermodynamic data (Table 4) the authors conclude that the additional linker stabilises a favourable arrangement of the two cyclopeptide rings. The results show that coordination of sulfate is entropically favourable but enthalpically disfavoured.
| H2O: CH3OH |
27 | 28 | ||||
|---|---|---|---|---|---|---|
| 35: 65 |
50: 50 |
65:35 |
35: 65 |
50: 50 |
65:35 |
|
| a Standard deviations of at least three independent measurements are specified in brackets. | ||||||
| log Kaa | 6.34 (±0.02) | 5.70 (±0.03) | 5.19 (±0.05) | 5.67 (±0.02) | 4.96 (±0.05) | 4.26 (±0.02) |
| ΔG [kJ mol−1]a | −36.2 (±0.1) | −32.6 (±0.2) | −29.6 (±0.3) | −32.3 (±0.1) | −28.4 (±0.3) | −24.3 (±0.1) |
| ΔH [kJ mol−1]a | 13.3 (±0.1) | 10.1 (±0.1) | 6.9 (±0.3) | −12.4 (±0.1) | −13.5 (±0.3) | −11.8 (±0.2) |
| TΔS [kJ mol−1]a | 49.5 (±0.1) | 42.7 (±0.2) | 36.5 (±0.1) | 19.9 (±0.2) | 14.9 (±0.5) | 12.5 (±0.3) |
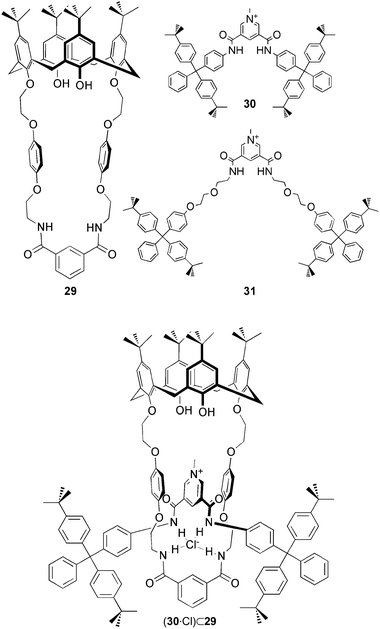
Amides have also been employed in the formation of anion templated interlocked molecular assembles. Beer and co-workers reported the first example of the calix[4]arene based rotaxane host for anion recognition.26 The Cl−-templated ring closure reaction of macrocycle 29 with isophthaloyl chloride in the presence of threads 30 and 31 results in rotaxanes (30·Cl)⊂29 and (31·Cl)⊂29. The structure of the interlocked assembly is confirmed by the X-ray crystal structure of the calix[4]arene based rotaxane anion host. The Cl− ion is bound by NH⋯Cl interactions between 3.222 and 3.544 Å, with the amide groups of the macrocycle and the thread. In addition close aryl CH⋯Cl contacts between 3.276 and 3.366 Å are observed. Following the removal of the chloride anion template, the anion binding properties of the resulting rotaxanes (30·PF6)⊂29 and (31·PF6)⊂29 have been studied by 1H NMR titration experiments in CDCl3/CD3OD (1:1). The calculated binding constants Ka [M−1] show preferred uptake of Cl− (Ka = 3820) over Br− (Ka = 1560), H2PO4− (Ka = 1140) and AcO− (Ka = 530) by rotaxane (30·PF6)⊂29. Significantly reduced binding constants were found for rotaxane (31·PF6)⊂29 (Ka [M−1] = 650 for Cl−, 630 for Br−, 420 for H2PO4− and 140 for AcO−), which incorporates a conformationally flexible thread 31. Comparable binding experiments of the thread 31 only show preferred binding of H2PO4− (Ka = 710) over Br− (Ka = 440), Cl− (Ka = 300) and AcO− (Ka = 160).
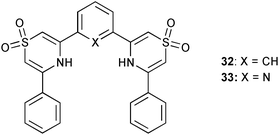
The novel thiazine-1,1-dioxide based receptors 32 and 33 were employed by Wisner and co-workers as hosts for anions.27 Proton NMR titrations in acetone-d6 show 1:1 (L:A) stoichiometry in solution and higher stability constants for anion complexes with 32 than for receptor 33, presumably due to the repulsion of the anionic guest by the lone pair of the pyridine nitrogen. The binding of tetrabutylammonium chloride by 32 includes a much weaker 2:1 (L:A) component. The order of anion affinity for 32 is Cl− (K21 = 300 M−1, K11 = 59000 M−1) over AcO− (Ka = 12500 M−1), H2PO4− (Ka = 540 M−1), Br− (Ka = 380 M−1), HSO4− (Ka = 220 M−1) and I− (Ka = 53 M−1). Whereas 33 shows a preferred binding of AcO− (Ka = 480 M−1) over H2PO4− (Ka = 360 M−1), Cl− (Ka = 300 M−1), HSO4− (Ka = 130 M−1) and Br− (Ka = 83 M−1).
Urea containing receptors
Urea based receptors have proven very effective anion hosts, particularly in the recognition of oxo-anions. Tris-urea receptor 34 has recently been reported by Wu and co-workers for the complexation of tetrahedral oxo-anions such as phosphate and sulfate.28 Single crystals, obtained form a DMSO/25% water solution of n-Bu4NH2PO4 and 34, revealed deprotonation of the anion upon coordination by two ligand molecules. The urea groups chelate the anion on the edge of the tetrahedron and form twelve N–H⋯O hydrogen bonds with N⋯O distances between 2.752 and 2.901 Å. Proton NMR studies in DMSO-d6/25% water in the presence of PO43− (as Na+ salt) and Et3N show the same 2:1 (L:A) stoichiometry in solution. The addition of H3PO4 or HClO4 results in proton transfer to the coordinated anion and subsequently complex dissociation. Extended NMR studies show that this process is reversible upon addition of Et3N (Fig. 5). The same reversible binding of the deprotonated anion was observed for SO42−, whilst HSO4− was not coordinated. UV-Vis titration experiments in DMSO/25% water were used to assess stability constants of the formed complexes. Fitting of the titration curve to a 2:1 (L:A) model results in log K11 = 5.0 and log K21 = 12.0 for PO43− (as Na+ salt). Job plot experiments reveal a 1:1 binding mode of SO42− (as the tetrabutylammonium salt) under comparable experimental conditions. The calculated binding constant in this case was log Ka = 4.72.
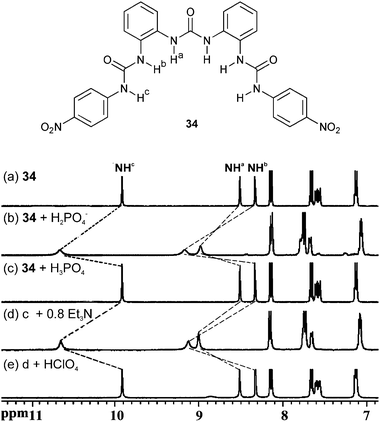 | ||
| Fig. 5 HClO4/Et3N modulated reversible binding of phosphate by 34 in DMSO-d6/25% water (H2PO4− added as tetrabutylammonium salt, H3PO4 as DMSO-d6 solution diluted from 85% H3PO4). Chem. Commun. 2010, 46, 5376–5378. Reproduced with permission of The Royal Society of Chemistry. | ||
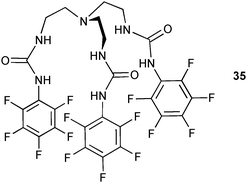
The strong affinity for carbonate ions of the known pentafluorophenyl-tren-based receptor 3529 has been demonstrated Ghosh and co-workers.30 Aerial CO2 is fixed in basic DMSO solutions and the resulting carbonate complex of 35 crystallised as 2:1 (L:A) complex. The anion is coordinated via sixteen N–H⋯O bonds from the urea groups with N⋯O distances between 2.705 and 3.346 Å. A closer examination reveals that thirteen out of the sixteen hydrogen bonds are in the strong hydrogen bonding interaction region with N⋯O distances <3.2 Å and N–H⋯O angles >140°. Proton NMR titration experiments of 35 with tetraethylammonium bicarbonate show the formation of the 2:1 (L:A) complex in solution with a stability constant log β21 of 4.04. The authors demonstrate further that the free receptor 35 can be regenerated and collected by filtration upon addition of CH3OH/water (1:4) to the complex [CO3⊂352]2−.
The same group has reported the first example of (n-Bu4N+)2SO4 bound as an ion pair by the pentafluorophenyl tripodal host 36 in polar solvents and in the solid state (Fig. 6).31 The cyanuric acid based receptor coordinates the sulfate ion via six N–H⋯O hydrogen bonds with N⋯O distances between 2.733 and 2.994 Å. On the exposed side the anion is bound by C–H⋯O interaction towards the tetrabutylammonium counterion. The C⋯O distances range from 3.304 to 3.523 Å. Extensive 2-dimensional diffusion ordered spectroscopy (2D-DOSY) and proton NMR studies show the presence of the ion pair complex in DMSO-d6 at 298 K. Where comparable experiments at 333 K point to a separation of [SO4⊂36]2− and the n-Bu4N+ cation, which reassemble after the solution is cooled down to 298 K. However no evidence for the coordination of an ion pair was observed with the tetrabutylammonium salts of dihydrogen phosphate, acetate or chloride at 298 K. Proton NMR titrations in DMSO-d6 show the strong binding of sulfate (log Ka = 5.74) over dihydrogen phosphate (log Ka = 4.39), acetate (log Ka = 3.41) and chloride (log Ka = 3.38) in 1:1 binding stoichiometry.
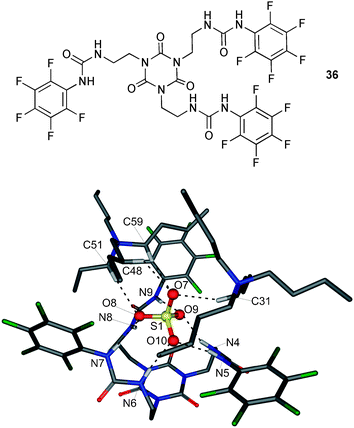 | ||
| Fig. 6 The X-ray structure of ((n-Bu4N)2·SO4)⊂36 with selected atom labels and hydrogen bonds as dotted lines, co-crystallised solvent molecules and non-acidic hydrogen atoms omitted for clarity. | ||
Moyer and co-workers32 have carried out extensive solution studies with receptor 37 which binds sulfate, selenate, sulfite and carbonate strongly forming a 2:1 (L:A) capsule upon the coordination of Mg(H2O)62+.33 These types of capsule often do not persist in aqueous solution. The determination of the solubility product Ksp in borax buffered aqueous solution by inductively coupled plasma (ICP) determinations of the magnesium concentration give a value Ksp of 2.0 × 10−17 at 25 °C. Comparable experiments at 35, 45 and 52 °C result in ΔH° = −99.1 kJ mol−1 and TΔS° = −3.8 kJ mol−1 for the sulfate complex [SO4⊂372]Mg(H2O)6, Ksp of 5.5 × 10−16 with ΔH° = −108.5 kJ mol−1 and TΔS° = −21.4 kJ mol−1 for the selenite complex [SeO4⊂372]Mg(H2O)6 and for the sulfite complex [SO3⊂372]Mg(H2O)6Ksp = 6.6 × 10−16, ΔH° = −64.6 kJ mol−1 and TΔS° = 22.0 kJ mol−1 were obtained. Further, the group carried out potentiometric titrations to obtain the protonation constants of 37 and stability constants of the SO42− binding. The four observed protonation steps of 37 (in 75 mM KNO3) correspond to the three pyridine groups and the tertiary amine group with pKa values of 3.58, 4.37, 5.23 and 7.11. The stability constants for a 1:1 binding mode of the protonated species Hn37(n+) (n = 2–4) for sulfate are log Ka = 1.8 for SO4⊂H237, log Ka = 2.45 for SO4⊂H337+ and log Ka = 2.62 for SO4⊂H4372+. These values are comparable to the stability constants of the SO42− complexes observed for the mono-, di-, and triprotonated parental tren ligand (log Ka for SO42− = 1.66 (mono-), 1.72 (di-), and 2.22 (triprotonated), respectively),34 and give evidence for the dominance of the electrostatic effect in anion recognition in aqueous media, as observed in crystal structure of ([SO4⊂H437] SO4)2 (Fig. 7).
![Schematic draw of X-ray structure of ([SO4⊂H437]SO4)2 with selected atom labels and hydrogen bonds as dotted lines, co-crystallised solvent molecules and non-acidic hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f7.gif) | ||
| Fig. 7 Schematic draw of X-ray structure of ([SO4⊂H437]SO4)2 with selected atom labels and hydrogen bonds as dotted lines, co-crystallised solvent molecules and non-acidic hydrogen atoms omitted for clarity. | ||
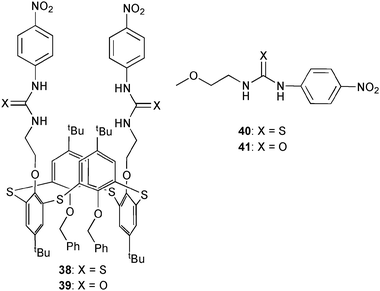
Yatsimirsky and co-workers have compared the anion binding abilities of thiacalix[4]arenes functionalised with thiourea 38 and urea 39 moieties and with model compounds 40 and 41.35 The crystal structures of all receptors reveal the cis-orientation of the urea N–H groups in both ligands where the N–H groups in the thiourea derivatives are present in the trans-orientation in the solid state. Nevertheless, spectrophotometric titrations in CHCl3 show a stronger binding of the anion by thiourea derivatives 38 and 40 over the urea receptors 39 and 41. Further, the thiacalix[4]arene receptors give higher stability constants than model compounds 40 and 41 due to the greater number of urea/thiourea groups. Overall a preferred binding of acetate over fluoride, dihydrogen phosphate and chloride was observed as shown in Table 5.
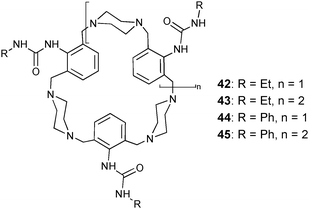
A series of urea functionalised cyclophanes (42–45) was prepared by Rissanen and co-workers.36 Crystal structures of the tetramethylphosphonium chloride and phosphoric acid complexes of ligand 44 illustrate the anion binding ability of these compounds. The chloride ion is bound by one urea N–H⋯Cl interaction (N⋯Cl distance of 3.307 Å) from the ligand and two O–H⋯Cl interactions from co-crystallised CH3OH (O⋯Cl distances of 3.026 and 3.200 Å). The treatment of 44 with phosphoric acid and subsequent crystallisation results in the triprotonated ligand, with one ammonium group present in each of the three piperazine linkers, coordinated to a hydrogen bonded H2PO4−–HPO42− anion pair. Two P–O–H⋯OP hydrogen bonds (O⋯O distances of 2.583 and 2.679 Å) link the anion pair. The H2PO4− anion is bound by one urea N–H⋯O interaction (N⋯O distance of 2.881 Å) to the ligand, whereas the HPO42− anion is coordinated by the ammonium groups of three different ligands with N⋯O distances between 2.590 and 2.668 Å.
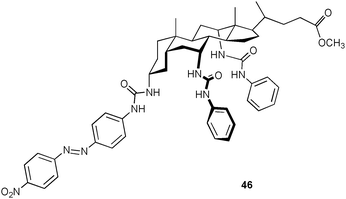
Davis and co-workers employed steroid-based receptor 46 for the recognition of unprotected amino acids.37 Proton NMR titration experiments in DMSO-d6 reveal a preferred binding of L-tryptophan (Trp) over D-Trp in a 1:1 binding mode. The calculated stability constants Ka are 480 M−1 for L-Trp and 260 M−1 for D-Trp, whereas the interactions towards D/L-Phe (phenylalanine), D/L-Leu (leucine) and D/L-Ala (alanine) are too weak to determine stability constants using this method. NOESY and computer simulation (Density functional theory (DFT) (B3LYP/6-31G(d)) studies of the complexes [L-Trp⊂46] and [D-Trp⊂46] suggest the presence of CH–π interactions between both the L- and D-Trp guest and the receptor. Thus, differences in the hydrogen bond pattern between the carboxylate group of the two enantiomers and the host presumably results in the observed enhance binding of the L-enantiomer.
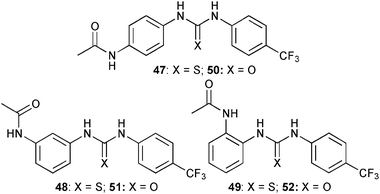
Gunnlaugsson and co-workers38 have synthesised a series of diaryl thiourea receptors 47–49 with acetamide moieties in the para-, meta- and ortho-positions and compared their anion binding properties to the known urea counterparts 50–52.39 UV-Vis titration experiments in CH3CN showed a shift in the absorption band upon addition of the tetrabutylammonium salts of H2PO4−, AcO−, HP2O73−, F− and Cl−. The dominant species for all receptors is a 1:2 (L:A) complex. The expected 1:1 complex due to cooperative binding of the amide and thiourea group in 49 (ortho) was not obtained. However 1:1 complexes were formed by the urea counterpart 52.39 In general the ligands bind acetate more strongly than hydrogen pyrophosphate, fluoride, dihydrogen phosphate and chloride. A summary of the stability constants is listed in Table 6. Compared to their urea counterparts, the thioureas 47 and 48 show a higher anion affinity than 50 and 51.
 :1 and 1:2 (L:A) binding mode in CH3CN at room temperature.a Anions added as their tetrabutylammonium salts
:1 and 1:2 (L:A) binding mode in CH3CN at room temperature.a Anions added as their tetrabutylammonium salts
| Anion | 47 | 48 | 49 | |||
|---|---|---|---|---|---|---|
| log K11 | log K12 | log K11 | log K12 | log K11 | log K12 | |
| a Standard deviations are specified in brackets. | ||||||
| AcO− | 6.31 (±0.17) | 5.12 (±0.37) | 5.61 (±0.11) | 2.93 (±0.23) | 5.46 (±0.05) | 3.44 (±0.28) |
| F− | 4.84 (±0.1) | 4.3 (±0.28) | 5.39 (±0.07) | 3.71 (±0.25) | 5.30 (±0.05) | 3.47 (±0.21) |
| H2PO4− | 5.34 (±0.24) | 4.66 (±0.46) | 4.42 (±0.19) | 4.945 (±0.39) | 4.81 (±0.01) | 4.60 (±0.14) |
| HP2O73− | 5.76 (±0.007) | 1.52 (±0.13) | 5.46 (±0.15) | 2.77 (±0.30) | 6.33 (±0.06) | 2.13 (±0.19) |
| Cl− | 4.1 (±0.13) | 3.26 (±0.31) | 3.69 (±0.05) | — | 3.20 (±0.07) | 2.0 (±0.01) |
The remote electronic effect on the binding of F− and AcO− by the urea group was studied by Ghosh and co-workers.40 Density functional theory (B3LYP/6-311+G**) studies of NH⋯F and NH⋯O-COCH3 hydrogen bond lengths in the 1:1 complex of 53–61 reveal shorter distances for the NH binding side adjacent the substituent containing the electron withdrawing nitro or trifluoromethane groups. Closer examination of the positional isomers 58–60 show binding in the order 60 > 59 > 58 for both anions highlighting the important role of the relative positions of the substituents.
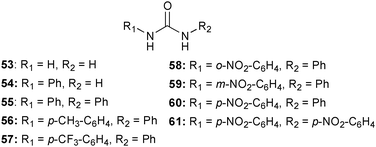
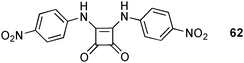
Fabbrizzi and co-workers have synthesised simple squaramide receptor 6241 and compared its anion binding properties to those of known urea receptor 61.42 Spectrophotometric titrations with tetraalkylammonium salts were performed in CH3CN to assess the stability constants. A 1:1 binding mode was found for the studied anions, except for F− and CH3COO−. These anions showed more complex behaviour and the authors suggest deprotonation of the receptor and subsequent formation of hydrogen bonded anion pairs ([F–H⋯F]− and [CH3COO–H⋯OOCCH3]−, respectively) in the presence of excess of these anions, as observed previously.42,43 Overall, both receptors show decreasing complex stability along the series F− > Cl− > Br− > I− and CH3COO− > H2PO− > NO2− > HSO4− > NO3−. The observed stability constants are higher for the squaramide receptor 62 than the urea analogue 61 (Table 7), except for HSO4− and AcO−. Interestingly, the enhanced affinity of 62 towards the halide ions is more pronounced than for the oxo-anions. The authors point out the presence of additional C–H⋯anion interactions from the Cα aryl group in the crystal structure of the Cl− and Br− complexes of 62 to explain this behaviour. The observed C⋯Cl distances are 3.784 and 3.862 Å, whereas the C⋯Br distances are 3.868 and 3.930 Å.
 :1 binding mode in MeCN at 298 K.a Anions added as their tetraalkylammonium salts
:1 binding mode in MeCN at 298 K.a Anions added as their tetraalkylammonium salts
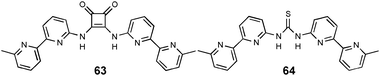
In a similar fashion Al-Sayah and Branda have compared the anion binding ability of thiourea and squaramide groups.44 Proton NMR titration studies with tetrabutylammonium acetate in CD3CN/CDCl3, fitted to a 1:1 binding mode, reveal a higher stability constant for the squaramide receptor 63 (Ka = 7390 M−1) than for the thiourea 64 (Ka = 5790 M−1). Calorimetric titration studies show that the binding is both enthalpically and entropically driven. An enthalpy change ΔH of −505 cal mol−1 and entropy change ΔS of 8.7 cal mol−1 K−1 were calculated for 64 in DMSO, whereas a ΔH of −2209 cal mol−1 and a ΔS of 10.3 cal mol−1 K−1 were obtained for 63 in CH3CN/CHCl3.
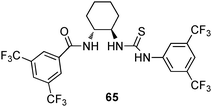
The application of a series of chiral thiourea-amide ligands such as compound 65 as co-catalyst in combination with 4-(dimethylamino)-pyridine was investigated by Seidel and co-workers.45 The authors observed significantly improved discrimination rates between the reaction rate of faster and slower reacting enantiomers (s-factor) of up to 56 times for the conversion of various propargylic amines with benzoic anhydride.
Another series of catalysts for asymmetric introduction based on the formation of hydrogen bonds have been reported by Takenaka and co-workers.46 The 2-aminopyridinium ion based catalysts of the general type 66 and 67 were tested against the addition of 4,7-dihydroindole to nitroalkenes.
Amine, ammonium and pyridinium based receptors
Anion binding by amine based receptors requires protonation of the host. Thus, the receptor's binding ability is pH dependent and can be tuned by the substituents close to the ammonium groups and the type of amine present in the molecule (e.g. primary, secondary, tertiary). The determination of the protonation and stability constants of triphosphate with a series of tripodal tris(tetraamine) ligands described by Tripier and co-workers illustrates this strikingly.47 The protonation constants were investigated by potentiometric titrations in aqueous solution at 25 °C and summarised in Table 8. The calculated species distribution diagrams (Fig. 8) illustrate the presence of the higher protonated species for 68, which contains primary, secondary and tertiary ammonium groups, over 69 and 70. The last (70) contains tertiary ammonium groups only and is present as H3703+, H4704+ and H5705+ at significant lower pH only. For example at pH 6 the major species for 70 is H3703+, for 69 H6696+ and for 68 H6686+ and H7687+ are present at approximately 50% each. At pH 3 the major species of 70 is H5705+, where 69 is present as H7697+ and H6696+ (approximately 50% each) and 68 as H9699+. Due to this different level of protonation a comparison of the triphosphate complex stability log Kalh is not practical and the authors utilise a plot of the logarithmic conditional constants log K over pH to illustrate the order of anion binding at a given pH (Fig. 9).47 For example, the affinity towards triphosphate at pH 6 was found to decrease from 68 > 69 > 70 whereas at pH 3 the stability constants increase in the order is 70<68<69.
| Equilibriuma | 68 | 69 | 70 |
|---|---|---|---|
| a Charges omitted for clarity. b Values in parentheses are standard deviation in the last significant digit. | |||
| L + HLH |
10.55(3) | 11.00(1) | — |
| LH + HLH2 |
9.85(2) | 9.80(1) | — |
| LH2 + HLH3 |
9.38(2) | 9.21(1) | — |
| LH3 + HLH4 |
8.71(3) | 8.30(1) | 4.60(1) |
| LH4 + HLH5 |
8.26(1) | 8.20(1) | 4.26(1) |
| LH5 + HLH6 |
6.63(2) | 6.92(2) | 3.23(2) |
| LH6 + HLH7 |
5.93(2) | 3.00(2) | |
| LH7 + HLH8 |
4.30(2) | ||
| LH8 + HLH9 |
4.25(2) |
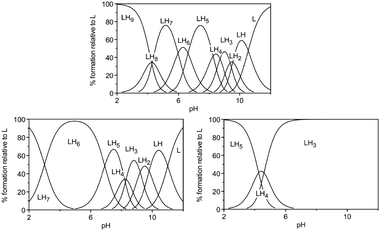 | ||
| Fig. 8 Species distribution diagrams for 68 (top), 69 (bottom left) and 70 (bottom right) as a function of pH. Tripier et al., J. Org. Chem. 2010, 5380–5390. Reproduced with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
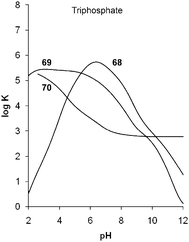 | ||
| Fig. 9 Logarithms of the conditional constant for triphosphate versus pH for 68, 69 and 70. Tripier et al., Eur. J. Org. Chem., 2010, 5380–5390. Reproduced with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
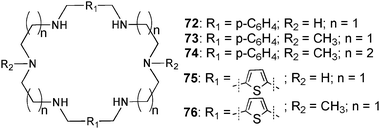
Numerous hexa-aza-macrocycles (72–76) containing secondary and tertiary ammonium groups for binding halides and oxo-anions, have been described by Hossain and co-workers.48–52 Proton NMR titrations carried out in D2O with the hexatosylated salt of 73 at pH 2.149 and the tetratosylated salt of 74 at pH 1.748 show a selectivity for sulfate over perchlorate, nitrate and halides. The binding studies fitted to a 1:1 binding mode result in stability constants Ka [M−1] with 73 and 74 of 3600 and 580 for sulfate, >20 and 120 for perchlorate, 120 and 40 for nitrate, 15 each for chloride, 90 and 20 for bromide and 25 and 30 for iodide, respectively. Crystal structures of the sulfate complexes of p-xylyl49 (72) and 1,5-thiophene50 (76) linked macrocycles agree with the 1:1 binding stoichiometry. The anion is bound by N–H⋯O and C–H⋯O interactions of the ammonium groups and the ligand backbone with N⋯O distance between 2.688 and 3.366 Å and C⋯O distance between 3.100 and 3.207 Å.
In contradistinction to these results, the perchlorate48,52 complex of 72, 74 and 75 as well as the chloride51 complexes of 73 were isolated with a 1:2 ligand to anion ratio with the anion bound inside the macrocycle. The latter example is a complex with complete encapsulation of two chloride anions within the protonated aza-macrocycle 73 (Fig. 10).51 The intramolecular chloride-chloride distance is 4.433 Å and each of the chloride atoms is bound by three hydrogen bonds with NH⋯Cl distances between 3.067 and 3.146 Å.
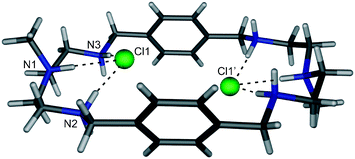 | ||
| Fig. 10 Crystal structure of Cl6⊂H673 with selected atom labels and hydrogen bonds as dotted lines, Cl− atoms located in ligand periphery, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
In a related octa-protonated aza-cryptand, compound 77, an unusual bridged binding mode of three nitrate anions was observed in the complex in the solid state (Fig. 11).53 The three anions are located in close proximity in a triangular arrangement between the two tertiary bridgehead N–H groups and each nitrate anion is coordinated via one oxygen atom. The intermolecular NH⋯O distances are between 3.099 and 3.370 Å and show no particular order. The bound O atoms of the three nitrate anions are separated by 2.494, 2.602 and 3.000 Å. Proton NMR titrations in D2O at pH 3.3 revealed a 1:1 binding mode in solution.54 The calculated stability constant are log Ka = 4.23 for NO3− and log Ka = 3.75 for NO2−.
![Schematic draw of 77 (left) and crystal structure of [(NO3)3⊂H877]5+ (right), hydrogen bonds as dotted line, external NO3−, H2O and selected hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f11.gif) | ||
| Fig. 11 Schematic draw of 77 (left) and crystal structure of [(NO3)3⊂H877]5+ (right), hydrogen bonds as dotted line, external NO3−, H2O and selected hydrogen atoms omitted for clarity. | ||
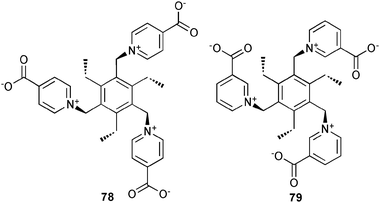
To avoid the formation of a highly charged hosts for the binding of anions in water Steed and co-workers55 have reported the synthesis of two tripodal zwitterionic receptors 78 and 79 based on the combination of pyridinium and carboxylate subunits on a 2,4,6-triethylbenzene platform. Both receptors show no affinity towards NaCl, NaI, NaAcO, MgATP and K[Al(oxalate)] in NMR titration experiments in high competitive solvents such as D2O and CD3OD. However, the PF6− complex (HPF6⊂79) shows moderate binding of chloride (log K11 = 2.17) and bromide (log K11 = 1.98) in DMSO-d6. In contrast, the para isomer 78 was not isolated as a halide-free receptor, due to the strong coordination of bromide in the self-assembled capsule. The mutual interdigitation of the six pendant arms of two ligand molecules in a sandwiched arrangement of the carboxylate functions of one ligand between the pyridinium groups of the other results in the formation of a stable capsule such that even the addition of AgPF6 was not sufficient to remove bromide completely.
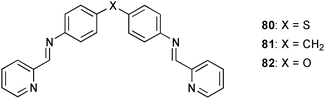
An example of anion controlled self-assembly of Cu(II) complexes of the simple bis-pyridylimine ligands 80–82 has been reported by Gloe and co-workers.56 Crystallisation in the presence of SO42− results in the formation of hexanuclear circular meso-helicates with all three ligands, wherein the coordination sphere of the metal centres are saturated by a chelating interaction to the anion. The observed Cu–Osulfate distances range from 1.959 to 2.792 Å. The authors observed the formation of non-cyclic triple helicate complexes when SO42− was replaced with ClO4− or NO3− and used the different coordination mode of the metal centre as strong evidence for influence of the nature of the anion on the topology of the formed assembly.
Pyrrole based anion receptors
Pyrrole based anion receptors have been shown to be effective and selective receptors for a variety of anionic species.57–59 The anion binding ability of macrocyclic 83 and open chain dipyrrolylmethane receptor 84 were investigated by Mani and co-workers.60 Proton NMR titration experiments in acetone-d6 reveal a strong binding for F− (Ka > 104 M−1) over Cl− (Ka = 4763 M−1) and Br− (Ka = 317 M−1) by 83 in a 1:1 binding mode. Slow exchange processes in acetone-d6 and CDCl3 were observed upon the titration of 83 with n-Bu4NHSO4. Comparable experiments with open chain receptor 84 in CDCl3 result in stability constants log Ka > 4 in a 1:2 (host to guest) binding mode. However, crystallisation of both receptors with two equivalents of n-Bu4NHSO4 results in the formation of the 1:1 complexes [SO4⊂H283] and [SO4⊂H284], respectively (Fig. 12). The crystal structures reveal that both receptors are doubly protonated. The sulfate ion in [SO4⊂H283] is coordinated by N–H⋯O hydrogen bonds to the pyrrole groups (N⋯O distances of 2.992 and 3.076 Å) and the formed ammonium group (N⋯O distances of 2.740 and 2.833 Å). In [SO4⊂H284] the anion is bound by two N–H⋯O hydrogen bonds from the pyrrole groups (N⋯O distances of 2.800 and 2.821 Å) and by only one of the two ammonium groups with an N⋯O distance of 2.773 Å.
![Crystal structure of [SO4⊂H283] (left) and [SO4⊂H284] (right), hydrogen bonds as dotted line, co-crystallised solvent molecules and non-acidic hydrogen atoms are omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f12.gif) | ||
| Fig. 12 Crystal structure of [SO4⊂H283] (left) and [SO4⊂H284] (right), hydrogen bonds as dotted line, co-crystallised solvent molecules and non-acidic hydrogen atoms are omitted for clarity. | ||
The same group have studied the anion binding of amine linked pyrrole based macrocycles 85 and 86 by proton NMR titration in acetone-d6.61 The smaller macrocycle 85 binds F− in a 1:1 binding mode with a stability constant Ka [M−1] of 1138. A 2:1 (L:A) stoichiometry was obtained for the binding of Cl− and Br− with stability constants K21 [M−1] of 3182 and 243, respectively. The larger macrocycle 86 forms 1:1 complexes with Cl− and Br− and shows enhanced binding of halide anions (Ka [M−1] = 13586 for Cl− and 1181 for Br−).
Proton NMR titrations of a new tripyrrole-methane based cryptand 87 reveal a preferred binding of F− over Cl−, NO3−, HSO4−, CH3COO− and H2PO4−, added as their tetrabutylammonium salts.62 Complex formation is kinetically slow in CD2Cl2. However, the integration of the resonances of free and complexed cryptand results in a calculated stability constant of 1562 M−1 in a 1:1 binding mode.63
The ability to assemble anion complexes of the known meso-octamethylcalix[4]pyrrole 88 by bis-cations in the solid phase and in solution was reported by Gale, Sessler and co-workers.64 Crystallisation experiments of 88 in the presence of dibromide salt 89 and 90 results in assemblies where the bromide ions are coordinated by four N–H⋯Br hydrogen bonds (N⋯Br distances between 3.410 and 3.503 Å) of the calixpyrrole. The pyridinium or imidazolium rings of the bis-cation reside in the bowl shaped calixpyrrole-anion complex cavity with C-centroid distances of 3.640 and 3.679 Å for (Br⊂88)289 and 3.537 Å for (Br⊂88)290 (Fig. 13). Isothermal titration calorimetry experiments in acetonitrile point at the formation of similar 1:2 (bis-cationic salt to calixpyrrole) complexes in solution. The stability constant for the association of bis-bromide salt 90 and 88 was calculated to 2220 M−1.
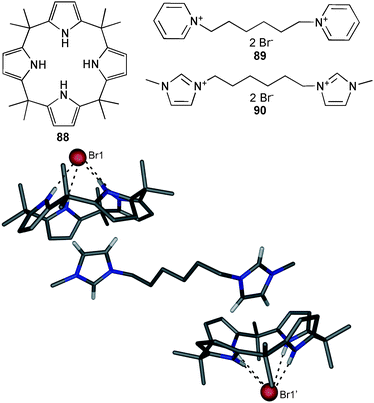 | ||
| Fig. 13 Crystal structure of the ion pair assembly of (Br⊂88)290, hydrogen bonds as dotted line, selected hydrogen atoms omitted for clarity. | ||
Sessler and co-workers have reported additional binding studies with bromide and benzoate of a series of known strapped calix[4]pyrroles6591–93 and have demonstrated the pronounced anion selectivity of these compounds.66 Proton NMR titrations of 93 and isothermal titration calorimetry studies of 91 and 92 and the parent calix[4]pyrrole 88 where performed in acetonitrile solutions. The calculated stability constants, employing a 1:1 binding mode, are summarized in Table 9 and reveal selective binding of Cl− over Br− and PhCOO− for all receptors, with a pronounced Cl− selectivity for the strapped system 91–93. The highest binding for all anions was obtained with the pyrrole-strapped system 92.
Sessler and co-workers also reported the use of anion templates to synthesise the large macrocycle cyclo[8]pyrrole (94) by electrochemical oxidation of bipyrrole 96.67 The electrolyses were carried out in dichloromethane in the presence of F−, Cl−, Br−, NO3−, BF4−, HSO4− and ClO4−. The highest reaction yield of 68% was achieved in the presence of HSO4−. Further, the authors successfully synthesised cyclo[7]pyrrole 95 and cyclo[8]pyrrole 94 from the simple 3,4-diethylpyrrole by the same method, which was not achieved before by chemical oxidation.
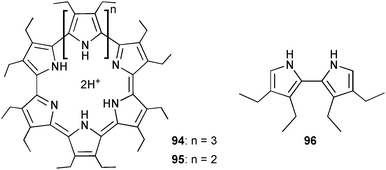
The same group have also continued their work to apply various calix[4]pyrroles as liquid–liquid extractants to aid sulfate/nitrate exchange selectivity.68
Indole based receptors
The relatively new area of indole based anion receptor systems69,70 continued to grow in 2010. Anion receptors containing a 7-indole-ureido moiety may be used to provide additional hydrogen bond donors in a favourable position for the multidentate coordination of anions. Detailed proton NMR studies of the C2 and C7 substituted receptors 97–100 by Plavec and co-workers illustrate the conformational change of the ligand upon the addition of spherical halides and oxo-anions.71 NOE studies reveal the dominance of the anti–anti conformation of the free receptor in acetone, whereas the addition of chloride, bromide and acetate results in the syn–syn conformation of 97–100. No conformational changes were observed upon the addition of nitrate to all receptors, which also exhibits very weak binding.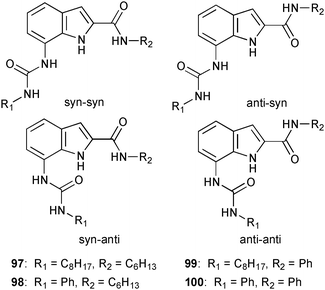
Diindolyurea based receptors 101 and 102 were employed by Gale and co-workers to bind various oxo-anions in the flexible ligand cleft.72 Crystal structures reveal the coordination of SO42− and HPO42−via 8 and 9 N–H⋯O hydrogen bonds from all the N–H groups in 101, respectively (Fig. 14). The observed N⋯O distances in the SO42− complex range from 2.788 to 2.953 Å, whilst the N⋯O distances are between 2.736 and 3.109 Å in [HPO4⊂101]2−. However the structure of the benzoate complex reveals a 1:3 (L:A) stoichiometry (Fig. 14). One anion is bound by four hydrogen bonds of the urea and the two adjacent indole groups in the central ligand cleft (N⋯O distances range from 2.717 to 2.820 Å) where the two amidoindole moieties coordinate to remaining anions by N–H⋯O interactions with N⋯O distances between 2.766 and 3.144 Å. Solution studies with carboxylates show that the first equivalent of anion binds to the central diindolylurea group, whilst the second and third equivalents bind to the terminal amidoindole groups. However, upon addition of one equivalent of tetrabutylammonium sulfate to the receptor all the NH groups bind the anion in a 1:1 stoichiometry. Addition of further equivalents of sulfate results in the formation of higher order complexes. Proton NMR studies in DMSO-d6/0.5% water reveal deprotonation of H2PO4− and HCO3− by free anions in solution when complexed by the receptors.
![Crystal structure of [SO4⊂101]2− (top) and [(PhCOO)3⊂101]3− (bottom) with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f14.gif) | ||
| Fig. 14 Crystal structure of [SO4⊂101]2− (top) and [(PhCOO)3⊂101]3− (bottom) with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
More detailed studies of anion–anion proton transfer of coordinated H2PO4− were performed by the same group.73 Proton NMR titration experiments of 103 in DMSO-d6/0.5% water and DMSO-d6/10% water show a new set of signals after the addition of more than one equivalent of n-Bu4NH2PO4 with a considerable downfield shift. Comparable experiments where n-Bu4NH2PO4 was added up to 1.4 equivalent followed by the addition of n-Bu4NOH yield the same set of signals in the proton NMR spectra (Fig. 15). The authors explain their observation with a reduced pKa of the bound oxo-anion and subsequent proton transfer to the unbound anion. The solid-state structure of [HPO4⊂104]2− supports the observed proton transfer (Fig. 16). Single crystals were isolated by slow evaporation of a DMSO solution of 104 in the presence of excess n-Bu4NH2PO4. The coordinated anion is HPO42−, which is bound by six N–H⋯O hydrogen bonds with N⋯O distances between 2.660 and 2.842 Å.
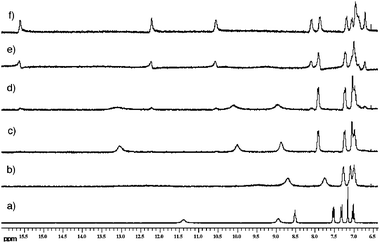 | ||
| Fig. 15 Proton NMR titration of 104 in DMSO-d6/0.5% water. (a) Free receptor; (b) 0.6 equivalents n-Bu4NH2PO4; (c) 1.0 equivalents n-Bu4NH2PO4; (d) 1.4 equivalents n-Bu4NH2PO4; (e) 1.4 equivalents n-Bu4NH2PO4 + 0.7 equivalents n-Bu4NH2PO4; (f) 1.4 equivalents n-Bu4NH2PO4 + 1.4 equivalents n-Bu4NH2PO4. Gale et al., Chem. Asian J. 2010, 5, 555–561. Reproduced with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
![Crystal structure of [HPO4⊂104]2− with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation, co-crystallised water molecules and non-interacting hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f16.gif) | ||
| Fig. 16 Crystal structure of [HPO4⊂104]2− with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation, co-crystallised water molecules and non-interacting hydrogen atoms omitted for clarity. | ||
The same receptor 103 amongst others of the same type was also employed to recognise alkylcarbamate anions in solution and the solid state.74 A crystal structure reveals that the alkylammonium-alkylcarbamate (AAAC) salt, the reaction product of 1,3-diaminopropane and carbon dioxide, is coordinated by 103 (binding the carbamate group) and 18-crown-6 (binding the ammonium group) (Fig. 17). Both functional groups are bound by multiple hydrogen bonds with N⋯O distances ranging from 2.677 to 3.163 Å between the receptor and carbamate and N⋯O distances between 2.738 and 3.073 Å for the ammonium⋯18-crown-6 interactions. Significant proton NMR shifts indicate the interaction of the receptors and the carbamate substrates in DMSO-d6 solutions.
![Crystal structure of [AAAC⊂103]·18-crown-6 with selected atom labels, hydrogen bonds as dotted line, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f17.gif) | ||
| Fig. 17 Crystal structure of [AAAC⊂103]·18-crown-6 with selected atom labels, hydrogen bonds as dotted line, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
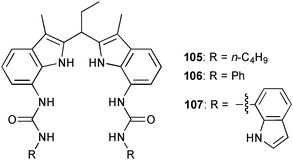
Jurczak and co-workers have reported the anion binding ability of a series of 7,7′-diureido-2,2′-diindolylmethane based receptors 105–107.75 Proton NMR titrations in CD3OD were employed to calculate the stability constants of the 1:1 anion complexes. The three receptors show selective binding of H2PO4− over HSO4−, PhCOO−, Cl− and Br− (Table 10). Interestingly, the additional 7-indole NH donor functions in 107 do not result in an enhanced binding of the anion. The obtained stability constants are evidence of a pronounced influence of n-butyl and phenol substituent in 105 and 106, respectively. The crystal structure of the phosphate complex of 106 shows a deprotonation of the anion upon crystallisation. The anion is bound via five N–H⋯O hydrogen bonds with N⋯O distances between 2.818 and 2.942 Å. Closer examination reveals the formation of a [H3PO4·PO4]3− dimer, that is coordinated by two ligands 106. The phosphate anions are bridged by three protons with O⋯O distances between 2.496 and 2.676 Å and a P-P distance of 3.677 Å.
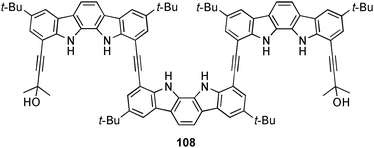
The indolocarbazole oligomer 108 was employed by Jeong and co-workers for the recognition of sulfate in solution and the solid state.76 Quantitative NMR analysis of 108 in the presence of one equivalent tetrabutylammonium sulfate shows significant shifts of the NH and OH signals, resulting from hydrogen bonding between the anion and the six N–H and two O–H donor groups. NOE cross-peaks of non-adjacent protons indicate folding of 108 upon the coordination of the anion in solution. This is in agreement to the observed arrangement in the solid state. The crystal structure of [SO4⊂108]2− shows a folded structure of 108 and the coordination of the SO42− anion by eight hydrogen bonds in the helical cavity. The six N–H⋯O hydrogen bonds range from 2.780 to 2.844 Å where the distances of the two O–H⋯O hydrogen bonds are 2.810 and 2.883 Å. Nonlinear fitting analyses were performed on the fluorescence emission change of 108 (excited at 320 nm) upon the addition of tetrabutylammonium salts in 10% CH3OH–CH3CN to determine the stability constants. The calculated constants (Ka [M−1]) show preferential binding of SO42− (640000) over Cl− (8800), AcO− (5700), H2PO4− (3600), Br− (2800), CN− (1600), N3− (790) and I− (< 100).
Yan and co-workers have reported a series of crystal structures illustrating the binding modes of known indolocarbazole-quinoxaline receptor 10977 with various anions such as F−, Br−, AcO−, PhCOO−, NO3−, HSO4−, H2PO4− and SiF62− in the solid state.78 The crystals structures reveal a 1:1 stoichiometry for F−, Br−, AcO− and PhCOO− complex, whilst a 2:1 (L:A) complex is observed with SiF62− and a 2:2 dimer with HSO4−. In the latter structure, two anions are bridged by two protons with an O⋯O distance of 2.527 Å to form the dimer (Fig. 18). The two indolocarbazole NH donor atoms are involved in two hydrogen bonds to the HSO4− anion with N⋯O distances of 2.748 and 2.821 Å. The NH⋯F interactions in [SiF6⊂1092]2− range from 2.770 to 3.120 Å. The F− ion in [F⊂109]− is bound by N⋯F distances of 2.611 and 2.633 Å where N⋯Br distances of 3.281 and 3.383 Å were observed for [Br⊂109]−. The N⋯O distances of 2.676 and 2.676 Å in [AcO⊂109]− are comparable to the observed N⋯O distances of 2.643 and 2.701 Å in [PhCOO⊂109]−.
![Crystal structure of [(HSO4)2⊂1092]2− with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation and non-interacting hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f18.gif) | ||
| Fig. 18 Crystal structure of [(HSO4)2⊂1092]2− with selected atom labels, hydrogen bonds as dotted line, tetrabutylammonium cation and non-interacting hydrogen atoms omitted for clarity. | ||
The same group reported the anion binding properties of the pyrazino[2,3-g]quinoxaline linked indole based receptors 110–112.79 Significant absorption spectral changes upon addition of AcO− and H2PO4− as their tetrabutylammonium salts were employed to determine the stability constants for the 1:2 (L:A) complexes in DMSO. The stability constants reveal enhanced binding by the biindolocarbazole receptor 112 (for AcO−: log β11 = 5.0, log β12 = 9.2; for H2PO4−: log β11 = 4.9, log β12 = 9.1) over the indole-indolocarbazole receptor 111 (for AcO−: log β11 = 4.7, log β12 = 8.3; for H2PO4−: log β11 = 4.5, log β12 = 8.2) and the biindole receptor 110 (for AcO−: log β11 = 3.7, log β12 = 7.5; for H2PO4−: log β11 = 3.3, log β12 = 7.2). The stronger binding may be due to the rigid indolocarbazole group possessing a higher degree of preorganisation than the flexible biindole receptor. The crystal structures of the H2PO4− complexes of the three receptors 110–112 result in distinct multi-dimensional networks as a result of the different binding modes of the indolocarbazole and biindole moieties towards the anion in the solid state. The biindole side tends to bind two respective H2PO4− ions, whereas the indolocarbazole moiety prefers only one (Fig. 19). Thus, in [(H2PO4)2⊂1102]2− each anion is bound by one N–H⋯O hydrogen bond from two ligands and two alternating O–H⋯O hydrogen bonds toward another H2PO4− ion to form an anion dimer. This results in a two-dimensional network in the crystal packing. In [(H2PO4)2⊂1112]2− the anion dimer is bound by two indole N–H⋯O and four indolocarbazole N–H⋯O hydrogen bond of four ligands to form a one-dimensional 2+2 dimeric ribbon. In [(H2PO4)2⊂112]2− the anion forms an infinite chain, which is alternate coordinated by the double N–H⋯O hydrogen bonds of the indolocarbazole moiety of 112 to form a two-dimensional coordination polymer.
 | ||
| Fig. 19 Schematic representation of the preferred binding mode towards H2PO4− by the biindole and the indolocarbazole moiety. | ||
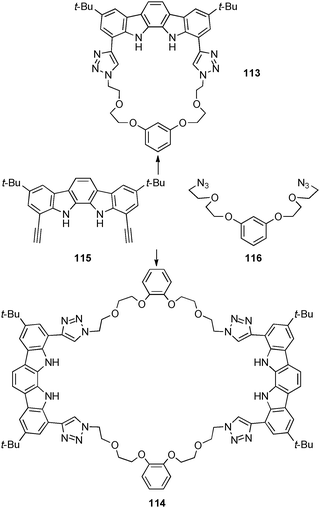
Li and co-workers have reported the anion binding ability of macrocyclic indolocarbazole based receptors 113 and 114 in solution.80 The ring closure of the macrocycles was achieved by ‘click chemistry’ on the 1:1 and 2:2 assemblies of the precursors 115 and 116, tuned by the reaction conditions. Proton NMR titration experiments fitted to a 1:1 binding model in CDCl3 reveal a preferred binding of the spherical halide ions by the smaller macrocycle 113, even through only one indolocarbazole moiety is present in this system. Further, the stability constants show preferred binding of F− (Ka [M−1] 11600 for 113, 3010 for 114) over Cl− (Ka [M−1] 1500 for 113, 120 for 114), Br− (Ka [M−1] 325 for 113, 16 for 114) and I− (Ka [M−1] 106 for 113, 4 for 114) for both macrocyclic receptors.
The asymmetric tripodal amine bridged indole receptor 117 was employed by Fujita and co-workers for the binding of halide ions or alkali metal ions in solution.81 Proton NMR titration experiments in CD3CN fitted to a 1:1 binding mode reveal a preferred binding of Cl− (Ka [M−1] = 1042) over Br− (Ka [M−1] = 157) and I− (Ka [M−1] = 12). No shift of the protons was observed if n-Bu4NF was added.
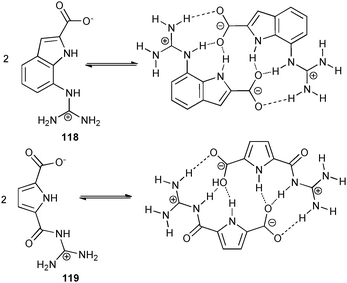
Schmuck and co-workers have continued their work on the dimerisation of zwitterionic guanidinium–carboxylates.82 This group have recently reported the synthesis of an indole-containing analogue 118 of their self-assembling guanidiniocarbonylpyrrole systems (e.g.119). Proton NMR studies reveal a stability constant Kass > 105 M−1 in DMSO-d6 for the indole based ligand 118, which is significantly smaller than the estimated value Kass > 1010 M−1 for the pyrrole based analogue 119.83 The crystal structure of the dimer of 118 reveals a steric hindrance between the guanidinium group and the proton in the 6-position of the indole group. Consequently the guanidinium group is twisted out of the plane and the attractive nature of the additional hydrogen bonds is reduced.
Imidazole and triazole base receptors
Imidazole and triazole based receptors provide different binding modes for the recognition of anions. Where imidazole derivatives require protonation and subsequently anion binding is accomplished by charged N+–H⋯X−-type hydrogen bonds, triazole based receptors form C–H⋯X−-type hydrogen bonds. Substitution of both functional groups results in imidazolium and triazolium based receptors, which form (C–H)+⋯X− type hydrogen bonds with dominant electrostatic interactions.The nitrate complexes of a series of imidazole and pyrazole arene capped tripodal receptors 120–122 were investigated by Arunachalam and Ghosh.84 Previous reports show that the solid state complex of protonated benzimidazole substituted receptor 120 forms a discrete dimeric capsule including all six NO3−.85 In contradistinction, the reaction product of the imidazole substituted receptor 121 and nitric acid crystallised in an infinite disordered capsular structure. Each nitrate anion is bound by one N–H⋯O hydrogen bond with N⋯O distances between 2.750 and 2.806 Å. The anions are linked by hydrogen bonds towards a co-crystallised water molecule. The replacement of imidazole with dimethyl pyrazole in 122 and subsequent crystallisation with nitric acid results in the formation of an anion bridged macrocycle (Fig. 20). The anion bridge is formed by two NO3− ions coordinated by the arms of two different ligand molecules with N⋯O distances between 2.730 and 2.803 Å. The three pyrazolium groups of the ligand are approximately planar and occupy the corners of the triangle. This results in a 1-D sheet like assembly of anion bridged macrocycles.
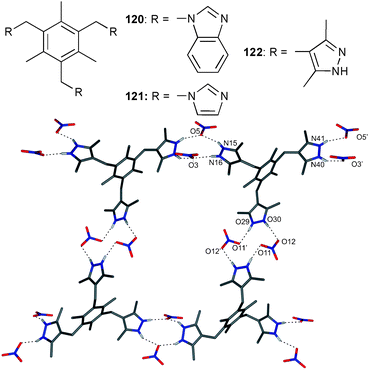 | ||
| Fig. 20 Single macrocyclic unit formed in the crystal structure of (NO3)3⊂H3122 with selected atom labels, hydrogen bonds as dotted line, non-interacting hydrogen atoms omitted for clarity. | ||
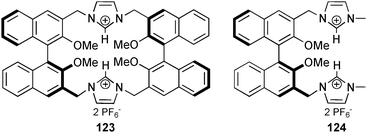
The anion binding ability of imidazolium receptors 123 and 124, based on the 1,1′-bi-2-naphthol (BINOL) scaffold, were investigated by Yu and co-workers.86 The treatment of the ligands with various anions as their tetrabutylammonium salts in CH3CN-DMSO (9:1 v/v) results in a significant change in the emission spectra upon excitation at 290 nm. The calculated stability constants show an enhanced anion binding in a 1:1 binding mode by the macrocyclic receptor 123 (Ka [M−1] ∼ 2.6 × 106 for F−) over the monomer 124 (Ka [M−1] = 2.6 × 105 for F−). Furthermore, the titration experiments reveal a selectivity of 123 for F− (Ka [M−1] ∼ 2.6 × 106) over AcO− (Ka [M−1] = 2.8 × 105), H2PO4− (Ka [M−1] = 1.4 × 104), HSO4− (Ka [M−1] = 1.1 × 103), Cl− (Ka [M−1] = 341), Br− (Ka [M−1] = 180) and I− (Ka [M−1] = 75). The obtained stability constants of 124 point at a reduced selectivity of the more flexible receptor: Ka [M−1] = 1.4 × 105 for AcO−, 8.7 × 103 for H2PO4−, 1.4 × 103 for HSO4−, 6.2 × 103 for Cl−, 933 for Br− and 154 for I−. Preliminary titration experiments towards the chiral recognition of amino acids were carried out for 123 with the tetrabutylammonium salts of t-Boc-L-phenylalanine and t-Boc-D-phenylalanine. The calculated stability constants of 64200 and 43000 M−1 respectively, point at a moderate selectivity of 1.5.
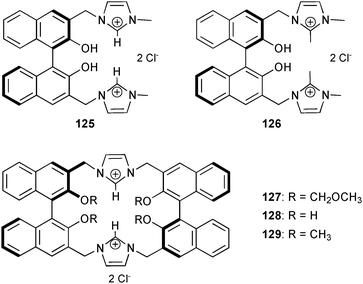
More detailed studies towards the binding of α-amino acids by 1,1-binaphthyl based imidazolium receptors 125–129 were reported by You and co-workers.87 Fluoresence titration experiments in aqueous solution (HEPES buffer, pH 7.4) of 125 reveal a pronounced binding of L-Trp (Ka [M−1] = 1.73 × 104) over L-Phe (Ka [M−1] = 7.28 × 103), L-His (Ka [M−1] = 3.79 × 103), L-Tyr (Ka [M−1] = 2.96 × 103), L-Val (Ka [M−1] = 6.06 × 103), Gly (Ka [M−1] = 2.17 × 103) and L-Cys (Ka [M−1] = 1.45 × 103). No spectral changes were observed upon the addition of L-Pro, L-Glu, L-Ala and L-Ser. The chiral recognition of the two enantiomers of tryptophan (L-Trp and D-Trp) was investigated for 125–129. The results are summarized in Table 11 and reveal a remarkable chiral recognition capability KD/KL of 6.2 by macrocyclic receptor 127.
| Receptor | K L-Trp/M−1 | K D-Trp/M−1 | K D-Trp/KL-Trp |
|---|---|---|---|
| a Error values were obtained by the result of Benesi–Hildebrand plots and the correlation coefficient of lininear fitting is over 0.99. | |||
| 125 | (1.73 ± 0.01) × 104 | (1.09 ± 0.03) × 104 | 0.6 |
| 126 | (8.04 ± 1.62) × 10 | (8.34 ± 2.20) × 10 | 1.0 |
| 127 | (2.89 ± 0.12) × 103 | (1.79 ± 0.03) × 104 | 6.2 |
| 128 | (4.78 ± 0.07) × 103 | (3.38 ± 0.13) × 103 | 0.7 |
| 129 | (2.59 ± 0.12) × 103 | (5.42 ± 0.02) × 103 | 2.1 |
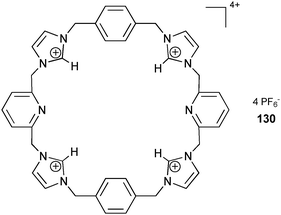
Sessler and co-workers have employed the tetracationic macrocycle 1304+, containing four imidazolium moieties, for the formation of oligo-pseudorotaxanes with a mono-terephthalate anion.88 The crystal structure shows a chair-like conformation for the macrocycle, with the mono-terephthalate anion bound within the central cavity. Hydrogen bonds between the head-to-head stacked mono-terephthalate anion result in a 1-D arrangement in the solid state, where electrostatic and C–H⋯π interactions stabilize the pseudorotaxane arrangement. Detailed NOESY NMR experiments in DMSO-d6 reveal the presence of various aggregates of the pseudorotaxane in solution. Stability constants of 1.3 × 102 M−1 (Ka1) and 8.8 × 101 M−1 (Ka2) were calculated for the formation of the dimeric and trimeric oligomers [(1304+)·(mono-terephthalate anion)]2 and [(1304+)·(mono-terephthalate anion)]3, respectively. The 1:1 complex formation of the free host 1304+ and mono-terephthalate anion gives a stability constant Ka of 2.1 × 103 M−1.
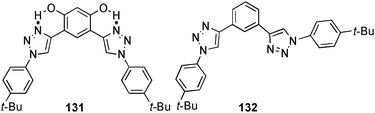
The influence of preorganisation, due to intramolecular hydrogen bonding in the receptor backbone, on the chloride binding ability of aryl-triazole based receptors was investigated by Flood and co-workers.89 Proton NMR titration experiments in CD2Cl2 reveal the expected significant enhanced affinity of the chloride anion for 131 (Ka [M−1] = 46800) compared to 132 (Ka [M−1] = 1000). The coordination of anions in both receptors is based on multiple C–H⋯Cl interactions only.
The same group reported the chloride binding of a photo-switchable triazole foldamer 133.90 The cis–trans photoisomerisation of azobenzene end groups was used to enable the wavelength dependent uptake and release of the anion. Electronic absorption spectra titration experiments with n-Bu4NCl in CH3CN reveal stability constants of Ka = 3000 M−1 for the trans and Ka = 380 M−1 for the cis isomer. The authors demonstrate the reversible photo-switchable conformation of the receptor and the subsequent influence on the anion binding in conductivity experiments. Alternate radiation of the receptor in the presence of n-Bu4NCl in CH3CN with light at 365 and 436 nm results in significant changes in the observed conductivity (Fig. 21).
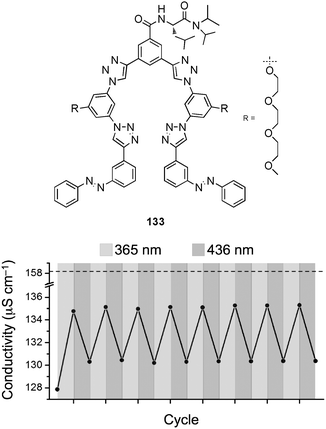 | ||
| Fig. 21 Solution conductivity of n-Bu4NCl in the presence of 133 (1 equivalent, 1mM, CH3CN, 298 K) obtained upon exposure to UV (365 nm) and visible (4367 nm) light. Dashed line corresponds to the conductivity in the absence of 133. Reprinted with permission from J. Am. Chem. Soc. 2010, 132, 12838–12840. Copyright 2011 American Chemical Society. | ||
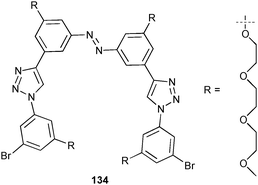
The anion binding ability of a similar photo-switchable triazole based receptor 134 was investigated by Jiang and co-workers.91 Proton NMR titration experiments in acetone-d6 fitted to a 1:1 binding mode show the enhanced binding of the cis isomer (Ka [M−1] = 290 for Cl−) compared to the trans isomer (Ka [M−1] = 70). All other anions result in reduced stability constants of 66 M−1 for HSO4−, 87 M−1 for Br−, 31 M−1 for I− and 21 M−1 for NO3− with the cis isomer and Ka = 38 M−1 for HSO4−, 22 M−1 for Br−, 11 M−1 for I− and 10 M−1 for NO3− with the trans isomer.
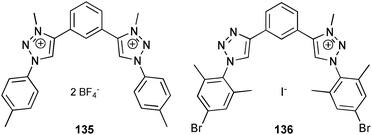
The coordination of sulfate by the triazolium based receptors 135 and 136 was investigated by Schubert and co-workers.92 A different complex composition was obtained depending on the degree of methylation of the receptor. Proton NMR titration experiments in CD3CN/CH3OH 4:1 show the formation of a 1:1 complex by 135 with a stability constant log Ka = 4.39. On the other hand, the coordination of sulfate by the mono-methylated receptor 136 results in the formation of a 2:1 (L:A) complex. The stability constants in CD3CN were determined to be log K11 = 3.73 and log K21 = 3.1.
Hydroxy and Lewis acid based receptors
The anion binding abilities of two D-ribose based epimers 137 and 138 in solution were reported by Kondo and co-workers.93 Significant shifts of both OH-signals in proton NMR titration experiments in CDCl3 and CD3CN indicate the formation of O–H⋯anion hydrogen bonds. The calculated stability constants fitted to a 1:1 binding mode reveal a preferred anion binding by 138 over 137, presumably due to the electrostatic and/or steric repulsion between the anomeric O atom in 137 and the anion. Both receptors show an enhanced binding of AcO− over H2PO4−, Cl− and Br−. A stronger affinity was observed in CD3CN with respect to CDCl3 for both receptors. A summary of the obtained stability constants is given in Table 12.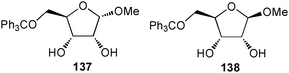
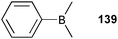
Competitive studies to investigate the F−/CN− selectivity of the simple Lewis acid dimethyl(phenyl)borane 139 were reported by Aldridge and co-workers.94 The absorption change at 351 nm of 139 in UV-Vis titration experiments in CH2Cl2 were employed to calculated the stability constants Ka [M−1] of 1.9 × 105 for CN− and 8.9 × 104 for F−. Competitive studies in CD2Cl2, monitored by multinuclear NMR experiments, reveal a replacement of F− from [F⊂139]− by an excess of CN−, whereas no spectral change was observed if [CN⊂139]− was treated with an excess of F−. The crystal structures [F⊂139](n-Bu4N) and [CN⊂139]·[K(18-crown-6)] show the formation of covalent bonds between the borane receptor and the anion. The B–F distance is 1.482 Å, whereas CN− is bound by the C atom with a B–C distance of 1.631 Å and a B–C–N angle of 173.4°.
The same group also reported the F−/CN− binding ability of 1,2-diborylferrocene 140 and 141.95 UV-Vis titration experiments of 140 in THF give a stability constant of 3.7 × 104 M−1 for CN− whereas the binding of F− appears to be very slow. Competitive experiments, monitored by 11B NMR show the quantitative formation of [CN⊂140]− upon the addition of 140 to a equimolar solution of F− and CN−. The crystal structures [F⊂141]·[K(18-crown-6)] and [CN⊂141]·[K(18-crown-6)] reveal different binding modes for the anions. The CN− anion is C-bound with a B–C distance of 1.648 Å and coordinated in the exo position to one borane atom. Whereas F− is bound by one borane centre with a B–F distance of 1.471 Å, but located in the endo position (Fig. 22).
![Crystal structure of [CN⊂141]·[K(18-crown-6)] (left) and [[F⊂141]·[K(18-crown-6)] (right) with selected atom labels, hydrogen atoms and [K(18-crown-6)]+ cation in [[F⊂141]·[K(18-crown-6)] omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f22.gif) | ||
| Fig. 22 Crystal structure of [CN⊂141]·[K(18-crown-6)] (left) and [[F⊂141]·[K(18-crown-6)] (right) with selected atom labels, hydrogen atoms and [K(18-crown-6)]+ cation in [[F⊂141]·[K(18-crown-6)] omitted for clarity. | ||
The anion binding ability of the sulfonium fluorosilane Lewis acid receptor 142 was reported by Gabbai and co-workers.96 The addition of tetrabutylammonium salts of Cl−, Br− or I− to 142 in CDCl3 results in the formation of neutral silane 143, whereas the presence of n-Bu4NF gives the stable complex [F⊂142]. Changes in the absorption spectrum were employed to determine the stability constant Ka of 7 × 106 M−1 for the formed 1:1 complex in CHCl3. The crystal structure of [F⊂142] reveals a trigonal-bipyramidal geometry of the Si atom (Fig. 23). The F-atoms occupy the axial positions with a F–Si–F angle of 177.3° and Si–F distances of 1.706 and 1.732 Å. The F atom with the longer Si–F bond is involved in interactions towards the S-atom with a S⋯F distance of 2.741 Å.
![Crystal structure of [F⊂142] with selected atom labels, S⋯F interaction represented as dotted line, hydrogen atoms omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f23.gif) | ||
| Fig. 23 Crystal structure of [F⊂142] with selected atom labels, S⋯F interaction represented as dotted line, hydrogen atoms omitted for clarity. | ||
Armand and co-workers employed the simple boron esters 144–147 to form Lewis acid anion complexes from LiF and to dissolve the salts as an electrolyte in battery solvents such as ethylene carbonate/dichloromethane (EC/CH2Cl2) or dimethylformamide (DMF).97
Anion recognition by halogen bonds
Halogen bonding is an emerging field in the recognition of anions. Crystallisation of the chloride complexes of triamino (148) and tetraamino (149) piperazine cyclophane in the presence of 1,4 diiodotetrafluorobenzene (F4DIB) results in the formation of C–I⋯Cl halogen bonds in the solid state.98 Each of the three chloride ions in Cl3⊂H3148·(F4DIB)6 is coordinated by two C–I⋯Cl halogen bonds with I⋯Cl distances ranging from 3.129 to 3.289 Å and C–I⋯Cl− angles between 168.7 and 175.6°. Where in Cl6⊂H6149·(F4DIB)2 only four Cl− ions are bound by one C–I⋯Cl interaction each with a I⋯Cl distance of 3.087 and 3.117 Å and a C–I⋯Cl− angle of 179.8 and 178.2°, respectively (Fig. 24).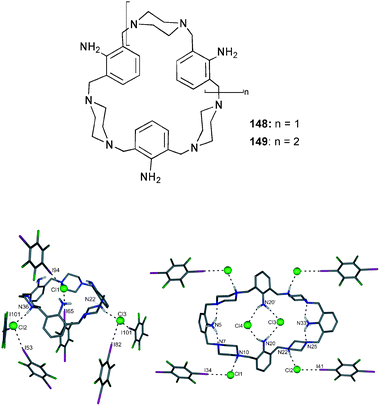 | ||
| Fig. 24 Crystal structure of Cl3⊂H3148·(F4DIB)6 (left) and Cl6⊂H6149·(F4DIB)2 (right) with selected atom labels, hydrogen and halogen bonds represented as dotted lines, Cl− atoms located in ligand periphery, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
Taylor and co-workers investigated the possibility to coordinate a single anion by multiple halogen bonds in solution.99 Fluorine NMR titrations were performed with the mono-, bi- and tridentate ligands 150–153 at 295 K in acetone-d6. The addition of tetrabutylammonium salts results in a downfield shift of the 19F NMR signals and can be fitted to a 1:1 binding model. The calculated stability constant Ka [M−1] for Cl− increases considerably with the number of available iodine atoms in the receptor from 70 for 150, 1.1 × 103 for 151, 1.8 × 103 for 152 and 1.9 × 104 for 153 and demonstrate the chelating effect of the halogen bond. Additional 19F NMR titrations with 153 reveal an anion binding selectivity in the order Cl− (1.9 × 104 M−1) > Br− (3.8 × 103 M−1) > I− (7.6 × 102 M−1) ≫ TsO− (10 M−1) > HSO4− (<10 M−1) > NO3− (<10 M−1).
The same group examined the influence of the type of donor atom and the number and position of adjacent electron withdrawing fluorine atoms on the strength of the halogen bonds.100 The Cl− stability constants were calculated employing a 1:1 binding mode from 19F NMR titration experiments in acetone-d6 of the tridentate ligands 153–163. The obtained results of the series of tetrafluoro-phenyl ligands 153–156 point at the presence of significant halogen bonding for the iodine-based receptor only. The stability constant Ka [M−1] for 153 is 1.9 × 104, where negligible values were obtained for the bromide-based receptor 154 (Ka [M−1] = < 10), CH-anion hydrogen bond receptor 156 (Ka [M−1] = <5) and pentafluoro-phenyl receptor 155 (Ka [M−1] = <5). The investigation of ligands 157–163 demonstrates a striking correlation between the complex stability and the number and position of fluorine atoms present in the iodo-phenyl-moiety. A stability constant Ka [M−1] of 18 was calculated for 157, whereas Ka = 38 for 160, Ka = 2.8 × 102 for 163 and Ka = 1.9 × 104 for 153. Positional isomers 158 and 159 reveal stability constants of Ka = 3 M−1 and Ka = 7 M−1, respectively, and the bi-fluorinated 161 and 162 give values of 2.2 × 102 M−1 and 15 M−1, respectively.
Significant smaller effects on the complex stability were observed for a series of para-substituted iodoperfluoroarenes 164–168.101 Fluorine NMR titrations, fitted to a 1:1 binding mode in cyclohexane-d12 at 298 K with tri-n-buthylphosphine oxide, reveal stability constants Ka [M−1] of 12 for 164, 5.6 for 165, 3.9 for 166, 3.3 for 167 and 1.3 for 168.
Beer and co-workers reported the first example of an imidazolium-based pseudorotaxane that employs halogen bonds in order to assemble.102 The interpenetration of a series of bromo- and methyl-functionalized imidazolium derivatives 169–173 with known macrocycle 174 were studied by 1H NMR spectroscopy in CDCl3 at 293 K. The calculated stability constants from titration experiments reveal a pronounced association with the 2-bromo-imidazolium chloride thread 172 (Ka [M−1] = 254) over the value obtained for the hydrogen bonded equivalent 170 (Ka [M−1] = 97) and point at halogen bond mediated anion templated interpenetration. The almost identical behaviour of 169 (Ka [M−1] = 92) and 170 is consistent with chloride ion hydrogen bonding in unsubstituted imidazolium systems occurring primarily through the acidic 2-position. The obtained stability constant with the 2-methyl-imidazolium thread 171 (Ka [M−1] = 245) was explained by the authors with an altered coordination of the thread towards the anion template. Detailed analysis of the NMR spectra point at a chelating coordination of the chloride ion by hydrogen bonds originating from the 4- and 5-positions of the imidazolium subunit. In contradistinction the strongly linear nature of halogen bonds prevents a chelating effect with thread 173 and results in no noticeable interpenetration.
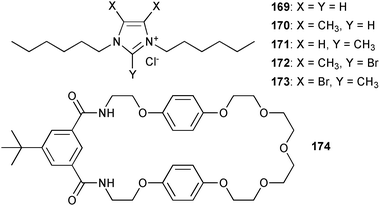
Anion recognition by anion–π interactions
A more recent objective in anion receptor chemistry is the use of anion–π interactions to bind anionic guests and is a topic of some controversy.103 Frontera and co-workers synthesised the hydrochloride salts of N9- and N6-decyladenine 175 and 176, respectively, in order to investigate the influence of the substitution pattern on the formation of anion–π contacts.104 The crystals structures of both salts reveal the presence of Cl−⋯π interactions with N6-decyladenine 176 only (Fig. 25). The shortest chloride-carbon distances are 3.482 and 3.550 Å. Further, the Cl− ion is bound by two N–H⋯Cl− hydrogen bonds of other ligand molecules with N⋯Cl− distances of 2.892 and 2.784 Å.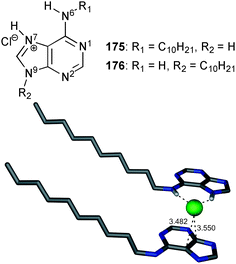 | ||
| Fig. 25 Crystal structure of Cl⊂176, hydrogen and anion–π interactions represented as dotted line, non-interacting hydrogen atoms omitted for clarity. | ||
Anion–π interactions of the pentafluorobenzyl-substituted ammonium salt 177 and the two pyridinium salts 178 and 179 were reported by Albrecht and co-workers.105 All crystal structures of the Cl−, Br−, and I− salts of 177 show the position of the anion on top of the π-system of the pentafluorophenyl ring (Fig. 26), while no anion–π attractions were observed in the PF6− salt of 177. The observed C–anion distances are in the range of C⋯Cl = 3.757–3.951 Å (Cl⊂177), C⋯Br = 3.809–4.046 Å (Br⊂177) and C⋯I = 3.833–4.207 Å (I⊂177) and point at only a slight off-centre location of all three anions. On the other hand, the Br⋯π binding mode in the complexes of (Br⊂178) and (Br⊂179) depend on the substitution of the receptor. CH-anion hydrogen bonding in Br⊂178 forces the anion to a location towards the rim of the pentafluorophenyl group. This results in C⋯Br− distances of 3.756 and 3.609 Å for the η2 type interaction to the C6F5 unit. The additional methyl group in 179 and subsequent steric effects result in an orthogonal orientation of the pyridinium ring with respect to the C6F5 group. Thus the methyl substituent points over the centre of the aromatic ring and fixes the Br− ion in a η6 fashion with C⋯Br distances ranging from 3.492 to 3.976 Å.
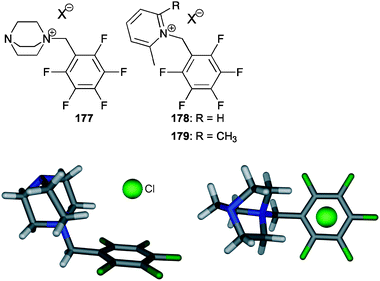 | ||
| Fig. 26 Side (left) and top view (right) of the crystal structure of Cl⊂177, Br− and I− are very similar, structures not shown. | ||
The same group explored the formation of anion–π interactions between pentafluorophenyl substituted receptors 180 and 181 and polyhalide anions, such as Br3−, I3− and I42−.106 Crystal structure analysis reveals a η3 and a slightly off-centre η6 type interaction of the tribromide anion towards the C6F5 units of two quinoline receptors 180 (Fig. 27). The C⋯Br interactions in the η3 interaction of Br1 range from 3.596 to 3.917 Å. Where C⋯Br3 distances are between 3.430 to 4.015 Å. The tribromide anion is unsymmetrical with a Br1–Br2 distance of 2.644 Å and a Br2–Br3 distance of 2.469 Å. The anion is bound to the protonated receptor by an N–H⋯Br hydrogen bond to Br1 with a N⋯Br distance of 3.624 Å. In contrast, the crystal structure of the bromide complex of 180 reveals a η1 anion–π motive with a C⋯Br distance of 3.876 Å. The diffusion of HI into a solution of pentafluorobenzyldibenzylamine 181 in toluene and subsequent crystallisation results in the anion complex containing four protonated ligands, two triiodide ions and one tetraiodide dianion (Fig. 27). Both triiodide anions are linear with I–I bond lengths of 2.896 and 2.921 Å. The observed I–I distances of the symmetrical dianion I42− are 2.762 Å (I–Iinternal) and 3.363 Å (I–Iterminal). Anion–π interactions were observed for the triiodide anion, which is positioned in a side-on fashion to the C6F5 moiety of the receptor. The C⋯I distances to the central iodine atom I2 range from 3.804 to 4.082 Å. The tetraiodide dianion is involved in N–H⋯I hydrogen bonds (N⋯I distance 3.496 Å) and in anion–π interactions to two electron-poor phenyl rings with C⋯I distances of 3.661 and 4.068 Å.
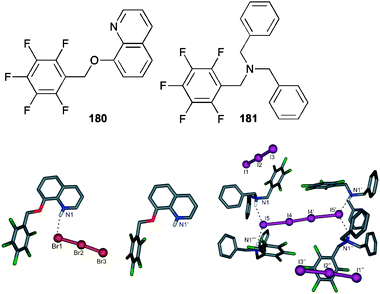 | ||
| Fig. 27 Crystal structure of (Br3)⊂H180 (left) and (I4)·(I3)2⊂(H181)4 (right) with selected atom labels, hydrogen bonds represented as dotted lines, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
Anion–π interactions of the extended π-electron deficient receptor 182 were investigated in the solid state and in solution by Chifotides and co-workers.107 The crystal structure of the Br− complex reveals a stacked assembly of [Br3⊂1822]3− units linked by one Br− ion as observed previously for the iodide complex of 182 (Fig. 28).108 In both structures the three equivalent halide ions are bound to the pyrazine external carbon atoms Cext in a η2, η2 fashion, with distances up to 0.35 Å shorter than the sum of the van der Waals radii. The observed C⋯Br distances range from 3.245 to 3.280 Å where C⋯I distances are between 3.337 and 3.419 Å. UV-Vis, 13C and halogen NMR studies are evidence for the formation of the similar [X3⊂1822]3− assembly of the tetrabutylammonium salts of Cl−, Br−, I− and 182 in CD3NO2 and THF-d8 solution. Job plot experiments provide evidence for a 2:3 (or 1:1.5) (L:A) stoichiometry of the present complex. Downfield shifts of the single 13Cext resonances by 1–3 ppm upon the addition of the halides point at comparable interactions between the anions and the receptor in solution as observed in the solid state. Additional 35Cl, 81Br and 127I NMR studies in CD3NO2 and THF-d8 exhibit a single resonance with a considerable downfield shift with respect to the corresponding free halide ions and support the formation of similar [X3⊂1822]3− assemblies in solution. Quantitative analyses in THF of the new absorption bands at 630, 419 and 408 nm for I−, Br− and Cl−, respectively, as a function of the respective concentration of the anion were performed to determine the KCT [M−1] values of 3780 for Cl−, 2200 for Br− and 940 for I−. Comparable experiments in CH3NO2 reveal KCT [M−1] values of 71 for Cl−, 48 for Br− and 20 for I− employing the absorption bands at 557, 406 and 390 nm, respectively. The observed order KCT,Cl > KCT,Br > KCT,I in both solvents is in accordance with the order of Lewis basicity and thus the electron donating ability of the anion in the CT complex.
![Schematic draw of 182 (left) and diagram of the stacked [X3⊂1822]3− adduct (right).](/image/article/2012/CS/c1cs15257b/c1cs15257b-f28.gif) | ||
| Fig. 28 Schematic draw of 182 (left) and diagram of the stacked [X3⊂1822]3− adduct (right). | ||
The anion–π interactions of the conformationally rigid triazine ring containing cage 183 were studied by Wang and co-workers in both solid state and solution.109 Isothermal titration calorimetry in acetonitrile reveals the formation of 1:1 complexes with the tetrabutylammonium salts of F−, Cl− and Br−. The calculated stability constants Ka [M−1] are 361 for F−, 146 for Cl− and 95 for Br−. Proton NMR studies of the complex formation give no evidence for the anion binding being originated by hydrogen bonding in solution. The C3 symmetric crystal structure of chloride complex shows a 1:3 (host:guest) stoichiometry with one Cl− ion located in each of the three ligand clefts. The anion forms η1 interaction to a carbon atom of one triazine ring with a C⋯Cl distance of 3.342 Å and is further bound by a C–H⋯Cl (C⋯Cl distance 3.541 Å) and an O–H⋯Cl (O⋯Cl distance 3.541 Å) hydrogen bond to the ligand backbone and a co-crystallised water molecule.
Lippert and co-workers observed the presence of anion–π interactions towards the oxo-anions NO3−110 and SO42−111 encapsulated in the cationic triangular Pt3-tris(2,2′-bipyrazine) based metallo-macrocycles 184 and 185, respectively. Two O atoms of a sandwiched NO3− anion in 184 form two O⋯π interaction with the pyrazine rings with O⋯π distances of 3.270 and 3.587 Å. Where one O atom of the positionally disordered SO42− atom in 185 is involved in two anion–π interactions with O⋯π distances between 2.898 and 3.063 Å.
The anion–π interactions towards the anion encapsulated in the 2 × 2 grid like metallo-macrocycles formed by four Cu(I) centres linked by the four aminopyrimidine based ligands 186 and 187 were reported by Manzano and co-workers.112 The crystal structures of the ClO4− complex of 186 and 187 as well as the BF4− and OTf− (trifluoromethanesulfonate anion) complex of 187 reveal close contacts between the encapsulated anion and the centroid of the aminopyrimidine moiety of the ligand. The shortest O⋯centroid distance in the ClO4− complexes is 3.084 Å in 186 and 2.848 Å in 187, respectively. The OTf− O⋯centroid distance is 2.868 Å in 187, where 2.848 Å is the shortest observed F⋯centroid distance in the BF4− complex of 187.
Ion pair recognition
Ditopic receptors for ion pair recognition continue to be developed by a number of groups world-wide. Such systems provide a distinct binding site for cationic and anionic species and enable an enhanced binding due to electrostatic attraction. This was demonstrated by Sessler and co-workers in their continued work on calix[4]arene strapped calix[4]pyrroles such as 188.113 Quantitative proton NMR studies in 10% CD3OD/CDCl3 show no significant proton shift when CsClO4 or n-Bu4NF is added. However, when CsF or CsClO4 and n-Bu4NF are added simultaneously a significant change in the proton signals of both the calix[4]arene and the calix[4]pyrrole moiety is observed. ITC experiments in the same solvent mixture reveal a stability constant of Ka = 1.3 × 104 M−1 for a 1:1 binding mode of CsF and 188. No indication of similar ion pair binding was observed in proton NMR studies with other metal ions, such as Li+, Na+, K+, Rb+ or NH4+. This suggests that anion binding by 188 occurs only in the presence of Cs+. Comparable experiments with CsClO4 and tetrabutylammonium salts of Cl−, Br− and NO3− show ion pair binding for all studied anions, with CsF as the preferred ion pair. In the crystal structure, the CsF ion pair is solvent-bridged by a co-crystallised water molecule (Fig. 29). As expected, the F− anion is bound by four NH⋯F bonds (N⋯F distance between 2.793 to 2.809 Å) in the calix[4]pyrrole moiety and the Cs+ ion is bound by ion-dipole interactions with two oxygen atoms and by cation–π interactions involving the calix[4]arene moiety in the 1,3-alternate conformation.
 | ||
| Fig. 29 Schematic drawing of 188 (left) and crystal structure of (Cs·H2O·F)⊂188 (right) with selected atom labels, hydrogen bonds represented as dotted line, co-crystallised solvent molecules and non-interacting hydrogen atoms omitted for clarity. | ||
In a similar fashion, Jabin and Menand114 employed the known tren-capped calix[6]arene 189115 for the recognition of anions and contact ion pairs. Proton NMR experiments in CD3CN of a 1:1 mixture of the host 189 and the tetrabutylammonium salts of Cl−, Br−, CN− and AcO− reveal a slow exchange at 243 K, which enables the calculation of the stability constants from the integration of the appropriate signals. Where the constants for N3− and NO3− were calculated from titration experiments performed at 298 K. A preferred binding of Cl− (Ka = 48300 M−1) over Br− (Ka = 1930 M−1), CN− (Ka = 640 M−1), N3− (Ka = 215 M−1), AcO− (Ka = 160 M−1) and NO3− (Ka = 98 M−1) was observed. Comparable experiments in the presence of PrNH3Cl reveal a significant enhanced stability constant of Ka > 1.6 × 109 M−2 (Ka = [PrNH3Cl⊂189]/[189][PrNH3+][Cl−]) for the ion pair in CDCl3 at 258 K.
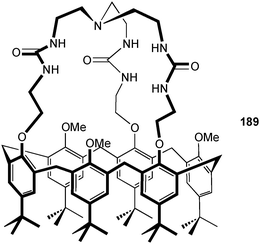
Gattuson and co-workers employed the (1,3)-bridged calix[5]arene-crown-3 receptors 190 and 191 for the recognition of ion pairs.116 Proton NMR titration experiments were performed in CD2Cl2 with n-butylammonium (n-BuNH3+) hexafluorophosphate, tetrabuthylammonium halides and n-butylammonium halides to assess stability constants of the n-BuNH3+ cation and the anions Cl−, Br− and I−, both independent and as ion pair. The stability constants calculated using a 1:1 binding mode are summarised in Table 13 and show clearly an enhanced binding for the ion pair.
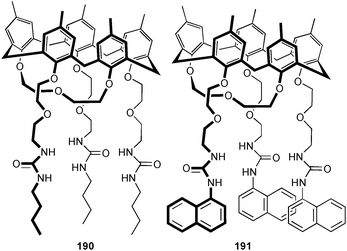
The recognition of amino acid ammonium salts (tetrabutyl- and tetramethylammonium) by heteroditopic chiral uranyl-salen receptor substitueted with 1-oxypyrenyl groups (192) reported Tomaselli and co-workers.117 UV-Vis spectral studies indicate a 1:1 binding stoichiometry in CHCl3 solution. High binding affinities were noted along with good enantiomeric selectivity for L-Phe over D-Phe with binding constants of 2.50 × 106 and 7.36 × 104 M−1, respectively. T-ROESY NMR experiments show cation–π and CH–π interactions of the pyrene moieties and the ammonium cations and result in the formation of stable complexes with chiral carboxylate guests.
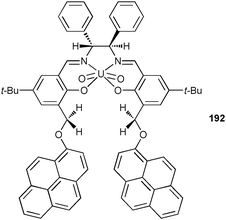
The same group also employed the chiral uranyl-salen macrocycles 193 and 194 for the recognition of chiral and achiral ammonium halide salts (tetramethylammonium, tetraethylammonium, tetrabutylammonium, acetycholine, trimethylanilinium, benzyltrimethylammonium and (a-methylbenzyl)trimethylammonium).118 Detailed T-ROESY NMR experiments show that the binding of the cation occurs by CH–π and π–π interactions of the binaphthyl, or the salicylaldehyde moiety, or the phenyl rings of thediimine bridge, where the halide ion is bound on the uranyl side.
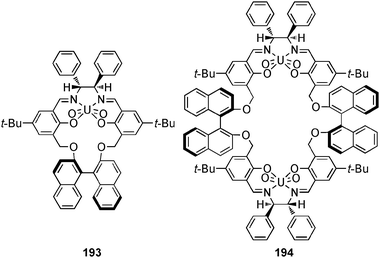
Tasker and co-workers continued their work on the use of polytopic ligands for the extraction of metal salts. The authors show a significant enhanced metal-to-ligand loading and extraction ability (≈250% Zn to receptor) by the coordination of Zn2+ and anionic chlorozincate species, such as ZnCl42−, Zn2Cl62− or ZnCl3−, in the polytopic salen based ligand system 195.119 Comparable dual host extraction experiments with 1:2 mixture of the simple salen ligand 196 and the tertiary amine 197 result in a reduced Zn extraction power (loading up to 150% Zn to receptor 196). Proton NMR shifts of the azomethine and the 3-aminomethyl signals of 195 at ZnCl2 loading >100% indicate the coordination of Zn2+ in the salen moiety and the interaction of a chlorozincate species with the anion binding sides.
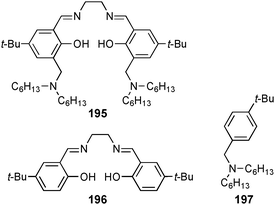
On the other hand, Plieger and co-workers used an in situ formed metallo-macrocycle based on the 2:2 coordination of Cu(II) and the salicyl-aldimine ligand 198 for the binding of anions (Fig. 30).120 Crystal structures of the ClO4− and NO3− complexes show the uptake and coordination of the anion by multiple NH and CH hydrogen bonds in the metallo-macrocycle. In addition, the ClO4− ion is coordinated by direct Cu–O–ClO3− interactions as previously observed for BF4− and SO42− complexes.121 Changes in the UV-Vis absorption of the cationic Cu(II)-complex [Cu21982]4+ upon the addition of acids in isopropanol were employed to calculate the stability constants. Job plot experiments show the formation of 1:1 complexes. The calculated stability constants show a preferred uptake of SO42− (log Ka = 5.07) over H2PO4− (log Ka = 4.34), ClO4− (log Ka = 3.38), BF4− (log Ka = 3.03) and NO3− (log Ka = 2.95) and reflect the influence of the anion charge, size and ability to form coordinative bonds to the metal centres in the complex.
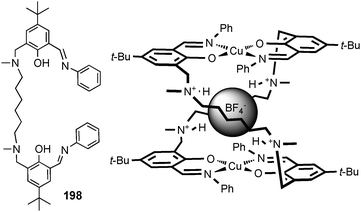 | ||
| Fig. 30 Schematic draw of 198 (left) and diagram of the anion encapsulated 2:2 metallo-macrocycle formed by 198 and two Cu(II) metal centres (right). | ||
Huang and co-workers synthesised the unsymmetric diamide bis(m-phenylene)[32]crown-10 cryptands 199 and 200 and studied their ion pair binding of various dipyridinium dication salts 201–203.122 Single crystal X-ray diffraction and proton NMR titrations in CD3CN show the interaction of the amide groups and the CH2 crown ether backbone with the Cl− ions and dipyridinium cations. Job plot experiments based on UV-Vis absorption data in CHCl3/CH3OH show a 1:1 stoichiometry. The highest stability constant for the binding of the 4,4′-bispyridinium chloride salt 201 was obtained for cryptand 200 (Ka = 1.7 × 104 M−1 for 200 and Ka = 4.8 × 103 M−1 for 199), presumably due to the additional anion binding site supplied by the acidic phenyl hydrogen atom. The effect of the nature of the anion on the ion pair binding in 199 and 200 was investigated by comparing experiments with the Cl− and PF6− salts of the bishexyl-dipyridinium dication salts 202 and 203. Stability constants of 7.8 × 102 M−1 with 199 and 6.3 × 102 M−1 with 200 for the PF6− salt 202 are significantly smaller that those obtained for the Cl− salt 203 (Ka = 2.4 × 103 M−1 for 199 and Ka = 3.4 × 103 M−1 for 200).
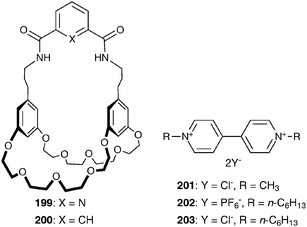
Another example of the recognition of ion pairs was reported by Dalcanale and co-workers.123 Proton NMR titrations of the phosphate triester functionalized cavitand 204 in acetone-d6 with the tetrabutylammomium salts of Cl−, Br− and I− result in significant shifts of the aromatic CH (Hα) and the ArCH2CH2 protons (Hβ). The calculated stability constants Ka [M−1] are 150 for Cl−, 140 for Br− and 73 for I−. Phosphorous NMR experiments show a negligible interaction towards the tetrabutylammonium cation (Δδ = −0.005 ppm). Comparable titration experiments with the n-octylammonium salts in CDCl3 reveal stability constants of 1.5 × 103 M−1 for Cl− and 2.1 × 104 M−1 for Br−. Significant shifts of both the aromatic CH proton (Hα) in 1H experiments and the phosphate triester signal in 31P experiments are evidence that the ion pair is bound in the cavitand, resulting in higher stability constants for the binding of halide anions.
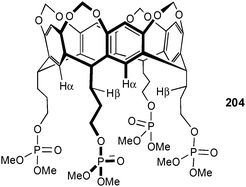
The recognition of carboxylate anions, such as oxalate (ox2−), malonate (mal2−), phthalate (ph2−), isophthalate (iph2−) and terephthalate (tph2−), in cascades formed between two Cu(II) centres in the hexaazamacrocycles 205 and 206 was reported by Delgardo and co-workers.124 The crystal structure reveals the cascade coordination of two phthalate anions by the two Cu(II) centres in the macrocycle 205, where each metal centre is coordinated by three carboxylate oxygen atoms of the anions and three N-donor atoms of the ligand. The Cu–O bond distance of the single O-atom is 1.925 Å where the Cu–O distances of the two chelating carboxylate oxygen atoms are 2.387 and 2.866 Å. UV-Vis absorption titration experiments were utilized to obtain the stability constants of the 1:1 and 1:2 (host:guest) association of the dianions and the dicopper(II) complexes of 205 and 206. In general, higher values were observed for the larger macrocycle 206, except for mal2− where log K11 = 4.50 and log K12 = 2.53 for 205, as opposed to log K11 = 4.06 and log K12 = 2.96 for 206. The anions with a shorter distance between the two binding sites are preferred by both receptors, with stability constants for ox2− of log K11 = 4.54 and log K12 = 2.73 for 205 and log K11 = 5.08 and log K12 = 2.88 for 206 and log Ka values for ph2− of 4.01 and 2.75 for 205 and 4.08 and 3.13 for 206. Consequently smaller values were obtain for iph2− (log K11 = 3.43 and log K12 = 1.82 for 205 and log K11 = 3.78 and log K12 = 2.81 for 206) and tph2−. For the latter only the formation of 1:1 complex was observed with log K11 = 3.07 for 205 and log K11 = 3.61 for 206.
The simple monopyridylurea receptor 207 was employed by Wu and co-workers for the coordination of ZnSO4.125 Depending on the synthetic route two different complexes were isolated, both by slow evaporation of methanol. [Zn(SO4)2073]·CH3OH was obtained when Zn(SO4)·7H2O dissolved in CH3OH was added to a solution of 207 in CH3OH, whereas {[Zn(μ2-SO4)2072]·0.5CH3OH}n was isolated when the ligand was added to the metal salt. The anion is coordinated by multiple N–H⋯O hydrogen bonds in both complexes (with N⋯O distances between 2.931 and 3.069 Å in [Zn(SO4)2073]·CH3OH and between 2.775 and 3.017 Å in {[Zn(μ2-SO4)2072]·0.5CH3OH}n) and by direct metal anion interactions. The disordered tetrahedral coordination spheres of the metal centres are saturated by interactions to the Npy atom of the ligand.
Similarly, Dastidar and co-workers126,127 employed a variety of bis-pyridylurea receptors for the separation of sulfate over other oxo-anions, such as nitrate and perchlorate by selective crystallisation in metal–organic frameworks. Upon the coordination of Zn(II) by 208 a 3-D framework is formed that coordinates a sulfate anion via six hydrogen bonds with three ligand molecules with N⋯O distances between 2.621 and 2.940 Å and a Zn(II)–O–SO3 bond of 2.111 Å.126 On the other hand, the treatment of 209 with CdSO4 results in a framework where the sulfate anion is bound by four N–H⋯O hydrogen bonds with N⋯O distances between 2.835 and 3.200 Å originating from two ligands and a Cd(II)–O–SO3 interaction of 2.339 Å.127 Both crystallisations were repeated in the presence of mixtures of sulfate, perchlorate, nitrate and triflate as Zn(II) salts and a mixture of CdSO4, Cd(NO3)2, Cd(ClO4)2, Cd(OAc)2, CdCl2 and CdBr2, respectively. The isolated material shows comparable FT-IR, XRPD and elemental analysis data to the results obtained for their sulfate salts, respectively and demonstrates the selective uptake of sulfate into the crystals. The same behaviour was observed by Wu and co-workers128 for the pentyl linked bis-pyridylurea receptor 210. Again, a selective uptake of sulfate was observed upon crystallisation of the Zn(II) linked framework.
Others receptors
The novel halide selective macrocycle 211, that was directly prepared by the condensation of 2,4-dimethylglycoluril with formaldehyde in the presence of HCl, was reported by Sindelar and co-workers.129 Quantitative proton NMR studies in CD3OD/CDCl3 (2:1) show a preferred uptake of I− over Br−, Cl− and F−. The crystal structures of the Cl− complex shows the uptake of the anion in the cavity and coordination by 12 weak C–H⋯anion bonds originating from the methine carbon atoms on the convex face of the glycoluril unit. The average C⋯anion distance in the Cl− complex is 3.707 Å.
In contradistinction, Holman and co-workers encapsulated larger anions such as BF4− and PF6− in continued work on cryptophane cavitands of the general type 212.130
Commercially available amine terminated dendrimers (AT-PAMAM) have been employed by Anslyn and co-workers as anion receptors.131 UV-Vis studies employing anionic indicators such as fluorescein, pyrogallol red (PR) and pyrocatechol violet (PV) were performed in methanol/water (1:1 and 95:5 CH3OH/H2O for fluorescein, pH = 7 buffered with 25 mM HEPES) to monitor the level of anionic uptake by generation 3 to 7 AT-PAMAM. Bathochromic shifts observed upon the addition of the AT-PAMAM were found to be due to the deprotonation of the indicator as the interaction of the dendrimer lowers the pKa of the indicator. The estimated stoichometries were determined by extrapolating the binding isotherms saturation point. Stoichiometries by the generations 5, 6 and 7 of AT-PAMAM was found to be 3, 6 and 14 PV molecules respectively for each dendrimer while the PR uptake by generations 4, 5 and 6 was found to be significantly higher at 8, 16 and 32 molecules of PR per respective dendrimer. This showed the high level of anionic guests accommodated in the commercially available AT-PAMAM dendrimers.
Anion transport
Another application of anion receptor chemistry is the development of compounds that can bind and transport anions across lipid membranes, either as synthetic channels or as mobile carriers.4 J.T. Davis and co-workers facilitated sphingolipid C2-ceramide 213 as a mobile carrier for Cl− and HCO3− transmembrane transport.132 Proton NMR studies show a significant shift of the NH and OH protons in water saturated CD2Cl2 upon addition of Cl− or HCO3− anions and confirm that the ceramide head-group is a potent anion binder. Titration experiments reveal a stability constant Ka [M−1] of 1734 for n-Bu4NCl for a 1:1 binding mode. Transport ability was studied in egg-yolk phosphatidylcholine (EYPC) vesicles loaded with 100 mM NaCl and 2 mM lucigenin, a chloride-sensitive dye, and dispersed in a 100 mM solution of either NaNO3, NaHCO3 or Na2SO4. The addition of 213 (1 mol%) results in an increase in fluorescence and indicates the efflux of Cl− when NO3− or HCO3− is present as external anion (Fig. 31). Comparable experiments with doubly charged SO42− present as the anion in the extravesicular solution show minimal Cl− transport. This evidence suggests an anion-exchange mechanism and provides evidence against the formation of large pores by 213, as observed in previous studies at high concentrations (>10 mol%).133
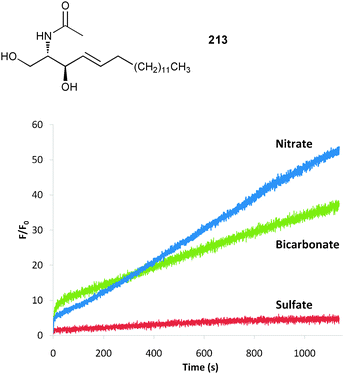 | ||
| Fig. 31 Observed relative fluorescence in Cl− transport assays promoted by 213 at 1 mol% receptor to EYPC liposomes (100 nm, 200 μM) with various external anions in 20 mM HEPES buffer (pH = 7.4). Chem. Commun. 2010, 46, 3950–3952. Reproduced with permission of The Royal Society of Chemistry. | ||
Judd and A. P. Davis continued their research on cyclosteroids such as 214 and 215, containing a macrocyclic unit for anion recognition.134 Transport studies were performed in 1-palmitoyl-2-oleoylphosphatidylcholine (POPC)/cholesterol (7:3 ratio) vesicles with pre-incorporated receptors in a 1:2500 ratio. The vesicles were loaded with 225 mM NaNO3 and 1 mM lucigenin and dispersed in 225 mM NaNO3. The addition of NaCl (final concentration 25 mM) results in a Cl− influx, indicated by quenched fluorescence (Fig. 32). Comparable experiments with the acyclic analogues 216 and 217 illustrate strikingly an enhanced Cl−/NO3− transport activity of the macrocyclic receptors 214 and 215.
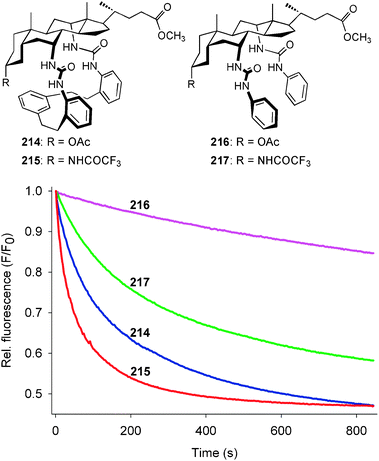 | ||
| Fig. 32 Observed relative fluorescence promoted by 214–217 at 1:2500 receptor to lipid ratio in Cl− transport assays into vesicles (7:3 POPC/cholesterol, 04 mM, 200 nm diameter). Vesicles loaded with 225 mM NaNo3 and 1mM lucigenin, final external NaCl concentration 25 mM. Chem. Commun. 2010, 46, 2227–2230. Reproduced with permission of The Royal Society of Chemistry. | ||
The tren based urea and thiourea receptors 218–221 were employed by Gale and co-workers for the transmembrane transport of Cl− by either Cl−/NO3− or Cl−/HCO3− antiport mechanisms.135 Therefore POPC vesicles loaded with 489 mM NaCl were dispersed in either NaNO3 (489 mM) or Na2SO4 (167 mM) and upon the addition of the receptor the Cl− efflux was monitored using a Cl− selective electrode. For experiments investigating HCO3− transport, NaHCO3 was added to the external Na2SO4 solution as a pulse after 120 s to give an overall HCO3− concentration of 40 mM. The observed Cl− efflux reveals a significant activity of the thiourea receptors 220 and 221 (>90% for Cl−/NO3− and >75% for Cl−/HCO3− over the time of the experiment), whereas the urea analogues 218 and 219 are less active and point at a pronounced influence of the substituent (about 30% Cl− efflux with phenyl substituted 218 and negligible transport for 219 with n-Bu moieties). Furthermore, the authors provide direct evidence of transmembrane transport of HCO3− by comparable 13C NMR experiments. Distinct 13C NMR signals for intravesicular (δ ≈ 161 ppm) and extravesicular H13CO3− (δ ≈ 160 ppm) are observed in the presence of 220 and 221 and the subsequent addition of Mn2+ results in a broadened signal of the extravesicular H13CO3− into the baseline, illustrating the transport of HCO3− by 220 and 221.
Gale, Sessler and co-workers continued their work on meso-octamethylcalix[4]pyrroles as ion transporters through lipid bilayers.136 An extended series of POPC vesicles studies reveal that the presence of electron-withdrawing fluorine substituents in 222 results in a change in the transport mechanism. Fluorinated 222 is capable of acting as a Cl−/NO3− and Cl−/HCO3− antiport agent, whereas the parent calix[4]pyrrole 88 functions as a membrane transporter for the cesium chloride ion pair only.
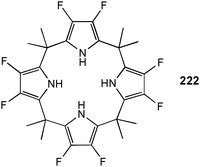
Gale and co-workers observed the same switch in transport mechanism for a series of strapped calix[4]pyrroles 223–225.137 The new receptors contain two triazole groups and show a decreasing dependence of the Cl− transport activity on the type of group one metal counterion. This suggests that the predominant cesium chloride ion pair co-transport process for 223, is replaced by a Cl−/NO3− antiport process with compound 225. The transport efficiency of the previous generation compound 226138 lies between those of 224 and 225 for Cl−/NO3− antiport.
A switch between transmembrane anion transport mechanism from mobile carriers to synthetic channels was observed by Gokel and co-workers in their study of the dipicolineamides 227 and 228.139 Fluorescence quenching in dioleoylphosphatidyl choline (DOPC) vesicles loaded with 225 mM NaNO3 and 1 mM lucigenin shows an influx of Cl− mediated by both receptors. Hill plot analysis of the transport mediated by 228 at various concentrations suggests that both mobile carrier and channel mechanisms may be present. Planar bilayer conductance technique140 reveals the formation of channels for 228, whereas no conductance behaviour was obtained for 227.
Matile and co-workers continued their work on synthetic channels formed by naphthalenediimides (NDI) derivatives to prove the functional relevance of anion–π interactions for the transmembrane transport of anions.141 Laser-induced ESI-MS and charge-transfer absorption spectra illustrate the formation of anion complexes of the simple NDI derivatives 229 and 230, whereas transport studies using different fluorescent probes in EYPC vesicles prove anion transport facilitated by 231. Therefore vesicles experiments were repeated with 8-hydroxy-1,3,6-pyrenetrisulfonate (HPTS), 5(6)-carboxyfluorescein (CF) and lucigenin (LG) encapsulated and the ‘effective’ monomer concentration needed to achieve 50% activity (EC50) was determined for each experiment. The authors use a quantitative comparison of the obtained EC50 values for 231 for the transport of Cl− (0.75 μM for LG, 0.33 μM for HPTS and >100 for CF) to conclude a pronounced anion over cation or dye transport due to the formed channels of the receptor. The authors explain their observations due to the presence of anion–π interactions.
The same group also employed a series of sulfur substituted NDI derivatives for the transmembrane transport by channels employing anion–π interactions.142 The stepwise oxidation of the sulfide donors in 232 or 235 to the sulfoxides 233 and 236 and the more powerful sulfones 234 and 237 results in an increase in the π-acidity of the NDI core and subsequently a higher anion transport activity was observed.
Bulk lipid membrane transport experiments were performed by Mutihac and co-workers to probe the acid and amido functionalized calix[4]arenes 238–242 as transporters for the amino acid esters L-tryptophane methylester (L-TrpOMe), L-phenylalanine methylester (L-PheOMe) and L-tyrosine methylester (L-TyrOMe).143 A pH gradient was applied (source phase pH = 5.5, receiving phase pH = 1.5) across the CHCl3 membrane and UV-Vis absorption spectroscopy was used to determine the amino acid concentration in the source phase and receiving phase after 24 h. The obtained transport yield shows a transport ability in the order L-TrpOMe > L-PheOMe ≫ L-TyrOMe for all receptors, with the acid 240 and the amido calix[4]arene 242 as the most efficient transporters.
Another carrier for amino acids in bulk CHCl3 lipid membranes was reported by Urban and Schmuck.144 The low pKa value of approximately 6–7 of the guanidiniocarbonyl pyrrole group in 243 enables a pH dependent binding of amino acid under ambient conditions.145 Thus, proton driven symport of the N-acetylated L-amino acid of alanine (Ala), valine (Val) tyrosine (Tyr), phenylalanine (Phe) and tryptophan (Trp) was observed in simple U-tube experiments from a source phase (pH = 6) to the receiving phase with a pH of 8. The transport studies reveal a decreasing flux for Val over Phe, Ala, Trp and Tyr for single amino acid experiments, while competitive experiments show a reduced flux and an order of Trp > Phe > Val > Tyr > Ala. The authors explain this observation using the binding strength of 243 towards the amino acids. The lower stability constant for Val (Kass [M−1] = 4.3 × 103) favours the release of the substrate into the receiving phase and result in the highest flux of Val (1.1 × 10−6 mol m−2 s−1) in the single amino acid experiments. In contradistinction, the stronger binding of Trp (Kass [M−1] = 1.5 × 104) leads to preferred transport in the competitive setup (flux: 2.1 × 10−7 mol m−2 s−1).
Anion-controlled gelation
Another interesting application of anions is in the field of organogels, in which they may be used to trigger gel–sol transitions. Chen and co-workers reported this process, along with a significant colour change, when H2PO4−, AcO− and F− are added to the gel formed by 244 in CHCl3, whereas Cl−, Br− and I− do not cause a gel–sol transition.146 Closer examination in UV-Vis and proton NMR experiments in DMSO-d6 reveal a deprotonation of the hydrazine NH group in 244 upon the addition of H2PO4−, AcO− and F−.
A novel anion induced sol–gel transition of an organogel based on the rotaxane formed from the macrocycle 245 and the thread 246 were investigated by Chiu and co-workers.147 The addition of either AcO−, Cl− or Br− as their tetrabutylammonium salts to the gel formed in 1-pentanol results in the gel becoming sol. Subsequent treatment with NaClO4 or AgPF6 restores the original gel after sonication. Comparable proton NMR studies in CD3NO2 show a switch of the interlocked macrocycle to the urea station upon the addition of n-Bu4NAcO, presumably due to stronger interaction of the AcO− ion with the ammonia group. Thus, the lack of the available urea group for intermolecular hydrogen bonding results in a gel–sol transition. The subsequent addition of NaClO4 gives the original set of signals in the NMR spectra, presumably due to precipitation of NaOAc. When Cl− (or Br−) is added the change in the NMR spectra is less pronounced and the authors suspect a coordination of the anion by the amide groups of the macrocycle and the ammonia group of the thread (Scheme 1). Consequently the conformational change in the rotaxane results in the switch to the solution of the organogel.
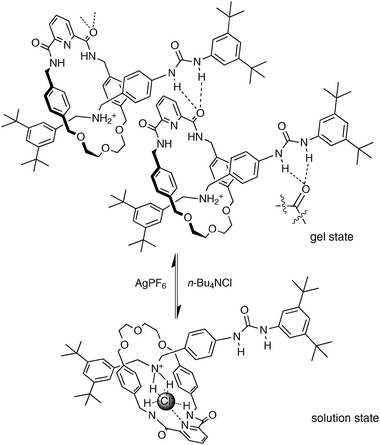 | ||
| Scheme 1 Proposed interactions in the gel and solution by 245 and 246. | ||
Steed and co-workers utilized bis(urea) gelators 247–250 to aid the crystallisation of various organic compounds, including pharmaceuticals such as carbamazepine.148 The inert gel matrix results in a slower crystallisation process and subsequently larger, more uniform crystals are formed. Furthermore, the gel inhibits the conversion of metastable polymorphs. After crystallisation the addition of AcO− results in a gel–sol transition of the matrix and allows facile recovery of the formed crystals. The same group also reported metal-induced gel–sol transitions, where the metal ion competes with intramolecular urea-pyridine and urea-urea hydrogen bonds.149
Anion sensing
Anion receptors can be combined with reporter groups to produce sensors that respond to the presence of anions via changes in their optical or electrochemical properties. Alternatively receptors may be used as part of a selective membrane in an electrode to produce device that responds selectively to analytes.Fluorescent and luminescent sensors
In elegant work, Steed and co-workers have used the symmetric urea substituted ridged receptor 251 for sensing chloride.150 The free receptor in CHCl3/DMSO solution (95/5) solution adopts a twisted conformation which upon binding to one equivalent of chloride, adopts a planar conformation. The formation of the planar complex switches-on fluorescence with an emission maximum at 395 nm (λex = 340 nm). The most significant response was obtained with chloride over the other anions tested (Br−, I−, F−, NO3−, AcO−, H2PO4− and HSO4−) due to the complementarity of chloride for the receptor site formed by the two urea groups in this receptor.
Severin and co-workers have developed a method for sensing of chloride anions in aqueous solution at pH = 7.0 (100 mM MOPS buffer) by fluorescence spectroscopy using turn-on chemosensing assemblies.151 Two closely related chloride sensors were synthesised by mixing the rhodium complexes 252 and 253 together and the fluorescent dyes 254 or 255 in buffered aqueous media. The authors proposed from extensive ESI-MS and proton NMR studies in H2O/D2O 95/5 (v/v) an association of the fluorescent dye with the metal complexes via π–π interactions between the Cp* ligand and the aromatic ring system of the fluorophore producing an non-emissive ground state structure similar to that observed in the crystal structure (Fig. 33). Stability constants for a 2:1 receptor:fluorescent dye complex of 254 were calculated; log K11 = 3.53 and log K21 = 2.86 for receptor 252 and log K11 = 3.89 and log K21 = 3.20 for receptor 253. The chloride recognition in the formed sensor is achieved by replacement of bound water molecule at the rhodium centre with stability constants log Ka = 2.82 and 2.80 for free complexes 252 and 253. Fluorescence studies of a solution of 252 (500 μM) and 254 (50 μM) results in a 325% enhancement of the emission intensity at 510 nm (λex = 480 nm) upon the addition of NaCl (30 mM). In contrast, only minor spectral changes were observed upon the addition of NaF, NaNO3, Na2SO4, NaH2PO4, Na4P2O7, NaHCO3 and Na·salicylate, whereas NaAcO gives an IF/I0 value of 2. Interference problems were noted with the heavier halides Br− and I− as well as the pseudohalide CN−. Comparable experiments with 253 and 254 show an enhanced sensitivity of an II/I0 value of 6.0 due to enhanced association of the dicationic rhodium complex 253 and the fluorescent dye.
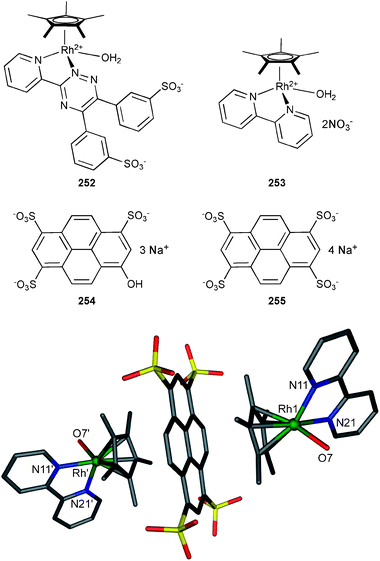 | ||
| Fig. 33 Crystal structure of 255⊂2532 with selected atom labels, hydrogen atoms and co-crystallised water omitted for clarity. | ||
The same group increased the sensitivity and selectivity for Cl− of their sensor design by employing a micelle-base approach.152 The fluorophore 254 was encapsulated in micelles formed by cetyltrimethylammonium hydrogensulfate in aqueous solution at pH 7 containing 252. The Cl− binding of 252 reduces the net positive charge of the Cp*Rh(III) fragment and the chloro-adduct may be considered as more amphiphilic. Subsequently the micelle/water partition constant Kp for Cl⊂(252-H2O) increases to 3700 compared to 7 for 252 allowing the complex to migrate into the micelle. Thus, quenching of 254 (λex = 360 nm, λem = 528 nm) was obtained in the presence of Cl− in a millimolar range, whereas no spectral change was observed upon the addition of AcO−, H2PO4−, HP2O73−, NO3−, SO42− and HCO3−.
Reversible sensing of chloride has also been the focus of recent work by Smith and co-workers, through the synthesis of a squaraine based rotaxane 256.153 The interaction of the macrocycle with the squaraine dye in 256 results in a threefold decrease in fluorescence. Upon the addition of n-Bu4NCl to 256 in CHCl3 the thread is displaced from its original position and the fluorescence returns at 655 nm (λex = 365 nm). Detailed 1H NMR studies suggest a Cl− triggered translocation of the macrocycle from the central squaraine portion of the thread to the triazole functionalities (Scheme 2). This is reversed when the chloride is precipitated out of solution with a source of sodium ions. Fluorescence titration studies showed a stability constant of 340 M−1 for the 1:1 chloride rotaxane complex in CHCl3. This is considerably lower than that of the free macrocycle (Ka > 105 M−1 in CH2Cl2) and reflects the energy cost of translocation of the macrocycle away from the squaraine station in order to bind the anion. To investigate the practical potential of this system the rotaxane was immobilised on dipsticks that were shown to be able to reversibly detect chloride fluorometrically and colorimetrically in aqueous solution.
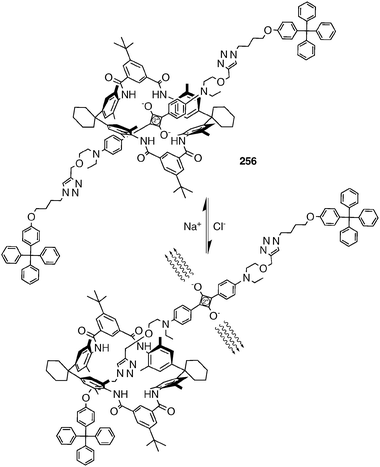 | ||
| Scheme 2 Chloride triggered translocation in the rotaxane 256. | ||
Matthews and co-workers have reported the synthesis of the new pyrenyl-appended triazole-based calix[4]arenes 257 and 259154 and compared them to the known fluorescent sensor 258.155 The fluorescent properties of these receptors were studied in CH3CN with anions added as their tetrabutylammonium salts. The receptors were excited at 343 nm with receptor 257 displaying monomer and excimer bands at 395 and 475 nm respectively. However the fluorescence spectrum of receptors 258 and 259 showed the monomer band at 395 nm only, while 259 had the weakest emission due to photo-induced electron transfer (PET) caused by the addition of the pendant amide group. Compound 257 was found to exhibit fluorescent selectivity for iodide, with the monomer emission of the pyrene moiety increasing as the excimer emission decreases. This ratiometric change was due to the encapsulation of iodide in the receptors cavity, causing a conformational change resulting in the two pyrene subunits moving apart from one another. Upon addition of over 200 equivalents of iodide both signals from the monomer and excimer were quenched due to heavy atom effect imparted by I−. A stability constant of 979 M−1 was determined for the 1:1 complex of receptor 257 with iodide.
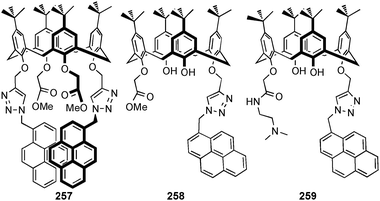
Lee and co-workers have synthesised calix[1]pyreno-[3]pyrrole 260, that uses fluoride induced conformational change to become a sensitive fluorescence sensor with a preferential affinity for C70 in toluene/acetonitrile (5%) solution.156 The [F⊂260]− complex adopts a cone-like conformation that enable binding of electron deficient spherical guests. The binding affinity for fullerenes was found to increase with the addition of fluoride with the greatest affinity for C60 and C70 observed when three equivalents of fluoride were present. Minor fluorescence quenching was noted in the absence of fluoride.
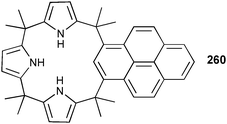
Deprotonation of the lactam group in the diketopyrrolopyrrole based receptor 261 was employed by Qu, Hua and Tian to produce a colorimetric and ratiometric red fluorescent sensor for fluoride.157 Comparable UV-Vis absorption (λ = 496 and 594 nm) and fluorescent (λex = 435 nm, λem = 563 nm) experiments in CH2Cl2 reveal a high fluoride selectivity over chloride, bromide and iodide as their tetrabutylammonium salts.
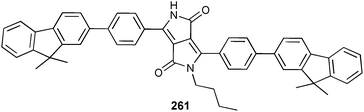
Gunnlaugsson and Elmes have synthesised a novel naphthalimide-ruthenium(II)-polypyridyl complex 262 for sensing fluoride via a change in luminescence caused by the modulation of the metal to ligand charge transfer (MLCT) emission.158 The presence of tetrabutylammonium fluoride in CH3CN could be detected by the naked eye as a colour change from yellow to red. Initial hydrogen bonding interactions of the anion with 262, using the amine residue as the hydrogen bond donor, followed by deprotonation results in the observed spectral changes. Only minor changes were observed in the UV-Vis spectra in the presence of AcO−, Cl− and HSO4−. Fluorescence studies (λex = 435 nm) in CH3CN show a quenching of the emission at 615 nm upon the addition of F− (log K11 = 4.20, log K12 = 3.7). Similar fluorescence quenching was observed with AcO− (log K11 = 4.25, log K12 = 3.11), Cl− (log K11 = 2.90) and H2PO4− (log K11 = 4.52), where the addition of tetrabutylammonium hydrogensulfate did not result in a significant spectral change. Competitive experiments show further quenching of the MLCT emission if F− is added to a solution of 262 in the presence of an excess of H2PO4−, whereas the other studied anions do not result in any spectral changes.
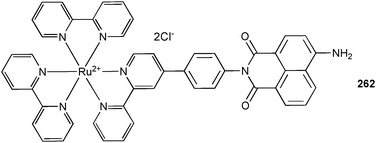
A carbazole and triarylboron containing iridium(III) complex 263 was synthesised by Huang and co-workers and has been shown to act as a ratiometric fluoride probe upon complex formation.159 The addition of up to two equivalents of F− to 263 in CH3CN results in large change in the emission maxima from λem = 584 to λem = 458 nm if excited at λex = 379 nm. The authors explained their observation with a triplet–singlet emission switch due to the coordination of the F− by the boron centres. Subsequently the triplet emission of the metal centre is quenched and only singlet emission at λem = 458 nm of the carbazole moiety was detected. Selectivity studies with anions such as Cl−, Br−, I−, ClO4−, NO3− present in 4 times excess gave no noticeable change in the emission intensity at 584 nm in CH3CN, if 263 was excited at λex = 480 nm. Minor quenching was observed in the presence of H2PO4− and AcO−, whereas F− results in significant quenching (II/I0 of 0.1).
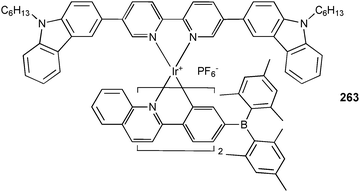
The spectroscopic response of tri(indolyl)methane receptors 264 and 265 upon addition of anions was investigated by Shoa and co-workers.160 Fluorescence studies (λex = 283 nm) in CH3CN of 264 reveal no significant change in the emission intensity at 356 nm upon addition of Cl−, Br−, HSO4− and ClO4−, added as their tetrabutylammonium salts, whereas the addition of tetrabutylammonium fluoride, acetate or dihydrogen phosphate, results in quenching of the fluorescence emission. The authors explain their observation as being due to the loss of planarity of the receptors upon complex formation with the latter anions. Titration experiments were employed to determine stability constants Ka of 3.39 × 105 M−1 for F−, 2.22 × 105 M−1 for AcO− and 1.66 × 105 M−1 for H2PO4− for a 1:1 binding mode. Comparable experiments with 265 reveal no significant interactions with any of the anions examined in this study. The authors suggest that in 265 the location of the link in the 3-position may prevent the formation a convergent hydrogen bonding array.
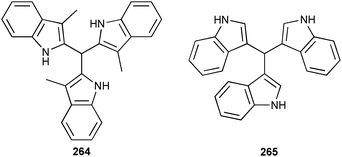
Steed and co-workers have utilized the quinoline group in the tripodal receptor 266 for detection of anions. The strongest fluorescence quenching in CH3CN at λem = 408 nm (λex = 317 nm) was observed for AcO− over Br−, Cl−, I− and NO3−, presence as their tetrabutylammonium salts. Extensive NMR studies in CD3CN reveal the formation of 1:1 and 2:1 (L:A) as well as the presence of host dimer (log K = 1.57) in solution. The obtained stability constants are summarised in Table 14.
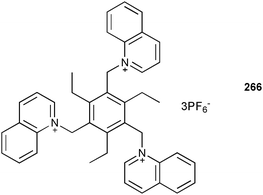
Naito and co-workers have employed a series of helical fluorinated poly(dialkylsilanes) (267–269) as fluorescent sensors for monovalent anions.161 The observed quenching of the emission maxima between 330 and 340 nm for 267–269 (λex = 300 nm) in THF decreases from F− > NO3−, Br− > Cl− > HSO4− > PF6− > I3− and is in correlation to the surface charge density of this anions (surface charge density (mCm−3) F−: −389, Cl−: −254, Br−: −227, NO3−: −207, HSO4−: −166, PF6−: −146 and I3−: −108). Furthermore, the sensitivity of these systems could be modified by changing the length of the alkyl chains. The authors explain their observations as being due to the stronger electrostatic interaction between compact anions, such as F− with the positively charged Si polymer chain in the fluorinated helical poly(dialkylsilanes). Comparable experiments with the unfluorinated poly(dialkylsilane) 270 show a minor quenching of the fluorescence in the presence of F− only. Due to the non-ionic character of the Si main chain in the helix no electrostatic interactions are present between the polymer 270 and monovalent anions.
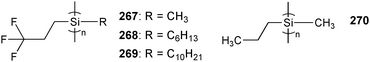
The sensing of phosphate nucleotides in the tweezer-like ferrocene linked pyrene functionalised Zn(II)-1,4,7-triazacyclononane (TACN) complexes 271 has been reported by Bond and co-workers.162 The binding of pyrophosphate, di- and triphosphate nucleotides between the two Zn(II) centres promotes a rearrangement of the pendant arms to a cis-orientation and results in an off-on fluorescence due to an enhanced excimer emission at 475 nm in 1:9 acetonitrile:tris/HCl buffer (pH 7.4, 10 mM) (Scheme 3). A summary of the obtained stability constants if given in Table 15. Phosphorous NMR studies in 1:9 CD3CN:D2O (tris/HCl buffer pD = 7.6, 10 mM, 293 K) with pyrophosphate (PPi) and adenosine tri-, di- and mono-phosphate (ATP, ADP and AMP) reveals the formation of 1:1 complexes for all anions in 271 and supports the formation of the proposed host–guest complex.
![Proposed mode of polyphosphate anion (XTP) binding by [271-4(ClO4)].](/image/article/2012/CS/c1cs15257b/c1cs15257b-s3.gif) | ||
| Scheme 3 Proposed mode of polyphosphate anion (XTP) binding by [271-4(ClO4)]. | ||
| Anion | K a [M−1] | Anion | K a [M−1] |
|---|---|---|---|
| a Pyrophosphate. b Adenosine tri-, di- and mono-phosphate (ATP, ADP and AMP). c Cytidine tri-, di- and mono-phosphate (CTP, CDP and CMP). d Guanosine tri-, di- and mono-phosphate (ATP, ADP and AMP), e Thymine tri-, di- and mono-phosphate (ATP, ADP and AMP). f Not determined due to small change in fluorescence emission. | |||
| PPia | (4.45±0.41) × 106 | TDPe | (2.03±0.18) × 104 |
| ATPb | (9.31±0.84) × 104 | AMPb | —f |
| CTPc | (2.32±0.19) × 105 | CMPc | —f |
| GTPd | (2.13±0.29) × 105 | GMPd | —f |
| TTPe | (5.05±0.46) × 105 | TMPe | —f |
| ADPb | (9.63±0.93) × 103 | H2PO4− | —f |
| CDPc | (1.55±0.14) × 104 | F− | —f |
| GDPd | (1.41±0.12) × 104 | AcO− | —f |
Ghosh and co-workers have used the pyridinium amide-urea conjugate anthracene appended receptor 272 to sense the presence of the L-N-acetylvaline and L-N-acetylalanine as their tetrabutylammonium salts.163 Fluorescence studies of 272 in CH3CN (λex = 370 nm) reveal a moderate quenching of the emission at 415 nm upon the addition of L-N-acetylproline, (S)-mandelate, pyruvate and L-N-acetylphenylglycine, whereas L-N-acetylalanine and L-N-acetylvaline result in a broad emission observed at 492 nm of moderate intensity. Comparable experiments with amide-amide analogue 273 do not show the broad emission band at 492 nm highlighting the significance of the electron deficient urea motif in 272. Titration experiments reveal selective binding of L-N-acetylvaline (Ka = 1.87 × 104 M−1) over L-N-acetylalanine (Ka = 2.60 × 103 M−1), acetate (Ka = 2.20 × 103 M−1), (S)-mandelate (Ka = 1.60 × 103 M−1), L-N-acetylproline (Ka = 1.38 × 103 M−1) and L-N-acetylphenylglycine (Ka = 1.38 × 103 M−1) in CH3CN in a 1:1 binding mode.
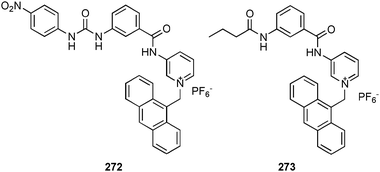
Wang and co-workers have employed the red luminescent Eu(III) complex 274 for the detection of F− and AcO− in DMSO solution.164 Hydrogen bonding between the imidazole moiety and F− (one equivalent, added as tetrabutylammonium salt) results in a change from red Eu(III) to a green emission (λex = 500 nm), where the presence of tetrabutylammonium acetate gave rise to yellow-red to green emission change. Comparable experiments with HSO4− show minor spectral changes, whereas Cl−, Br− and I−, added as their tetrabutylammonium salts, show no indication for hydrogen bonds towards 274. Further the authors manufactured transparent poly-methyl methacrylate (PMMA) films containing 0.5 mg of 274 in 5 mg MMA. Recognition tests with F− and AcO− in DMSO reveal a slow response of the film upon presence of F− only presumably due to the anion's higher basicity.
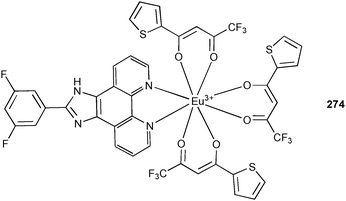
An electro-generated chemiluminescent (ECL) sensor based upon a BODIPY group in 275 that exhibits pyrophosphate selectivity has been reported by Hong and co-workers.165 The ECL emission maxima at 540 nm was quenched upon the coordination of the pyrophosphate by the dipicolylamine zinc(II) moiety in 275. Competitive assays in CH3CN (10 μM of 275, 10 mM tri-n-propylamine, 0.1 M n-Bu4NPF6) with a 10-fold excess of I−, Cl−, F−, NO3−, ClO4− and AcO− show a slight change in the ECL intensity only, which significantly decreases if pyrophosphate (10 μM) was added to each solution. Comparable experiments show the same pronounced response for pyrophosphate in the presence of 10 μM of H2PO4−, ATP, ADP or AMP.
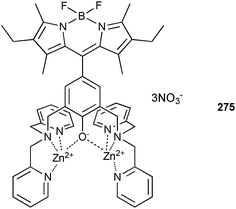
Smith and co-workers have employed the squaraine rotaxane 278 formed by the mono-oxidised macrocycle 276 and the squaraine thread 277 as chemiluminescent dye for effective imaging in tissue.166 The temperature triggered reduction of the 9,10-anthracene endoperoxide moiety in 278 results in singlet oxygen release, which initiates a near infrared emission of the squaraine group at 733 nm. This light emission is used as an efficient in vivo imaging method in mice with a high target to background ratio at tissue depth of more than 2 cm. Radiation with red light in the presence of air regenerates the oxidised rotaxane 278, which can be stored indefinitely at −20 °C.
Anzenbacher and co-workers have developed a simple sensor array capable of recognising ion pairs in water employing weak interactions of six pH indicators (279–285).167 Only one of these indicators (284) has a hydrogen bonding thiourea group. The indicators were doped into a polyether-urethane matrix 285 which contained amide groups that interact with anions and an oligo-ethylene glycol which interacts with the cations. Multiple studies were carried out with combinations of five different cations namely lithium, sodium, potassium, ammonium and tetrabutylammonium and seven different anions fluoride, chloride, iodide, bromide, acetate, nitrate and dihydrogen phosphate at five different pH levels (5, 6, 7, 8, and 9). The responses of the different pH indicator displacement events upon the addition of the different ion pairs were reported using a UV-excitation scanner with four different emission filters. These simple chemosensors were shown to be capable of sensing ion pairs over this wide pH range with a discrimination capacity and recognition efficiency of (6:35) and an accuracy of ≥93%, this is normally observed with very complex sensors.
Colorimetric sensors
A dinuclear copper(II) bromide complex of 73 synthesised by Hossain and co-workers gives a selective response to iodide (23900, 125 M−1) over fluoride (2270, 20 M−1), chloride (3200, 25 M−1) and bromide (no binding event observed) in 1:3 water:acetonitrile and water solutions buffered to pH 7.168 The stability constants were calculated from UV-Vis titration experiments with the anions added as tetrabutylammonium salts (and bound in 1:1 stoichiometry). This macrocyclic receptor forms a bowl shape with metal cations at either end of the cavity separated by 7.101(4) Å in the solid state. The introduction of iodide anions to a solution of the receptor is thought to result in the replacement of groups previously linked to the copper(II) centres. This could be responsible for the colour change from blue to green and a unique λmax absorption value.
Saha and Guha have synthesised two receptors based on the π-electron deficient naphthalene diimide (NDI) group that interacts with the anion through anion–π interactions.169 This results in charge/electron transfer events that trigger a two-step optical response towards fluoride with receptors 286 and 287. The simple NDI receptor 286 was tested with a wide variety of anions added as their tetrabutylammonium salts in a variety of solutions (DMSO, DMF, DMAc, CH3CN, (CH3)2CO and THF) containing up to 15% water. The first colour change exhibited was from colourless to orange which occurred when there were ≤5 equivalents of fluoride present. This colour change was triggered by the first electron transfer event between the anion and NDI forming the NDI radical anion. When the quantity of fluoride was increased the solutions turned pink. This was due to the reduction of the NDI radical anion to NDI2− by the fluoride anion. No other colour changes were noted with any other anion combinations even with up to 30 equivalents of the anions added. In order to test these compounds for any potential applications the more selective pincer scaffold of receptor 287 was exposed to anticavity toothpaste containing 0.24% w/v of NaF and fluoride free toothpaste in DMSO/water. A colour change was noted from colourless to a light orange with the former toothpaste but not with the fluoride free toothpaste.
Xie and co-workers have produced two pyrrole-hemiquinone based sensors for the fluoride anions through a colorimetric response via deprotonation of the receptor (an internal charge transfer process).170 Upon the addition of forty equivalents of tetrabutylammonium fluoride to a DMSO solution of 288 a colour change was observed from dark orange (496 nm) to blue (568 nm); with 289 showing a colour change from wine red (459 and 571 nm) to grey (740 nm). With > 40 equivalents of anion a further colour change is noted to green (624 nm), this is due to a second deprotonation process. This doubly deprotonated species did not form upon addition of other basic anions such as cyanide, acetate and dihydrogen phosphate allowing differentiation between the anions. The double deprotonation is possible because of the increased acidity of the NH groups of 289 due to the additional hemiquinone electron withdrawing group.
Lees and co-workers have reported a bis thiourea-based sensor 290 that gives a colorimetric response with cyanide.171 The red orange colour which is observed upon the addition of the cyanide to a DMSO solution of the receptor is due to the intramolecular charge transfer transition from the deprotonated thiourea nitrogen to the para-nitrophenyl substituent. UV-Vis and naked eye experiments with a variety of tetrabutylammonium salts showed a selective response to cyanide over other basic anions such as fluoride, acetate, and benzoate and dihydrogen phosphate although some colour changes were noted upon the addition of these anions.
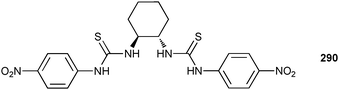
Urea based polymers for anion sensing have been prepared by Kakuchi and co-workers.172 The affinity of polymer 291 for a variety of anions was tested in THF via UV-Vis spectroscopy, using the tetrabutylammonium salts of the anions studied. Selectivity was shown for acetate (11500 M−1), benzoate (11700 M−1), azide (12300 M−1) and hydrogen sulfate (11500 M−1) over nitrate (4900 M−1), chloride (9100 M−1), bromide (5300 M−1), and fluoride (6500 M−1), with stability constants calculated using the Hill equation. The addition of anions such as acetate, benzoate, fluoride, chloride triggered a colour change from yellow through orange to red was observed depending on the anion added. This colour change was found to be a direct result of complexation between the anion and receptor and was verified by 1 H NMR experiments.
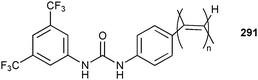
Taylor and co-workers have synthesised a variety of N,N′-diarylsquaramides and investigated the colorimetric response to anionic species of the p-nitrophenyl derivative 292.173 UV-Vis studies of the free receptor show a red shift in the absorption maxima by about 100 nm from approximately 380 nm (yellow) in CH3CN to about 480 nm (red) in DMSO. The authors explain this observation with an ionization of free receptor and the formation of deprotonated species [292-H]− due to the different solvent polarity. Titration experiments with n-Bu4NF in DMSO result in second deprotonation and subsequent colour change to blue, whereas no spectral change was observed upon the addition of AcO− and H2PO4−. Comparable experiments in CH3CN show a similar colour change from yellow to red in the presence of AcO− and H2PO4− due to deprotonation of 292. Surprisingly titrations with even weakly basic anions such as p-toluenesulfonate results in the same colour change in CH3CN due to the high acidity of the NH groups in 292. However, detailed UV-Vis and 1H NMR investigations with p-toluenesulfonate (added as the tetrabutylammonium salt) in DMSO are evidence for a an unique re-protonation process of the receptor in presence of an anion excess (10-30 equivalent). The observed UV-Vis absorption shifts back to yellow (395 nm) and points at presence of the free receptor 292. Additional evidence for this re-protonation process are the appearance of sharp proton signals at about 10.6 ppm, which is very broad for free 292.
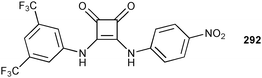
Continuing their work on anion binding biindole scaffolds Jeong and co-workers have produced receptor 293 capable of acting as a colorimetric anion sensor for the more basic anions such as acetate, dihydrogen phosphate, fluoride and cyanide.174 Addition of these anions causes a colour change from yellow to orange/red due to complex formation or the deprotonation of the receptor in a 10% DMSO/CH3CN solution with anions added as the tetrabutylammonium salts. The addition of two further hydrogen bonding groups creates receptor 294 capable of binding anions strongly with a stability constant of 2 × 105 M−1 for fluoride binding in 1:1 stoichiometry determined via UV-Vis spectroscopy. Receptor 293 exhibits deprotonation under the same conditions.
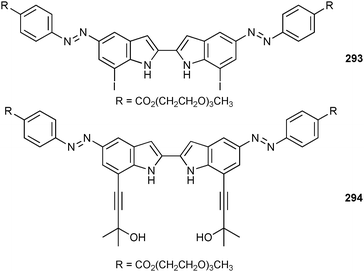
Maeda and co-workers continued their work with dipyrrolyldiketone boron based anion receptors 295–306 by tuning the electronic and optical properties with modifications of the substitution pattern on the pyrrole backbone.175 Anion complexation was explored by 1H NMR titration techniques in CD2Cl2 with the anions added as their tetrabutylammonium salts and confirmed a conformational change of the receptor to enable anion interactions through both pyrrole NH groups and the central CH hydrogen bond donor. The same binding pattern was observed in the solid phase (Fig. 34) with the pyrrole NH groups inverted compared to the free receptor. A combination of various observations showed that the substituents modulate the electronic, optical and fluorescent efficiencies as well as the solid state X-ray crystal structures. The UV-Vis titration data revealed that acetate is selectively bound over dihydrogen phosphate, chloride, bromide and hydrogen sulfate by all twelve receptors due to the higher basicity of this anion. Both the fluorescence and UV-Vis spectra of these receptors are perturbed upon the addition of anions. It was also shown that the singlet oxygen generation behaviour of these boron complexes was tuneable by anions. The combination of these properties is useful for both sensing anions and for agents to be used in photodynamic therapy.
![Crystal structure of 299 (left) and [Br⊂299]+ (right) with selected atom labels, hydrogen bonds represented as dotted lines, tetrabutylammonium cation and non-interacting hydrogen atoms in [Br⊂299]+ omitted for clarity.](/image/article/2012/CS/c1cs15257b/c1cs15257b-f34.gif) | ||
| Fig. 34 Crystal structure of 299 (left) and [Br⊂299]+ (right) with selected atom labels, hydrogen bonds represented as dotted lines, tetrabutylammonium cation and non-interacting hydrogen atoms in [Br⊂299]+ omitted for clarity. | ||
Shores and co-workers utilised the anion triggered spin state switching of a series of heteroleptic iron(II) 2,2′-bi-1,4,5,6-tetrahydropyrimidine complexes such as 307 for the detection of Br−.176 Electronic absorption spectroscopy of 307 in CH2Cl2 with tetrabutylammonium bromide results in a distinct colour change from indigo to teal. Titration experiments give stability constants of log K11 = 6.2 and log K12 = 5.6, while quantitative experiments reveal an order of affinity of Br− > Cl− > I− > NO3− > ClO4− as observed for the previously reported for the symmetric homoleptic complex cation 308.177
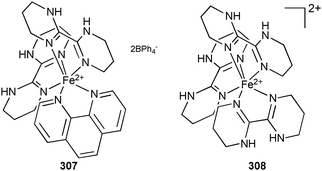
Redox sensors
A number of new redox active sensors for anions have been reported in 2010. Sessler and co-workers employed this strikingly in the tetrathiafulvalene based receptor 309, which also possesses an optical response due to the diindolylquinoxaline moiety.178 Both, UV-Vis and cyclic voltammometric studies show selective detection of H2PO4−. A summary of the obtained stability constants for a 1:1 binding mode in CH2Cl2 and ΔE of the first redox potential of the receptor are shown in Table 16. As expected results the addition of tetrabutylammonium dihydrogen phosphate in a cationic shift of 112 mV in the first oxidation wave ascribed to the tetrathiafulvalene unit. Comparable experiments with Cl−, PhCOO− and HSO4− produce only minor changes in E1/21, while the addition of F− caused a broadening and splitting in the cyclic voltammogram features. Extensive NMR studies in CD2Cl2 and DMSO-d6 are evidence for a deprotonation of 309 upon the addition of F−.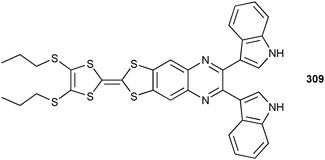 :1 binding mode obtained by UV-Vis titrations and ΔE of the first redox potential of 309·X (X = anion) in CH2Cl2C at 298 K
:1 binding mode obtained by UV-Vis titrations and ΔE of the first redox potential of 309·X (X = anion) in CH2Cl2C at 298 K
| Anion | K a [M−1] | ΔE1b [mV] |
|---|---|---|
| a Errors < ±10%. Anions used as their tetrabutylammonium salts. b ΔE1 calculated again Ag/AgCl in the presence of 2 equivalent of anion and 0.3 M n-Bu4NPF6. c Value after suspected deprotonation. | ||
| F− | 3.6 × 103 | −278c |
| H2PO4− | 6.5 × 103 | −112 |
| PhCOO− | 9.5 × 102 | −15 |
| HSO4− | 3.7 × 102 | +19 |
| Cl− | 1.1 × 103 | 0 |
Bachas and co-workers have employed the known triazolophane 310 for the selective potentiometric detection of halides in an ion-selective electrode.179 Incorporation of 310 into a poly(vinyl chloride) based membrane (2% m/m of 310 in a membrane based on 60% m/m tridodecylmethylammonium chloride plasticised with 2-nitrophenyl octyl ether) afforded an electrode with a near-Nernstian response of −54.6 mV/decade with chloride detection limit of 5.6 × 10−6 M, whereas bromide is detected at a concentration of 8.5 × 10−6 M with a near-Nernstian response of −57.1 mV/decade.
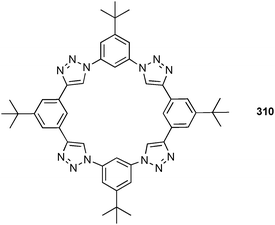
Conclusions
In 2010 we have seen advances in a number of areas related to the supramolecular chemistry of anionic species. Research in the transmembrane transport of anionic species has demonstrated that very simple receptors can function as effective carriers. Halogen bonding is becoming increasingly important as a new non-covalent interaction with which to form complexes with halide anions and new systems containing triazole groups have extended this chemistry into new molecular architectures including systems switchable by light. We can anticipate further exciting developments in the coming years.Acknowledgements
PAG thanks the EPSRC/NSF and the EU for funding.References
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- P. A. Gale, Chem. Soc. Rev., 2010, 39, 3746–3771 RSC.
- P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC.
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CAS.
- P. A. Gale, S. E. Garcia-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- P. A. Gale and R. Quesada, Coord. Chem. Rev., 2006, 250, 3219–3244 CrossRef CAS.
- A. J. Baer, B. D. Koivisto, A. P. Côté, N. J. Taylor, G. S. Hanan, H. Nierengarten and A. V. Dorsselaer, Inorg. Chem., 2002, 41, 4987–4989 CrossRef CAS.
- S. L. Jain, P. Bhattacharyya, H. L. Milton, A. M. Z. Slawin, J. A. Crayston and J. D. Woollins, Dalton Trans., 2004, 862–871 RSC.
- A. Dorazco-Gonzalez, H. Höpfl, F. Medrano and A. K. Yatsimirsky, J. Org. Chem., 2010, 75, 2259–2273 CrossRef CAS.
- Y. Xia, B. Wu, S. Li, Z. Yang, Y. Liu and X.-J. Yang, Supramol. Chem., 2010, 22, 318–324 CrossRef CAS.
- D. E. Gross, V. Mikkilineni, V. M. Lynch and J. L. Sessler, Supramol. Chem., 2010, 22, 135–141 CrossRef CAS.
- M. Arunachalam and P. Ghosh, Org. Lett., 2010, 12, 328–331 CrossRef CAS.
- M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943–951 CrossRef CAS.
- I. Ravikumar, P. S. Lakshminarayanan and P. Ghosh, Inorg. Chim. Acta, 2010, 363, 2886–2895 CrossRef CAS.
- G. Ozturk, M. Colak and M. Togrul, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 49–54 CrossRef CAS.
- M. Arunachalam and P. Ghosh, Chem. Commun., 2009, 5389–5391 RSC.
- P. S. Lakshminarayanan, E. Suresh and P. Ghosh, Inorg. Chem., 2006, 45, 4372–4380 CrossRef CAS.
- G. Ozturk, B. Gumgum, M. Kizil and S. Emen, Synth. Commun., 2007, 37, 3981–3988 CrossRef.
- S. O. Kang, V. W. Day and K. Bowman-James, J. Org. Chem., 2010, 75, 277–283 CrossRef CAS.
- M. A. Hossain, S. O. Kang, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2003, 42, 5043–5045 CrossRef CAS.
- S. O. Kang, V. W. Day and K. Bowman-James, Inorg. Chem., 2010, 49, 8629–8636 CrossRef CAS.
- J. Eckelmann, V. Saggiomo, F. D. Sonnichsen and U. Luning, New J. Chem., 2010, 34, 1247–1250 RSC.
- S. Colombo, C. Coluccini, M. Caricato, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2010, 66, 4206–4211 CrossRef CAS.
- L. Molina, E. Moreno-Clavijo, A. J. Moreno-Vargas, A. T. Carmona and I. Robina, Eur. J. Org. Chem., 2010, 4049–4055 CrossRef CAS.
- T. Fiehn, R. Goddard, R. W. Seidel and S. Kubik, Chem. Eur. J., 2010, 16, 7241–7255 CAS.
- A. J. McConnell, C. J. Serpell, A. L. Thompson, D. R. Allan and P. D. Beer, Chem.–Eur. J., 2010, 16, 1256–1264 CrossRef CAS.
- H.-B. Wang, J. A. Wisner and M. C. Jennings, Beilstein J. Org. Chem., 2010, 6, 50 Search PubMed.
- C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC.
- P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Chem. Commun., 2007, 5214–5216 RSC.
- I. Ravikumar and P. Ghosh, Chem. Commun., 2010, 46, 1082–1084 RSC.
- I. Ravikumar and P. Ghosh, Chem. Commun., 2010, 46, 6741–6743 RSC.
- R. Custelcean, A. Bock and B. A. Moyer, J. Am. Chem. Soc., 2010, 132, 7177–7185 CrossRef CAS.
- R. Custelcean, P. Remy, P. V. Bonnesen, D.-e. Jiang and B. A. Moyer, Angew. Chem., Int. Ed., 2008, 47, 1866–1870 CrossRef CAS.
- C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, C. Giorgi and B. Valtancoli, Inorg. Chem., 2009, 48, 2391–2398 CrossRef CAS.
- C. Pérez-Casas, H. Höpfl and A. Yatsimirsky, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 387–398-398 CrossRef.
- K. Raatikainen, N. K. Beyeh and K. Rissanen, Chem.–Eur. J., 2010, 16, 14554–14564 CrossRef CAS.
- A. Sirikulkajorn, T. Tuntulani, V. Ruangpornvisuti, B. Tomapatanaget and A. P. Davis, Tetrahedron, 2010, 66, 7423–7428 CrossRef CAS.
- E. M. Boyle, T. McCabe and T. Gunnlaugsson, Supramol. Chem., 2010, 22, 586–597 CrossRef CAS.
- C. M. G. dos Santos, T. McCabe, G. W. Watson, P. E. Kruger and T. Gunnlaugsson, J. Org. Chem., 2008, 73, 9235–9244 CrossRef CAS.
- A. Ghosh, D. Jose, A. Das and B. Ganguly, J. Mol. Model., 2010, 16, 1441–1448-1448 CrossRef CAS.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem.–Eur. J., 2010, 16, 4368–4380 CrossRef CAS.
- M. Boiocchi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507–16514 CrossRef CAS.
- D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495–1500 Search PubMed.
- M. H. Al-Sayah and N. R. Branda, Thermochim. Acta, 2010, 503–504, 28–32 CrossRef CAS.
- E. G. Klauber, C. K. De, T. K. Shah and D. Seidel, J. Am. Chem. Soc., 2010, 132, 13624–13626 CrossRef CAS.
- N. Takenaka, J. Chen, B. Captain, R. S. Sarangthem and A. Chandrakumar, J. Am. Chem. Soc., 2010, 132, 4536–4537 CrossRef CAS.
- A.-S. Delépine, R. Tripier, M. Le Baccon and H. Handel, Eur. J. Org. Chem., 2010, 5380–5390 CrossRef.
- K. R. Dey, T. Horne, F. R. Fronczek and M. A. Hossain, Inorg. Chem. Commun., 2010, 13, 1515–1518 CrossRef CAS.
- J. S. Mendy, M. L. Pilate, T. Horne, V. W. Day and M. A. Hossain, Chem. Commun., 2010, 46, 6084–6086 RSC.
- M. A. Saeed, D. R. Powell, F. R. Fronczek and M. A. Hossain, Tetrahedron Lett., 2010, 51, 4233–4236 CrossRef CAS.
- M. A. Hossain, M. A. Saeed, F. R. Fronczek, B. M. Wong, K. R. Dey, J. S. Mendy and D. Gibson, Cryst. Growth Des., 2010, 10, 1478–1481 CAS.
- M. A. Saeed, J. J. Thompson, F. R. Fronczek and M. A. Hossain, CrystEngComm, 2010, 12, 674–676 RSC.
- M. A. Saeed, F. R. Fronczek, M.-J. Huang and M. A. Hossain, Chem. Commun., 2010, 46, 404–406 RSC.
- H. Wu, M. A. Saeed, H.-M. Hwang, S. Zhao, Y.-M. Liu and M. A. Hossain, J. Phys. Org. Chem., 2005, 24, 1–5 CrossRef CAS.
- A. L. Cresswell, M. O. M. Piepenbrock and J. W. Steed, Chem. Commun., 2010, 46, 2787–2789 RSC.
- H. B. Tanh Jeazet, K. Gloe, T. Doert, O. N. Kataeva, A. Jager, G. Geipel, G. Bernhard, B. Buchner and K. Gloe, Chem. Commun., 2010, 46, 2373–2375 RSC.
- H. Maeda, Top. Heterocycl. Chem., 2010, 24, 103–144 CrossRef CAS.
- P. A. Gale and C.-H. Lee, Top. Heterocycl. Chem., 2010, 24, 39–73 CrossRef CAS.
- J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef CAS.
- G. Mani, T. Guchhait, R. Kumar and S. Kumar, Org. Lett., 2010, 12, 3910–3913 CrossRef CAS.
- G. Mani, D. Jana, R. Kumar and D. Ghorai, Org. Lett., 2010, 12, 3212–3215 CrossRef CAS.
- G. Cafeo, H. M. Colquhoun, A. Cuzzola, M. Gattuso, F. H. Kohnke, L. Valenti and A. J. P. White, J. Org. Chem., 2010, 75, 6263–6266 CrossRef CAS.
- K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS.
- C. Caltagirone, N. L. Bill, D. E. Gross, M. E. Light, J. L. Sessler and P. A. Gale, Org. Biomol. Chem., 2010, 8, 96–99 CAS.
- D.-W. Yoon, D. E. Gross, V. M. Lynch, J. L. Sessler, B. P. Hay and C.-H. Lee, Angew. Chem., Int. Ed., 2008, 47, 5038–5042 CrossRef CAS.
- D. Gross, D.-W. Yoon, V. Lynch, C.-H. Lee and J. Sessler, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 81–85 CrossRef CAS.
- M. Buda, A. Iordache, C. Bucher, J.-C. Moutet, G. Royal, E. Saint-Aman and J. L. Sessler, Chem.–Eur. J., 2010, 16, 6810–6819 CrossRef CAS.
- B. A. Moyer, F. V. Sloop, C. J. Fowler, T. J. Haverlock, H.-A. Kang, L. H. Delmau, D. M. Bau, M. A. Hossain, K. Bowman-James, J. A. Shriver, N. L. Bill, D. E. Gross, M. Marquez, V. M. Lynch and J. L. Sessler, Supramol. Chem., 2010, 22, 653–671 CrossRef CAS.
- P. A. Gale, Chem. Commun., 2008, 4525–4540 RSC.
- H. Juwarker, J.-m. Suk and K.-S. Jeong, Top. Heterocycl. Chem., 2010, 24, 177–204 CrossRef CAS.
- D. Makuc, M. Albrecht and J. Plavec, Supramol. Chem., 2010, 22, 603–611 CrossRef CAS.
- P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC.
- P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem.–Asian J., 2010, 5, 555–561 CrossRef CAS.
- P. R. Edwards, J. R. Hiscock, P. A. Gale and M. E. Light, Org. Biomol. Chem., 2010, 8, 100–106 CAS.
- P. Dydio, T. Zielinski and J. Jurczak, Org. Lett., 2010, 12, 1076–1078 CrossRef CAS.
- J.-i. Kim, H. Juwarker, X. Liu, M. S. Lah and K.-S. Jeong, Chem. Commun., 2010, 46, 764–766 RSC.
- T. Wang, Y. Bai, L. Ma and X.-P. Yan, Org. Biomol. Chem., 2008, 6, 1751–1755 CAS.
- T. Wang, H.-F. Wang and X.-P. Yan, CrystEngComm, 2010, 12, 3177–3182 RSC.
- T. Wang and X.-P. Yan, Chem.–Eur. J., 2010, 16, 4639–4649 CrossRef CAS.
- Y. Zhao, Y. Li, Y. Li, C. Huang, H. Liu, S.-W. Lai, C.-M. Che and D. Zhu, Org. Biomol. Chem., 2010, 8, 3923–3927 CAS.
- K. N. Skala, K. G. Perkins, A. Ali, R. Kutlik, A. M. Summitt, S. Swamy-Mruthinti, F. A. Khan and M. Fujita, Tetrahedron Lett., 2010, 51, 6516–6520 CrossRef CAS.
- C. Rether, W. Sicking, R. Boese and C. Schmuck, Beilstein J. Org. Chem., 2010, 6, 3 CrossRef.
- C. Schmuck, Eur. J. Org. Chem., 1999, 2397–2403 CrossRef CAS.
- M. Arunachalam and P. Ghosh, CrystEngComm, 2010, 12, 1621–1627 RSC.
- M. Arunachalam and P. Ghosh, Chem. Commun., 2009, 3184–3186 RSC.
- Q.-S. Lu, J. Zhang, L. Jiang, J.-T. Hou and X.-Q. Yu, Tetrahedron Lett., 2010, 51, 4395–4399 CrossRef CAS.
- L. Yang, S. Qin, X. Su, F. Yang, J. You, C. Hu, R. Xie and J. Lan, Org. Biomol. Chem., 2010, 8, 339–348 CAS.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS.
- S. Lee, Y. Hua, H. Park and A. H. Flood, Org. Lett., 2010, 12, 2100–2102 CrossRef CAS.
- Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS.
- Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630–3633 CrossRef CAS.
- B. Schulze, C. Friebe, M. D. Hager, W. Gunther, U. Kohn, B. O. Jahn, H. Gorls and U. S. Schubert, Org. Lett., 2010, 12, 2710–2713 CrossRef CAS.
- S.-i. Kondo, Y. Kobayashi and M. Unno, Tetrahedron Lett., 2010, 51, 2512–2514 CrossRef CAS.
- C. Bresner, C. J. E. Haynes, D. A. Addy, A. E. J. Broomsgrove, P. Fitzpatrick, D. Vidovic, A. L. Thompson, I. A. Fallis and S. Aldridge, New J. Chem., 2010, 34, 1652–1659 RSC.
- I. R. Morgan, A. E. J. Broomsgrove, P. Fitzpatrick, D. Vidovic, A. L. Thompson, I. A. Fallis and S. Aldridge, Organometallics, 2010, 29, 4762–4765 CrossRef CAS.
- Y. Kim, M. Kim and F. P. Gabbai, Org. Lett., 2010, 12, 600–602 CrossRef CAS.
- D. Shanmukaraj, S. Grugeon, G. Gachot, S. Laruelle, D. Mathiron, J.-M. Tarascon and M. Armand, J. Am. Chem. Soc., 2010, 132, 3055–3062 CrossRef CAS.
- K. Raatikainen and K. Rissanen, Cryst. Growth Des., 2010, 10, 3638–3646 CAS.
- M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem. Int. Ed., 2010, 49, 1674–1677 CAS.
- E. Dimitrijevic, O. Kvak and M. S. Taylor, Chem. Commun., 2010, 46, 9025–9027 RSC.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS.
- C. J. Serpell, N. L. Kilah, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2010, 49, 5322–5326 CrossRef CAS.
- B. P. Hay and R. Custelcean, Cryst. Growth Des., 2009, 9, 2539–2545 CAS.
- A. Garcia-Raso, F. M. Albertí, J. J. Fiol, Y. Lagos, M. Torres, E. Molins, I. Mata, C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Eur. J. Org. Chem., 2010, 5171–5180 CrossRef CAS.
- M. Albrecht, M. Müller, O. Mergel, K. Rissanen and A. Valkonen, Chem.–Eur. J., 2010, 16, 5062–5069 CrossRef CAS.
- M. Müller, M. Albrecht, V. Gossen, T. Peters, A. Hoffmann, G. Raabe, A. Valkonen and K. Rissanen, Chem.–Eur. J., 2010, 16, 12446–12453 CrossRef.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202–7207 CrossRef CAS.
- P. S. Szalay, J. R. Galán-Mascarós, B. L. Schottel, J. Bacsa, L. M. Pérez, A. S. Ichimura, A. Chouai and K. R. Dunbar, J. Cluster Sci., 2004, 15, 503–530 CrossRef CAS.
- D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem.–Eur. J., 2010, 16, 13053–13057 CrossRef CAS.
- A. Galstyan, P. J. Sanz Miguel and B. Lippert, Dalton Trans., 2010, 39, 6386–6388 RSC.
- A. Galstyan, P. J. Sanz Miguel and B. Lippert, Chem. Eur. J., 2010, 16, 5577–5580 CAS.
- M. I. Ortiz, M. L. Soriano, M. P. Carranza, F. A. Jalon, J. W. Steed, K. Mereiter, A. M. Rodriguez, D. Quinonero, P. M. Deya and B. R. Manzano, Inorg. Chem., 2010, 49, 8828–8847 CrossRef CAS.
- S. K. Kim, J. L. Sessler, D. E. Gross, C.-H. Lee, J. S. Kim, V. M. Lynch, L. H. Delmau and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 5827–5836 CrossRef CAS.
- M. Ménand and I. Jabin, Chem.–Eur. J., 2010, 16, 2159–2169 CrossRef.
- M. Ménand and I. Jabin, Org. Lett., 2009, 11, 673–676 CrossRef.
- C. Capici, R. De Zorzi, C. Gargiulli, G. Gattuso, S. Geremia, A. Notti, S. Pappalardo, M. F. Parisi and F. Puntoriero, Tetrahedron, 2010, 66, 4987–4993 CrossRef CAS.
- F. P. Ballistreri, A. Pappalardo, G. A. Tomaselli, R. M. Toscano and G. T. Sfrazzetto, Eur. J. Org. Chem., 2010, 3806–3810 CrossRef CAS.
- M. E. Amato, F. P. Ballistreri, S. Gentile, A. Pappalardo, G. A. Tomaselli and R. M. Toscano, J. Org. Chem., 2010, 75, 1437–1443 CrossRef CAS.
- T. Lin, V. Gasperov, K. J. Smith, C. C. Tong and P. A. Tasker, Dalton Trans., 2010, 39, 9760–9762 RSC.
- M. Wenzel, S. R. Bruere, Q. W. Knapp, P. A. Tasker and P. G. Plieger, Dalton Trans., 2010, 39, 2936–2941 RSC.
- P. G. Plieger, S. Parsons, A. Parkin and P. A. Tasker, J. Chem. Soc., Dalton Trans., 2002, 3928–3930 RSC.
- K. Zhu, L. Wu, X. Yan, B. Zheng, M. Zhang and F. Huang, Chem. Eur. J., 2010, 16, 6088–6098 CAS.
- F. Tancini, T. Gottschalk, W. B. Schweizer, F. Diederich and E. Dalcanale, Chem.–Eur. J., 2010, 16, 7813–7819 CrossRef CAS.
- F. Li, S. Carvalho, R. Delgado, M. G. B. Drew and V. Felix, Dalton Trans., 2010, 39, 9579–9587 RSC.
- X. Huang, Z. Yang, X.-J. Yang, Q. Zhao, Y. Xia and B. Wu, Inorg. Chem. Commun., 2010, 13, 1103–1107 CrossRef CAS.
- N. N. Adarsh and P. Dastidar, Cryst. Growth Des., 2010, 10, 483–487 CAS.
- S. Banerjee, N. N. Adarsh and P. Dastidar, Eur. J. Inorg. Chem., 2010, 3770–3779 CrossRef CAS.
- B. Wu, J. Liang, Y. Zhao, M. Li, S. Li, Y. Liu, Y. Zhang and X.-J. Yang, CrystEngComm, 2010, 12, 2129–2134 RSC.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem. Int. Ed., 2010, 49, 2378–2381 CAS.
- K. T. Holman, S. D. Drake, J. W. Steed, G. W. Orr and J. L. Atwood, Supramol. Chem., 2010, 22, 870–890 CrossRef CAS.
- J. C. Rainwater and E. V. Anslyn, Chem. Commun., 2010, 46, 2904–2906 RSC.
- W. A. Harrell, M. L. Bergmeyer, P. Y. Zavalij and J. T. Davis, Chem. Commun., 2010, 46, 3950–3952 RSC.
- L. J. Siskind and M. Colombini, J. Biol. Chem., 2000, 275, 38640–38644 CrossRef CAS.
- L. W. Judd and A. P. Davis, Chem. Commun., 2010, 46, 2227–2229 RSC.
- N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and J. W. A. Harrell, Chem. Commun., 2010, 46, 6252–6254 RSC.
- P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS.
- M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 CAS.
- M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC.
- C. R. Yamnitz, S. Negin, I. A. Carasel, R. K. Winter and G. W. Gokel, Chem. Commun., 2010, 46, 2838–2840 RSC.
- B. Hill, Ionic Pores of Excitable Membranes, Mass: Sinauer Associates, Sunderland, 3 edn, 2001 Search PubMed.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533–538 CrossRef CAS.
- J. Míšek, A. Vargas Jentzsch, S.-i. Sakurai, D. Emery, J. Mareda and S. Matile, Angew. Chem., Int. Ed., 2010, 49, 7680–7683 CrossRef.
- L. Kim, A. Hamdi, A. Stancu, R. Souane, L. Mutihac and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 55–59 CrossRef CAS.
- C. Urban and C. Schmuck, Chem.–Eur. J., 2010, 16, 9502–9510 CrossRef CAS.
- S. Niebling, S. K. Srivastava, C. Herrmann, P. R. Wich, C. Schmuck and S. Schlucker, Chem. Commun., 2010, 46, 2133–2135 RSC.
- J.-W. Liu, Y. Yang, C.-F. Chen and J.-T. Ma, Langmuir, 2010, 26, 9040–9044 CrossRef CAS.
- S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170–9173 CrossRef CAS.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS.
- P. Byrne, G. O. Lloyd, L. Applegarth, K. M. Anderson, N. Clarke and J. W. Steed, New J. Chem., 2010, 34, 2261–2274 RSC.
- A. N. Swinburne, M. J. Paterson, A. Beeby and J. W. Steed, Chem.–Eur. J., 2010, 16, 2714–2718 CrossRef CAS.
- T. Riis-Johannessen, K. Schenk and K. Severin, Inorg. Chem., 2010, 49, 9546–9553 CrossRef CAS.
- T. Riis-Johannessen and K. Severin, Chem.–Eur. J., 2010, 16, 8291–8295 CrossRef CAS.
- J. J. Gassensmith, S. Matthys, J. J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem.–Eur. J., 2010, 16, 2916–2921 CrossRef CAS.
- J. S. Kim, S. Y. Park, S. H. Kim, P. Thuery, R. Souane, S. E. Matthews and J. Vicens, Bull. Korean Chem. Soc., 2010, 31, 624–629 CrossRef CAS.
- S. Y. Park, J. H. Yoon, C. S. Hong, R. Souane, J. S. Kim, S. E. Matthews and J. Vicens, J. Org. Chem., 2008, 73, 8212–8218 CrossRef CAS.
- J. Yoo, Y. Kim, S. J. Kim and C. H. Lee, Chem. Commun., 2010, 46, 5449–5451 RSC.
- Y. Qu, J. L. Hue and H. Tian, Org. Lett., 2010, 12, 3320–3323 CrossRef CAS.
- R. B. P. Elmes and T. Gunnlaugsson, Tetrahedron Lett., 2010, 51, 4082–4087 CrossRef CAS.
- W. J. Xu, S. J. Liu, X. Y. Zhao, S. Sun, S. Cheng, T. C. Ma, H. B. Sun, Q. A. Zhao and W. Huang, Chem. Eur. J., 2010, 16, 7125–7133 CAS.
- W. Wei, Y. Guo, J. A. Xu and S. J. Shao, Spectrochim. Acta, Part A, 2010, 77, 620–624 CrossRef.
- M. Naito, M. Nakamura, K. Terao, T. Kawabe and M. Fujiki, Macromolecules, 2010, 43, 7919–7923 CrossRef CAS.
- Z. H. Zeng, A. A. J. Torriero, A. M. Bond and L. Spiccia, Chem.–Eur. J., 2010, 16, 9154–9163 CrossRef CAS.
- K. Ghosh, T. Sarkar and A. P. Chattopadhyay, Beilstein J. Org. Chem., 2010, 6, 1211–1218 CrossRef CAS.
- J. T. Lin, Q. M. Wang, C. L. Tan and H. Y. Chen, Synth. Met., 2010, 160, 1780–1786 CrossRef CAS.
- I. S. Shin, S. W. Bae, H. Kim and J. I. Hong, Anal. Chem., 2010, 82, 8259–8265 CrossRef CAS.
- J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS.
- Y. L. Liu, M. A. Palacios and P. Anzenbacher, Chem. Commun., 2010, 46, 1860–1862 RSC.
- J. S. Mendy, M. A. Saeed, F. R. Fronczek, D. R. Powell and M. A. Hossain, Inorg. Chem., 2010, 49, 7223–7225 CrossRef CAS.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS.
- Q. G. Wang, Y. S. Xie, Y. B. Ding, X. Li and W. H. Zhu, Chem. Commun., 2010, 46, 3669–3671 RSC.
- M. O. Odago, D. M. Colabello and A. J. Lees, Tetrahedron, 2010, 66, 7465–7471 CrossRef CAS.
- R. Sakai, S. Okade, E. B. Barasa, R. Kakuchi, M. Ziabka, S. Umeda, K. Tsuda, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7406–7411 CrossRef CAS.
- A. Rostami, A. Colin, X. Y. Li, M. G. Chudzinski, A. J. Lough and M. S. Taylor, J. Org. Chem., 2010, 75, 3983–3992 CrossRef CAS.
- G. W. Lee, N. K. Kim and K. S. Jeong, Org. Lett., 2010, 12, 2634–2637 CrossRef CAS.
- H. Maeda, M. Takayama, K. Kobayashi and H. Shinmori, Org. Biomol. Chem., 2010, 8, 4308–4315 CAS.
- Z. P. Ni, A. M. McDaniel and M. P. Shores, Chem. Sci., 2010, 1, 615–621 RSC.
- Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS.
- C. Bejger, J. S. Park, E. S. Silver and J. L. Sessler, Chem. Commun., 2010, 46, 7745–7747 RSC.
- E. M. Zahran, Y. R. Hua, Y. J. Li, A. H. Flood and L. G. Bachas, Anal. Chem., 2010, 82, 368–375 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |