Structure, energy, vibrational spectrum, and Bader's analysis of π⋯H hydrogen bonds and H−δ⋯H+δ dihydrogen bonds
Received 28th May 2012, Accepted 5th September 2012
First published on 5th September 2012
Abstract
In this paper, the intermolecular structural study asserted by the vibrational analysis in the stretch frequencies of hydrogen bonds (π⋯H) and dihydrogen bonds (H−δ⋯H+δ) have definitively been revisited by means of calculations carried out by Density Functional Theory (DFT) and topological parameters derived from the classic treatise of the Quantum Theory of Atoms in Molecules (QTAIM). As a matter of fact the π⋯H hydrogen bond is formed between the hydrofluoric acid and the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of the acetylene, but the QTAIM calculations revealed a distortion in this interaction due to the formation of the ternary complex C2H2⋯2(HF). Although the π bonds of ethylene (C2H4), propylene (C2H3(CH3)), and t-butylene (C2H2(CH3)2) are considered proton acceptors, two hydrogen-bond types—π⋯H and C⋯H—can be observed. Over and above the analysis of the π hydrogen bonds, theoretical arguments also were used to discuss the red-shifts in the stretch frequencies of the binary dihydrogen complexes formed by BeH2⋯HX with X = F, Cl, CN, and CCH. Although a vibrational blue-shift in the stretch frequency of the H–C bond of HCF3 due to the formation of the BeH2⋯HCF3 dihydrogen complex was obtained, unmistakable red-shifts were detected in LiH⋯HCF3, MgH2⋯HCF3, and NaH⋯HCF3. Moreover, the alkali–halogen bonds were identified in relation to the formation of the trimolecular systems NaH⋯2(HCF3) and NaH⋯2(HCCl3). At last, theoretical calculations and QTAIM molecular integrations were used to study a novel class of dihydrogen-bonded complexes (mC2H5+ ⋯nMgH2 with m = 1 or 2 and n = 1 or 2) based in the insight that MgH2 can bind with the non-localized hydrogen H+δ of the ethyl cation (C2H5+). In an overview, QTAIM calculations were applied to evaluate the molecular topography, charge density, as well as to interpret the shifted frequencies either to red or blue caused by the formation of the hydrogen bonds and dihydrogen bonds.
C bond of the acetylene, but the QTAIM calculations revealed a distortion in this interaction due to the formation of the ternary complex C2H2⋯2(HF). Although the π bonds of ethylene (C2H4), propylene (C2H3(CH3)), and t-butylene (C2H2(CH3)2) are considered proton acceptors, two hydrogen-bond types—π⋯H and C⋯H—can be observed. Over and above the analysis of the π hydrogen bonds, theoretical arguments also were used to discuss the red-shifts in the stretch frequencies of the binary dihydrogen complexes formed by BeH2⋯HX with X = F, Cl, CN, and CCH. Although a vibrational blue-shift in the stretch frequency of the H–C bond of HCF3 due to the formation of the BeH2⋯HCF3 dihydrogen complex was obtained, unmistakable red-shifts were detected in LiH⋯HCF3, MgH2⋯HCF3, and NaH⋯HCF3. Moreover, the alkali–halogen bonds were identified in relation to the formation of the trimolecular systems NaH⋯2(HCF3) and NaH⋯2(HCCl3). At last, theoretical calculations and QTAIM molecular integrations were used to study a novel class of dihydrogen-bonded complexes (mC2H5+ ⋯nMgH2 with m = 1 or 2 and n = 1 or 2) based in the insight that MgH2 can bind with the non-localized hydrogen H+δ of the ethyl cation (C2H5+). In an overview, QTAIM calculations were applied to evaluate the molecular topography, charge density, as well as to interpret the shifted frequencies either to red or blue caused by the formation of the hydrogen bonds and dihydrogen bonds.
 Boaz Galdino de Oliveira | Boaz Galdino de Oliveira studied industrial chemistry at the State University of Paraíba (1999), obtained his D.Sc in chemical physics at the Federal University of Paraíba (2005) under the guidance of Professor Regiane C.M.U. Araújo, and spent four years in postdoctoral researches at the Federal University of Pernambuco (2009). Today he is adjunct professor at the Federal University of Bahia. He has experience in theoretical chemistry, acting on the following topics: hydrogen bonds, halogen bonds, dihydrogen bonds, stacking interactions, quantum interactions, quantum theory of atoms in molecules, chemometric investigations of density functional theory and ab initio methods. |
Introduction
It is widely known that the atomic properties determine a specific condition by which the bonded contacts or non-bonded interactions exist or coexist in a molecular system.1–5 As such, it should be mentioned that the hydrogen bond is not only a feasible model of non-bonded interaction,6–13 but its properties play a crucial role in studies of chemistry, biology and physics,14–27 and thereby ascribe it as one of the most important interactions of nature. Moreover, the origin of the hydrogen bond is a singular topic and its application is mightily useful, such as for example in engineering,28 medicinal chemistry with drug design,29–31 polymers,32–35 macromolecular research,36–38 understanding of biochemical processes39–42 such as enzymatic and organometallic catalysis,43,44 synthesis,45–47 molecular recognition,48 development of raw materials to be used in industrial scale,49 and molecular modelling of biological systems aided by spectroscopy parameters.50,51 Nevertheless, some attention also has been dedicated to the applicability of hydrogen bonds in molecular modelling of cooperative systems,52–63 wherein the origin of life based on DNA structure certainly is the most remarkable, incisive, and thorough investigation focus.64–69 Notoriously all these researches provide a summary of the ‘state of the art for hydrogen bonds’,70 although it must be remembered that its evidence is the greatest goal in any investigation type.71–74 Over all these years, a lot of definitions for hydrogen bond75–83 have been documented by famous scientists, such as Lewis,84 Pauling,85 Huggins,86 Pimentel and McClellan,87 Latimer and Rodebush88 as well as Coulson and Danielson.89 In the past, many other researchers designed empirical studies in order to understand the phenomena inherent to the hydrogen bond.90–94 Although the experimental assays have helped decisively to characterize the formation of hydrogen bonds,95–97 its first experimental evidence was documented only with the discovering of the X-ray technique at the beginning of the 20th century.98,99In order to capture the chemical genesis of the intermolecular interactions, the energy decomposition projected by Kitaura, Umeyama, Morokuma and co-authors,100–104 Divide and Conquer (DC) elaborated by Merz Jr. et al.,105–109 or even the Symmetry Adapted Perturbation Theory (SAPT) developed by Szalewicz and others,110–117 are routinely applied in theoretical investigations aimed at examining the hydrogen bond through the quantification of the Coulomb energy, exchange term, dispersion forces, and charge transfer,118,119 albeit it should be cited that the valence bond theory or quantum-chemical methods are traditional and important tools to investigate the non-bonded phenomenology.120–126 In agreement with these assertions, there are three excellent study guides, namely are a book edited by Grabowski,127 a review produced by Kojić-Prodić and Molćanov,128 and a paper elaborated by Gilli and Gilli.129 These works provide a detailed overview of hydrogen bonds, mainly the relationship between a proton donor and a region containing high electron density, and in addition the ideal condition to interact one with another is also stressed.130–134 Such a condition has been crucial to define the hydrogen bonds, although, according to a IUPAC project entitled ‘categorizing hydrogen bonding and other intermolecular interactions’,135,136 the hydrogen bonds are generated by two electronegative atoms (X and Y) interacting by means of a hydrogen atom. In other words, the interaction model is symbolized by X–H⋯Y with X and Y having been established to be N, O, and F,137–139 in spite of some other elements that are also able to interact with proton donors.140,141 Undoubtedly, that the hydrogen bond represents the most traditional noncovalent interaction profile ever seen,142–148 but the property of proton donors (H+δ) for binding with charge density sites,149–151 such as lone electron pairs,152–157 π or pseudo-π bonds,158–161 surely is the guideline attributed to the elucidation of intermolecular interactions.162–175
Some time ago, the Crabtree research group176,177 discovered a remarkable intermolecular interaction type. One peculiar feature observed in this contact is the stable interaction between hydrides (H−δ) and protons (H+δ),178,179 which intriguingly bind themselves and form the dihydrogen bond H−δ⋯H+δ.180–185 This discovery led to the appearing of a new era in intermolecular systems researches with great prominence for the dihydrogen-bonded complexes, e.g., BH3NH3 dimer.186 In this insight, it has been reported that BeH2 operates well as proton receptor187 by which the formation of complexes with rare gases188 became reliable, or even in other situations such as reactions with transition metals,189,190 for instance. In a research paper, Alkorta et al.191 have declared some tenuous differences between hydrogen bonds, dihydrogen bonds, and hydride bonds.192–203 Namely, dihydrogen bonds are considered apart or even a special case of hydrogen bonds, whereas hydride bonds are formerly an opposite hydrogen bond. In order to ensure some theoretical insights about these interaction types, it seems to be fundamental that two basic features should be compulsorily obeyed in any kind of analysis:
(i) Vibrational characterization of the interaction.
(ii) Choice of an appropriate theoretical approach to be used in the spectroscopy analysis.
Thus, this current review is guided by these two features, whereby the Quantum Theory of Atoms in Molecules (QTAIM)204–210 is used to interpret accurately the red- and blue-shifts on the stretch frequencies of the proton donors upon the formation of intermolecular interactions formed by lone pairs of electrons211–214 or π clouds as proton acceptors,215 dihydrogen bonds,216 and other similar interactions.217–223 In this view, once again it is to be hoped that all the concepts and conclusions brought together here can be useful for further studies not only of the aforementioned intermolecular interactions, but other types should be also included.224–227
The wide chemical range of the π⋯H hydrogen bonds and H−δ⋯H+δ dihydrogen bonds
As reported by Kim and collaborators,228–230 both hydrogen bonds and dihydrogen bonds are widely studied and their properties have been clearly unveiled. Regarding to π⋯H hydrogen bonds, in particular, it is worth mentioning that the p clouds are accessible from the spectroscopy viewpoint. It is worth mentioning that the π clouds are accessible from the spectroscopy viewpoint, particularly as a result of their ability to form conjugated systems and chromospheres.231 In the case of dihydrogen bonds, the interaction between the hydride and the boron or B–H groups provides important insights into interactions in the solid state of matter.232 In a review elaborated by Singh and co-authors,233 it was demonstrated that not only classic hydrogen bonds and ionic interactions,234 but also π⋯H hydrogen bonds are relevant for the design of nanomaterials, such as those formed by fullerenes and ionospheres. The π⋯H hydrogen bonds are currently characterized in nanoscience by their weak interaction strengths, where the contribution of the dispersion energy is dominant upon the formation and stabilization of the analyzed system.235 Regarding the chemistry of materials, the dihydrogen bond function also has been established. In a recent paper, Chu et al.236 reported an alternative procedure for hydrogen storage widely useful in fuel cell technologies. Similar to dihydrogen bonds where alkaline earth elements or metals bind with hydrogen, the interaction observed was H−δ⋯Ca+2 in a series of calcium borohydride diammoniates. Furthermore, the properties of alkali-metal aminoborane have been also effectively used for hydrogen storage in a proposal called ‘hydrogen economy’, but with a typical dihydrogen bond between LiH−δ237,238 and H+δNH2BH3.239π⋯H hydrogen bonds
Since the first publications of Pauling85,240–241 until some more recent works,242,243 the electronegativity is still being considered a reliant parameter to investigate the formation of atomic interactions.244 In more recent years, elements with higher electronegativity than hydrogen have been used in the proton acceptor framework.245,246 In addition to the three atoms N, O, and F,247 recent theoretical studies have shown that some regions containing high levels of electron density are potent proton acceptors248 such as the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CC bonds. As is widely known, hydrogen-bonded complexes formed by π bonds and proton donors are thus stabilized by the formation of π⋯H hydrogen bonds.249–257 However, this statement was not entirely accepted at first, and thereby various works were elaborated in order to discover whether individual atoms or π-electrons can be really considered protons acceptors. In this scenery the neutron diffraction analysis has shown that π⋯H interactions are aligned directly at the midpoint of the π bond.258 Indeed, this phenomenon has also been studied theoretically by means of ab initio259–262 and Density Functional Theory (DFT) calculations263–267 and also observed through the analysis of rotational268,269 and infrared spectra,270,271 as well as by the association of experimental procedures and theoretical calculations compatible to those abovementioned.272 In the electrophilic addition reaction of hydrofluoric acid to the CC bond of the acetylene with formation of vinylfluoride (4), an important intermediary is identified as the hydrogen-bonded complex C2H2⋯HF,273 which is formed by the π charge transfer toward the σ* anti-bonding orbital of the hydrogen on the hydrofluoric acid. The hydrogen complex C2H2⋯HF (2) is characterized by a T-shape structure with C2v symmetry wherein the π⋯H interaction is linked at the midpoint of the CC bond of the acetylene,274,275 as can be seen in Fig. 1. In opposition to the conformation analysis for the hydrogen bond symmetry proposed by Perrin,276 note that the main focus here is dedicated just to structural evidence.
C and CC bonds. As is widely known, hydrogen-bonded complexes formed by π bonds and proton donors are thus stabilized by the formation of π⋯H hydrogen bonds.249–257 However, this statement was not entirely accepted at first, and thereby various works were elaborated in order to discover whether individual atoms or π-electrons can be really considered protons acceptors. In this scenery the neutron diffraction analysis has shown that π⋯H interactions are aligned directly at the midpoint of the π bond.258 Indeed, this phenomenon has also been studied theoretically by means of ab initio259–262 and Density Functional Theory (DFT) calculations263–267 and also observed through the analysis of rotational268,269 and infrared spectra,270,271 as well as by the association of experimental procedures and theoretical calculations compatible to those abovementioned.272 In the electrophilic addition reaction of hydrofluoric acid to the CC bond of the acetylene with formation of vinylfluoride (4), an important intermediary is identified as the hydrogen-bonded complex C2H2⋯HF,273 which is formed by the π charge transfer toward the σ* anti-bonding orbital of the hydrogen on the hydrofluoric acid. The hydrogen complex C2H2⋯HF (2) is characterized by a T-shape structure with C2v symmetry wherein the π⋯H interaction is linked at the midpoint of the CC bond of the acetylene,274,275 as can be seen in Fig. 1. In opposition to the conformation analysis for the hydrogen bond symmetry proposed by Perrin,276 note that the main focus here is dedicated just to structural evidence.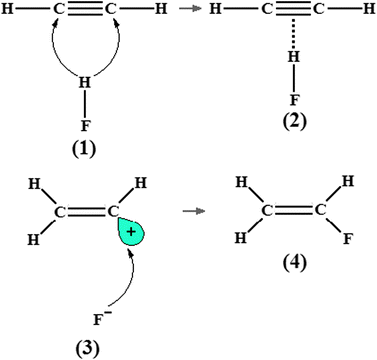 |
| | Fig. 1 Electrophilic addition reaction of one hydrofluoric acid to the CC bond of the acetylene and the formation of the C2H2⋯HF hydrogen-bonded complex (2). | |
Nevertheless, according to studies documented by van der Veken et al.277,278 concerning the π hydrogen complexes formed by ethylene and cyclopropane as proton acceptors, non-elementary reactions composed by more than two systems are favoured because the second acid molecule improves the intermolecular stability.279,280 In fact, the reaction mechanism between ethylene and hydrofluoric acid was divulged on the basis of a structural analysis of the complex C2H4⋯2(HF), in which the double acid addition is energetically favoured by 38 kJ mol−1 in comparison with systems formed with only one acid as proton donor.281 A ternary hydrogen-bonded complex (2′) would thus also be formed by the hydrofluoric acid and acetylene, as predicted in Fig. 2. As for isolated species, it is often argued that their molecular properties are significantly affected by the formation of hydrogen complexes.120,282–286 Indeed, several studies have exploited the molecular identities of the hydrogen-bonded complexes C2H2⋯HF and C2H2⋯2(HF) by analyzing structural arrangements, electronic data, and spectroscopy parameters,287,288 but the application of a methodology capable of describing the charge density distribution and thereby characterizing the π⋯H interaction289 should be worthwhile. To the best of our knowledge, according to Grabowski290 and Falvello291 the ‘different faces of hydrogen bonding’ and ‘the hydrogen bond, front and center’ are great points of view that should be taken into account. Ideally, it is prudent to presume that the interaction between the hydrocarbon ion and its neutral component also may be resultant of electrostatic interactions and charge transference. With the progress in the study of intermolecular interactions, a lot of researches have been conducted in order to elucidate the nature of the proton systems formed exclusively of hydrocarbons, as already highlighted and revised by Meot-Ner.292 The main aim of such researches is to develop a theoretical investigation of systems composed of neutral and ionic hydrocarbons, through which the interaction types ‘cation⋯π’293–298 and ‘anion⋯π’299–304 are formed.
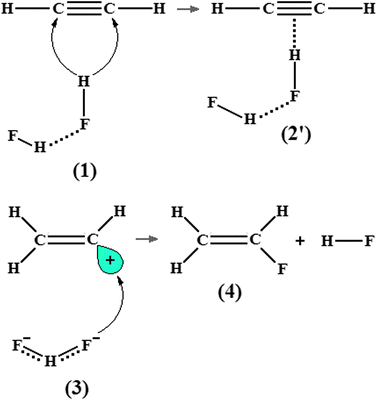 |
| | Fig. 2 Electrophilic addition reaction of two hydrofluoric acid molecules to the CC bond of the acetylene and the formation of the C2H2⋯2(HF) hydrogen-bonded complex (2′). | |
In spite of the large number of hydrocarbons existent in nature, some researches were developed on a model system whose chemical entity and properties are well known: the ethyl cation (C2H5+) (7). This cationic hydrocarbon is frequently chosen for study because its electron structure displays an interesting phenomenon so-called the hyperconjugation effect305 (see Fig. 3). By definition, the hyperconjugation effect of the ethyl cation occurs when the non-localized hydrogen H+ interacts with the carbons of the ethane. This H+ proton donor can be used to demonstrate the interaction ability of the ethyl cation towards the π bonds of hydrocarbons,306,307e.g., the triple (CC), double (CC), and σ (C–C) bonds of the acetylene (C2H2), ethene (C2H4), and cyclopropane (C3H6), respectively. In this context, an objective was projected to confirm whether the hyperconjugation effect of the ethyl cation continues after the formation of the C2H5+⋯C2H2, C2H5+⋯C2H4, and C2H5+⋯C3H6 hydrogen complexes. Finally, an intimate comparison between the molecular properties of these systems and those whose electronic sites are formed by pseudo-π308 and π309 bonds must be worthwhile.
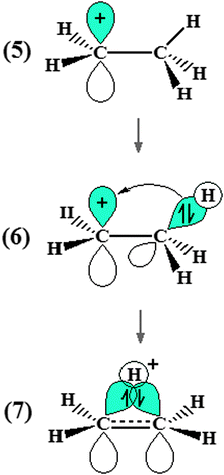 |
| | Fig. 3 Schematic drawing of the electronic delocalization on the ethyl cation. Hyperconjugation effect can be visualized in (7). | |
H+δ⋯H−δ dihydrogen bonds
Even taking into account modern studies of intermolecular interactions based on the electronegative criterion,310,311 amazingly other ones highlight uncommon hydrogen interactions formed by elements (M) with higher electropositivity bound to hydrogen (H), by which an ambiguous character to this atom is revealed.312 It is through the formation of M+δ–H−δ bond and the improved ‘electronegative power’ of the hydrogen that the so-called dihydrogen bonds (M+δ–H−δ⋯H+δ) are formed.313 In practice, M+δ–H−δ need not be the result of a dihydrogen bond, can be a metal–hydrogen bond if M is an alkaline earth or else a transition metal,314 for instance. Nevertheless, after the dihydrogen bonds were discovered,176 intermolecular chemistry has expanded to new horizons, such as studies of solid state, catalysis, crystal engineering, and materials chemistry.315–317 Among these, many other works about dihydrogen bonds180,318,319 are conducted by means of theoretical analysis in small systems,320–322 generally those where M is beryllium, aluminium, gallium, silicon, or germanium.323a However, the literature reports that beryllium hydride (BeH2) is the most efficient proton acceptor to be used in the formation of dihydrogen-bonded systems.323b From a structural viewpoint, the Be–H bond length is slightly increased upon the formation of the BeH2⋯HX complex with X = F, Cl, CN, and CCH, although the H–X distance is drastically elongated when the strength of the dihydrogen bond H−δ⋯H+δ is taken into account.324 This is widely established for traditional hydrogen-bonded systems formed by the Y⋯H+δ interaction.325Vibrational red- and blue-shifts in intermolecular interactions
The applicability of spectroscopy techniques to interpret bound systems is greatly useful and constantly utilized by the most renowned research groups all over the world.326–344 This great importance occurs due to the constant eminence of discovering hydrogen bond evidences on the most diversified systems.345–349 In the specific case of the formation of traditional hydrogen-bonded complexes, the variations in charge density provoke the appearance of red- and blue-shifts in the infrared spectrum often related to the proton donors.350–357 Hitherto, the quantification of the charge transfer is fundamental in this regard358–364 because these vibrational phenomena are immediate consequences of the drastic structural deformations in both proton donors and acceptors following complexation.365–367 One interesting example of deformation on monoprotic acids is related to their bond lengths,368–369 whereby the red-shift effect is observed when the stretch frequencies are shifted to the lowest values followed by large increases in the absorption intensities370,371 and thereby the formation of red-shifting hydrogen bonds are manifested.In regards to the blue-shift effect, this vibrational event was detected firstly by Pinchas,372 Adcock and Zhang,373 and Sandorfy et al.374 Some period later, however, Hobza et al.375–377 have studied this blue-shift effect in the context of the intermolecular interactions, which were then named as blue-shifting hydrogen bonds. By definition, the strengthening of the bonds of the proton donors is characteristic of blue-shifting hydrogen bonds because their stretching frequencies are displaced to upward values in the vibrational infrared spectrum.378,379 Although it is very common to interpret blue-shifts in specific proton donors, such as those found in haloform complexes,380 weakly bound complexes with π proton acceptors,381 amino acid fragments,382 or even the C–H bond of the fluoroform (HCF3)383 by means of the formation of the C2H4O⋯HCF3 complex, it is also interesting to admit the possibility to form intermolecular π complexes between fluoroform and ethylene (C2H4), propylene (C2H3(CH3)), and t-butylene (C2H2(CH3)2).384 Instead, by taking into account the hydrides BeH2, LiH, MgH2, and NaH, as well as HCF3, it is also important to know whether the molecular properties of these systems are drastically affected upon the formation of dihydrogen-bonded complexes (alkaline interactions)385 and blue-shifting systems (fluoroform as a proton donor),386 respectively. In light of this, a theoretical investigation into the possibility of these monomers forming intermolecular complexes, such as BeH2⋯HCF3, LiH⋯HCF3, MgH2⋯HCF3, and NaH⋯HCF3 was documented.387 Of course, it is already established that these systems are formed by the dihydrogen bonds H−δ⋯H+δ, but it is worthwhile to be aware whether the blue-shift of the C–H bond of the HCF3 can be identified by the analysis of the harmonic infrared spectrum.388–391
Furthermore, it is not obligatory that intermolecular complexes should be formed through only one hydrogen bond.392–395 Bifurcate hydrogen bonds or even those possessing multiple sites with donors (Lewis acid)396 and acceptors (lone-electron pairs or π clouds) of protons can lead to the formation of ternary systems.350,371,378,397 Thus, it is also important to investigate the formation of ternary dihydrogen-bonded complexes,398,399 such as NaH⋯2(HCF3) and NaH⋯2(HCCl3), and a comparative analysis of their properties with those obtained for the binary complexes NaH⋯HCF3 and NaH⋯HCCl3387 must be carried out.
The use of the QTAIM topology for intermolecular modelling
On a purely theoretical chemistry basis, there are many methods400–408 implemented to investigate the electronic structure and its intrinsic phenomena.409–414 The use of ab initio415–420 or DFT calculations,421–434 semiempirical methods,435–440 molecular dynamics simulations441–445 or even the discrete analysis of the solvent effect,446–451 the nature of the chemical bond, in particular the hydrogen bond, still is an essentially hazy question.452–454 Surely, the appearance of quantum mechanics has helped many research groups to develop quantum chemical methods,455,456 principally the QTAIM formalism designed by Bader in the 1960s.457–459 In a brief comment, Bader adopted the pure principles of quantum mechanics, whose great objective was the description of atomic behaviour within the molecular environment,460–462i.e., molecular fragments in chemical bonds.463,464 Moreover, Bader's insight was also based on Daltonian atomic theory,465,466 according to which the molecular structure is shaped into a cooperative ensemble.467 Thus, Bader has defined cooperative atoms in a molecule as open systems capable of exchanging charge and momentum with their neighbours, meaning that atoms can be treated as quantum mechanics entities within the molecules.468 Through the historical background of the QTAIM, several novel approaches have been idealized, such as the Two-Component Quantum Theory of Atoms in Molecules (TC-QTAIM),469 Quantum Division Basins (QDB),470 and Quantum Theory of Atoms in Positronic Molecules (QTAIPM),471 for instance.Nevertheless, the QTAIM architecture consists of mathematical conceptions derived from the theories of Dirac, Feynman, and Schwinger,472–474 whose contributions give the foundation for the principle of the least action for particle motion. In classical terms, the principle of least action states that a quantity (q) derived from the wave function is minimized in space and time (t1 to t2):
| | 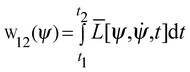 | (1) |
where
![[L with combining macron]](https://www.rsc.org/images/entities/i_char_004c_0304.gif)
is the Lagrangian differential operator defined by the contributions of the kinetic (
K) and potential (
U) energies as follows:
L =
K −
U. In a review containing the basic principles of QTAIM, Bader
475 and Nasertayoob and Shahbazian
476 have refined some topological concepts. For instance, the atomic frontier of an open system is defined as a zero-flux surface, whereby:
(i) The time variation at the end point is zero.
(ii) The surface is zero when the functions are at the extreme.
These two arguments provide support for the QTAIM explanation of how atoms bond to form molecules.477 By taking over that the two propositions given above are obeyed in concordance with the integration of the eqn (1), by which is obtained:
| | 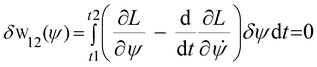 | (2) |
This equation states that minimization of action occurs if a differential equation is solved for the surface. In other words, δW12 vanishes according to the Euler–Lagrange equation,478 and the Schrödinger equation for a normalized wave function can be determined as ĤΨ* − EΨ* = 0 and ĤΨ − EΨ = 0. In his article published in 1926,479 Schrödinger concluded that the state function ψ and the Hamiltonian operator Ĥ for the hydrogen atom are related as follows:
| | 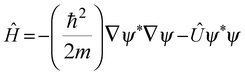 | (3) |
Whose expression for the Hamiltonian operator is
Ĥ = −
ℏ2/2
m∇
2 +
Û. There are two forms to express the kinetic energy (
K and
G), which jointly furnish the Laplacian for one-electron according to
eqn (4):
| | 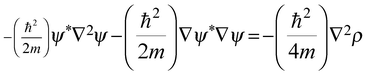 | (4) |
Integrating
eqn (4) over a spatial region
S(Ω) gives:
| | 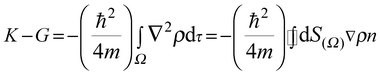 | (5) |
where
K and
G represent the kinetic energy densities
480 which are equivalent to the Laplacian of the charge density, (∇
2ρ).
481,482 If the zero-flux is one at any point of the surface
S(Ω) where
n is a normal vector,
483 then
K =
G and the condition to be satisfied is:
| | | ∇ρn = 0 for all points on the surface S(Ω) | (6) |
This equation defines the surface by which the atom is delimited as zero-flux charge density.484–486 For an ordinary quantum system, it is necessary to impose the vanishing of the wave functions ψ and ψ* at the quantum boundary. Well, according to the Born postulates,487 the product ψψ*dτ represents a probability distribution (eqn (7)), which, on the surface, should be zero.
The relation between surface conditions and high and low electron density sites is governed by the virial theorem.488,489 By considering the contributions of kinetic and potential energies, molecular regions with reduction and increase of charge density are represented by the positive (kinetic energy density G is positive) and negative (potential energy density U is negative) values of the Laplacian490,491 as demonstrated by eqn (8) and (9):
| | 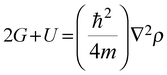 | (8) |
or
| | 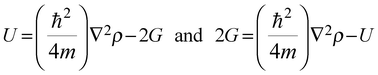 | (9) |
In general, this short overview shows that QTAIM492–500 is a useful approach to the study of chemical bonds and intermolecular interactions, and furthermore, it identifies Bond Critical Points (BCP)501 between interactive atoms502–506 through the calculations of topological parameters related to the molecular stability, from which the electron density profile and its Laplacian field are extracted.507 Based on the kinetic and potential energy operators given above,508,509 the QTAIM theory parameters identify maxima and minima of the electron density on the surface of the molecule and classify the chemical bonds as closed shells guided by ∇2ρ > 0, or additionally as shared interactions if ∇2ρ < 0.510,511 This theoretical argument has been used successfully to establish the intermolecular topology of several systems,512–531e.g., compounds with π bonds532 and binary complexes,533,534 including those formed by acetylene and hydrofluoric acid.535 It is not common to study ternary hydrogen complexes from the theoretical viewpoint.281 However, even if it can be done, the QTAIM results should be derived either from the equilibrium geometry536 or from fitting parameters derived from the energy.537
Likewise for Rozas et al.,538 the inherent qualities of the QTAIM warrants its application in the identification of the interactions in the C2H2⋯2(HF) complex, but it should be remembered that the result of the interaction between the CC bond of the acetylene and two hydrofluoric acid molecules reveals an unusual π hydrogen bond.539 The parameters related to the C2H2⋯HF binary complex thus represent a starting point to discuss the formation of π⋯H, not only in the C2H2⋯2(HF) ternary complex, but also in other systems formed by novel interactions.540–547 Besides the π⋯H hydrogen bonds, the QTAIM method has also been very useful for studying H−δ⋯H+δ dihydrogen bonds. For example, Popelier186a and later Grabowski and Leszczynski548 have carried out theoretical studies of systems stabilized by dihydrogen bonds by means of ab initio calculations and QTAIM integrations. As it is widely known that suitable analysis of vibrational modes is a decisive factor for describing adequately the formation of intermolecular interactions,549–551 our group reported a theoretical study of dihydrogen-bonded complexes formed by BeH2⋯HX with X = F, Cl, CN, and CCH, in which was proposed the investigation not only of their structural and electronic parameters, but also the characterization of their stretch frequencies and red-shift effects.552,553
According to Caballero and Jalón,554 the ‘electronegative capacity’ of H−δ in BeH2 can be demonstrated by a theoretical analysis of the ternary dihydrogen-bonded complex, BeH2⋯2(HCl).555 This observation led us to invest in a computational study in order to confirm the capability of HCN and HNC to form this ternary dihydrogen-bonded complex, not only by evaluating the terminal hydride (8) as a proton acceptor in BeH2⋯2(HX), but also simultaneously two (9) hydrides in XH⋯BeH2⋯HX, in this case X = CN and NC, as illustrated in the Fig. 4.556 Ideally, the characterization of intermolecular interactions, in particular dihydrogen bonds formed by multiple sites either of donors or acceptors of protons or any other ones can be performed by computing the molecular charge density, according to which the explanation of the cooperative effect related to molecular energy becomes possible557,558 besides the elucidation of the dynamism of the infrared vibrational modes, e.g., the redshifts on the stretch frequencies of the proton donors.559–566 The questions regarding the red- and blue-shifting hydrogen bonds are debated constantly in the literature.567–574 One of the most important topics is the presentation of new theoretical approaches, such as the analysis of the induced dipole proposed by Hermansson575 for investigating the origin of the vibrational blue shift. In line with this, the magnitude and direction of derivatives related to the dipole enhancements and bond lengths576,577 corroborate and reinforce the vibrational changes, either red or blue, in the proton donors.578 According to some studies,579–581 the applicability of this electropotential method has been proven. However, the main aim of this review is to demonstrate the QTAIM capability to interpret the vibrational red- and blue-shift events after the formation of hydrogen bonds (π⋯H) and dihydrogen bonds (H−δ⋯H+δ) by the quantification of their electronic densities (ρ) and Laplacians (∇2ρ), as well as by the whole topological analysis.582–585
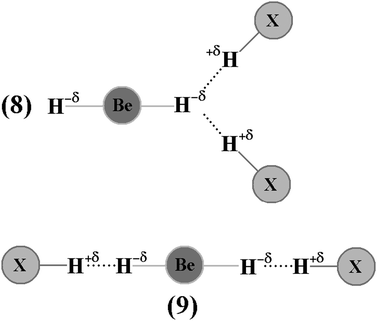 |
| | Fig. 4 Illustration of the BeH2⋯HX bifurcate (8) and XH⋯BeH2⋯HX linear (9) ternary dihydrogen systems (X=CN and NC). | |
The π⋯H hydrogen bonds: structure and vibrational spectrum
It should be pointed out that exploring theoretically the minimum of the potential energy surface,586 the optimized geometries of π hydrogen complexes were determined through the execution of the B3LYP/6-311++G(d,p) calculations. By this insight, the structures of the hydrogen complexes C2H2⋯HF (10), C2H2⋯2(HF) (10′), C2H4⋯HF (11), and C2H4⋯2(HF) (11′) obtained by this B3LYP/6-311++G(d,p) quantum level are shown in Fig. 5 (acetylenic complexes) and Fig. 6 (ethylenic complexes). Some studies reported by the specialized literature587–590 have shown that the formation of these complexes can be interpreted by the charge transfer591 flux from the bonding orbital (π) towards the antibonding orbital (σ*) of the unsaturated bonds (CC and CC) and proton donors (HF, HCl, HCN, or HCCH),273,538 respectively. Thereby, a series of drastic structural deformations on the molecular subunits are consequently observed, such as the weakness on the C1C2, Hc–Fd, and He–Ff bonds, whose values are listed in Table 1. Once this charge transfer phenomenon is taken into account, it can be clearly seen that, in almost all the complexes, C2H4⋯HF, C2H4⋯2(HF), C2H2⋯HF, and C2H2⋯2(HF), the most affected bonds are CC and H–F, while the C–H bonds remain almost unchanged.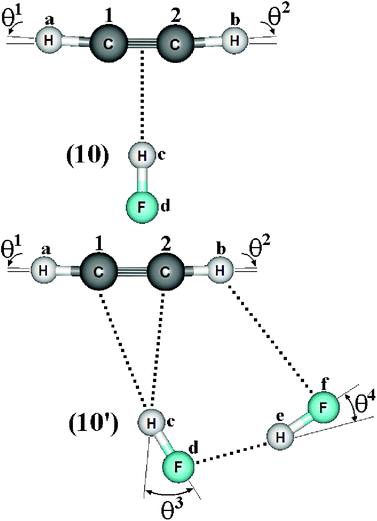 |
| | Fig. 5 Geometries of the C2H2⋯HF (10) and C2H2⋯2(HF) (10′) hydrogen complexes using the B3LYP/6-311++G(d,p) calculations. | |
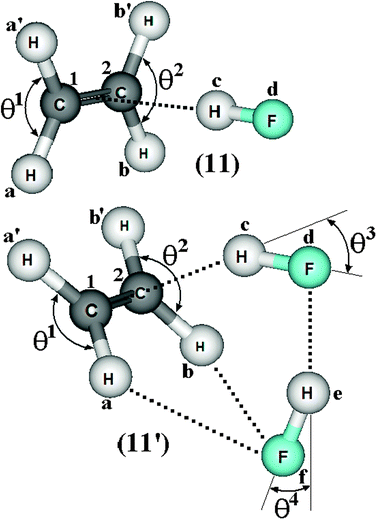 |
| | Fig. 6 Geometries of the C2H4⋯HF (11) and C2H4⋯2(HF) (11′) hydrogen complexes using the B3LYP/6-311++G(d,p) calculations. | |
Table 1 Values of the structural enhancements (Δr), hydrogen bond distances (R), and linearity deviation (θ) for the 2(HF), C2H2⋯HF (10), C2H2⋯2(HF) (10′), C2H4⋯HF (11), and C2H4⋯2(HF) (11′) complexes obtained from B3LYP/6-311++G(d,p) calculationsa
| Parameters | Hydrogen complexes |
|---|
| 2(HF) | (10) | (10′) | (11) | (11′) |
|---|
| Values of Δr and R are given in Å. Values of θ are given in degrees. |
|---|
| ΔrC1–Ha | — | 0.001 | 0.001 | −0.000(5) | 0.003 |
| ΔrC1–Ha′ | — | — | — | −0.000(5) | 0.003 |
| ΔrC2–Hb | — | 0.001 | 0.004 | −0.000(5) | 0.003 |
| ΔrC2–Hb′ | — | — | — | −0.000(5) | 0.003 |
| ΔrC1C2 | — | — | — | 0.004 | 0.008 |
| ΔrC1C2 | — | 0.002 | 0.002 | — | — |
| ΔrHc–Fd | 0.003 | 0.009 | 0.020 | 0.011 | 0.024 |
| ΔrHe–Ff | 0.007 | — | 0.012 | — | 0.018 |
| R(Hc⋯C1) | — | 2.234 | 2.176 | 2.273 | 2.141 |
| R(Hc⋯C2) | — | 2.234 | 2.105 | 2.273 | 2.141 |
| R(Fd⋯He) | 1.831 | — | 1.777 | — | 1.680 |
| R(Ff⋯Hb) | — | — | 2.547 | — | 2.458 |
| θ1 | — | 0.65 | 1.45 | 116.70 | 117.80 |
| θ2 | — | 0.65 | 1.93 | 116.70 | 117.80 |
| θ3 | — | — | 32.10 | — | 27.80 |
| θ4 | 12.10 | — | 24.10 | — | 22.30 |
| R(π⋯H) | — | 2.151 | — | 2.173 | 2.034 |
| R(p-π⋯H) | — | — | — | — | — |
| R(un-π⋯H) | — | — | 2.105 | — | — |
As justified by Del Bene,539 the π hydrogen bond in the C2H2⋯HF complex is formed exactly at the midpoint of the CC bond of the acetylene, whose distance value is 2.151 Å. The T-shape geometry of the C2H2⋯HF complex can thus be fully described by the C2v symmetry because the distances between Hc and both C1 and C2 (Hc⋯C1 and Hc⋯C2) are precisely equal to 2.234 Å.592 In the C2H2⋯2(HF) complex, however, the dimer of the hydrofluoric acid forms one hydrogen bond which is not aligned in the midpoint of the CC bond of the acetylene. This can be clearly seen due to the distances of R(Hc⋯C1) and R(Hc⋯C2), and precisely by their correspondingly distinct values of 2.176 and 2.105 Å the formation of one asymmetrical hydrogen bond profile is favoured.593 Indeed, the interaction R(Hc⋯C2) is stronger once its length is shorter, as highlighted by Parra and Zeng594 as well as by Remer and Jensen,595 and thereby indicates an interaction between the Hc hydrogen atom of the hydrofluoric acid and the C2 carbon of the acetylene molecule in the C2H2⋯2(HF) ternary system. Despite the π hydrogen bond aforementioned, a similar result of shortening of hydrogen bond in halothane:ammonia complexes has been reported by Michielsen, Dom, van der Veken, Herrebout et al.596 In the C2H2⋯HF binary, the charge transfer from the CC bond of the acetylene to the HF molecule causes elongation of the Hc–Fd bond. This is also confirmed by the 2(HF) dimer along the structure of C2H2⋯2(HF), where the shorter distance of the hydrogen bond Fd⋯He provokes enlargement in the H–F bond lengths as follows: r(Hc–Fd) > r(He–Ff). In spite of this, some important structural modifications in 2(HF) also have been observed, such as the contraction from 1.831 to 1.777 Å related to the distance of R(Fd⋯He).
In acetylene, the distance between C2 and Hb is longer than the distance between C1 and Ha. This occurs due to a possible interaction between the Hb hydrogen of the acetylene and the Ff fluorine of the second molecule of hydrofluoric acid (He–Ff). Thus, the Ff⋯Hb interaction is likely to exist, because its distance of 2.547 Å corroborates the values of the van der Waals radii of hydrogen (H = 1.2 Å) and fluorine (F = 1.35 Å), which together sum 2.55 Å.597–602 Another important indication of a possible interaction between Hb and Ff are the variations in the bond angles θ1, θ2, and θ4. It should be noted that θ1 = θ2 in the C2H2⋯HF complex, while, in the C2H2⋯2(HF) hydrogen complex, θ1 > θ2. Furthermore, the θ4 angle in 2(HF) undergoes drastic enlargement owing to the formation of the C2H4⋯2(HF) and C2H2⋯2(HF) hydrogen-bonded complexes, which varies from 12.10 up to 22.30 and 24.10°, respectively. The θ4 angle deviation is yet another structural confirmation that R(Ff⋯Hb) exists in the C2H4⋯2(HF) and C2H2⋯2(HF) complexes.603
The main spectroscopy event in the structures of hydrogen complexes is the displacement in the stretch frequencies of the proton donors to downward energy regions of the vibrational spectrum, an effect widely known as red-shift.274,604,605 So, the red-shifts of the C2H2⋯HF and C2H2⋯2(HF) complexes were computed at the B3LYP/6-311++G(d,p) level of theory, whose values are listed in Table 2. The formation of the dimer 2(HF) confirms that the stretch frequencies of Hc–Fd and He–Ff are shifted in −37.9 and −138.8 cm−1 to downward values, which are in good agreement with the respective experimental data of −31.0 and −93.0 cm−1.273a,b,369 Likewise, the greater increase in infrared intensity can also be found in the He–Ff bond, whose ratio is 3.7, whereas that in Hc–Fd is only 1.1. Nevertheless, the red-shift of −31 cm−1 on the stretch frequency 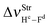 in 2(HF) is shorter than the respective value for C2H2⋯HF, which in turn, is also shorter in comparison with the C2H2⋯2(HF) complex. In other words, the
in 2(HF) is shorter than the respective value for C2H2⋯HF, which in turn, is also shorter in comparison with the C2H2⋯2(HF) complex. In other words, the 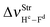 value is −438.0 cm−1 in C2H2⋯2(HF), whereas in C2H2⋯HF is −231.0 cm−1, which accords with the available experimental value of −215.0 cm−1. On the other hand, it can also be seen in Fig. 7 that, in the C2H2⋯2(HF) complex,
value is −438.0 cm−1 in C2H2⋯2(HF), whereas in C2H2⋯HF is −231.0 cm−1, which accords with the available experimental value of −215.0 cm−1. On the other hand, it can also be seen in Fig. 7 that, in the C2H2⋯2(HF) complex, 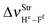 is displaced much more than
is displaced much more than 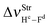 . The formation of the C2H4⋯2(HF) and C2H4⋯HF hydrogen-bonded systems provokes the arising of a more accentuated red-shift on the ternary ones, in the region of −463.1 and −259.6 cm−1, respectively, as explained above for C2H2⋯2(HF) and C2H2⋯HF.
. The formation of the C2H4⋯2(HF) and C2H4⋯HF hydrogen-bonded systems provokes the arising of a more accentuated red-shift on the ternary ones, in the region of −463.1 and −259.6 cm−1, respectively, as explained above for C2H2⋯2(HF) and C2H2⋯HF.
Table 2 Vibrational shift frequencies (ΔνStrHF), intensities ratios for the hydrofluoric acid bonds after complexation (IStrHF,c/IStrHF,m), stretching frequencies of the hydrogen bonds (νStrR), and absorption intensities (IStrR) for the 2(HF), C2H2⋯HF (10), C2H2⋯2(HF) (10′), C2H4⋯HF (11), and C2H4⋯2(HF) (11′) complexes obtained at the B3LYP/6-311++G(d,p) level of theory. Experimental data in parenthesesc
| Parameters | Hydrogen complexes |
|---|
| 2(HF) | (10) | (10′) | (11) | (11′) |
|---|
Ref. 273a. Ref. 273b. 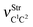 (monomers): C2H4 = 1683.9 cm−1 and C2H2 = 2062.5 cm−1; (monomers): C2H4 = 1683.9 cm−1 and C2H2 = 2062.5 cm−1; 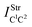 (monomers): C2H4 = 0.0 km mol−1 and C2H2 = 0.0 km mol−1; νStrHF and IStrHF (monomer): 4096 cm−1 and 130 km mol−1 respectively. (monomers): C2H4 = 0.0 km mol−1 and C2H2 = 0.0 km mol−1; νStrHF and IStrHF (monomer): 4096 cm−1 and 130 km mol−1 respectively. |
|---|
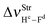 | −37.9 (−31)a | −231.0 (−215)b | −438 | −259.6 | −463.1 |
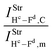 | 1.1 | 5.6 | 7.3 | 6.1 | 4.7 |
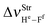 | −138.8 (−93)a | — | −246 | — | −292.5 |
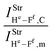 | 3.7 | — | 3.8 | — | 4.3 |
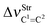 | — | — | — | −8.5 | 3.7 |
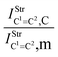 | — | — | — | 1.3 | 1.4 |
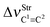 | — | −6.1 | −12.9 | — | — |
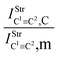 | — | 1.2 | 1.8 | — | — |
| νStr(π⋯H) | — | 137.8 | — | 133 | 183.8 |
| IStr(π⋯H) | — | 1.0 | — | 0.95 | 4.0 |
| νStr(un-π⋯H) | — | — | 181.5 | — | — |
| IStr(un-π⋯H) | — | — | 3.6 | — | — |
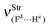 | — | — | 209 | — | 253.3 |
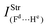 | — | — | 8.1 | — | 0.6 |
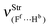 | — | — | 70.2 | — | 143.8 |
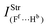 | — | — | 7.9 | — | 8.6 |
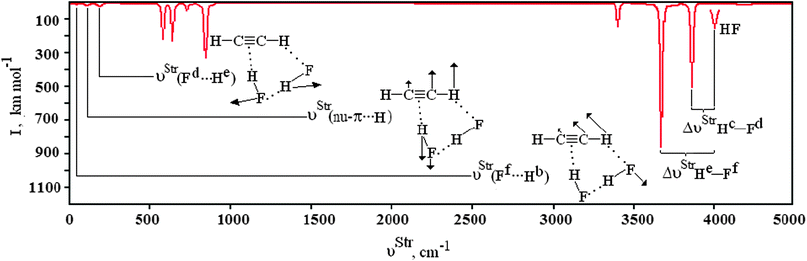 |
| | Fig. 7 Simulation of the vibrational harmonic infrared spectrum for the C2H2⋯2(HF) hydrogen-bonded bond complex using the B3LYP/6-311++G(d,p) calculations. The displacement vectors indicate the stretches of the new vibrational modes νStr(π⋯H), 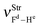 , and , and 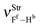 . . | |
The formation of the C2H4⋯HF, C2H4⋯2(HF), C2H2⋯HF, and C2H2⋯2(HF) hydrogen complexes gives rise to the new vibrational modes known as hydrogen bond stretch frequencies.606 This kind of vibrational mode exhibits very interesting features, such as low stretch frequency and weak absorption intensity.607,608 In comparison with the π complexes, the hydrogen bond stretch frequencies, νStr(π⋯H) and νStr(un-π⋯H), of the C2H4⋯2(HF) and C2H2⋯2(HF) hydrogen complexes are strongest, and the values of 183.8 and 181.5 cm−1 are identified in accordance with the directional arrows illustrated in Fig. 7. In concordance with the foregoing results of the analysis of the C2H4⋯HF and C2H2⋯HF T-shaped complexes, the oscillator νStr(π⋯H) is characterized as a stretch mode at the midpoint in both acetylene and ethylene.609 The C2H2⋯2(HF) ternary complex is considered a system apart because the stretch frequency of its hydrogen bond does not appears as π⋯H. Amazingly this stretch frequency is identified as unusual (un), un-π⋯H, once its harmonic oscillator is composed by the hydrogen (Hc) of the first hydrofluoric acid (Hc–Fd) and the carbon (C2) of the acetylene, respectively. Moreover, the oscillator of the additional interaction 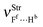 is also observed, although at a much lower frequency, in the region of 70.2 cm−1 with an absorption intensity of 7.9 km mol−1. Besides the new vibrational modes, the formation of the C2H4⋯HF, C2H4⋯2(HF), C2H2⋯HF, and C2H2⋯2(HF) hydrogen complexes exhibits a slight vibrational variation in the CC, CC, and C–H bonds, and therefore these results were not considered.
is also observed, although at a much lower frequency, in the region of 70.2 cm−1 with an absorption intensity of 7.9 km mol−1. Besides the new vibrational modes, the formation of the C2H4⋯HF, C2H4⋯2(HF), C2H2⋯HF, and C2H2⋯2(HF) hydrogen complexes exhibits a slight vibrational variation in the CC, CC, and C–H bonds, and therefore these results were not considered.
Fig. 8 illustrates the optimized geometries of the complexes C2H4⋯HCF3 (12), C2H3(CH3)⋯HCF3 (13), and C2H2(CH3)2⋯HCF3 (14) obtained through the B3LYP/6-311++G(d,p) calculations. As depicted in Table 3, one important initial observation was the slight contraction of −0.000(2) and elongation of 0.000(3) Å in the lengths of the C3–Hc bonds of HCF3 in the 12 and 14 complexes, respectively. In a middle term, C3–Hc in 13 remained unchanged. Investigations of the hydrogen bond distances have shown that 13 and 14 are the respective shortest and longest bonded complexes, whereas 12 is of intermediate length. However, it is not possible to establish any relationship between the interaction strength and the variation on the C3–Hc bond length. Most likely this unsystematic trend is caused by the hydrogen bond profile in 13, where, instead of a contact between the π bond of propylene and the hydrogen of HCF3, a direct interaction on the carbon can be seen wherein this element is acting as a proton acceptor. The same trends determined for hydrogen bond distance also are applied to their vibrational stretching frequencies, which are 50.5, 58.4, and 47.1 cm−1 for the 12, 13, and 14 complexes, respectively.
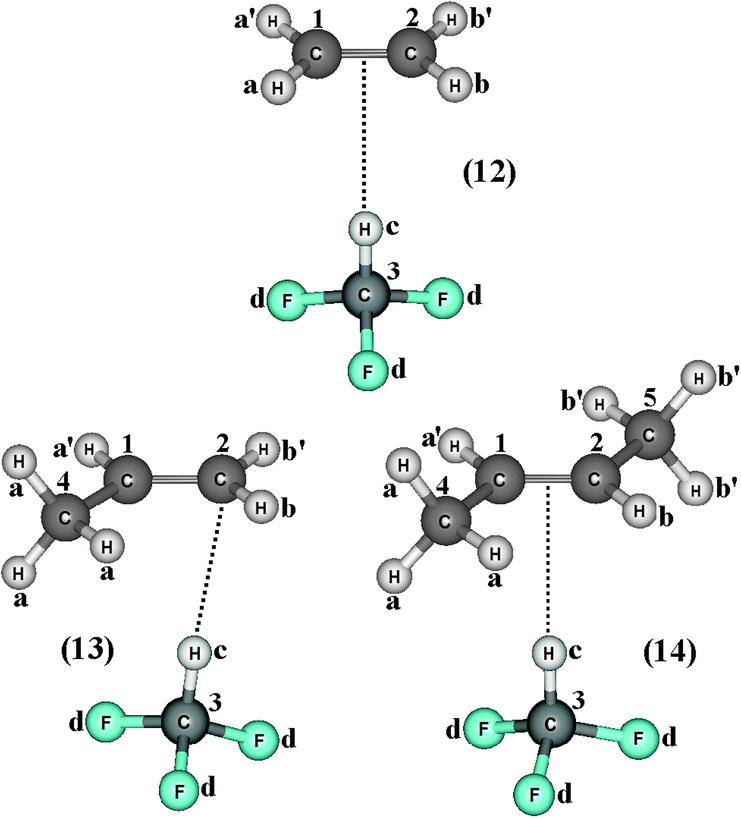 |
| | Fig. 8 Optimized geometries of the blue-shifting hydrogen-bonded complexes C2H4⋯HCF3 (12), C2H3(CH3)⋯HCF3 (13), and C2H2(CH3)2⋯HCF3 (14) by using the B3LYP/6-311++G(d,p) calculations. | |
Table 3 Values of the structural parameters and infrared modes of the HCF3 monomer, as well as of the C2H4⋯HCF3 (12), C2H3(CH3)⋯HCF3 (13), and C2H2(CH3)2⋯HCF3 (14) complexes obtained by using the B3LYP/6-311++G(d,p) calculationsa
| Parameters | Hydrogen complexes |
|---|
| HCF3 | (12) | (13) | (14) |
|---|
| Values of R and r are given in Å. Values of νStr and IStr are given in cm−1 and km mol−1 respectively. |
|---|
| R(Hc⋯π) | — | 2.878 | — | 3.120 |
| R(C2⋯Hc) | — | — | 2.801 | — |
| r(C3–Hc) | 1.089(6) | 1.089(4) | 1.089(6) | 1.089(9) |
| Δr(C3–Hc) | — | −0.000(2) | 0.000 | 0.000(3) |
| r(C1C2) | — | 1.3300 | 1.3329 | 1.3347 |
| (1.328) | (1.331) | (1.333) |
| Δr(C1C2) | — | −0.0012 | −0.0017 | −0.0017 |
| νStr(π⋯H) | — | 50.5 | — | 47.1 |
| IStr(π⋯H) | — | 0.1 | — | 0.1 |
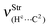 | — | 49.2 | 58.4 | — |
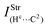 | — | 0.09 | 0.5 | — |
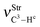 | 3141.2 | 3144.4 | 3140.7 | 3136.8 |
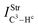 | 32.8 | 4 | 2.9 | 0.98 |
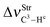 | — | 3.2 | −0.5 | −5.0 |
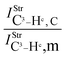 | — | 0.12 | 0.08 | 0.03 |
In regards to the structure and spectroscopy of the proton donors,610 some of them closely similar to those revisited here, an interesting observation is the direct relationship between the variations of their bond lengths, Δr(C3–Hc), versus the shifting of the stretch frequencies, 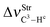 . It should be noted that the blue-shift of 3.2 cm−1 in 12 is justified by the decrease of −0.0002 Å, and the red-shift of −5.0 cm−1 in 14 can be explained by the slight increase of 0.0003 Å, just as that found by Hippler611 in spectroscopy studies of hydrogen-bonded complexes in the gas phase. The invariant C3–Hc bond in 13 also remains non-shifted in the infrared spectrum, corroborating the structural and vibrational parameters. Nevertheless, the red-shift values of −0.5 (13) and −5.0 cm−1 (14) are followed of a decrease in their absorption intensities. These can be contradictory results, but theoretical and experimental evaluations of Herrebout et al.612 have shown that not all red-shifts are accompanied by a reduction in the absorption intensities, when π bonds of hydrocarbons bind with trifluoro(halo)methanes, for instance. Of course these spectroscopy profiles may be useful as a guide for experimental investigations,369,613–616 once all the results were obtained using the B3LYP functional, which is considered an efficient method for conducting studies of intermolecular interactions617 and infrared spectra of hydrogen bonds.618–622
. It should be noted that the blue-shift of 3.2 cm−1 in 12 is justified by the decrease of −0.0002 Å, and the red-shift of −5.0 cm−1 in 14 can be explained by the slight increase of 0.0003 Å, just as that found by Hippler611 in spectroscopy studies of hydrogen-bonded complexes in the gas phase. The invariant C3–Hc bond in 13 also remains non-shifted in the infrared spectrum, corroborating the structural and vibrational parameters. Nevertheless, the red-shift values of −0.5 (13) and −5.0 cm−1 (14) are followed of a decrease in their absorption intensities. These can be contradictory results, but theoretical and experimental evaluations of Herrebout et al.612 have shown that not all red-shifts are accompanied by a reduction in the absorption intensities, when π bonds of hydrocarbons bind with trifluoro(halo)methanes, for instance. Of course these spectroscopy profiles may be useful as a guide for experimental investigations,369,613–616 once all the results were obtained using the B3LYP functional, which is considered an efficient method for conducting studies of intermolecular interactions617 and infrared spectra of hydrogen bonds.618–622
The H−δ⋯H+δ dihydrogen bonds: structure and vibrational spectrum
Spectroscopy studies of dihydrogen bonds have attracted much attention in recent years.623–632 Besides the crystallography as one of the most used techniques in studies of dihydrogen bonds, the contribution of the infrared spectrum is also a powerful tool because there are weak and sensitive stretch frequencies to be unveiled. Here the results of the infrared spectrum of the BeH2⋯HX dihydrogen complexes (with X = F, Cl, CN, and CCH) are one of the main points of discussion. However, firstly it is essential to discuss of the equilibrium geometries of these systems from the theoretical viewpoint. Fig. 9 exhibits the optimized geometries of the dihydrogen-bonded complexes BeH2⋯HF (15), BeH2⋯HCl (16), BeH2⋯HCN (17), and BeH2⋯HCCH (18) obtained by using the B3LYP/6-311++G(3d,3p) calculations. In Table 4 are listed the results for the dihydrogen bond distances, R(H−δ⋯H+δ), as well as the increases in the bond lengths, Δr(Be–H) and Δr(H–F), related to the beryllium hydride and hydrofluoric acid, respectively. Initially, the interaction strength in the BeH2⋯HX complexes (with X = F, Cl, CN, and CCH) with regard to the values of the distances R(H−δ⋯H+δ) is represented by the following trend: F > Cl > CN > CCH. About BeH2 and HF, however, Δr(Be–H) exhibits small variations in comparison to Δr(H–F). The main results of the infrared spectra analysis for the dihydrogen-bonded complexes BeH2⋯HX are presented in Table 5. The values of the vibrational mode 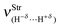 , understood as stretch frequencies of the dihydrogen bond or new vibrational modes varying between 69.5–173.4 cm−1, are very similar to those observed for the classical hydrogen-bonded complexes.326–329 Although the vibrational modes must be identified here as stretch frequencies of the dihydrogen bonds, their absorption intensities demonstrate the weakness of the interactions formed in the BeH2⋯HX complexes, e.g., the values of 0.7 and 0.3 km mol−1 for BeH2⋯HCN and BeH2⋯HCCH respectively. However, examination of the red-shift on the stretch frequencies of the Lewis’s acids reveals a synchronism related to the displacement. It can be noted that the longer downward stretch value of −240.0 cm−1 is related to the hydrofluoric acid of the BeH2⋯HF complex, whereas the shorter result of −15.3 cm−1 is related to the acetylene of the BeH2⋯HCCH complex.
, understood as stretch frequencies of the dihydrogen bond or new vibrational modes varying between 69.5–173.4 cm−1, are very similar to those observed for the classical hydrogen-bonded complexes.326–329 Although the vibrational modes must be identified here as stretch frequencies of the dihydrogen bonds, their absorption intensities demonstrate the weakness of the interactions formed in the BeH2⋯HX complexes, e.g., the values of 0.7 and 0.3 km mol−1 for BeH2⋯HCN and BeH2⋯HCCH respectively. However, examination of the red-shift on the stretch frequencies of the Lewis’s acids reveals a synchronism related to the displacement. It can be noted that the longer downward stretch value of −240.0 cm−1 is related to the hydrofluoric acid of the BeH2⋯HF complex, whereas the shorter result of −15.3 cm−1 is related to the acetylene of the BeH2⋯HCCH complex.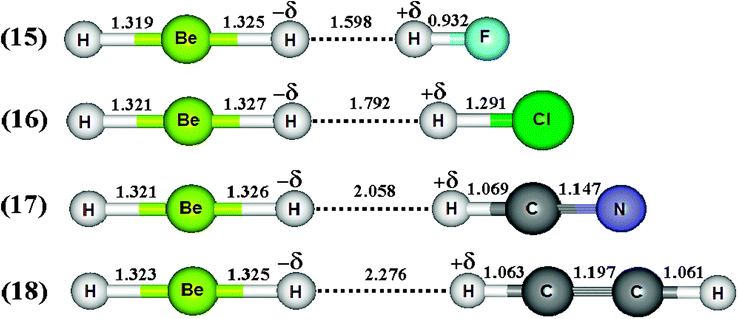 |
| | Fig. 9 Optimized geometries of the BeH2⋯HX dihydrogen-bonded complexes with X = F (15), Cl (16), CN (17), and CCH (18) using the B3LYP/6-311++G(3d,3p) calculations. | |
Table 4 Values of the structural parameters for the dihydrogen-bonded complexes BeH2⋯HX with X = F, Cl, CN, and CCH obtained from the B3LYP/6-311++G(3d,3p) calculationsa
| Parameters | Dihydrogen complexes |
|---|
| X = F (15) | X = Cl (16) | X = CN (17) | X = CCH (18) |
|---|
| All values are given in Å. |
|---|
| R(H−δ⋯H+δ) | 1.598 | 1.792 | 2.058 | 2.276 |
| Δr(Be–H) | 0.001 | 0.028 | 0.003 | 0.001 |
| Δr(H–X) | 0.010 | 0.010 | 0.004 | 0.002 |
Table 5 Profiles of the vibrational harmonic spectrum of the dihydrogen-bonded complexes BeH2⋯HX with X = F, Cl, CN, and CCH by using the B3LYP/6-311++G(3d,3p) calculationsa
| Parameters | Dihydrogen complexes |
|---|
| X = F (15) | X = Cl (16) | X = CN (17) | X = CCH (18) |
|---|
| νStr values are given in cm−1. IStr values are given in km mol−1. |
|---|
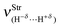 | 173.4 | 115.5 | 97.7 | 69.5 |
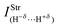 | 1.9 | 3.0 | 0.7 | 0.3 |
| ΔνStrH–X | −240.0 | −148.7 | −62.5 | −15.3 |
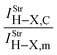 | 6.2 | 8.9 | 3.2 | 2.6 |
Similarly to the π⋯H hydrogen bonds mentioned previously, such as those with three centers603commonly known as ternary systems, it is very interesting that dihydrogen bonds can be formed in this same context, i.e., ternary dihydrogen systems.633,634 Thus, the formation of the BeH2⋯2(HCl) ternary dihydrogen-bonded complex is related to the possibility of forming the three isomers 19, 20, and 21, whose optimized geometries, accompanied by the BeH2⋯HCl binary complex 22 illustrated in Fig. 10, are based on the scheme outlined in Fig. 4. Exhibiting a symmetric geometry, the bond lengths of the hydrochloric acid are identical upon the formation of the isomers 20 and 21. For the isomer 19, all its bond lengths are dissimilar, especially the values of 1.741 and 3.262 Å attributed to R(H−δ⋯H+δ) and R(Cl⋯H+δ), respectively. Moreover, the structure of isomer 19 is shaped by both the dihydrogen bonds (H−δ⋯H+δ)-(19)-A and hydrogen bonds (Cl−δ⋯H+δ)-(19)-B, suggesting the formation of a mixed interaction system: a dihydrogen-bonded and hydrogen-bonded complex.635,636 The values of the main vibrational modes of the dihydrogen complexes 19, 20, and 21 are listed in Table 6. It can be seen that the hydrochloric acids have their stretch frequencies (νStr) shifted downwards (ΔνStr) and their absorption intensities (IStrC) are substantially increased (IStrC/IStrm), and it is due to these phenomena that the red-shift effects arise.636 It should be noted that the 20 and 21 undergo similar and slighter modifications in ΔνStr(H–Cl), while ΔνStr(H–Cl)-(22) and ΔνStr(H–Cl)-(19)-A are the greatest variations, suggesting that the 19 isomer tends to form a binary complex instead of ternary. In other words, the stretch frequency of the proton donor (H–Cl)-(19)-B is slightly affected by the formation of BeH2⋯2(HCl). Thereby, the weakest interaction is formed between the BeH2⋯HCl binary complex and the extra molecule of hydrochloric acid, (H–Cl)-(19)-B. The results of the intermolecular stretch frequencies presented in Table 6 demonstrate a low value of 13.8 cm−1 followed by a lowest intensity of 0.1 km mol−1 for (Cl⋯H+δ)-(19)-B, which means that the vibrational detection of the dihydrogen bond in the infrared spectrum is not an easy task, as can be seen in Fig. 11.
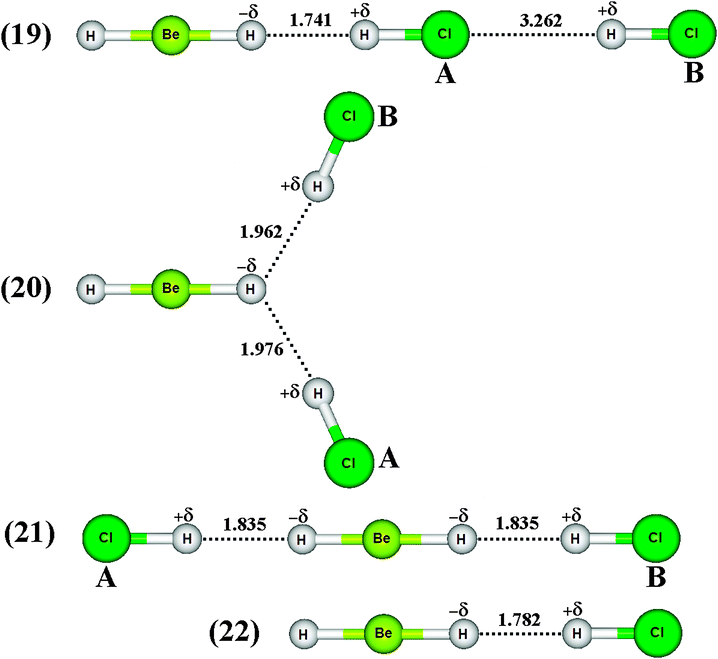 |
| | Fig. 10 Optimized geometries of BeH2HCl binary dihydrogen-bonded complex (22), as well as ternary dihydrogen-bonded isomers (19), (20), and (21) obtained from B3LYP/6-31++G(3d,3p) calculations. The H–Be and H–Cl bond lengths on free monomers are 1.324 and 1.2807 Å respectively. | |
Table 6 Values of the stretch frequencies for the BeH2⋯HCl binary dihydrogen complex (22) and ternaries dihydrogen complexes (19), (20), and (21) obtained at the B3LYP/6-31++G(3d,3p) theoretical levela
| Modes | Vibrational parameters |
|---|
| νStr | ΔνStr | IStrC | IStrC/IStrm |
|---|
| Values of νStr and IStr are given in cm−1 and km mol−1 respectively. Values of νStrHCl and IStrHCl (monomer) 2943 cm−1 and 44.9 km mol−1 obtained by using the B3LYP/6-31++G(3d,3p) calculations. |
|---|
| (H–Cl)-(22) | 2796 | −146.3 | 392 | 8.7 |
| (H–Cl)-(19)-A | 2764 | −179.0 | 514 | 11.4 |
| (H–Cl)-(19)-B | 2939 | −4.0 | 73.8 | 1.6 |
| (H–Cl)-(20)-A | 2842 | −100.0 | 408 | 9.0 |
| (H–Cl)-(20)-B | 2842 | −100.0 | 408 | 9.0 |
| (H–Cl)-(21)-A | 2821 | −122.0 | 698 | 15.5 |
| (H–Cl)-(21)-B | 2821 | −122.0 | 698 | 15.5 |
| (H−δ⋯H+δ)-(22) | 116.1 | — | 3.0 | — |
| (H−δ⋯H+δ)-(19)-A | 125.6 | — | 4.0 | — |
| (Cl⋯H+δ)-(19)-B | 13.8 | — | 0.1 | — |
| (H−δ⋯H+δ)-(20)-A | 74.8 | — | 0.6 | — |
| (H−δ⋯H+δ)-(20)-B | 74.8 | — | 0.6 | — |
| (H−δ⋯H+δ)-(21)-A | 52.4 | — | 0.0 | — |
| (H−δ⋯H+δ)-(21)-B | 52.4 | — | 0.0 | — |
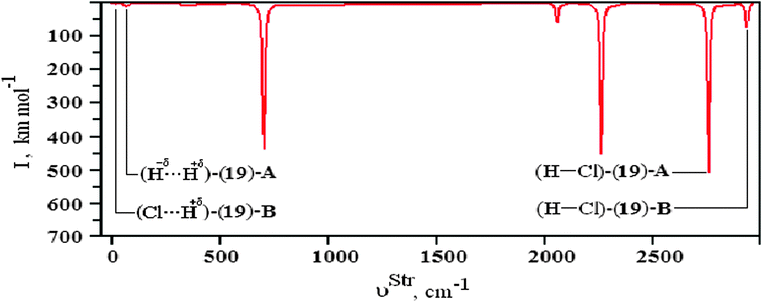 |
| | Fig. 11 Illustration of the infrared spectrum of the (19) isomer of the BeH2⋯2HCl ternary dihydrogen-bonded complex using the B3LYP/6-31++G(3d,3p) theoretical level. | |
By assuming the possibility of the formation of ternary dihydrogen complexes due to the attack of two proton donors on the beryllium hydride, the optimized geometries of the BeH2⋯2(HCN) (23) and BeH2⋯2(HNC) (24) bifurcate dihydrogen complexes as well as the HCN⋯BeH2⋯HCN (25) and CNH⋯BeH2⋯HNC (26) linear ones are illustrated in Fig. 12 and 13, respectively. First, it can be seen that the bifurcate complexes present a difference in their dihydrogen bond distances, e.g., 2.340 and 2.322 Å, when HCN (A) and HCN (B) interact with BeH2 in 23. On the other hand, the dihydrogen bond distances are practically identical in the linear complexes, as demonstrated by the results of 1.859 and 1.857 Å for the 25 complex. With regard to H–C and H–N bond lengths, there are not significant differences between the acids (A and B) in all ternary complexes. However, the distortions in the structure of BeH2 have yielded interesting results, which should be carefully investigated. In the bifurcate dihydrogen complexes, it should be noted that the value of 1.325 Å for the Be–H bond length is decreased to 1.316 Å (−δH–Be) as well as increased to 1.338 Å (Be–H−δ) after the formation of 24. Because the formation of the dihydrogen bonds in linear complexes occurs simultaneously on H−δ, there is a slight reduction on their bond lengths −δH–Be and Be–H−δ, whose respective values for the complexes 25 and 26 are 1.323 and 1.322 Å.
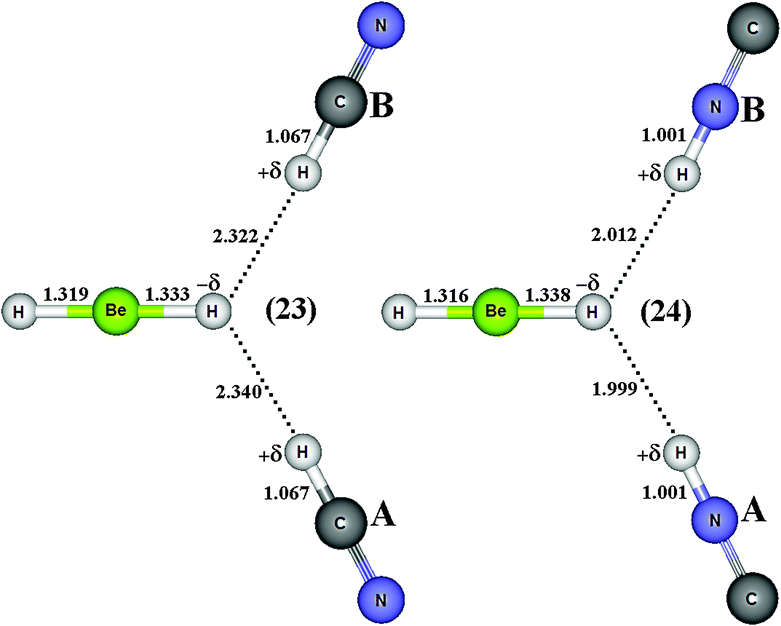 |
| | Fig. 12 Optimized geometries of bifurcate (23–24) dihydrogen-bonded complexes BeH2⋯2HX (with X = CN and NC) obtained by using the B3LYP/6-31++G(3d,3p) calculations. H–Be (BeH2), H–C (HCN) and H–N (HNC) bond lengths on free monomers are 1.3255, 1.0672, and 1.0007 Å, respectively. | |
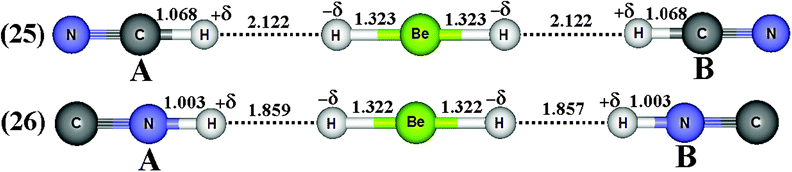 |
| | Fig. 13 Optimized geometries of linear (25–26) dihydrogen-bonded complexes XH⋯BeH2⋯HX (with X = CN and NC obtained by using the B3LYP/6-31++G(3d,3p) calculations. H–Be (BeH2), H–C (HCN) and H–N (HNC) bond lengths on free monomers are 1.3255 Å, 1.0672 Å and 1.0007 Å, respectively. | |
The spectroscopy techniques and DFT calculations have been used conjointly for investigating the vibrational modes of bifurcate dihydrogen bonds,637 as reported in the pioneering study conducted by Nikonov et al.638 The values for the main stretching frequencies and absorption intensities of the bifurcate dihydrogen complexes BeH2⋯2(HCN) (23) and BeH2⋯2(HNC) (24), and linear ones HCN⋯BeH2⋯HCN (25) and CNH⋯BeH2⋯HNC (26) are listed in Table 7. In agreement with other studies,639 it can be seen that the stretch frequencies of the bonds (H+δ–C)-A and (H+δ–C)-B are displaced downward in value. However, the H+δ–N bonds of 24 (bifurcate) and 26 (linear) provide the largest red-shifts, with values of −37.4 and −75.3 cm−1 respectively. In bifurcate complexes, red-shifts of −8.5 and −16.9 cm−1 have been found for the bond (Be–H−δ) of the 23 and 24 complexes. On the other hand, blue-shifts of +23.6 and +45.8 cm−1 have been computed for the bond Be–H−δ of the 25 and 26 complexes.640,641 Briefly, the vibrational blue-shifts of (Be–H−δ) are caused by the shortening of its bond length. The absorption intensity (IStrm) of the bond Be–H−δ in the BeH2 monomer is zero, although the complexes 23 and 24 have furnished the most evident ratios (IStrc/IStrm), whose values are 43.9 and 90.0 respectively. Regardless the geometric shape, either linear or bifurcate, the red- and blue-shifts on the stretch frequencies of the bond (Be–H−δ) were observed in 23–24 and 25–26, indistinctly of whether they are the longest and shortest bonded complexes, respectively.
Table 7 Values of the stretch frequencies of the BeH2⋯2HX and XH⋯BeH2⋯HX ternary dihydrogen complexes obtained using the B3LYP/6-31++G(3d,3p) theoretical levela
| Modes | Vibrational parameters |
|---|
| νStr | ΔνStr | IStrC | IStrC/IStrm |
|---|
| Values of νStr and IStr are given in cm−1 and km mol−1 respectively. |
|---|
| (H+δ–C)-(23)-A | 3422.0 | −13.2 | 84.70 | 1.2 |
| (H+δ–C)-(23)-B | 3422.0 | −13.2 | 84.70 | 1.2 |
| (Be–H−δ)-(23) | 2041.8 | −8.5 | 43.9 | 43.9 |
| (H+δ–N)-(24)-A | 3727.1 | −37.4 | 312.3 | 1.2 |
| (H+δ–N)-(24)-B | 3727.1 | −37.4 | 312.3 | 1.2 |
| (Be–H−δ)-(24) | 2033.4 | −16.9 | 90.0 | 90.0 |
| (H+δ–C)-(25)-A | 3405.4 | −29.8 | 18.2 | 3.7 |
| (H+δ–C)-(25)-B | 3405.4 | −29.8 | 18.2 | 3.7 |
| (Be–H−δ)-(25) | 2073.9 | +23.6 | 0.0 | 0.0 |
| (H+δ–N)-(26)-A | 3689.2 | −75.3 | 1.4 | 1.4 |
| (H+δ–N)-(26)-B | 3689.2 | −75.3 | 1.4 | 1.4 |
| (Be–H−δ)-(26) | 2096.1 | +45.8 | 0.0 | 0.0 |
Through the application of the B3LYP/6-311++G(3df,3pd) calculations, Fig. 14 presents the optimized geometries of the BeH2⋯HCF3 (27), MgH2⋯HCF3 (28), LiH⋯HCF3 (29), and NaH⋯HCF3 (30) dihydrogen-bonded complexes. The deformations in the C–H+δ bonds of the fluoroform can be clearly seen by means of the results of −0.001, 0.0002, 0.003, and 0.004 Å related to the 27, 28, 29, and 30 dihydrogen-bonded complexes respectively. Although the bond lengths on the hydrides are altered due the complexation, we focused here on the structural changes occurring in the C–H+δ bonds. A shortening of −0.001 Å in C–H+δ of the fluoroform was thus computed only in 27. In contrast to this, slight elongations in the bond lengths of C–H+δ with variations between 0.0002–0.004 Å were obtained after the formation of 28, 29, and 30. It can be seen that reduction trends in C–H+δ have been observed when longer R(H−δ⋯H+δ) dihydrogen bonds occur, specifically with the value of 2.215 Å in 27. On the other hand, enlargements in the bond lengths of C–H+δ were observed in the 28, 29, and 30 systems, which are stabilized by the shortest dihydrogen bond distances.642
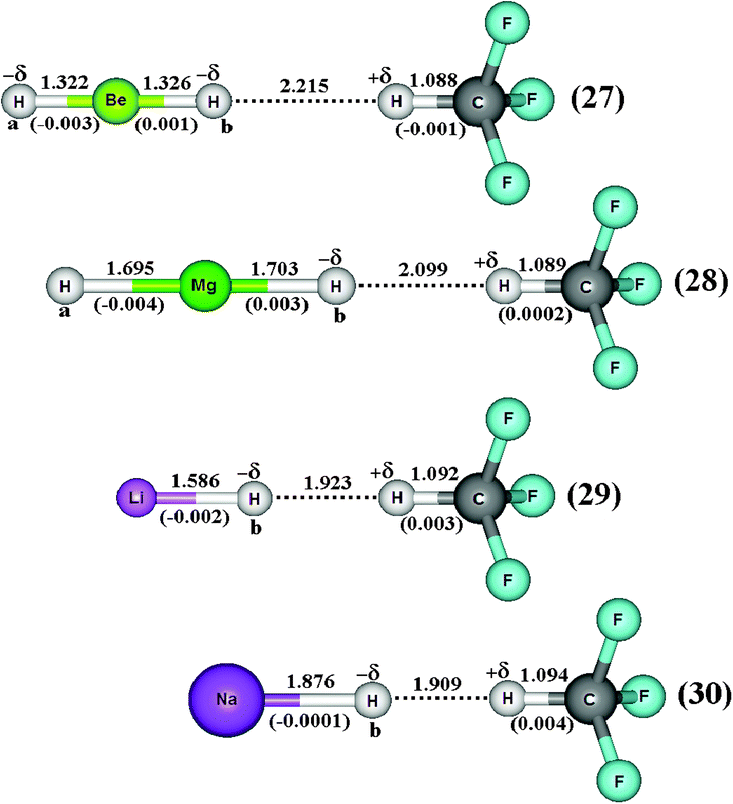 |
| | Fig. 14 Optimized geometries of the BeH2⋯HCF3 (27), MgH2⋯H CF3 (28), LiH⋯HCF3 (29), and NaH⋯HCF3 (30) dihydrogen-bonded complexes using B3LYP/6-311++G(3df,3pd) calculations. Values of the bond length enhancements are given in parentheses. The values of the C–H, Mg–H, Be–H, Li–H, and Na–H bond lengths of the monomers (HCF3, MgH2, LiH and NaH) are 1.089 Å, 1.700 Å, 1.325 Å, 1.589 Å and 1.876 Å, respectively. | |
In Table 8 are listed the results of the stretch frequencies and absorption intensities computed for the dihydrogen-bonded complexes 27, 28, 29, and 30 with all calculations performed at the B3LYP/6-311++G(3df,3pd) level of theory. As is well-known, the new infrared vibrational modes emerge by means of the formation of hydrogen-bonded complexes,643 and in addition, these vibrational modes are weak stretch bands and they are often interpreted as being the frequencies of the intermolecular interaction.644 As such, but taking into account the revisited dihydrogen complexes in this current work, it should be assumed that these kinds of vibrational modes are related to the dihydrogen bond frequencies, 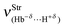 . In this regard, it can be reported that, according to the values of the dihydrogen bond energies (2.31–19.66 kJ mol−1),387 in general, the
. In this regard, it can be reported that, according to the values of the dihydrogen bond energies (2.31–19.66 kJ mol−1),387 in general, the 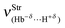 results obtained at the B3LYP/6-311++G(3df,3pd) level of theory do not corroborate these values for intermolecular energy. In other words, the weakly bonded dihydrogen complex 27, which has the lowest interaction energy ΔEC(BSSE + ΔZPE) of 2.31 kJ mol−1, also presents a low dihydrogen bond frequency
results obtained at the B3LYP/6-311++G(3df,3pd) level of theory do not corroborate these values for intermolecular energy. In other words, the weakly bonded dihydrogen complex 27, which has the lowest interaction energy ΔEC(BSSE + ΔZPE) of 2.31 kJ mol−1, also presents a low dihydrogen bond frequency 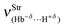 , though not the weakest. On the contrary, the stretch frequency
, though not the weakest. On the contrary, the stretch frequency 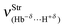 of the dihydrogen bond in the complex 28 is the weakest, but its intermolecular energy ΔEC(BSSE + ΔZPE) of 6.67 kJ mol−1 is of medium-strength, although higher than the 2.31 kJ mol−1 found in 27. It is greatly important to emphasize that 29 furnishes a stronger stretch frequency
of the dihydrogen bond in the complex 28 is the weakest, but its intermolecular energy ΔEC(BSSE + ΔZPE) of 6.67 kJ mol−1 is of medium-strength, although higher than the 2.31 kJ mol−1 found in 27. It is greatly important to emphasize that 29 furnishes a stronger stretch frequency 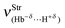 of the dihydrogen bond than that observed in 30, whose values are 148.2 and 91.7 cm−1 respectively. On the other hand, their energies ΔEC of 19.43 and 19.66 kJ mol−1 are equivalent.
of the dihydrogen bond than that observed in 30, whose values are 148.2 and 91.7 cm−1 respectively. On the other hand, their energies ΔEC of 19.43 and 19.66 kJ mol−1 are equivalent.
Table 8 Values of the stretch frequencies of the BeH2⋯HCF3 (27), MgH2⋯HCF3 (28), LiH⋯HCF3 (29), and NaH⋯HCF3 (30) dihydrogen complexes obtained by using the B3LYP/6-311++G(3df,3pd) calculationsa
| Parameters | Dihydrogen complexes |
|---|
| (27) | (28) | (29) | (30) |
|---|
| Values of νStr and IStr are given in cm−1 and km mol−1 respectively. Values of νStr and IStr (HCF3 monomer) in parentheses. |
|---|
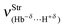 | 73.2 | 66.0 | 148.2 | 91.71 |
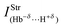 | 0.30 | 0.22 | 0.33 | 0.02 |
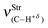 | 3137.9 (3121) | 3116.7 (3121) | 3062.3 (3121) | 3039.0 (3121) |
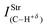 | 3.99 (26.9) | 0.57 (26.9) | 45.8 (26.9) | 57.5 (26.9) |
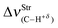 | +16.3 | −4.9 | −59.3 | −82.6 |
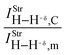 | 0.15 | 0.02 | 1.70 | 2.14 |
Nevertheless, if the changes in the C–H+δ bonds of the fluoroform are taken into account, there is an interesting observation with regard to the 27, 28, 29, and 30 dihydrogen-bonded complexes. As debated above, no satisfactory correlation between dihydrogen bond energy and vibrational modes has been obtained, but in terms of modifications in C–H+δ, distinct vibrational shifts have been verified on this bond. In accordance with recent studies published in the specialized literature,387 it was extremely unexpected to observe the red-shifts of −4.9, −59.3, and −82.6 cm−1 in the 28, 29, and 30 dihydrogen-bonded complexes, of which the last two showed an increase in absorption intensity, whose 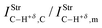 ratio values are 1.70 and 2.14 respectively. Meanwhile, a blue-shift of +16.3 cm−1 was detected in 27, although a reduction in absorption intensity of 0.15 has been also observed. In terms of intermolecular strength, it can be confirmed that the higher values of dihydrogen bond energy in 28, 29, and 30 are associated with larger red-shifts, whereas the weak intermolecular contacts are not related either to longer or shorter red-shifts. In fact, it is evident that the dihydrogen-bonded complex 27, in addition to complexes 28, 29, and 30, they coexist under the formation of red- and blue-shifted dihydrogen bonds387 respectively.
ratio values are 1.70 and 2.14 respectively. Meanwhile, a blue-shift of +16.3 cm−1 was detected in 27, although a reduction in absorption intensity of 0.15 has been also observed. In terms of intermolecular strength, it can be confirmed that the higher values of dihydrogen bond energy in 28, 29, and 30 are associated with larger red-shifts, whereas the weak intermolecular contacts are not related either to longer or shorter red-shifts. In fact, it is evident that the dihydrogen-bonded complex 27, in addition to complexes 28, 29, and 30, they coexist under the formation of red- and blue-shifted dihydrogen bonds387 respectively.
Regarding the ternary dihydrogen complexes (32 and 33), their dihydrogen bond distances are longer in comparison with the binary complexes. In a contrasting reflection on this but already discussed here, it is well-known that ternary hydrogen complexes formed by π or lone-electron pairs as proton acceptors are much more stable than binary complexes in terms of their intermolecular distances.213,378,397,398,639,645,646 Unfortunately, this observation is not verified here, but it is common because increases in double or bifurcate dihydrogen bond distances are well-known,555,556,647e.g., dihydrogen-bonded systems formed by ethyl cation and beryllium hydride.398 Nevertheless, it can be seen that the values of 2.1404 Å (32) and 1.7934 Å (33) for H−δ⋯H+δ are shorter than the sum of the van der Waals radii for the hydrogen atom, whose tabulated datum is 1.09 Å. Moreover, a novel interaction, the so-called alkali–halogen bond (the alkali being sodium), occurs between sodium and fluorine (Na⋯F) or chlorine (Na⋯Cl). Once again, this statement can be demonstrated by the van der Waals radii of Na (2.27 Å), F (1.47 Å), and Cl (1.75 Å), whose summed values of 3.74 Å (Na + F) and 4.02 Å (Na + Cl) are longer than the corresponding results of 2.4345 Å (32) and 2.9000 Å (33) shown in Fig. 15. In line with these findings, significant enhancements of 0.0386 Å and 0.0461 Å in the C–W bonds (W = F or Cl) were noted only in the ternary systems. On comparing binary with ternary complexes, however, a change in the sodium hydride could be evidenced. The Na–H bond shows a reduction in its length due to the formation of the binary complexes NaH⋯HCF3 (30) and NaH⋯HCCl3(31). Otherwise, an increase in the Na–H bond length was detected in the ternary systems 32 and 33.
 |
| | Fig. 15 Optimized geometries of the ternary complexes NaH⋯2(HCF3) (32) and NaH⋯2(HCCl3) (33) obtained from B3LYP/6-311++G(3df,3pd) calculations. | |
These contrasting structural profiles certainly lead to very distinct vibrational effects, which if hydrogen bonds, dihydrogen bonds,190,648–651 or even the alkali–halogen bonds are independently formed, the characterization of the chemical shifts either from red or blue origin is the spectroscopy cornerstone for identifying the formation of intermolecular interactions. There is a trend toward weakness in the bonds of the proton donors (H–X) of HCCl3 from NaH⋯HCCl3 up to the formation of 33. Thus, the stretch frequency of the oscillator νC–H is downward shifted by −81.00 cm−1 (δνC–H,33 − δνC–H,31) followed by an absorption intensity ratio of 2.4, whose values are also given in Table 9. On the other hand, HCF3 behaves as a blue-shifted proton donor because the value of its stretch frequency νC–H is upward shifted by +42 cm−1 (δνC–H,32 − δνC–H,30), which is a spectroscopic characteristic widely-established for fluoroform.376,377 In total corroboration with recent investigations,652 it is demonstrated that intermolecular complexes with shorter distances such as those observed in 33 are identified by red-shift events on the stretch frequencies of their proton donors. In contrast to this, stretch frequencies of longer intermolecular distances are shifted to blue, e.g., complex 31.
Table 9 Structural and vibrational parameters of the NaH⋯HCF3 (30) and NaH⋯HCCl3 (31) binary dihydrogen complexes, as well as for of the NaH⋯2(HCF3) (32) and NaH⋯2(HCCl3) (33) ternary alkali–halogen complexes obtained through the B3LYP/6-311++G(3dp,3df) calculationsa
| Parameters | Hydrogen complexes |
|---|
| (30) | (31) | (32) | (33) |
|---|
| Values of ν and I are given in cm−1 and km mol−1, respectively. Values of R and r are given in Å. X = CF3 or CCl3; W = F or Cl. Values of ν(Na–H−δ) and I(Na–H−δ) for the sodium hydride monomer are 1167.93 cm−1 and 215.93 km mol−1, respectively. Values of ν(H–X) and I(H–X) for the fluoroform monomer are 3121.66 cm−1 and 26.95 km mol−1, respectively. Values of ν(H–X) and I(H–X) for the chloroform monomer are 3187.48 cm−1 and 1.67 km mol−1, respectively. |
|---|
| δr(Na–H−δ) | −0.0006 | −0.0020 | 0.0785 | 0.0925 |
| δr(H–X) | 0.0046 | 0.0166 | 0.0027 | 0.0218 |
| δr(C–W) | 0.0063 | 0.0048 | 0.0386 | 0.0461 |
| R(H⋯H) | 1.9090 | 1.7250 | 2.1404 | 1.7934 |
| R(Na⋯W) | — | — | 2.4345 | 2.9000 |
| ν(Na–H−δ) | 1245.80 | 1265.62 | 1079.76 | 1031.31 |
| I(Na–H−δ) | 351.7 | 539.35 | 565.80 | 233.62 |
| δν(Na–H−δ) | +77.87 | +97.69 | −88.17 | −136.62 |
| I(Na–H−δ),c/I(Na–H−δ),m | 1.63 | 2.49 | 2.62 | 1.08 |
| ν(H–X) | 3039.01 | 2929.71 | 3080.95 | 2848.7 |
| I(H–X) | 57.52 | 510.93 | 52.47 | 1228.03 |
| δν(H–X) | −82.65 | −257.77 | −40.71 | −338.77 |
| I(H–X),c/I(H–X),m | 2.13 | 305.94 | 1.94 | 735.34 |
| ν(C–W) | 686.51 | 366.74 | 683.46 | 361.29 |
| I(C–W) | 20.91 | 0.68 | 43.60 | 0.61 |
| δν(C–W) | −6.69 | +1.03 | −9.74 | −3.42 |
| I(C–W),c/I(C–W),m | 1.70 | 4.53 | 3.55 | 4.06 |
| ν(H−δ⋯H+δ) | 91.71 | 97.68 | 69.16 | 81.28 |
| I(H−δ⋯H+δ) | 0.02 | 0.12 | 0.34 | 0.00(1) |
| ν(Na⋯W) | — | — | 47.54 | 35.89 |
| I(Na⋯W) | — | — | 1.06 | 0.65 |
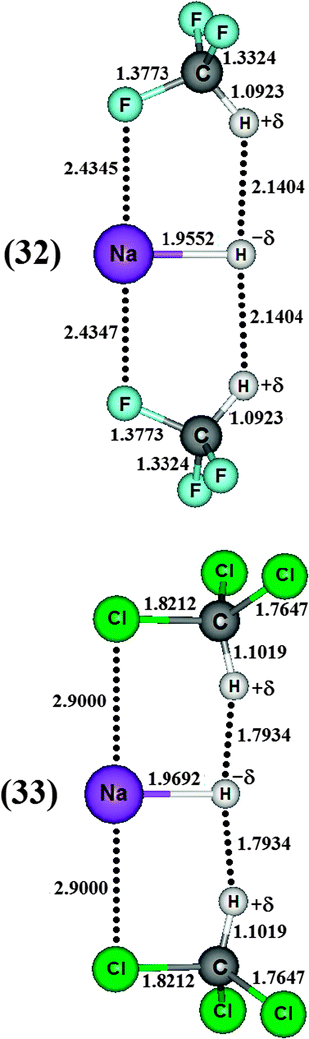
Besides the chemical shifts, Table 9 also lists the values for the new vibrational modes, commonly referred as intermolecular infrared stretch frequencies 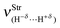 and their absorption intensities
and their absorption intensities 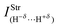 for the systems 30, 31, 32 and 33. Applying the same comparative procedure of analysis used for the binary and ternary systems, blue-shifts in sodium hydride were determined, whose values for 30 and 31 were +77.87 cm−1 and +97.69 cm−1, respectively. In contrast, red-shifts were identified in the H–X oscillator for which, although the value of −257.7 cm−1 in 31 is large, the result of −82.65 cm−1 for 30 is unusual because fluoroform is a standard proton donor model with blue-shift characteristic.381 Hence, some chemical shifts, such as the red of −6.69 cm−1 and the blue of +1.03 cm−1, are inconsistent with the observation of the systematic increase in the C–F and C–Cl bond lengths of 30 and 31. Although the C–F and C–Cl bond lengths are those with the largest variations in the ternary complexes, their stretch frequencies are slightly downward shifted, corresponding to lower red-shift values of −9.74 and −3.42 cm−1 for 32 and 33, respectively. These red shifts are associated with those observed for Na–H, in which the computed values of −88.17 and −136.62 cm−1 for 32 and 33 are surprising because blue-shift effects of +77.87 and +97.69 cm−1 have been previously observed for this oscillator upon the formation of the binary systems 30 and 31. All of these red- and blue-shift effects can be explained through structural analysis, i.e., elongation and reduction of the corresponding bond length. For both sodium hydride and halocarbon bonds (C–F or C–Cl) it should be noted that no correlation between the magnitude of the red- and blue-shifts and the absorption intensity ratios was found. Besides the downward shift in the stretch frequencies, the specialized literature reports that red-shift effects for proton donors are often characterized by an increase in the absorption intensities, which in this study may also occur in the case of blue-shift effects. In other words, there is no tendency for chemical shifts, of red or blue origin, according to the absorption intensity.
for the systems 30, 31, 32 and 33. Applying the same comparative procedure of analysis used for the binary and ternary systems, blue-shifts in sodium hydride were determined, whose values for 30 and 31 were +77.87 cm−1 and +97.69 cm−1, respectively. In contrast, red-shifts were identified in the H–X oscillator for which, although the value of −257.7 cm−1 in 31 is large, the result of −82.65 cm−1 for 30 is unusual because fluoroform is a standard proton donor model with blue-shift characteristic.381 Hence, some chemical shifts, such as the red of −6.69 cm−1 and the blue of +1.03 cm−1, are inconsistent with the observation of the systematic increase in the C–F and C–Cl bond lengths of 30 and 31. Although the C–F and C–Cl bond lengths are those with the largest variations in the ternary complexes, their stretch frequencies are slightly downward shifted, corresponding to lower red-shift values of −9.74 and −3.42 cm−1 for 32 and 33, respectively. These red shifts are associated with those observed for Na–H, in which the computed values of −88.17 and −136.62 cm−1 for 32 and 33 are surprising because blue-shift effects of +77.87 and +97.69 cm−1 have been previously observed for this oscillator upon the formation of the binary systems 30 and 31. All of these red- and blue-shift effects can be explained through structural analysis, i.e., elongation and reduction of the corresponding bond length. For both sodium hydride and halocarbon bonds (C–F or C–Cl) it should be noted that no correlation between the magnitude of the red- and blue-shifts and the absorption intensity ratios was found. Besides the downward shift in the stretch frequencies, the specialized literature reports that red-shift effects for proton donors are often characterized by an increase in the absorption intensities, which in this study may also occur in the case of blue-shift effects. In other words, there is no tendency for chemical shifts, of red or blue origin, according to the absorption intensity.
In relation to the stretch frequencies 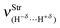 of the dihydrogen bonds, their values are in good agreement with the intermolecular distances. Note that R(H−δ⋯H+δ) is shorter in 31, although its
of the dihydrogen bonds, their values are in good agreement with the intermolecular distances. Note that R(H−δ⋯H+δ) is shorter in 31, although its 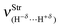 value of 97.68 cm−1 is slightly higher than the 91.71 cm−1 of 30. In satisfactory agreement with traditional bound complexes,630 the stretch frequencies
value of 97.68 cm−1 is slightly higher than the 91.71 cm−1 of 30. In satisfactory agreement with traditional bound complexes,630 the stretch frequencies 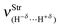 presented extremely low absorption intensities, which are spectroscopically inherent to any intermolecular interaction type. In conformity with the results of the intermolecular distances debated elsewhere, the ternary complexes presented weak dihydrogen bond stretch frequencies, whose values of 69.16 cm−1 (32) and 81.28 cm−1 (33) are accompanied by the lowest results for absorption intensity. With regard to the stretch frequencies of the alkali-halogen bonds, their values calculated by the B3LYP/6-311++G(3df,3pd) level associated with other levels show a direct correlation with the bond lengths,653 as can be seen by the linear coefficient R2 of 0.95 graphically illustrated in Fig. 16.
presented extremely low absorption intensities, which are spectroscopically inherent to any intermolecular interaction type. In conformity with the results of the intermolecular distances debated elsewhere, the ternary complexes presented weak dihydrogen bond stretch frequencies, whose values of 69.16 cm−1 (32) and 81.28 cm−1 (33) are accompanied by the lowest results for absorption intensity. With regard to the stretch frequencies of the alkali-halogen bonds, their values calculated by the B3LYP/6-311++G(3df,3pd) level associated with other levels show a direct correlation with the bond lengths,653 as can be seen by the linear coefficient R2 of 0.95 graphically illustrated in Fig. 16.
| | 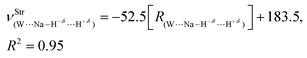 | (10) |
Since νStr(Na⋯F) and νStr(Na⋯Cl) are modes containing low absorption intensities in the range of 0.65–1.06 km mol−1, their localizations on the vibrational spectrum could only be obtained following preliminary structural identifications. Thus, although both alkali–halogen bonds Na⋯F and Na⋯Cl have been discovered, they are not sufficiently strongly bound to overestimate the stabilization of the ternary complexes. Therefore, the H−δ⋯H+δ dihydrogen bonds are still predominant in this regard.652
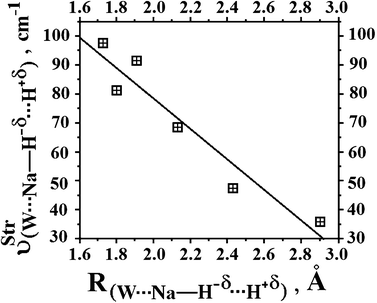 |
| | Fig. 16 Relationship between the values of the new vibrational modes and intermolecular distances obtained from B3LYP/6-311++G(3df,3pd) calculations. | |
The intermolecular QTAIM topology of the π⋯H hydrogen bonds
The bond paths (34–34′) presented in Fig. 17 show all the BCPs for the hydrogen complexes C2H2⋯HF and C2H2⋯2(HF). As mentioned previously, these BCPs are located by means of topological calculations,654–658 by which are computed the electron density (ρ) and its Laplacian field (∇2ρ),659 whose values are listed in Table 10. According to the ∇2ρ values, the H–F bonds are fully characterized as shared interactions in the 2(HF) dimer, as well as in the C2H2⋯HF and C2H2⋯2(HF) complexes. With regard to the hydrogen bonds, these interactions are defined as closed shell because the positive results of ∇2ρ are in the range of 0.026 up to 0.119 e ao−5. In terms of electronic density, the values obtained for all hydrogen bonds are very low. For instance, in the C2H2⋯2(HF) hydrogen complex, the respective values of electronic density for un-π⋯H and Fd⋯He are 0.023 and 0.029 e ao−3. However, the π⋯H interaction in the C2H2⋯HF hydrogen complex presents a lower electronic density of 0.018 e ao−3. This indicates that charge concentrations are located on the chemical bonds, whereas electronic depletion is related with the π⋯H interaction,660 as can be seen from the contour relief map (35) of C2H2⋯2(HF) shown in Fig. 18.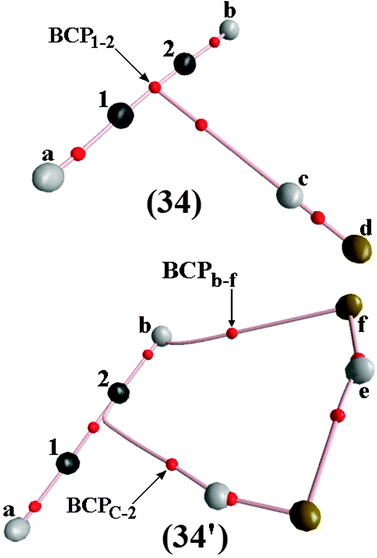 |
| | Fig. 17 QTAIM bond paths for the C2H2⋯HF (34) and the C2H2⋯2(HF) (34′) hydrogen-bonded complexes. | |
Table 10 Values of electronic densities ρ and Laplacians ∇2ρ for all interactions in the 2(HF), C2H2⋯HF (34), C2H2⋯2(HF) (34′), C2H4⋯HF (36), and C2H4⋯2(HF) (36′) hydrogen complexes obtained by using QTAIM calculationsa
| Parameters | Hydrogen complexes |
|---|
| 2(HF) | (34) | (34′) | (36) | (36′) |
|---|
| Values of ρ and ∇2ρ are defined as e ao−3 and e ao−5 respectively. |
|---|
| ρ(Hc–Fd) | 0.362 | 0.354 | 0.339 | 0.353 | 0.336 |
| ρ(He–Ff) | 0.358 | — | 0.349 | — | 0.345 |
| ρ(C1C2) | — | — | — | 0.342 | 0.343 |
| ρ(C1C2) | — | 0.411 | 0.410 | — | — |
| ρ(π⋯H) | — | 0.018 | — | 0.017 | 0.024 |
| ρ(un-π⋯H) | — | — | 0.023 | — | — |
| ρ(p-π⋯H) | — | — | — | — | — |
| ρ(Fd⋯He) | 0.024 | — | 0.029 | — | 0.040 |
| ρ(Ff⋯Hb) | — | — | 0.007 | — | 0.021 |
| ∇2ρ(Hc–Fd) | −2.776 | −2.65 | −2.51 | −2.63 | −2.30 |
| ∇2ρ(He–Ff) | −2.722 | — | −2.64 | — | −2.43 |
| ∇2ρ(C1C2) | — | — | — | −1.02 | −1.00 |
| ∇2ρ(C1C2) | — | −1.24 | −1.23 | — | — |
| ∇2ρ(π⋯H) | — | 0.053 | — | 0.050 | 0.019 |
| ∇2ρ(un-π⋯H) | — | — | 0.061 | — | — |
| ∇2ρ(p-π⋯H) | — | — | — | — | — |
| ∇2ρ(Fd⋯He) | 0.107 | — | 0.119 | — | 0.137 |
| ∇2ρ(Ff⋯Hb) | — | — | 0.026 | — | 0.141 |
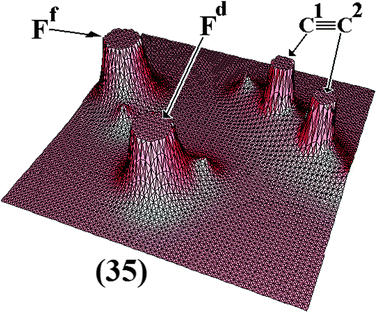 |
| | Fig. 18 Relief map of the C2H2⋯2(HF) (35) hydrogen-bonded complex. | |
The structural results and the infrared analysis for the C2H2⋯2(HF) complex suggest that a new hydrogen bond may be formed, R(Ff⋯Hb) and ν(Ff⋯Hb), but this interaction has not been confirmed yet. Nevertheless, the QTAIM calculations have categorically identified the Ff⋯Hb interaction, as can be seen by the BCPb–f (34′) illustrated in Fig. 17. Comparing the electron density of π⋯H, Fd⋯He, and Ff⋯Hb, the lowest value of 0.007 e ao−3 for Ff⋯Hb defines it as a closed shell interaction, as stated by Popelier, Bader, Matta and others.661–665 Moreover, the typical condition of the Laplacian field guarantees the formation of Ff⋯Hb, i.e., ∇2ρ > 0. For the formation of the complex C2H2⋯2(HF), the QTAIM calculations provide essential information regarding the π⋯H interaction, which thus requires special attention. In the discussion of the structure of the C2H2⋯HF hydrogen complex, it was noted that the π⋯H interaction is formed exactly at the midpoint of the C1C2 bond of the acetylene and QTAIM calculations, locating the bond path666 and BCP1–2 (34), as illustrated in Fig. 17, corroborates this.603 On the other hand, the QTAIM calculations for the C2H2⋯2(HF) hydrogen complex show that π⋯H seems not to be well defined in the T-shaped form. Surely, the uncommon un-π⋯H interaction, hitherto represented as Hc⋯C2, is characterized by an electronic density of 0.023 e ao−3 and a Laplacian field of 0.061 e ao−5, although it is formed by the interaction between the Hc hydrogen and the C2 atoms, as can seen in the BCPc–2 of Fig. 17 (34′). Some of these results have also been reported by Grabowski and Ugalde667 in an investigation in which it is explained whether carbon atoms or BCP can be proton acceptors. In confrontation with this finding, Tsuzuki et al.668 also have performed theoretical calculations at MP2 level as well as more sophisticated ones such as CCSD(T) in works elaborated to investigate the nature of these proton acceptor types. Since then the possibility of π charge transference to the σ* anti-bonding orbital of the HF molecule in the C2H2⋯2(HF) complex has been practically ruled out, instead there is a prominent redistribution of the electron density throughout the ‘intramolecular’ C2H2⋯2(HF) complex under the following conditions: σ → σ* (C2–Hc), σ → σ* (Fd–He), and σ → σ* (Ff–Hb).603
Fig. 19 shows the bond paths and BCP for the C2H4⋯HF (36) and C2H4⋯2(HF) (36′) hydrogen-bonded complexes. Once again, the results organized in Table 10 reveal that the electronic densities on the CC bonds are slightly changed, i.e., values of 0.342 and 0.343 e ao−3 for the C2H4⋯HF binary and C2H4⋯2(HF) ternary complex, respectively. On the other hand, there is evidence of a significant variation in the electronic density on the hydrofluoric acid, whose values are 0.353 and 0.336 e ao−3 for Hc–Fd from C2H4⋯HF to C2H4⋯2(HF), and 0.345 e ao−3 for He–Ff in the ternary complex. In terms of intermolecular interactions, the electronic density values are extremely low, which vary between 0.017 and 0.024 e ao−3. In association with the positive values of the Laplacian, these electronic densities indicate that a hydrogen bond is formed within each intermolecular BCP,669 as can be seen from the BCPc–π in 36 and 36′ shown in Fig. 19. Although, as mentioned previously, a ternary interaction between the second hydrofluoric acid (He–Ff) and hydrogen atoms (Ha and Hb) of ethylene may be structurally feasible, the identification of the BCPb–f and BCPa–f (see Fig. 19 once again) and therein the calculation of the unique Laplacian value of 0.141 e ao−3 characterizes both of the interactions Ff⋯Ha and Ff⋯Hb as hydrogen bonds. By taking into account the framework of the QTAIM atomic basis,670 the hydrogen bonds possess charge density concentrated in separate nuclei (donor and acceptor of protons),671 as can be seen in the relief map (see Fig. 20) of the C2H4⋯2(HF) (37) ternary complex.
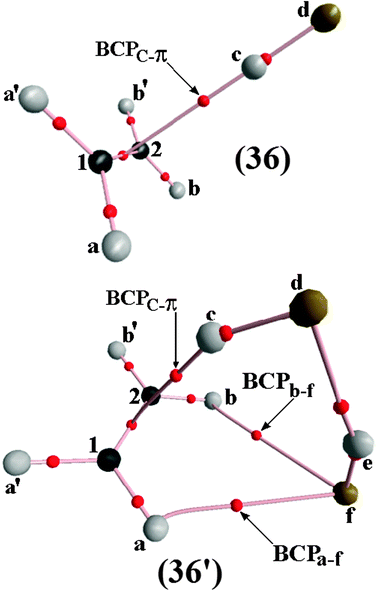 |
| | Fig. 19 Bond paths for the C2H4⋯HF (36) and C2H4⋯2(HF) (36′) hydrogen bond complexes. | |
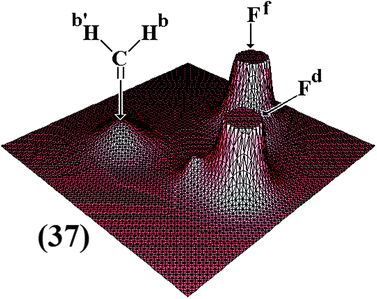 |
| | Fig. 20 Relief map for the C2H4⋯2(HF) (37) hydrogen-bonded complex. | |
The acquisition of the QTAIM protocol and its application is currently widespread in many scientific researches around the world.672–675 Precisely, the ability of the QTAIM protocol to model the chemical bonds via quantum mechanical arguments676,677 distinguish it as one of the most efficient theoretical procedures for studying intermolecular systems, such as hydrogen bond complexes and their spectral phenomena of shifted frequencies.678,679 In regards to the C2H4⋯HCF3, C2H3(CH3)⋯HCF3, and C2H2(CH3)2⋯HCF3 complexes, Table 11 gathers the results for their main QTAIM topological parameters: the electron density ρ and its Laplacian ∇2ρ. Very briefly, the low values of the electronic densities in the range of 0.006–0.007 e ao−3 and the positive Laplacian results explain the formation of hydrogen bonds. As for the hydrogen bond strengths whose structural parameters were previously discussed,680,681 the highest intermolecular electronic density was found in 40, whereas the lowest were computed for 38 and 39. In the latter case, the electronic density is related to the C2⋯Hc improper hydrogen bond, in which the carbon acts as a proton acceptor, a phenomenon that is not usually observed. Fig. 21 plots the BCPs and bond pathways of the electronic density of complexes 38, 39 and 40. It can be seen that π⋯Hc hydrogen bonds are formed in 38 and 40 through the characterization of the BCPc–π, whereas an improper C2⋯Hc interaction is typical of 39, when the BCPc–2 has been located. The chemical bond analyses with high charge concentration whose Laplacian results are negative or which, according to the QTAIM, are shared interactions show that the reduction in electronic density in the CaCb bond is one of the most intense charge transference effects upon the formation of the C2⋯Hc and π⋯Hc hydrogen bonds. The charge transference is understood as one of the electronic cornerstones for the formation of hydrogen bonds,682 and this can be demonstrated by the accumulation of electronic density on the C–Hc bonds of fluoroform (see Table 11), where, in contrast to the monomer state, an increase, of 0.303(3), 0.303(1), and 0.303(2) e ao−3, respectively, can be observed in (38), (39), and (40).
Table 11 Values of the electronic densities ρ and Laplacians ∇2ρ for the main bonds of the C2H4⋯HCF3 (38), C2H3(CH3)⋯HCF3 (39), and C2H2(CH3)2⋯HCF3 (40) complexes obtained by using the QTAIM calculationsa
| Parameters | Hydrogen complexes |
|---|
| (38) | (39) | (40) |
|---|
| Values of ρ and ∇2ρ are given in e ao−3 and e ao−5 respectively. Values of ρ and ∇2ρ for the CaCb bond of the ethylene, propylene, and t-butylene monomers are given in parentheses. Values of ρ and ∇2ρ for the H–C bond of the HCF3 monomer are 0.302 e ao−3 and −1.112 e ao−5 respectively. |
|---|
| ρ(Hc⋯π) | 0.006(0) | — | 0.007(0) |
| ∇2ρ(Hc⋯π) | 0.015(5) | — | 0.015(7) |
| ρ(Hc⋯C2) | — | 0.006(9) | — |
| ∇2ρ(Hc⋯C2) | — | 0.015(4) | — |
| ρ(CaCb) | 0.3430 (0.3437) | 0.3417 (0.3428) | 0.3410 (0.3419) |
| ∇2ρ(CaCb) | −1.025 (−1.028) | −1.0110 (−1.017) | −1.0921 (−1.004) |
| ρ(C–Hc) | 0.303(3) | 0.303(1) | 0.303(2) |
| ∇2ρ(C–Hc) | −1.138 | − 1.139 | −1.139 |
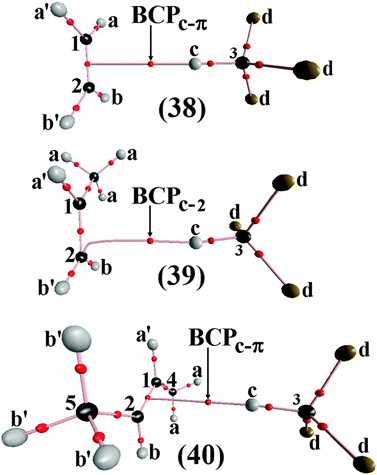 |
| | Fig. 21 BCP and bond paths of the C2H4⋯HCF3 (38), C2H3(CH3)⋯HCF3 (39), and C2H2(CH3)2⋯HCF3 (40) complexes. | |
The intermolecular QTAIM topology of the H−δ⋯H+δ dihydrogen bonds
By consulting the literature, the application of QTAIM to the studies of intermolecular interactions via hydrogen has yielded very interesting results,683–688 mainly about the interpretation of vibrational modes in halogen–hydride interactions as per a very recent work signed by Mohajeri et al.689 For the dihydrogen-bonded complexes re-examined here, being binary or ternary, the topological parameters should be ideally suitable for interpretation of their spectral shifts. Thus, Fig. 22 presents the bidimensional graphs of the Laplacian attributed to the electronic densities of the BeH2⋯HCl binary complex 41, as well as of all isomeric series 42, 43, and 44 of the BeH2⋯2(HCl) ternary complex. It can be seen that ∇2ρ > 0 for all intermolecular interactions, while ∇2ρ < 0 for H–Cl bonds. Thus, not only in terms either of the Laplacian or electronic density ρ, the QTAIM topology can also be discussed using through the atomic charges q(Ω), whose values are summarized in Table 12. In relation to 42, this complex is stabilized by (H−δ⋯H+δ)-(42)-A and (Cl−δ⋯H+δ)-(42)-B, by which the existence of a dihydrogen–hydrogen system type is unveiled. The values of the intermolecular electronic densities vary from 0.001 to 0.018 e ao−3, and in fact this is a wide relative variation, especially when the second contact is taken into account, because the R(Cl−δ⋯H+δ) distance of 3.262 Å is longer than the sum of the van der Waals radii,597,601,602,690–693 which for chlorine and hydrogen sums to 3.0 Å. |
| | Fig. 22 Illustration of the Laplacians of the BeH2⋯HCl binary dihydrogen complex (41), as well as of the ternaries (42), (43), and (44) dihydrogen isomers. | |
Table 12 QTAIM topological parameters for the binary (41) and ternary isomers (42), (43), and (44) of the BeH2⋯2(HCl) dihydrogen complexa
| BCP | QTAIM topological parameters |
|---|
| ρ | ∇2ρ | ΔQ(Ω),(H–Cl) |
|---|
| Values of ρ and ∇2ρ for the hydrochloric acid (monomer) are 0.252 e ao−3 and −0.775 e ao−5 respectively. Values of ΔQ(Ω) are given in atomic units (a.u.). |
|---|
| (H–Cl)-(41) | 0.246 | −0.761 | −0.030 |
| (H–Cl)-(42)-A | 0.245 | −0.756 | −0.034 |
| (H–Cl)-(42)-B | 0.252 | −0.777 | −0.002 |
| (H–Be)-(42) | 0.103 | 0.122 | — |
| (H–Be)-(42) | 0.100 | 0.127 | — |
| (H–Cl)-(43)-A | 0.249 | −0.767 | −0.023 |
| (H–Cl)-(43)-B | 0.249 | −0.767 | |
| (H–Be)-(43) | 0.104 | 0.123 | — |
| (H–Be)-(43) | 0.977 | 0.128 | — |
| (H–Cl)-(44)-A | 0.248 | −0.765 | −0.026 |
| (H–Cl)-(44)-B | 0.248 | −0.765 | −0.026 |
| (H–Be)-(44) | 0.101 | 0.126 | — |
| (H–Be)-(44) | 0.101 | 0.126 | — |
| (H−δ⋯H+δ)-(41) | 0.017 | 0.038 | — |
| (H−δ⋯H+δ)-(42)-A | 0.018 | 4.16 × 10−2 | — |
| (Cl⋯H+δ)-(42)-B | 0.001 | 4.16 × 10−2 | — |
| (H−δ⋯H+δ)-(43)-A | 0.013 | 0.030 | — |
| (H−δ⋯H+δ)-(43)-B | 0.013 | 0.030 | — |
| (H−δ⋯H+δ)-(44)-A | 0.015 | 0.035 | — |
| (H−δ⋯H+δ)-(44)-B | 0.015 | 0.035 | — |
However, the electronic density of (H−δ⋯H+δ)-(42)-A presents a very similar result to (H−δ⋯H+δ)-(41), indicating that 42 tends to form a binary complex, rather than a ternary one. With regard to the structure of 43, the formation of double interactions between the two molecules of hydrochloric acid directly aligned on the beryllium hydride demonstrates that H−δ can be an ‘electronegative hydrogen’.694 Although the electronic density value of 0.013 e ao−3 for both the (H−δ⋯H+δ)-(43)-A and (H−δ⋯H+δ)-(43)-B dihydrogen bonds are characteristic of van der Waals contacts (∼10−2–10−3 ao3),695,696 these interactions are essentially dihydrogen bonds.472 Thereby, system 43 is thus formed by two dihydrogen bonds, which provide the formation of three proton donor centres: H−δ(H–Be)-(43), H+δ(H–Cl)-(43)-A, and H+δ(H–Cl)-(43)-B. For this reason, a bifurcate dihydrogen bond is observed. In terms of the structure of 44, two dihydrogen bonds were formed, although in separate hydride elements (H−δ). Even though (H−δ⋯H+δ)-(44)-A and (H−δ⋯H+δ)-(44)-B are not the strongest interactions, the electronic density of 0.015 e ao−3 and Laplacian of 0.035 e ao−5 indicate that 44 is formed by four proton donor sites: H+δ(H–Cl)-(44)-A, H+δ(H–Cl)-(44)-B, H−δ(H–Be)-(44), and H−δ(H–Be)-(44).
The remaining proton donors, HCN and HNC, have been also used for the study of the ternary dihydrogen-bonded complexes, in which, the QTAIM calculations were performed for the purpose of topological characterization of the BeH2⋯2(HCN) (45) and BeH2⋯2(HNC) (46) bifurcate structures as well as the HCN⋯BeH2⋯HCN (47) and CNH⋯BeH2⋯HNC (48) linear ones illustrated in the Fig. 23. Governed by the virial theorem of the QTAIM methodology,697–699 by which the kinetic (G) and potential (U) energies are embodied into the electronic energy density (H) according to eqn (11), the topological integrations are carried out in order to model a surface map of ρ (see Fig. 24) with identification of the charge density pathways.7,502b,666,700–702
Analysis of the data gathered in Table 13 reveals that no variation has occurred in electronic density when the monomers and the dihydrogen complexes are compared; for example, the values of ρ for the H+δ–C and H+δ–N are 0.295 and 0.343 e ao−3, but on the bifurcate 45 and 46 these values are 0.295 and 0.343 e ao−3, as well as 0.294 and 0.341 e ao−3 on the linear 47 and 48, respectively. On the other hand, a reduction of the electronic density of 0.102 e ao−3 on the Be–H−δ bond was observed, mainly in the BeH2⋯2(HCN) and BeH2⋯2(HNC) bifurcate complexes, whose electronic density values are 0.098 and 0.096 e ao−3. Moreover, low quantities of ρ and positive values of ∇2ρ are observed for the whole series of H−δ⋯H+δ dihydrogen bonds. Thus, the BeH2⋯2(HCN), BeH2⋯2(HNC), HCN⋯BeH2⋯HCN, and CNH⋯BeH2⋯HNC complexes can be identified as intermolecular systems because charge density is accumulated in separate nuclei, which are the hydrogen atoms (H+δ) and the hydride elements (H−δ).703,704
Table 13 Values of the QTAIM topological parameters (ρ and ∇2ρ) for the BeH2⋯2(HX) and XH⋯BeH2⋯HX (X = CN and NC) ternary dihydrogen complexesa
| BCP | QTAIM topological parameters |
|---|
| ρ | ∇2ρ |
|---|
| Values of ρ and ∇2ρ are given in e ao−3 and e ao−5 respectively. |
|---|
| (H−δ⋯H+δ)-(45)-A | 0.006 | 0.016 |
| (H−δ⋯H+δ)-(45)-B | 0.005 | 0.015 |
| (H−δ⋯H+δ)-(46)-A | 0.010 | 0.028 |
| (H−δ⋯H+δ)-(46)-B | 0.010 | 0.028 |
| (H−δ⋯H+δ)-(47)-A | 0.008 | 0.021 |
| (H−δ⋯H+δ)-(47)-B | 0.008 | 0.021 |
| (H−δ⋯H+δ)-(48)-A | 0.013 | 0.034 |
| (H−δ⋯H+δ)-(48)-B | 0.013 | 0.034 |
| (H+δ–C)-(45)-A | 0.295 | −1.176 |
| (H+δ–C)-(45)-B | 0.295 | −1.176 |
| (Be–H−δ)-(45) | 0.098 | 0.125 |
| (H+δ–N)-(46)-A | 0.343 | −2.217 |
| (H+δ–N)-(46)-B | 0.343 | −2.217 |
| (Be–H−δ)-(46) | 0.096 | 0.129 |
| (H+δ–C)-(47)-A | 0.294 | −1.178 |
| (H+δ–C)-(47)-B | 0.294 | −1.178 |
| (Be–H−δ)-(47) | 0.101 | 0.124 |
| (H+δ–N)-(48)-A | 0.341 | −2.223 |
| (H+δ–N)-(48)-B | 0.341 | −2.223 |
| (Be–H−δ)-(48) | 0.101 | 0.128 |
 |
| | Fig. 23 Illustration of the bond paths of the bifurcate BeH2⋯2(HCN) (45) and BeH2⋯2(HNC) (46) as well as the linear HCN⋯BeH2⋯HCN (47) and CNH⋯BeH2⋯HNC (48) ternary dihydrogen-bonded complexes. | |
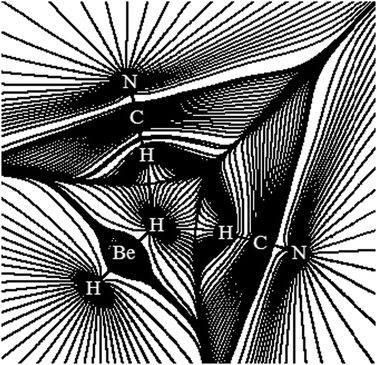 |
| | Fig. 24 Map of the gradient vector of the electron density for the BeH2⋯2(HCN) ternary dihydrogen-bonded complex. | |
One of the most expert and influential chemists in hydrogen bonding studies, Leland Cullen Allen,705 often has declaimed in its reports that the analysis of intermolecular properties should be performed by accuracy, careful criticism, prudent decision, and prospects.706 In order to improve this conjecture, several results point to a close relationship between the structural and topological properties.707,708 For example, one of the most important investigations, in either intramolecular hydrogen bonds,709–713 dihydrogen bonds,714 or π hydrogen bonds,715,716 is the prediction of intermolecular strength.133,300,717–721 Some time ago, vibrational shifts in proton donors were used to rationalize and improve the prediction of hydrogen bond strength.722,723 By the fact that the QTAIM electronic densities can be used to estimate the variations in the stretch frequencies,724 it is prudent to expound this statement in the analysis of red-shift effects in the systems reviewed here. Fig. 25 shows a graph illustrating the relationship between the red- and blue-shifts and the variations in electronic densities within the BCP of the Be–H−δ bond of the BeH2⋯2(HCN), BeH2⋯2(HNC), HCN⋯BeH2⋯HCN, and CNH⋯BeH2⋯HNC complexes. This relationship is validated by eqn (12) presented below:
| | 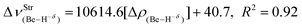 | (12) |
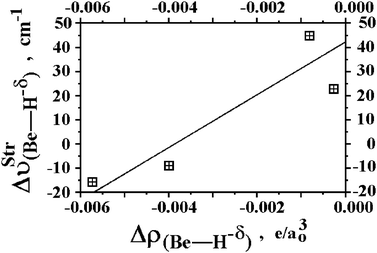 |
| | Fig. 25 Relationship between the harmonic vibrational shifts (red or blue) and variations on the QTAIM electronic densities for the (Be–H−δ) bonds upon the formation of the ternary dihydrogen-bonded complexes BeH2⋯2(HCN), BeH2⋯2(HNC), HCN⋯BeH2⋯HCN, and CNH⋯BeH2⋯HNC. | |
It is perceived that the more accentuated reductions on the electronic density of the proton donors of BeH2⋯2(HCN) and BeH2⋯2(HNC) are directly related to their red-shifts, whereas blue-shift events can be explained by slight changes on the charge density. Only for mention, HCN⋯BeH2⋯HCN and CNH⋯BeH2⋯HNC are representative complexes for this latter statement. The concentration of electron density is clearly the source of the bond strength, and in this case, small Δρ values explain the blue-shift effect, indicating that the concentration of charge density is not substantially perturbed or, in other words, the strength of the Be–H−δ bond is preserved. As for the structural parameters, a shortening of the Be–H−δ bond length occurs due to the formation of BeH2⋯2(HCN) and BeH2⋯2(HNC), which is in good agreement with the vibrational analysis and QTAIM topography results already discussed. However, despite the success of electronic density quantification and Laplacian analysis in studies of intermolecular systems, the atomic charge725 is another important QTAIM parameter in this regard, so much that it has been used in the structuring of methods employed to compute the atomic charge distribution, such as the Voronoi Deformation Density (VDD) approach,726 for instance. In contrast to the theoretical formalism,727–731 the rigorous concept of the QTAIM atomic charge is founded on a purely physical basis,732–740 namely as the own electronic density:741
| | 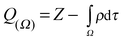 | (13) |
In a paper published in 1970,742 Hassel argued for the importance of charge transfer during the formation of interatomic interactions. Some years later, Orville-Thomas et al.743–746 have observed that the vibrational properties of hydrogen complexes could be predicted by measurement of charge transfer. Therefore, according to the values of the QTAIM atomic charges Q(Ω) for the series of BeH2⋯HX (X = F, Cl, CN, and CCH) complexes, the linear coefficient R2 of 0.99 obtained in concordance with eqn (14) would appear to describe a good association between the charge transfer and red-shifts of the HX molecules, as can be seen in Fig. 26. In comparison with the observations described by Solimannejad and Alkorta321 regarding the spectrum shifting and charge transference amounts of dihydrogen-bonded complexes, it can also be observed that the high values of Q(Ω) accord well with large displacements and vice-versa. In spite of this, similar results were obtained for π hydrogen bonded complexes formed by acetylene and monoprotic acids,273,274 albeit with the application of the Charge–Charge Flux-Overlap (CCFO)537,747–750 to compute the corrected charges.751
| | | ΔνStrH–X = 7324.4[ΔQ(Ω), H–X] + 61.1, R2 = 0.99 | (14) |
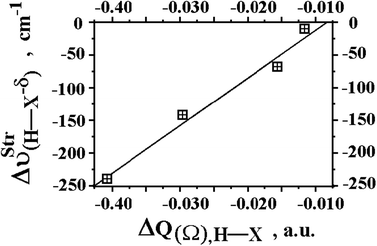 |
| | Fig. 26 Relationship between intermolecular charge transfer (ΔQ(Ω),H–X) and the red-shift effects (ΔνStrH–X) of the monoprotic acids for the BeH2⋯HX dihydrogen-bonded complexes with X = F, Cl, CN, and CCH using B3LYP/6-311++G(3d,3p) calculations. | |
At this current time, the QTAIM theory is considered one of the most used computational schemes in studies of electronic structure752–755 because the positive values of the Laplacians of ρ (∇2ρ) accompanied by low quantities of electron density bring the ideal information to characterize the chemical bond, in particular its strength and shape.756 By this insight, the results organized in Table 14 indicate that the H−δb⋯H+δ interaction can be identified as an intermolecular contact of a closed-shell, or, more specifically, a dihydrogen bond. The contour maps and BCPs of 49 and 50 can be seen in Fig. 27, which also illustrates the lines of electronic density and molecular pathway between the interactive atoms, such as for example the H−δb of BeH2 and MgH2, as well as the H+δ of the fluoroform. Since the magnitude of the charge density on the BCP provides a measure of the interaction strength on H−δb⋯H+δ, it is fair to say that 49 is the weakest bound dihydrogen complex, whereas 50, 51, and 52 are the strongest ones (see relief maps for 51 and 52 illustrated in the Fig. 28). In terms of shared interactions, it can be noted that the electronic charge density on the C–H+δ bond of the fluoroform is higher in comparison with the value calculated for the H−δb⋯H+δ dihydrogen bonds. The accumulation of charge density can be explained by the ∇2ρ results, which are less negative for the 49, 50, 51, and 52 complexes than for the fluoroform subunit, in which ∇2ρ is −2.124 e ao−5. Moreover, it is worthwhile to emphasize that the value of the charge density on C–H+δ is higher in comparison with the isolated fluoroform, in which the ρ value is 0.302 e ao−3.
Table 14 Values of the QTAIM topological parameters of the BeH2⋯HCF3 (49), MgH2⋯HCF3 (50), LiH⋯HCF3 (51), and NaH⋯HCF3 (52) dihydrogen complexesa
| Parameters | Dihydrogen complexes |
|---|
| (49) | (50) | (51) | (52) |
|---|
| Values of ρ and ∇2ρ are given in e ao−3and e ao−5 respectively. Values of ΔQ(Ω) are given in atomic units (a.u.). Values of ρ and ∇2ρ of the C–H+δ bond of the HCF3 monomer are given in parentheses. |
|---|
| ρ(Hb−δ ⋯H+δ) | 0.007 | 0.010 | 0.015 | 0.016 |
| ∇2ρ(Hb−δ⋯H+δ) | 0.018 | 0.023 | 0.032 | 0.033 |
| ρ(C–H+δ) | 0.313 (0.302) | 0.312 (0.302) | 0.311 (0.302) | 0.310 (0.302) |
| Δρ(C–H+δ) | 0.011 | 0.010 | 0.009 | 0.008 |
| ∇2ρ(C–H+δ) | −1.261 (−2.124) | −1.265 (−2.124) | −1.273 (−2.124) | −1.266 (−2.124) |
| ΔQ(Ω) | −0.014 | −0.025 | −0.046 | −0.056 |
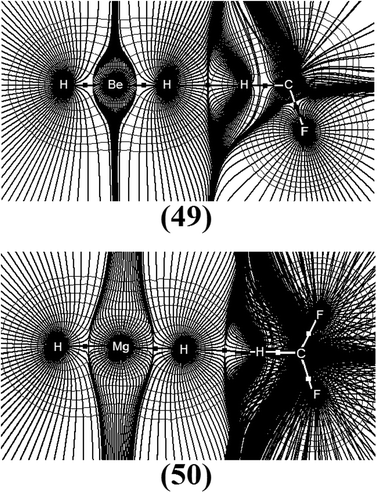 |
| | Fig. 27 Contour lines of the electronic density (thin), molecular pathways (bold) and BCPs (little squares) of the BeH2⋯HCF3 (49) and MgH2⋯HCF3 (50) dihydrogen-bonded complexes obtained from QTAIM calculations. | |
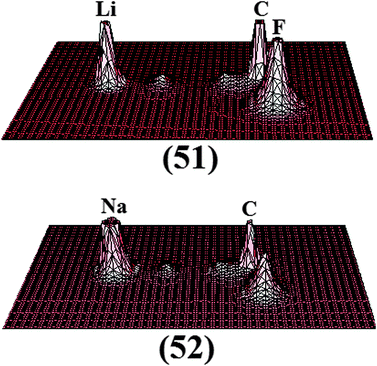 |
| | Fig. 28 Relief maps of the LiH⋯HCF3 (51) and NaH⋯HCF3 (52) dihydrogen-bonded complexes obtained from QTAIM integrations. | |
In Table 14 are listed the results of the total atomic charge transference ΔQ(Ω) in the HCF3 molecule. First, it can be seen that a systematic trend for ΔQ(Ω) has been found, where the values vary from −0.014 a.u. in 49 to −0.056 a.u. in 50. Although the strength of the H−δb⋯H+δ dihydrogen bond was initially measured using the attraction energies ΔEC(BSSE + ΔZPE), the computation of ΔQ(Ω) suggests that a good correlation between ΔQ(Ω) and ΔEC(BSSE + ΔZPE) may be obtained. In other words, the stronger bound the complex is, such as 52 for instance, the higher the quantity of charge density is, that is transferred from the HOMO orbital of NaH to the LUMO of HCF3.757 Although it is by means of the computation of charge transfer that a true description of how the 49, 50, 51 and 52 dihydrogen complexes are strongly or weakly bound, in terms of infrared vibrational modes, Fig. 29 can illustrate a close relationship between the charge transfer ΔQ(Ω) and the vibrational shifts 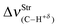 in the C–H+δ bond of the fluoroform.758 This result is supported by eqn (15):
in the C–H+δ bond of the fluoroform.758 This result is supported by eqn (15):
| | 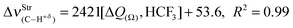 | (15) |
It can thus be assumed that the blue-shift of C–H+δ in 49 and red-shifts in 50, 51, and 52 can be explained by lower and higher charge transference amounts, whose values are −0.014, −0.025, −0.046 and −0.056 e.u., respectively. Surely, this accords well with a charge transfer study registered by Li,759 whose conclusion affirms that the elongation (red-shift) and contraction (blue-shift) of the bond lengths of the proton donors are caused by high and low quantities of charge transferred, respectively.
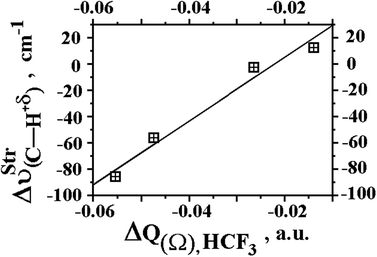 |
| | Fig. 29 Relationship between variation on stretch frequencies 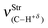 of fluoroform (HCF3) and ΔQ(Ω) charge transfers of the BeH2⋯HCF3 (49), MgH2⋯HCF3 (50), LiH⋯HCF3 (51), and NaH⋯HCF3 (52) complexes using B3LYP/6-311++G(3df,3pd) calculations. of fluoroform (HCF3) and ΔQ(Ω) charge transfers of the BeH2⋯HCF3 (49), MgH2⋯HCF3 (50), LiH⋯HCF3 (51), and NaH⋯HCF3 (52) complexes using B3LYP/6-311++G(3df,3pd) calculations. | |
Hybrid systems: unsaturated clouds, dihydrogen bonds, and alkali-halogen bonds
The universe of proton acceptors and donors for intermolecular interactions is immense, and even so the discovering of new hydrogen bond types is currently observed in experimental and theoretical researches.760–767 In a brief comment about vibrational displacements, there are some intermolecular systems presenting peculiar phenomena,641 such as rehybridization, structural reorganization, and hyperconjugation.768–772 For the last one, the structure of the ethyl cation (see Fig. 3) is a classical example, in which the theoretical modelling has demonstrated successfully its preferential configuration.773 In comparison with traditional hydrohalic acids,203,212,213,326,327 QTAIM calculations revealed that the ethyl cation acts as a typical proton donor upon the formation of the π⋯H+ cationic hydrogen bond.774 In accordance with Grabowski,775 the π-delocalization exerts a great influence upon hydrogen bond complexations, but unsurprisingly the ethyl cation also serves as proton donor in dihydrogen bond interactions.645,646 With this insight, the Fig. 30 shows the optimized geometries of the C2H5+⋯2(MgH2) (53) and 2(C2H5+)⋯MgH2 (54) dihydrogen-bonded complexes, in which it can be seen that two MgH2 molecules attack the non-localized hydrogen (H+) of the ethyl cation (C2H5+). It is clear that the length of 1.699 Å of the Mg–H−δ bond in MgH2 is drastically elongated after complexation, e.g., to 1.736 and 1.723 Å in the 53 and 54 complexes respectively. In terms of intermolecular interactions, or more specifically the distances of the R(H−δ⋯H+δ) dihydrogen bonds, although the (54) complex presents the shorter R(H−δ⋯H+δ) value of 1.807 Å, it is through this result that the most significant deformations of the MgH2 structure can be observed, as mentioned above in regards to the Mg–H−δ bond length.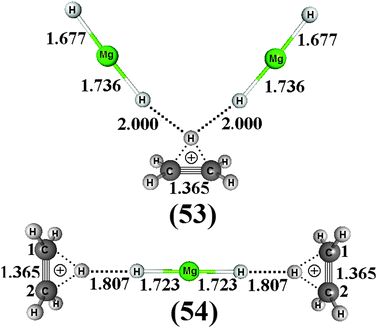 |
| | Fig. 30 Optimized geometries of the C2H5+⋯2(MgH2) (53) and 2(C2H5+)⋯MgH2 (54) dihydrogen-bonded complexes using the BHandHLYP/6-31G(d,p) level of theory. | |
It should be pointed out that not only the structural analysis, but also the vibrational spectrum is an useful criterion for indicating whether intermolecular systems are formed or not simply by analyzing the red- or blue-shifts in stretch frequencies. Thus, Table 15 lists the main vibrational modes of the 53 and 54 complexes. Initially, the argument that the Mg–H bond lengths are the most important alterations resulting from the formation of the 53 and 54 complexes is, in fact, also demonstrated by the analysis of the stretch frequencies (νStr) and absorption intensities (IStr). By comparing the 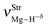 value of 1638.3 cm−1 for the MgH2 monomer, a red-shift of −66 cm−1 can be verified after complexation of 53, followed by a drastic increase in the absorption intensity ratio of 998.5. In 54, a red-shift of −80.2 cm−1 is firstly observed due the distorted polarizability on the MgH2 monomer, which is not active in the infrared spectrum, i.e., the value of the absorption intensity
value of 1638.3 cm−1 for the MgH2 monomer, a red-shift of −66 cm−1 can be verified after complexation of 53, followed by a drastic increase in the absorption intensity ratio of 998.5. In 54, a red-shift of −80.2 cm−1 is firstly observed due the distorted polarizability on the MgH2 monomer, which is not active in the infrared spectrum, i.e., the value of the absorption intensity 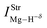 is zero. In view of this, it can be stated that the red-shifts of −66 and −80.2 cm−1 are in agreement with the distance values for the dihydrogen bonds, which reveal that the shorter distance of R(H−δ⋯H+δ) provides the appearance of the larger red-shifts, in particular when the modes of the Mg–H−δ bond in 54 are analyzed. Moreover, the stretch frequencies
is zero. In view of this, it can be stated that the red-shifts of −66 and −80.2 cm−1 are in agreement with the distance values for the dihydrogen bonds, which reveal that the shorter distance of R(H−δ⋯H+δ) provides the appearance of the larger red-shifts, in particular when the modes of the Mg–H−δ bond in 54 are analyzed. Moreover, the stretch frequencies 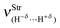 do not appear to accord with the dihydrogen bond distance strengths, i.e., the more weakly bound complex 53 has the larger
do not appear to accord with the dihydrogen bond distance strengths, i.e., the more weakly bound complex 53 has the larger 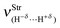 of 125.1 cm−1, whereas 71.9 cm−1 is related to the 54 complex.
of 125.1 cm−1, whereas 71.9 cm−1 is related to the 54 complex.
Table 15 Values of the stretch frequencies (νStr) and absorption intensities (IStr) of the C2H5+⋯2(MgH2) (53) and 2(C2H5+)⋯MgH2 (54) dihydrogen-bonded complexes by using the BHandHLYP/6-31G(d,p) calculationsa
| Parameters | Dihydrogen complexes |
|---|
| (53) | (54) |
|---|
| The values of νStr and IStr are given in cm−1 and km mol−1 respectively. The values of νStr and IStr of the MgH2 monomer are given in parentheses. |
|---|
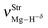 | 1572.3 (1638.3) | 1558.1 (1638.3) |
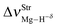 | −66 | −80.2 |
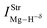 | 998.5 (0.0) | 0.0(0.0) |
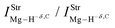 | 998.5 | 0.0 |
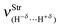 | 125.1 | 71.9 |
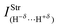 | 42.6 | 0.0 |
The QTAIM analysis has been currently embodied in studies of carbocation compounds.776 By taking into account the structural electronic analysis on the ethyl cation performed by Ren et al.,777 the QTAIM topological modelling applied in this situation can be used to interpret the formation of the 53 and 54 dihydrogen-bonded complexes. In this context, the values of the topological descriptors are listed in Table 16, and by analyzing the electronic densities computed by means of the QTAIM approach, the ρ(H−δ⋯H+δ) dihydrogen bonds of 53 and 54 were thus identified. However, the values of 0.036 and 0.029 e ao−5 of the Laplacian also indicate that these dihydrogen bonds are formed, or in other words, these interactions are seen as a closed-shell model. Taking this finding into account, it should be pointed out that the H−δ⋯H+δ dihydrogen bonds were also identified using the ρ results of 0.015 and 0.019 e ao−3. In practice, it can be said that the non-localized hydrogen (H+) of the ethyl cation binds with the hydrogen hydride H−δ of MgH2, but a very interesting finding is that H−δ can support a double dihydrogen bond interaction in 54, as can be seen in Fig. 31, which shows the QTAIM contour line maps for the electronic densities.778 Besides the properties of the ethyl cation and related systems have been well elucidated respectively by Kubas779 and Li et al.780 in an overview, it can be said that the ethyl cation is an efficient proton donor for dihydrogen bonds.
Table 16 Values of the QTAIM topological parameters of the C2H5+⋯2(MgH2) (53) and 2(C2H5+)⋯MgH2 (54) dihydrogen complexesa
| Parameters | Dihydrogen complexes |
|---|
| (53) | (54) |
|---|
| Values of ρ and ∇2ρ are given in e ao−3 and e ao−5 respectively. |
|---|
| ρ(H−δ⋯H+δ) | 0.015 | 0.019 |
| ∇2ρ(H−δ⋯H+δ) | 0.036 | 0.029 |
 |
| | Fig. 31 Visualization of the contour lines of the electronic densities of the 2(C2H5+)⋯MgH2 (54) dihydrogen-bonded complex using QTAIM calculations. | |
In addition, as QTAIM applicability reaches beyond the pure theoretical context, as reported by Abramov,495 Matta and Arabi,496 and others,781 once again it should be highlighted that the spectroscopic nature of the harmonic vibrational modes previously discussed herein is one of the greatest goals in this regards. In this scenery, Grabowski354 idealized an interpretation for the chemical shifts (red or blue) of proton donors (X–H with X = CF3 or CCl3) based on BCPs localized along their bonds. It was established that variations in the atomic radii of carbon and hydrogen are the reason for the red and blue shift effects as follows: δrX > δrH means red-shift while δrX < δrH means blue-shift. Meanwhile, a correlation with Natural Bond Orbital (NBO)782,783 analysis is suitable to explain the variation on these atomic radii quoted above. Based on the results listed in Table 17, all red-shift values for the H–X oscillator are explained by the increase in the carbon radius in comparison with hydrogen because the carbon s-character (see Table 18) of 30.52% is not considerably enhanced in comparison with the results of 32.63% (30), 32.94% (31), 32.72% (32), and 33.97% (33). Moreover, the carbon polarization of 56.78% is increased to 60.39% (30), 66.52% (31), 60.63% (32), and 67.40% (33). In other words, higher polarization values are associated with larger atomic radii, and hence are the cornerstone to explaining the red shift events in relation to the H–X bond. The blue-shifts in Na–H occur with δrNa > δrH, which is unusual since the very slight increase in the s-character from 99.18% (NaH monomer) to 99.48% in the 30 and 31 complexes cannot be associated with a significant decrease in polarization (% Na) in ranges of 13.23% (monomer) to 9.95% (30) and 9.23% (31). Therefore, the blue shift effect and length reduction are clearly evident for the Na–H bond.
Table 17 Values of the atomic radii determined from the location of the BCP of the monomers, binary dihydrogen complexes, and ternary alkali–halogen systemsa
| Systems | Atomic radii |
|---|
| rNa(Na–H) | rH(Na–H) | rC(C–W) | rW(C–W) | rC(C–H) | rH(C–H) |
|---|
| All values are given in Å. The atomic radii variations are listed in parentheses. |
|---|
| NaH | 0.8772 | 0.9969 | — | — | — | — |
| HCF3 | — | — | 0.4571 | 0.8816 | 0.7144 | 0.3563 |
| HCCl3 | — | — | 0.7790 | 0.9965 | 0.7043 | 0.3567 |
| 30 | 0.9965 (0.1192) | 0.8796 (−0.1172) | 0.4586 (0.0014) | 0.8865 (0.0048) | 0.7360 (0.0216) | 0.3581 (0.0017) |
| 31 | 0.9963 (0.1191) | 0.8783 (−0.1185) | 0.7751 (−0.0039) | 1.0049 (0.0084) | 0.7388 (0.0345) | 0.3577 (0.0010) |
| 32 | 1.0188 (0.1408) | 0.9343 (−0.0625) | 0.8963 (0.0171) | 1.7065 (0.0187) | 0.7373 (0.0229) | 0.3347 (−0.0216) |
| 33 | 1.0239 (0.1467) | 0.9432 (−0.0537) | 0.7838 (0.0047) | 1.0375 (0.0410) | 0.7463 (0.0420) | 0.3359 (−0.0208) |
Table 18 Values of the NBO parameters of the 30 and 31 binary dihydrogen complexes, as well as the estimated ones of the 32 and 33 ternary alkali–halogen complexes obtained through the B3LYP/6-311++G(3dp,3df) level of theorya
| Parameters | Complexes |
|---|
| 30 | 31 | 32 | 33 |
|---|
| p and s represent the polarization and s-character of the H–X bond, whose values are given in %. |
|---|
| p(H–X) | 60.39 | 66.52 | 60.63 | 67.40 |
| s(H–X) | 32.63 | 32.94 | 32.72 | 33.97 |
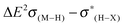 | 42.34 | 63.62 | 29.28 | 78.49 |
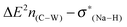 | — | — | 16.88 | 33.72 |
The values for the topography descriptors computed following the QTAIM procedure for the 30–33 systems are listed in Table 19. In agreement with the positive Laplacian values of 0.0328 and 0.0423 e ao−5 for the H⋯H dihydrogen bonds of the 30 and 31 binary systems, the Na–H bonds also present the same closed character, whose ∇2ρ values are 0.1304 and 0.1307 e ao−5. There are, thus, two simultaneous noncovalent interactions in 30 and 31, namely H−δ⋯H+δ and Na–H. Moreover, this contact leads to depletions in the electronic density, with values of 0.0165–0.0341 e ao−3 which are very low. In recent papers, Grabowski et al.527,784–786 predicted the covalent character of several binary and ternary system types by assuming a relationship (−G/U) between the positive values of the kinetic electronic energy density (G) and negative values of the potential electronic energy density (U) computed by the QTAIM routine through the localization of the BCP. Not surprisingly, considering the high dihydrogen bond energy of −23.10 kJ mol−1 and the shortest intermolecular distance of 1.7250 Å, it should be expected that the covalence partially dominates the intermolecular zoo of H−δ⋯H+δ in 31 instead of a noncovalent profile. Thus, it is stated that the dihydrogen bond systems formed by chloroform are not only more stable or strongly bound, but their intermolecular frameworks belong to the selective class of partially covalent systems.
Table 19 QTAIM (BCP) topological descriptors of the binary dihydrogen complexes (30 and 31), as well as of the ternary alkali-halogen ones (32 and 33) obtained through the Bader's calculations routinea
| Parameters | Complexes |
|---|
| 30 | 31 | 32 | 33 |
|---|
| Values of ρ and ∇2ρ are given in e ao−3 and e ao−5, respectively. Values of G and U are given in atomic units (a.u.). X = CF3 or CCl3; W = F or Cl. |
|---|
| ρ(Na–H) | 0.0341 | 0.0340 | 0.0301 | 0.0290 |
| ∇2ρ(Na–H) | 0.1304 | 0.1307 | 0.1094 | 0.1068 |
| G(Na–H) | 0.0314 | 0.0315 | 0.0263 | 0.0254 |
| U(Na–H) | −0.0303 | −0.0304 | −0.0252 | −0.0241 |
| –G(Na–H)/U(Na–H) | 1.0380 | 1.0399 | 1.043 | 1.0526 |
| ρ(H–X) | 0.3104 | 0.2978 | 0.3118 | 0.2943 |
| ∇2ρ(H–X) | −1.2660 | −1.1610 | −1.2834 | −1.1386 |
| G(H–X) | 0.0228 | 0.0307 | 0.0224 | 0.0301 |
| U(H–X) | −0.3622 | −0.3507 | −0.3657 | −0.3450 |
| −G(H–X)/U(H–X) | 0.0630 | 0.0863 | 0.0613 | 0.0874 |
| ρ(C–W) | 0.2817 | 0.1916 | 0.2576 | 0.1744 |
| ∇2ρ(C–W) | −0.4129 | −0.2553 | −0.3959 | −0.1979 |
| G(C–W) | 0.3229 | 0.0658 | 0.0270 | 0.0600 |
| U(C–W) | −0.7491 | −0.1956 | −0.0639 | −0.1695 |
| −G(C–W)/U(C–W) | 0.4311 | 0.3364 | 0.4225 | 0.3540 |
| ρ(H−δ⋯H+δ) | 0.0165 | 0.0240 | 0.0136 | 0.0243 |
| ∇2ρ(H−δ⋯H+δ) | 0.0328 | 0.0423 | 0.0244 | 0.0374 |
| G(H−δ⋯H+δ) | 0.0078 | 0.0114 | 0.0058 | 0.0104 |
| U(H−δ⋯H+δ) | −0.0075 | −0.0122 | −0.0054 | −0.0115 |
| −G(H−δ⋯H+δ)/U(H−δ⋯H+δ) | 1.047 | 0.9333 | 1.056 | 0.9036 |
| ρ(Na⋯W) | — | — | 0.0131 | 0.0101 |
| ∇2ρ(Na⋯W) | — | — | 0.0775 | 0.0452 |
| G(Na⋯W) | — | — | 0.0163 | 0.0092 |
| U(Na⋯W) | — | — | −0.0132 | −0.0071 |
| −G(Na⋯W)/U(Na⋯W) | — | — | 1.2200 | 1.2885 |
In the case of the ternary complexes, an extension of the parameters discussed in relation to the binary complexes is carried out, although there are four interactions of which two are dihydrogen bonds and two alkali-halogen bonds. In practice, one analysis is performed for the dihydrogen bonds and other to the alkali-halogen bonds. The H−δ⋯H+δ dihydrogen bonds were identified by means of the intermolecular electronic density values ρ(H−δ⋯H+δ) of 0.0136 e ao−3 (32) and 0.0243 e ao−3 (33) accompanied by their corresponding positive Laplacians ∇2ρ(H−δ⋯H+δ) of 0.0244 e ao−5 (32) and 0.0374 e ao−5 (33). In structural analysis, the presence of the Na⋯F and Na⋯Cl alkali-halogen bonds has been estimated empirically based on the van der Waals radii.787 Herein, these interactions are quantum-mechanically validated through the illustration of Fig 32, as well as by means of the ρ(Na⋯F) and ρ(Na⋯Cl) quantities of 0.0131 and 0.0101 e ao−3, as well as the positive ∇2ρ(Na⋯F) and ∇2ρ(Na⋯Cl) values of 0.0775 and 0.0452 e ao−5, respectively. The Na–H bonds remain noncovalent, as can be seen by the ρ(Na–H) values of 0.0301 (32) and 0.0290 e ao−3 (33). In addition, the Laplacian fields are positive, which suggests noncovalence in Na–H. Generally, if the −G/U relationship is taken into account, the interactions of dihydrogen bonds or sodium hydride bonds seem to be noncovalent in nature. As an exception to this, the H−δ⋯H+δ dihydrogen bond in 33 is partially covalent given the −G(H−δ⋯H+δ)/U(H−δ⋯H+δ) value of 0.9036. Even if dihydrogen bond pairing is formed in a hydride, its intermolecular partial covalent character is not lost in comparison with the binary system (31). Comparing the −G(H−δ⋯H+δ)/U(H−δ⋯H+δ) values of 0.9333 (31) and 0.9036 (33), clearly NaH⋯2(HCCl3) is intermolecularly more covalent than NaH⋯HCCl3, i.e., the contribution of the potential electronic energy density is greater in the dihydrogen bonds of the ternary complex. If the bound distances are evaluated, it can be affirmed that R(H−δ⋯H+δ) of 31 < R(H−δ⋯H+δ) of 30. For the alkali–halogen bonds, a similar tendency was observed, which is mainly related to the highly noncovalent character. The −G/U values of 1.2200 (32) and 1.2885 (33) indicate a definitive weakness for Na⋯F and Na⋯Cl. In this regard, Fig. 33 plots a linear relationship between the −G/U results and the intermolecular distances computed at the B3LYP/6-311++G(3df,3pd) level of theory.
| | | −G/U = 0.32[R(W⋯Na–H−δ⋯H+δ)] + 0.37, R2 = 0.96 | (16) |
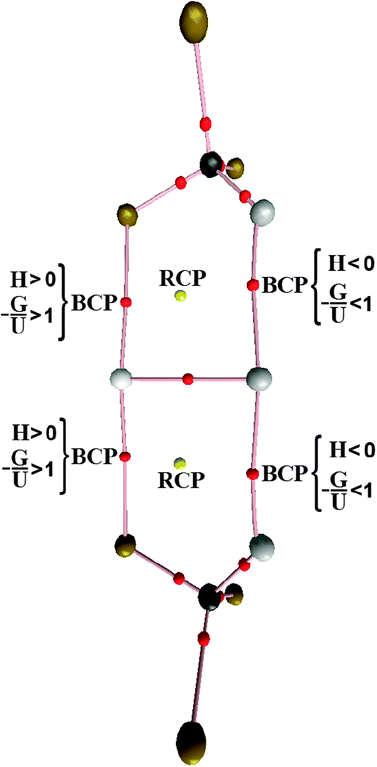 |
| | Fig. 32 Bond paths, BCP, RCP, and covalent characterization by means of H = G + U (eqn (11)) for the 33 complex. | |
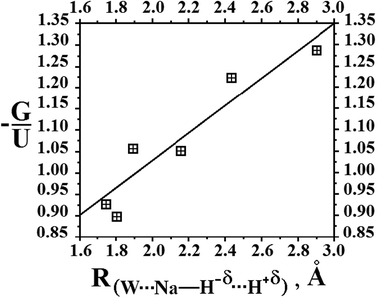 |
| | Fig. 33 Relationship between the −G/U ratios and intermolecular distances obtained from B3LYP/6-311++G(3df,3pd) calculations. | |
Although the interactions investigated herein presented very distinct energy values, individual examinations should be performed in order to verify their strengths. Some time ago, a new measure of the interaction strength based on structural data in association with the QTAIM topology was proposed.722 Subsequently, we have suggested that good relationships can also be modelled through spectroscopic parameters instead of structural ones.678 An equation to predict the interaction strength is conceived by means of the variation in the bond length or in the chemical shift (red or blue) of the proton donor (HX) combined with ρ and ∇2ρ.
| | 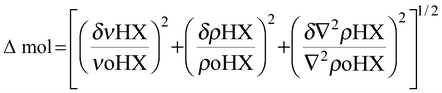 | (17) |
The molecular complex parameter (Δmol) can be fitted by structural (δr) or spectroscopy (δν) data. Besides the QTAIM parameters (δρHX = |ρoHX − ρHX| and δ∇2ρHX = |∇2ρoHX − ∇2ρHX|), it can be noted that Δmol was estimated via vibrational chemical shifts (δν = |νoHX − νHX|).788 Up to now, the interaction energies estimated herein can be considered indirect values, but this equation can validate them all. Fig. 34 illustrates the relationship between the values for the dihydrogen bond energies789 of 30, 31 and others, such as the dihydrogen-bonded complexes LiH⋯HCF3 (see Fig. 35), LiH⋯HCCl3, and NaH⋯HF.181,216
| | 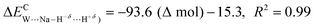 | (18) |
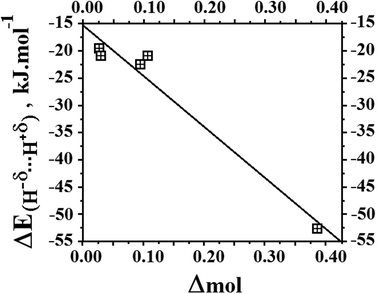 |
| | Fig. 34 Relationship between the values of the estimated intermolecular energies and molecular parameter (Δmol). | |
 |
| | Fig. 35 Illustration of the relief map of the electronic density of the LiH⋯HCF3 dihydrogen complex. | |
Although the ternary complexes are formed by a symmetrical (cooperative) energy distribution, the simultaneous presence of dihydrogen bonds and alkali–halogen leads to the quantification of their interaction energies through the application of eqn (17) being unapproachable. Thus, the binary systems are more suitable in this regard because their intermolecular energies are directly incorporated into a single interaction strength and, as such, other systems were inserted with the purpose to reinforcing the statement debated herein. As is well-known, the dihydrogen complex NaH⋯HF is strongly bonded and almost covalent, either based on its intermolecular energy of −52.15 kJ mol−1 or even the −G/U ratio of 0.5350. Among the complexes examined in this study, only those formed by chloroform yielded a partially covalent character, and therefore they are much less stabilized than NaH⋯HF. In other words, the covalent character is not systematic in terms of interaction energy, and a careful examination of the QTAIM topological parameters is mandatory in this situation. In summary, it can be reported that the remaining bonds (F–C, Cl–C, and C–H) are covalent or topologically shared. The Laplacian values are negative and vary between −0.3959 and −1.2845 e ao−5 in 32, and −1.1386 and −0.1979 e ao−5 in 33. In line with this, the −G/U values are significantly lower, thus indicating the pure covalence of these bonds. Finally, for the high and low electronic density centers located in BCPs along the intermolecular surfaces examined in this study, it may be necessary to discuss another topological descriptor, namely the Ring Critical Point (RCP), whose values for the ternary complex 33 are shown in Fig. 32. The algebraic difference between BCP and RCP is related to the Poincaré-Hopf relationship790–794 in terms of non-zero curvatures of ρ at the critical point, yielding the coordinates (3,−1) and (3,+1). According to the QTAIM framework, BCP is an interatomic pathway containing electronic density without chemical bond characteristics and RCP is defined by an intersection of interatomic surfaces that form a ring. According to Castillo et al.,526 from the lowest G values of 0.0049 a.u. and 0.0043 a.u. it can be easily noted that a slower charge density flux is observed for RCP compared with BCPs. Moreover, the electronic densities of 0.0070 and 0.0067 e ao−3 are smaller and the Laplacian fields tend toward annulment, i.e., ρBCP > ρRCP and ∇2ρBCP > ∇2ρRCP.
The relationship between QTAIM descriptors and quantum chemical calculations
In spite of the growth of the theoretical chemistry in the last decades,795 one of the benchmarks in this scientific area is absolutely the development of new and modern computational algorithms.796–804 Specifically in the context of the chemical bond and intermolecular interactions,55,805–815 a traditional task is to use a series of basis sets in order to reproduce accurately the available experimental data,816–817 in general those derived from structural and vibrational analysis.203 Regarding the QTAIM characterization of intermolecular interactions,212,818–829 in particular the hydrogen bonds, it has been demonstrated that Pople's split-valence double-ζ incremented by polarization functions830,831 with higher angular momentum are not appropriate for basis sets to describe the electronic density computed through the QTAIM protocols.832 Otherwise, valence and diffuse functions fairly do it well. Also, it must be important to emphasize that the variability of the basis sets is outweighed by the high-level quantum chemical calculations using Dunning's correlation-consistent basis sets,833 such as aug-cc-pVTZ834 that has been considered efficient to reproduce the characteristics of intermolecular interactions.810,835–840There are some works focusing on the application of several ab initio calculations and DFT functionals in order to improve the description of intermolecular interactions via QTAIM calculations,841 and currently B3LYP, X3LYP, MP2 and CCSD528,842,843 are equivalent to reproduce experimental results and predict topological properties, as highlighted by Varadwaj.605 Aiming to elucidate the relationship or even an explanation for whether QTAIM is seriously dependent on the theoretical level,672 Matta reported the following paper entitled: “How dependent are molecular and atomic properties on the electronic structure method? Comparison of Hartree-Fock, DFT, and MP2 on a biologically relevant set of molecules”.844 One of the greatest conclusions of this work is the agreement between HF and MP2, wherein the validation of the HF non-correlative results published as yet becomes reasonable. In fulfilment with this, it has been also documented that the QTAIM results obtained from both HF and DFT (depicted by the B3LYP functional) correlate well with those computed at the MP2 level. Of course even the intermolecular systems formed through van der Waals forces845–847 are appraised by the development of novel density-functional approaches, which have provided excellent results848–857 many of them better than MP2.617 In this assertion, Jabłoński and Palusiak858 have investigated the relationship between the basis sets and QTAIM descriptors used for the analysis of covalent bonds, wherein it has been demonstrated that HF and B3LYP levels give very similar QTAIM results with respect to the basis set, whereas 6-31G and DZ-quality yielded unsatisfactory results beyond than those obtained by aug-cc-pVTZ. Furthermore, complete basis sets containing high polarization character such as 6-311++G(2df,2pd) and 6-311++G(3df,3pd) are useful for predicting QTAIM parameters. About the hydrogen bonds formed in the F3CH⋯NH3, F3CH⋯NCH, and FCCH⋯NH3 complexes, however, another study revealed that QTAIM intermolecular properties computed at the BCP are practically independent from both the method and the basis set.859
Concluding remarks
Among several types of intermolecular interactions with their molecular properties clearly established,860–872 nowadays great progresses in hydrogen bonds studies have been published.873–881 The model Y⋯HX (where Y and X are elements with higher electronegativity than hydrogen) was evolved,882 but regardless of the features of the chemical elements, Y is usually assumed to be a region with high electron density, which may have π bonds instead of typical electronegative elements.883 Although it is widely known that the π⋯H hydrogen bond is formed exactly at the midpoint of the CC bond of the C2H2⋯HF complex, the application of the QTAIM theory to C2H2⋯2(HF) reveals an unusual characteristic of the π⋯H interaction. Instead of forming at the midpoint of the CC bond of the acetylene, the π⋯H hydrogen bond in the C2H2⋯2(HF) complex is formed by the interaction between the C2 (acetylene CC) and the Hc (first hydrofluoric acid molecule) atoms. Furthermore, it has been demonstrated that a new hydrogen bond, Ff⋯Hb, arises. Thus, the C2H2⋯2(HF) hydrogen complex can be considered an ‘intramolecular system’ formed by three hydrogen bonds, Hc⋯C2, Fd⋯He, and Ff⋯Hb. With regard to the theoretical study of the molecular properties and infrared spectra of the C2H4⋯HCF3, C2H3(CH3)⋯HCF3, and C2H2(CH3)2⋯HCF3 complexes, B3LYP/6-311++G(d,p) calculations reveal a shortening of the C–H bonds of the fluoroform. Besides the red-shifted hydrogen bonds, it has also been observed that the stretch frequencies of their C–H bonds are shifted to higher values after complexation. The charge transfer mechanism has been confirmed by QTAIM calculations, where the electronic density computation has shown a reduction in charge density on the π bonds accompanied by an increase in the H–C bond of the fluoroform.In terms of the dihydrogen bond, it has been demonstrated that its strength in the BeH2⋯HX complexes follows the order: BeH2⋯HF > BeH2⋯HCl > BeH2⋯HCN > BeH2⋯HCCH. This systematic interaction strength was evaluated by means of the molecular changes observed in the isolated subunits in comparison to those arising from the formation of the BeH2⋯HX dihydrogen-bonded complexes. Such changes have been described in terms of elongation of the bond lengths and vibrational red-shift effects on the monoprotic acids. A suitable explanation of these structural and vibrational phenomena has been found in the direct relationship between the QTAIM charge transfer and the red-shift effects of the proton donor molecules. The formation of ternary and binary dihydrogen-bonded complexes has been also re-examined. According to analysis of the optimized geometries, examination of the molecular topology by using QTAIM calculations, and the interpretation of the infrared spectrum, the most stable complexes derived from BeH2⋯2(HCl) are linear and formed by four locations of proton donors.
As for other proton donors, such as X = HCN and HNC, the geometries of the BeH2⋯2HX and XH⋯BeH2⋯HX dihydrogen complexes are well defined, with the NCH⋯BeH2⋯HCN and CNH⋯BeH2⋯HNC linear complexes exhibiting shorter dihydrogen bond distances, whereas the bifurcate BeH2⋯2(HCN) and BeH2⋯2(HNC) exhibit longer ones. More interesting deformations have thus been found in linear complexes, such as red-shift effects in the proton donor bonds H+δ–C and H+δ–N, as well as blue-shift in the hydride bonds Be–H−δ. These vibrational effects have been interpreted using the QTAIM parameters, where the stable electronic density conserved the strength of the Be–H−δ bonds and therefore caused an upward shift in the vibrational oscillator. Furthermore, the blue- and red-shifts in dihydrogen-bonded complexes formed by BeH2⋯HCF3, MgH2⋯HCF3, LiH⋯HCF3, and NaH⋯HCF3 have also been debated. The analysis of equilibrium geometries at the B3LYP/6-311++G(3df,3pd) level of theory has revealed that the main structural deformation occurring in these systems is related to the C–H+δ bond of fluoroform, which is considered a proton donor. Likewise, changes in hydride compounds have also been examined, although in the case of the infrared spectrum, significant frequency shifts were only observed on the C–H+δ bond, which took the form of blue-shifts in BeH2⋯HCF3, and red-shifts in the MgH2⋯HCF3, LiH⋯HCF3, and NaH⋯HCF3 dihydrogen-bonded complexes. Apart from characterizing the H−δb⋯H+δ dihydrogen bonds, the QTAIM approach has been used to complement analysis of the strength of the C–H+δ bond by using topological parameters and its relationship with the spectroscopy changes. It has been demonstrated that low and high charge transfer values correlate well with blue- and red-shift dihydrogen-bonded complexes respectively. This suggests that these vibrational effects are correctly interpreted whether the quantity of charge transfer between the HOMO and LUMO orbitals is measured, which, in this case, takes the form of a transfer from the hydride compounds (BeH2, MgH2, LiH, and NaH) to the fluoroform proton donor (HCF3). Regarding the ternary dihydrogen complexes, red-shift effects were verified for the more strongly bound complexes whereas the blue-shifts of weaker ones are caused by increases in the polarization of the carbon atom of the proton donors HCF3 or HCCl3. As such, and in agreement with the QTAIM foundations, the H⋯H dihydrogen bonds revealed partially covalent intermolecular profiles with strongly bonded complexes, such as NaH⋯HCCl3 and NaH⋯2(HCCl3) and, although being less stable than a pure intermolecular covalent complex like NaH⋯HF, the covalence evaluated by the kinetic and potential electronic energy densities plays a decisive role in the rationalization of the interaction strength of dihydrogen systems.
Finally, the structures of uncommon hydrogen-bonded complexes formed by a non-classical ethyl cation and BeH2 hydrides have been pointed out. The complexes formed were treated as a hybrid system owing to the existence of the π cloud and hydride bonds, Mg–H−δ, of the ethyl cation and magnesium hydride, respectively. As mentioned earlier, two dihydrogen complexes were investigated rather than one: C2H5+⋯2(MgH2) and 2(C2H5+)⋯MgH2. Analysis of the structural parameters and vibrational stretch modes revealed that the dihydrogen distances and the new vibrational modes not correlate. However, QTAIM calculations were applied to investigate the formation of bifurcate and linear dihydrogen bonds in competition with the hyperconjugation effect on the ethyl cation, a phenomenon that is responsible for keeping the structure in a stable condition.
Summary and outlook
In recent paper, Weinhold and Klein884 inquire: “What is a hydrogen bond?” Notwithstanding after a half-century of hydrogen bond studies,885–887 the scientific community elected this interaction as one of the most important issues in chemistry, physics, and biology.888–911 In specific studies of intermolecular interactions,912 it is also unanimously agreed that hydrogen bonds are a central topic,913 although recently dihydrogen bonds have also received a great deal of attention.914 The quantum chemical description of the hydrogen bonds915 has been efficiently performed using computational methods, in particular ab initio, DFT, and semiempirical. However, Blanco et al.916,917 adapted the quantum mechanics of QTAIM918–922 and its definition of atoms as open systems923 to interpret atomic interactions, in which the essence of the hydrogen bonding could be examined in terms of classical parameters, such as Coulomb and exchange potentials,924 as well as electronic correlation.925 In spite of all the quantum methods known, the high level researches on the most diversified systems make QTAIM a particular chapter of the theoretical chemistry, although Bader has stated that some reflections about the reality of the QTAIM analysis should be done.926 Independently, it is by the charge density distribution analysis that QTAIM is extremely useful in the study of the chemical bonds and molecular stability, as well as its application to estimate the interaction strength133,300,927–930 or to predict covalent features.788,931–934 For these reasons the representative group of π hydrogen complexes, dihydrogen complexes, as well as hybrid π dihydrogen complexes revisited here show that QTAIM can be used to study the vibrational shifts in the proton donor bonds of these systems.935,936Acknowledgements
I would like to thanks FAPESB, CAPES, and CNPq Brazilian Funding Agencies. This work is dedicated to the memory of Prof. Richard Frederick William Bader (1931-2012).†Notes and references
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- H. S. Rzepa, Nat. Chem., 2009, 1, 510–512 Search PubMed.
- A. I. Kitaygorodsky, Tetrahedron, 1961, 14, 230–236 Search PubMed.
- P. Chakrabarti and R. Bhattacharyya, Prog. Biophys. Mol. Biol., 2007, 95, 83–137 CrossRef CAS.
- N. J. Zondlo, Nat. Chem. Biol., 2010, 6, 567–568 Search PubMed.
- T. W. Martin and Z. S. Derewenda, Nat. Struct. Biol., 1999, 6, 403–406 CrossRef CAS.
- S. J. Grabowski, Annu. Rep. Prog. Chem., Sect. C, 2006, 102, 131–165 RSC.
- E. Arunan, Curr. Sci., 2007, 92, 17–18 Search PubMed.
- M. Jablonski and M. Solà, J. Phys. Chem. A, 2010, 114, 10253–10260 Search PubMed.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS.
- J. Overgaard and B. B. Iversen, Struct. Bonding, 2012, 146, 53–74 Search PubMed.
- D. Kaur and S. Khanna, Struct. Chem., 2012, 23, 755–764 Search PubMed.
- M. Hennemann, J. S. Murray, P. Politzer, K. E. Riley and T. Clark, J. Mol. Model., 2012, 18, 2461–2470 CrossRef CAS.
- E. Arunan, Curr. Sci., 1999, 77, 1233–1235 Search PubMed.
-
(a) S. Sarkhel and G. R. Desiraju, Proteins: Struct., Funct., Genet., 2004, 54, 247–259 Search PubMed;
(b) A.-N. Bondar and S. H. White, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 942–950 Search PubMed;
(c) T. Kajikawa, K. Kataoka and T. Sakurai, Biochem. Biophys. Res. Commun., 2012, 422, 152–156 Search PubMed;
(d) V. Agarwal, R. Linser, U. Fink, K. Faelber and B. Reif, J. Am. Chem. Soc., 2010, 132, 3187–3195 CrossRef CAS.
- J. J. Dannenberg, J. Am. Chem. Soc., 2010, 132, 3229–3229 Search PubMed.
- M. A. Saeed, B. M. Wong, F. R. Fronczek, R. Venkatraman and M. A. Hossain, Cryst. Growth Des., 2010, 10, 1486–1488 CrossRef CAS.
- M. A. Hossain, M. A. Saeed, F. R. Fronczek, B. M. Wong, K. R. Dey, J. S. Mendy and D. Gibson, Cryst. Growth Des., 2010, 10, 1478–1481 CrossRef CAS.
- J. U. Bowie, Curr. Opin. Struct. Biol., 2011, 21, 42–49 CrossRef CAS.
- M. Işiklan, M. A. Saeed, A. Pramanik, B. M. Wong, F. R. Fronczek and M. A. Hossain, Cryst. Growth Des., 2011, 11, 959–963 CrossRef CAS.
- A. Pramanik, D. R. Powell, B. M. Wong and M. A. Hossain, Inorg. Chem., 2012, 51, 4274–4284 CrossRef CAS.
- F. Musso, P. Mignon, P. Ugliengo and M. Sodupe, Phys. Chem. Chem. Phys., 2012, 14, 10507–10514 RSC.
- M. H. Abraham and W. E. Acree, Phys. Chem. Chem. Phys., 2012, 14, 7433–7440 RSC.
- A. Oleinikova and I. Brovchenko, Phys. Chem. Chem. Phys., 2012, 14, 5686–5694 RSC.
- J. Jadzyn and J. Swiergiel, Phys. Chem. Chem. Phys., 2012, 14, 3170–3175 RSC.
- C. C. Rich and J. L. McHale, Phys. Chem. Chem. Phys., 2012, 14, 2362–2374 RSC.
- L. Senthilkumar, T. K. Ghanty, P. Kolandaivel and S. K. Ghosh, Int. J. Quantum Chem., 2012, 112, 2787–2793 Search PubMed.
-
(a) D. Braga and F. Grepioni, Crystal Engineering with Hydrogen Bonds, Encyclopedia of Supramolecular Chemistry, Taylor & Francis, 2004 Search PubMed;
(b) C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC;
(c) T. Mes, M. M. J. Smulders, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2010, 43, 1981–1991 CrossRef CAS;
(d) H. Okuyama and I. Hamada, J. Phys. D: Appl. Phys., 2011, 44, 464004–464018 Search PubMed.
-
(a) B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS;
(b) C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS.
- T. R. Sharp, Int. J. Pharm., 2011, 418, 304–317 Search PubMed.
-
(a) H. Kubinyi, Hydrogen Bonding: The Last Mystery in Drug Design? in Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies, ed. B. Testa, H. van de Waterbeemd, G. Folkers and R. Guy, Verlag Helvetica Chimica Acta, Zürich, 2007 Search PubMed;
(b) S. Rey, P. A. Carrupt and B. Testa, Ann. Pharm. Fr., 2002, 60, 386–396 Search PubMed.
- Y. He, B. Zhu and Y. Inoue, Prog. Polym. Sci., 2004, 29, 1021–1051 CrossRef CAS.
- J. Borg, M. H. Jensen, K. Sneppen and G. Tiana, Phys. Rev. Lett., 2001, 86, 1031–1033 CrossRef CAS.
- A. J. Wilson, Soft Matter, 2007, 3, 409–425 RSC.
- C.-K. Lin, J.-F. Kuo, C.-Y. Chen and J.-J. Fang, Polymer, 2012, 53, 254–258 Search PubMed.
- G. D. Rose and R. Wolfenden, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 381–415 CrossRef CAS.
- S.-Y. Sheu, D.-Y. Yang, H. L. Selzle and E. W. Schlag, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12683–12687 CrossRef CAS.
- R. Flamia, G. Lanza, A. M. Salvi, J. E. Castle and A. M. Tamburro, Biomacromolecules, 2005, 6, 1299–1309 CrossRef CAS.
-
(a) P. Hobza and J. Šponer, Chem. Rev., 1990, 99, 3247–3276 Search PubMed;
(b) J. Šponer, P. Jurećka and P. Hobza, J. Am. Chem. Soc., 2004, 126, 10142–10151 CrossRef CAS.
- W. W. Cleland, Biochemistry, 1992, 31, 317–319 CrossRef CAS.
- W. W. Cleland and M. M. Kreevoy, Science, 1994, 264, 1887–1890 CrossRef CAS.
- T. Ose, K. Kuroki, M. Matsushima, K. Maenaka and I. Kumagai, J. Biochem., 2009, 146, 651–657 Search PubMed.
- J. P. Guthrie, Chem. Biol., 1996, 3, 163–170 CrossRef CAS.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- I. Alabugin, J. Am. Chem. Soc., 2010, 132, 6863–6864 Search PubMed.
- N. Nuñez-Dallos, A. Reyes and R. Quevedo, Tetrahedron Lett., 2012, 53, 530–534 Search PubMed.
-
(a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS;
(b) C. Dalvit and A. Vulpetti, ChemMedChem, 2012, 7, 262–272 CrossRef;
(c) K. Tanaka, M. Murakami, J.-H. Jeon and Y. Chujo, Org. Biomol. Chem., 2012, 10, 90–95 RSC.
- R. Molinari, M. G. Buonomenna, A. Cassano and E. Drioli, J. Chem. Technol. Biotechnol., 2004, 79, 361–368 Search PubMed.
- J. L. Atwood, F. Hamada, K. D. Robinson, G. W. Orr and R. L. Vincent, Nature, 1991, 349, 683–684 CrossRef CAS.
- C. A. Lesburg and D. W. Christianson, J. Am. Chem. Soc., 1995, 117, 6838–6844 CrossRef CAS.
- S. Suhai, J. Chem. Phys., 1994, 101, 9766–9782 CrossRef CAS.
- B. F. King and F. Weinhold, J. Chem. Phys., 1995, 103, 333–347 CrossRef CAS.
- C. E. Dykstra, THEOCHEM, 1996, 362, 1–6 CrossRef CAS.
- A. Karpfen, Molecular Interactions, ed. S. Scheiner, Wiley, New York, 1997 Search PubMed.
- R. D. Parra, M. Furukawa, B. Gong and X. C. Zeng, J. Chem. Phys., 2001, 115, 6030–6035 CrossRef CAS.
- C. Chen, M.-H. Liu and L.-S. Wu, THEOCHEM, 2004, 630, 187–204 Search PubMed.
- R. C. M. U. Araújo, V. M. Soares, B. G. Oliveira, K. C. Lopes, E. Ventura, S. A. do Monte, O. L. Santana, A. B. Carvalho and M. N. Ramos, Int. J. Quantum Chem., 2006, 106, 2714–2722 Search PubMed.
- S. J. Grabowski and E. Bilewicz, Chem. Phys. Lett., 2006, 427, 51–55 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, V. M. Soares and M. N. Ramos, J. Theor. Comput. Chem., 2008, 7, 247–256 Search PubMed.
- B. Gong, B. Jing, Q. Li, Z. Liu, W. Li, J. Cheng, Q. Zheng and J. Sun, Theor. Chem. Acc., 2010, 127, 303–309 CrossRef CAS.
- K. J. Tielrooij, N. Garcia-Araez, M. Bonn and H. J. Bakker, Science, 2010, 328, 1006–1009 CrossRef CAS.
- Q. Zhao, D. Feng and J. Hao, J. Mol. Model., 2011, 11, 2817–2823 Search PubMed.
- B. M. Powell and P. Martel, Biophys. J., 1981, 34, 311–323 Search PubMed.
- J. Šponer, J. Leszczynski and P. Hobza, J. Biomol. Struct. Dyn., 1996, 14, 117–135 Search PubMed.
- A. Hocquet, Phys. Chem. Chem. Phys., 2001, 3, 3192–3199 RSC.
- J. Šponer, J. Leszczynski and P. Hobza, Biopolymers, 2002, 61, 3–31 CrossRef CAS.
- J. A. Subirana and M. Soler-López, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 27–45 CrossRef CAS.
- A. Fuhrmann, S. Getfert, Q. Fu, P. Reimann, S. Lindsay and R. Ros, Biophys. J., 2012, 102, 2381–2390 Search PubMed.
- D. A. Smith, Modeling the Hydrogen Bond, American Chemical Society Series, Washingtin DC, 1994 Search PubMed.
- J. Thar and B. Kirchner, J. Phys. Chem. A, 2006, 110, 4229–4237 CrossRef CAS.
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- J. N. Latosinska, J. Seliger, V. Žagar and D. V. Burchardt, J. Mol. Model., 2012, 18, 11–26 Search PubMed.
- Y. Dai, Y. Wang, Z. Huang, H. Wang and L. Yu, J. Mol. Model., 2012, 18, 265–274 Search PubMed.
- L. Kellner, Rep. Prog. Phys., 1952, 15, 1–23 Search PubMed.
- R. Liminga, Acta Chem. Scand., 1966, 20, 2483–2496 Search PubMed.
- D. C. Rapaport, Mol. Phys., 1983, 50, 1151–1162 CAS.
- G. A. Jeffrey, Crystallogr. Rev., 1995, 4, 213–259 CrossRef CAS.
- P. Hobza, Annu. Rep. Prog. Chem., Sect. C, 2004, 100, 3–27 RSC.
- A. Vila and R. A. Mosquera, Int. J. Quantum Chem., 2006, 106, 928–934 Search PubMed.
- S. K. Sikka and S. M. Sharma, Phase Transitions, 2008, 81, 907–934 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52–59 CrossRef CAS.
- J. M. Andric, G. V. Janjic, D. B. Ninkovic and S. D. Zaric, Phys. Chem. Chem. Phys., 2012, 14, 10896–10898 RSC.
- G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762–785 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1935, 57, 2680–2684 CrossRef CAS.
- M. L. Huggins, Chem. Rev., 1943, 32, 195–218 CrossRef CAS.
- G. C. Pimentel and A. L. McClellan, Annu. Rev. Phys. Chem., 1971, 22, 347–385 CrossRef CAS.
- W. M. Latimer and W. H. Rodebush, J. Am. Chem. Soc., 1920, 42, 1419–1433 CrossRef CAS.
- C. A. Coulson and U. Danielson, Ark. Fys., 1954, 8, 239–244 Search PubMed.
- M. Faraday, Q. J. Sci., Lit., Arts, 1823, 15, 71–74 Search PubMed.
- E. N. Lassettre, Chem. Rev., 1937, 20, 259–303 Search PubMed.
- R. F. McGuire, F. A. Momany and H. A. Scheraga, J. Phys. Chem., 1972, 76, 375–393 Search PubMed.
- G. Brink and L. Glasser, J. Comput. Chem., 1981, 2, 14–19 Search PubMed.
- P. A. Frey and W. W. Cleland, Bioorg. Chem., 1998, 26, 175–192 CrossRef CAS.
- Y. E. Gorbaty and A. G. Kalinichev, J. Phys. Chem., 1995, 99, 5336–5340 CrossRef CAS.
- W. Zheng, M. Angelopoulos, A. J. Epstein and A. G. MacDiarmid, Macromolecules, 1997, 30, 2953–2955 CrossRef CAS.
- A. Varrot and G. J. Davies, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 447–452 CrossRef.
- I. K. McDonald and J. M. Thornton, Protein Eng., 1995, 8, 217–224 Search PubMed.
- L. H. R. Santos, B. L. Rodrigues, Y. M. Idemori and N. G. Fernandes, J. Mol. Struct., 2012, 1014, 102–109 Search PubMed.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1977, 99, 1316–1332 CrossRef CAS.
-
(a) H. Umeyama, K. Kitaura and K. Morokuma, Chem. Phys. Lett., 1977, 36, 11–15 Search PubMed;
(b) K. Morokuma and H. Umeyama, Chem. Phys. Lett., 1977, 49, 333–337 Search PubMed.
- D. G. Fedorov and K. Kitaura, J. Comput. Chem., 2007, 28, 222–237 CrossRef.
- C. Curutchet, J. M. Bofill, B. Hernández, M. Orozco and F. J. Luque, J. Comput. Chem., 2003, 24, 1263–1275 Search PubMed.
- A. van der Vaart and K. M. Merz Jr., J. Phys. Chem. A, 1999, 103, 3321–3329 CrossRef CAS.
- X. He and K. M. Merz Jr., J. Chem. Theory Comput., 2010, 6, 405–411 CrossRef CAS.
- M. Kobayashi, T. Touma and H. Nakai, J. Chem. Phys., 2012, 136, 084108–084117 CrossRef.
- M. Kobayashi and H. Nakai, Phys. Chem. Chem. Phys., 2012, 14, 7629–7639 RSC.
- M. Bosson, C. Richard, A. Plet, S. Grudinin and S. Redon, J. Comput. Chem., 2012, 33, 779–790 Search PubMed.
-
(a) B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS;
(b) J. Langlet, J. Bergès and P. Reinhardt, THEOCHEM, 2004, 685, 43–56 CrossRef CAS.
- A. J. Misquitta, R. Podeszwa, B. Jeziorski and K. Szalewicz, J. Chem. Phys., 2005, 123, 214103–214116 CrossRef.
- H. Cybulski and J. Sadlej, J. Chem. Theory Comput., 2008, 4, 892–897 CrossRef.
- M. Pitoňák, K. E. Riley, P. Neogrády and P. Hobza, ChemPhysChem, 2008, 9, 1636–1644 CrossRef CAS.
- J. Cukras and J. Sadlej, Chem. Phys. Lett., 2008, 459, 44–48 CrossRef CAS.
- H. M. Jaeger, H. F. Schaefer, E. G. Hohenstein and C. D. Sherrill, Comput. Theor. Chem., 2011, 973, 47–52 Search PubMed.
- E. G. Hohenstein, R. M. Parrish, C. D. Sherrill, J. M. Turney and H. F. Schaefer III, J. Chem. Phys., 2011, 135, 174107–174119 Search PubMed.
- E. G. Hohenstein and C. D. Sherrill, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 304–326 Search PubMed.
- F. B. van Duijneveldt and J. N. Murrell, J. Chem. Phys., 1967, 46, 1759–1767 Search PubMed.
- J. F. Beck and Y. Mo, J. Comput. Chem., 2007, 28, 455–466 CrossRef CAS.
- S. Yamabe and K. Morokuma, J. Am. Chem. Soc., 1975, 97, 4458–4465 CrossRef CAS.
- M. D. Newton, G. A. Jeffrey and S. Takagi, J. Am. Chem. Soc., 1979, 101, 1997–2002 CrossRef.
- M. S. Gordon and J. H. Jensen, Acc. Chem. Res., 1996, 29, 536–543 CrossRef.
- A. Famulari, M. Raimondi, M. Sironi and E. Gianinetti, Chem. Phys., 1998, 232, 289–298 CrossRef CAS.
-
(a) S. Humbel, J. Phys. Chem. A, 2002, 106, 5517–5520 CrossRef CAS;
(b) S. Humbel, J. Chem. Educ., 2007, 84, 1056–1061 Search PubMed.
- R. Mörtel and H. D. Lutz, J. Mol. Struct., 2003, 648, 171–176 Search PubMed.
- A. Ilnicka and J. Sadlej, Struct. Chem., 2012, 23, 1323–1332 Search PubMed.
- S. J. Grabowski, Hydrogen bonding – New insights, Springer, Dordrecht, 2006 Search PubMed.
- B. Kojić-Prodić and K. Molćanov, Acta Chim. Slov., 2008, 55, 692–708 CAS.
- P. Gilli and G. Gilli, J. Mol. Struct., 2010, 972, 2–10 CrossRef CAS.
-
(a) W. S. Koski, J. Am. Chem.
Soc., 1960, 82, 4120–4120 Search PubMed;
(b) D. C. Clary, D. M. Benoit and T. van Mourik, Acc. Chem. Res., 2000, 33, 441–447 CrossRef CAS.
- S. Scheiner, Phys. Chem. Chem. Phys., 2011, 13, 13860–13872 RSC.
- L. P. Wolters and F. M. Bickelhaupt, ChemistryOpen, 2012, 1, 96–105 Search PubMed.
- M. Huš and T. Urbic, J. Chem. Phys., 2012, 136, 144305–144311 Search PubMed.
- R. Vuilleumier and D. Borgis, Nat. Chem., 2012, 4, 432–433 Search PubMed.
- www.iupac.org/projects/2004/2004-026-2-100.html, accessed in July, 2010.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636.
- I. Alkorta and J. Elguero, Struct. Chem., 1999, 10, 157–159 CrossRef CAS.
- T. Steiner, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- K. Banerjee, S. De and D. Datta, J. Chem. Crystallogr., 2007, 37, 675–677 CrossRef CAS.
- A. Kovács and Z. Varga, Coord. Chem. Rev., 2006, 250, 710–727 CrossRef CAS.
- Y. Mori and K. Takano, Phys. Chem. Chem. Phys., 2012, 14, 11090–11098 RSC.
- A. A. Voityuk and A. A. Bliznyuk, J. Struct. Chem., 1993, 33, 899–924 Search PubMed.
- C. Trindle, J. Phys. Chem. A, 2000, 104, 5298–5301 Search PubMed.
- J. Grell, J. Bernstein and G. Tinhofer, Crystallogr. Rev., 2002, 8, 1–56 Search PubMed.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS.
-
(a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS;
(b) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC;
(c) M. Jabłoński, J. Phys. Chem. A, 2012, 116, 3753–3764 Search PubMed;
(d) U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2012, 116, 3487–3497 Search PubMed.
- G. S. Tschumper and N. I. Hammer, J. Am. Chem. Soc., 2010, 132, 9512–9512 Search PubMed.
- C. D. Keefe and Z. Istvankova, Can. J. Chem., 2011, 89, 34–46 Search PubMed.
- A. Ranganathan, G. U. Kulkarni and C. N. R. Rao, J. Mol. Struct., 2003, 656, 249–263 CrossRef CAS.
- M. Schmidtmann, L. J. Farrugia, D. S. Middlemiss, M. J. Gutmann, G. J. McIntyre and C. C. Wilson, J. Phys. Chem. A, 2009, 113, 13985–13997 CrossRef CAS.
- V. R. Hathwar and T. N. G. Row, J. Phys. Chem. A, 2010, 114, 13434–13441 CrossRef CAS.
- I. Olovsson, Z. Phys. Chem., 2006, 220, 963–978 Search PubMed.
- J.-P. Malrieu, N. Guihéry, C. J. Calzado and C. Angeli, J. Comput. Chem., 2007, 28, 35–50 CrossRef CAS.
- R. Ponec and J. Chaves, J. Comput. Chem., 2007, 28, 109–116 CrossRef CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- J. E. Del Bene, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2010, 114, 12958–12962 CrossRef CAS.
- T. H. Rehm and C. Schmuck, Chem. Soc. Rev., 2010, 39, 3597–3611 RSC.
- T.-H. Tang, W.-J. Hu, D.-Y. Yan and Y.-P. Cui, THEOCHEM, 1990, 207, 319–326 CrossRef.
- T.-H. Tang and Y.-P. Cui, Can. J. Chem., 1996, 74, 1162–1170 CAS.
- D.-L. Cao, F.-D. Ren, X.-Q. Feng, J.-L. Wang, Y.-X. Li, Z.-Y. Hu and S.-S. Chen, THEOCHEM, 2008, 849, 76–83 Search PubMed.
- F.-D. Ren, D.-L. Cao, W.-L. Wang, J. Ren, S.-Q. Hou and S.-S. Chen, J. Mol. Model., 2009, 15, 515–523 Search PubMed.
- G. R. Desiraju, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1992, 211, 63–74 Search PubMed.
- M. L. Webb and H. S. Rzepa, Chirality, 1994, 6, 245–250 CAS.
- J.-Y. Le Questel, G. Boquet, M. Berthelot and C. Laurence, J. Phys. Chem. B, 2000, 104, 11816–11823 CrossRef.
- T. Steiner and G. Koellner, J. Mol. Biol., 2001, 305, 535–557 CrossRef CAS.
- S. Furutaka and S.-I. Ikawa, J. Chem. Phys., 2002, 117, 751–756 Search PubMed.
- Y. Umezawa and M. Nishio, Biopolymers, 2005, 79, 248–258 CrossRef CAS.
- L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513–3560 CrossRef CAS.
- P. Lipkowski, S. J. Grabowski and S. J. Leszczynski, J. Phys. Chem. A, 2006, 110, 10296–10302 CrossRef CAS.
- T. Ozawa and K. Okazaki, J. Comput. Chem., 2008, 29, 2656–2666 CrossRef CAS.
- T. Ozawa, E. Tsuji, M. Ozawa, C. Handa, H. Mukaiyama, T. Nishimura, S. Kobayashi and K. Okazaki, Bioorg. Med. Chem., 2008, 16, 10311–10318 CrossRef CAS.
- C. Biswas, M. G. B. Drew, D. Escudero, A. Frontera and A. Ghosh, Eur. J. Inorg. Chem., 2009, 2238–2246 CrossRef CAS.
- D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, J. Comput. Chem., 2009, 30, 75–82 CrossRef CAS.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 12340–12347 Search PubMed.
- T. Richardson, S. de Gala, R. H. Crabtree and P. E. M. Siegbahn, J. Am. Chem. Soc., 1995, 117, 12875–12876 CrossRef CAS.
- T. B. Richardson, T. F. Koetzle and R. H. Crabtree, Inorg. Chim. Acta, 1996, 250, 69–73 CrossRef CAS.
- R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold and T. F. Koetzle, Acc. Chem. Res., 1996, 29, 348–354 CrossRef CAS.
- A. H. Pakiari and A. Mohajeri, THEOCHEM, 2003, 620, 31–36 Search PubMed.
-
(a) R. H. Crabtree, Hydrogen Bonding & Dihydrogen Bonding, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2011 Search PubMed;
(b) G. J. Kubas, Metal Dihydrogen and Sigma-Bonded Complexes: Structure, Theory and Reactivity, Springer, 2001 Search PubMed.
- D. Hugas, S. Simon and M. Duran, Struct. Chem., 2005, 16, 257–263 Search PubMed.
- D. Hugas, S. Simon, M. Duran, C. F. Guerra and F. M. Bickelhaupt, Chem.–Eur. J., 2009, 15, 5814–5822 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Spectrochim. Acta, Part A, 2010, 75, 563–566 Search PubMed.
- P. C. Singh, Chem. Phys., 2011, 389, 96–101 Search PubMed.
- Q. Li, X. Liu, R. Li, J. Cheng and W. Li, Spectrochim. Acta, Part A, 2012, 90, 135–140 Search PubMed.
-
(a) P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS;
(b) W. T. Klooster, T. F. Koetzle, P. E. M. Siegbahn, T. B. Richardson and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 6337–6343 CrossRef CAS.
- K. Eskandari, J. Mol. Model., 2012, 18, 3481–3487 Search PubMed.
- M. Solimannejad, L. M. Amlashi, I. Alkorta and J. Elguero, Chem. Phys. Lett., 2006, 422, 226–229 Search PubMed.
- P. G. Jessop and R. H. Morris, Coord. Chem. Rev., 1992, 121, 155–284 CrossRef CAS.
- R. H. Crabtree, Angew. Chem., Int. Ed. Engl., 1993, 32, 789–805 CrossRef.
- I. Alkorta, F. Blanco and J. Elguero, J. Phys. Chem. A, 2008, 112, 6753–6759 Search PubMed.
- M. G. Govender and T. A. Ford, THEOCHEM, 2003, 630, 11–16 Search PubMed.
- L. D. Speakman, J. M. Turney and H. F. Schaefer, J. Chem. Phys., 2005, 123, 204303–204314 Search PubMed.
- I. Alkorta, J. Elguero, M. Solimannejad and S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 201–210 CrossRef CAS.
- A.-Y. Li, L.-F. Xu and Z. Ling, Chin. J. Chem. Phys., 2009, 22, 57–62 Search PubMed.
- W.-L. Zhu, C. M. Puah, X.-J. Tan, H.-L. Jiang and K.-X. Chen, J. Phys. Chem. A, 2001, 105, 426–431 CrossRef CAS.
- M. Panigati, P. Mercandelli, G. D’Alfonso, T. Beringhelli and A. Sironi, J. Organomet. Chem., 2005, 690, 2044–2051 Search PubMed.
- K. N. Robertson, O. Knop and T. S. Cameron, Can. J. Chem., 2003, 81, 727–743 CrossRef CAS.
- S. J. Grabowski and J. Leszczynski, Pract. Asp. Comput. Chem., 2010, 255 Search PubMed.
- K. Abdur-Rashid, D. G. Gusev, A. J. Lough and R. H. Morris, Organometallics, 2000, 19, 834–843 CrossRef CAS.
- J. Haywood and A. E. H. Wheatley, Eur. J. Inorg. Chem., 2009, 5010–5016 CrossRef CAS.
- M. Jabloński and M. Palusiak, J. Phys. Chem. A, 2012, 116, 2322–2332 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, J. Mol. Model., 2009, 15, 421–432 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- P. L. A. Popelier, Coord. Chem. Rev., 2000, 197, 169–189 CrossRef CAS.
- F. Cortés-Guzmán and R. F. W. Bader, Coord. Chem. Rev., 2005, 249, 633–662 CrossRef CAS.
- R. F. W. Bader, Coord. Chem. Rev., 2005, 249, 3198–3198 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2011, 115, 12432–12435 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2011, 115, 12667–12676 Search PubMed.
- C. F. Matta, L. Massa and T. A. Keith, J. Phys. Chem. A, 2011, 115, 12427–12431 Search PubMed.
- B. G. Oliveira and M. L. A. A. Vasconcellos, THEOCHEM, 2006, 774, 83–88 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, J. Mol. Model., 2009, 15, 123–131 Search PubMed.
- B. G. Oliveira and R. C. M. U. Araújo, J. Mol. Model., 2012, 18, 2845–2854 Search PubMed.
- S. J. Grabowski, J. Phys. Chem. A, 2007, 111, 13537–13543 Search PubMed.
- S. J. Grabowski and J. Leszczynski, Chem. Phys., 2009, 355, 169–176 CrossRef CAS.
- S. J. Grabowski, J. Phys. Chem. A, 2000, 104, 5551–5557 CrossRef CAS.
- A. Robertazzi and J. A. Platts, J. Phys. Chem. A, 2006, 110, 3992–4000 CrossRef CAS.
- C. F. Matta, N. Castillo and R. J. Boyd, J. Phys. Chem. B, 2006, 110, 563–578 CrossRef CAS.
- T. I. Madzhidov and G. A. Chmutova, THEOCHEM, 2010, 959, 1–7 Search PubMed.
- C. D. M. Churchill, L. R. Rutledge and S. D. Wetmore, Phys. Chem. Chem. Phys., 2010, 12, 14515–14526 RSC.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, Quim. Nova, 2010, 33, 1155–1162 Search PubMed.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- J. Saßmannshausen, Dalton Trans., 2012, 41, 1919–1923 RSC.
- S. L. Capim, S. R. Santana, B. G. Oliveira, G. B. Rocha and M. L. A. A. Vasconcellos, J. Braz. Chem. Soc., 2010, 21, 1718–1726 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, E. S. Leite and M. N. Ramos, Int. J. Quantum Chem., 2011, 111, 111–116 Search PubMed.
- T. Yang, J.-J. An, X. Wang, D.-Y. Wu, W. Chen and J. S. Fossey, Phys. Chem. Chem. Phys., 2012, 14, 10747–10753 RSC.
- S. J. Grabowski, J. Phys. Chem. A, 2012, 116, 1838–1845 Search PubMed.
- P. Tarakeshwar, H. S. Choi and K. S. Kim, J. Am. Chem. Soc., 2001, 123, 3323–3331 CrossRef CAS.
- S. Vaupel, B. Brutschy, P. Tarakeshwar and K. S. Kim, J. Am. Chem. Soc., 2006, 128, 5416–5426 CrossRef CAS.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250–1258 CrossRef CAS.
- G. Grewer and M. Lösche, Makromol. Chem., Macromol. Symp., 1991, 46, 79–87 Search PubMed.
- T. Umegaki, J.-M. Yan, X.-B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 2303–2311 CrossRef CAS.
- N. J. Singh, H. M. Lee, S. B. Suh and K. S. Kim, Pure Appl. Chem., 2007, 79, 1057–1075 CrossRef CAS.
- H. M. Lee, A. Kumar, M. Kołaski, D. Y. Kim, E. C. Lee, S. K. Min, M. Park, Y. C. Choi and K. S. Kim, Phys. Chem. Chem. Phys., 2010, 12, 6278–6287 RSC.
- E. C. Lee, D. Kim, P. Jurečka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446–3457 CrossRef CAS.
- H. Chu, G. Wu, Z. Xiong, J. Guo, T. He and P. Chen, Chem. Mater., 2010, 22, 6021–6028 CrossRef CAS.
-
(a) D. Y. Kim, N. J. Singh, H. M. Lee and K. S. Kim, Chem.–Eur. J., 2009, 15, 5598–5604 CrossRef CAS;
(b) D. Y. Kim, H. M. Lee, J. Seo, S. K. Shin and K. S. Kim, Phys. Chem. Chem. Phys., 2010, 12, 5446–5453 RSC.
- D. J. Wolstenholme, J. T. Titah, F. N. Che, K. T. Traboulsee, J. Flogeras and G. S. McGrady, J. Am. Chem. Soc., 2011, 133, 16598–16604 CrossRef CAS.
- J. Li, S. M. Kathmann, H.-S. Hu, G. K. Schenter, T. Autrey and M. Gutowski, Inorg. Chem., 2010, 49, 7710–7720 CrossRef CAS.
- L. Pauling, Proc. Natl. Acad. Sci. U. S. A., 1928, 14, 359–362 CAS.
- L. Pauling and C. Niemann, J. Am. Chem. Soc., 1939, 61, 1860–1867 Search PubMed.
- K. B. Wiberg, in Pauling’s Legacy: Modern Modelling of the Chemical Bond, ed. Z. B. Maksić and W. J. Orville-Thomas, Elsevier, Amsterdam, 1999 Search PubMed.
- D. J. Klein, Theor. Comput. Chem., 1999, 6, 403–420 Search PubMed.
- W. B. Jensen, J. Chem. Educ., 2003, 80, 279–287 Search PubMed.
- P. W. Fowler, A. C. Legon, J. M. A. Thumwood and E. R. Waclawik, Coord. Chem. Rev., 2000, 197, 231–247 Search PubMed.
- S. Suzuki, P. G. Green, R. E. Bumgarner, S. Dasgupta, W. A. Goddard III and G. A. Blake, Science, 1992, 257, 942–945 CrossRef CAS.
- M. A. Fox and A. K. Hughes, Coord. Chem. Rev., 2004, 248, 457–476 CrossRef CAS.
- J. Lewiński, J. Zachara, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185–1199 CrossRef.
- S. J. Grabowski, J. W. Wilamowski, D. Osman, J. J. Sepiol and N. Rodier, Aust. J. Chem., 1996, 49, 951–954.
- C. R. Girija, N. S. Begum, A. A. Syed and V. Thiruvenkatam, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, 611–613 Search PubMed.
- M. Domagała and S. J. Grabowski, Chem. Phys., 2009, 363, 42–48 Search PubMed.
- M. N. Ali Mohamed, H. D. Watts, J. Guo, J. M. Catchmark and J. D. Kubicki, Carbohydr. Res., 2010, 345, 1741–1751 CrossRef CAS.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- Q. Zhao, D. Feng, Y. Sun, J. Hao and Z. Cai, J. Mol. Model., 2011, 17, 1935–1939 Search PubMed.
- S. J. Grabowski, Comput. Theor. Chem., 2012, 992, 70–77 Search PubMed.
- M. Nishio, J. Mol. Struct., 2012, 1018, 2–7 Search PubMed.
-
(a) M. J. Plevin, D. L. Bryce and J. Boisbouvier, Nat. Chem., 2010, 2, 466–471 CrossRef CAS;
(b) Q. Li, H. Sun, R. Li, W. Li and J. Cheng, Comput. Theor. Chem., 2012, 992, 150–155 Search PubMed.
- J. E. Bloor and J. Yu, J. Phys. Chem., 1990, 94, 5586–5589 CrossRef CAS.
- F. H. Allen, J. A. K. Howard, V. J. Hoy, G. R. Desiraju, D. S. Reddy and C. C. Wilson, J. Am. Chem. Soc., 1996, 118, 4081–4084 CrossRef CAS.
- R. M. Minyaev and D. J. Wales, Chem. Phys. Lett., 1994, 218, 413–421 CrossRef CAS.
- M. I. Menéndez, D. Suárez, J. A. Sordo and T. L. Sordo, J. Comput. Chem., 1995, 16, 659–666 CrossRef CAS.
- M. I. Menéndez, J. A. Sordo and T. L. Sordo, THEOCHEM, 1996, 371, 91–95 CrossRef CAS.
- J. B. P. da Silva, M. R. Silva Jr. and M. N. Ramos, J. Braz. Chem. Soc., 2005, 16, 844–850 Search PubMed.
- F.-D. Ren, D.-L. Cao, W.-L. Wang, J. Ren and S.-S. Chen, THEOCHEM, 2009, 896, 38–43 CrossRef CAS.
- K. S. Thanthiriwatte, E. G. Hohenstein, L. A. Burns and C. D. Sherrill, J. Chem. Theory Comput., 2011, 7, 88–96 CrossRef CAS.
- A. Kaczor, R. Almeida, A. Gómez-Zavaglia, M. L. S. Cristiano and R. Fausto, J. Mol. Struct., 2008, 876, 77–85 Search PubMed.
- S. Canneaux, B. Xerri, F. Louis and L. Cantrel, J. Phys. Chem. A, 2010, 114, 9270–9288 Search PubMed.
- W. G. Read and W. H. Flygare, J. Chem. Phys., 1982, 76, 2238–2246 CrossRef CAS.
- S. G. Kukolich, P. D. Aldrich, W. G. Read and E. J. Cambell, Chem. Phys. Lett., 1982, 90, 329–331 Search PubMed.
- K. Nauta and R. E. Miller, Chem. Phys. Lett., 2001, 346, 129–134 CrossRef CAS.
- G. E. Douberly, K. Nauta and R. E. Miller, Chem. Phys. Lett., 2003, 377, 384–390 Search PubMed.
- P. A. Block, K. W. Jucks, L. G. Pedersen and R. E. Miller, Chem. Phys., 1989, 139, 15–30 CrossRef CAS.
-
(a) M. Quack and M. A. Suhm, Theor. Chem. Acc., 1996, 93, 61–65 Search PubMed;
(b) R. C. M. U. Araújo, J. B. P. da Silva and M. N. Ramos, Spectrochim. Acta, Part A, 1995, 51, 821–830 CrossRef.
- R. C. M. U. Araújo and M. N. Ramos, THEOCHEM, 1996, 366, 233–240 CrossRef CAS.
- S. Scheiner and S. J. Grabowski, J. Mol. Struct., 2002, 615, 209–218 CrossRef CAS.
- C. L. Perrin, Pure Appl. Chem., 2009, 81, 571–583 CrossRef CAS.
- W. A. Herrebout, G. P. Everaert, B. J. van der Veken and M. O. Bulanin, J. Chem. Phys., 1997, 107, 8886–8898 CrossRef CAS.
- G. P. Everaert, W. A. Herrebout and B. J. van der Veken, J. Mol. Struct., 2000, 550–551, 399–411 Search PubMed.
- C. Clavero, M. Duran, A. Lledós, O. N. Ventura and J. Bertrán, J. Am. Chem. Soc., 1986, 108, 923–928 CrossRef CAS.
- C. Clavero, M. Duran, A. Lledós, O. N. Ventura and J. Bertrán, J. Comput. Chem., 1987, 8, 481–488 CrossRef CAS.
- B. S. Jursic, THEOCHEM, 1998, 434, 37–42 CrossRef CAS.
- F. Ramondo, L. Bencivenni, G. Portalone and A. Domenicano, Struct. Chem., 1994, 5, 1–7 CAS.
- P. Rath, F. Delange, W. J. Degrip and K. J. Rothschild, Biochem. J., 1998, 329, 713–717 Search PubMed.
- T. Steiner, Chem. Commun., 1998, 411–412 RSC.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Chem. Phys. Lett., 2007, 433, 390–394 Search PubMed.
-
(a) V. B. Delchev, Monatsh. Chem., 2004, 135, 249–260 Search PubMed;
(b) V. B. Delchev and H. Mikosch, J. Mol. Model., 2005, 11, 474–480 Search PubMed.
- F. Ortmann, K. Hannewald and F. Bechstedt, Phys. Rev. B, 2007, 75, 195219–195233 CrossRef.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2004, 108, 1806–1812 CrossRef CAS.
- S. J. Grabowski, Struct. Chem., 2005, 16, 175–176 Search PubMed.
- L. R. Falvello, Angew. Chem., Int. Ed., 2010, 49, 10045–10047 CrossRef CAS.
- M. Meot-Ner, Chem. Rev., 2005, 105, 213–284 CrossRef.
- J. D. Schmitt, C. G. V. Sharples and W. S. Caldwell, J. Med. Chem., 1999, 42, 3066–3074 CrossRef CAS.
- A. K. Bhattacharjee, THEOCHEM, 2000, 529, 193–201 Search PubMed.
- A. Ebrahimi, M. H. Khorassani and H. R. Masoodi, Chem. Phys. Lett., 2010, 493, 27–32 Search PubMed.
- J. Fernández-Recio, A. Romero and J. Sancho, J. Mol. Biol., 1999, 290, 319–330 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- S. E. Wheeler and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 3126–3127 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef CAS.
- E. D’Oria and J. J. Novoa, J. Phys. Chem. A, 2011, 115, 13114–13123 Search PubMed.
- C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2012, 14, 9738–9748 RSC.
- J. Valdespino-Saenz and A. Martínez, THEOCHEM, 2010, 939, 34–43 Search PubMed.
- Y. Lu, Y. Liu, H. Li, X. Zhu, H. Liu and W. Zhu, J. Phys. Chem. A, 2012, 116, 2591–2597 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2007, 438, 104–108 CrossRef CAS.
- T. Yonezawa, H. Nakatsuji and H. Kato, J. Am. Chem. Soc., 1968, 90, 1239–1245 Search PubMed.
- J. A. Pople, M. J. Frisch and J. E. Del Bene, Chem. Phys. Lett., 1982, 91, 185–189 CrossRef CAS.
- E. S. Stoyanov, I. V. Stoyanova and C. A. Reed, Chem.–Eur. J., 2008, 14, 7880–7891 Search PubMed.
- Y.-H. Zhang, J.-K. Hao, X. Wang, W. Zhou and T.-H. Tang, THEOCHEM, 1998, 455, 85–99 Search PubMed.
- L. Pejov, M. Solimannejad and V. Stefov, Chem. Phys., 2006, 323, 259–270 Search PubMed.
- T. M. Krygowski and H. Szatyłowicz, J. Phys. Chem. A, 2006, 110, 7232–7236 Search PubMed.
- H. Szatyłowicz and T. M. Krygowski, J. Mol. Struct., 2007, 844–845, 200–207 CrossRef CAS.
- R. H. Crabtree, Science, 1998, 282, 2000–2001 CrossRef CAS.
- J. Li, F. Zhao and F. Jing, J. Chem. Phys., 2002, 116, 25–32 CrossRef CAS.
- S. A. Kulkarni, J. Phys. Chem. A, 1998, 102, 7704–7711 CrossRef CAS.
- V. I. Bakhmutov, Dihydrogen Bond: Principles, Experiments, and Applications, John Wiley & Sons, New Jersey, 2008 Search PubMed.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS.
- Y. Liu, C. Hu, A. Comotti and M. D. Ward, Science, 2011, 333, 436–440 CrossRef CAS.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS.
- C. P. Lau, S. M. Ng, G. Jia and Z. Lin, Coord. Chem. Rev., 2007, 251, 2223–2237 CrossRef CAS.
- M. M. S. de Giambiagi and P. Bultinck, J. Braz. Chem. Soc., 2008, 19, 263–267 Search PubMed.
- M. Solimannejad and S. Scheiner, J. Phys. Chem. A, 2005, 109, 11933–11935 CrossRef CAS.
- M. Solimannejad and A. Boutalib, Chem. Phys., 2006, 320, 275–280 Search PubMed.
-
(a) M. Solimannejad and I. Alkorta, Chem. Phys., 2006, 324, 459–464 CrossRef CAS;
(b) S. J. Grabowski, J. Mol. Struct., 2000, 553, 151–156 CrossRef CAS.
- H. Cybulski, E. Tymińska and J. Sadlej, ChemPhysChem, 2006, 7, 629–639 CrossRef CAS.
- P. Lipkowski, S. J. Grabowski, T. L. Robinson and J. Leszczynski, J. Phys. Chem. A, 2004, 108, 10865–10872 CrossRef CAS.
- B. G. Oliveira, E. C. S. Santos, E. M. Duarte, R. C. M. U. Araújo, M. N. Ramos and A. B. Carvalho, Spectrochim. Acta, Part A, 2004, 60, 1883–1887 Search PubMed.
- B. G. Oliveira, E. M. Duarte, R. C. M. U. Araújo, M. N. Ramos and A. B. Carvalho, Spectrochim. Acta, Part A, 2005, 61, 491–494 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Spectrochim. Acta, Part A, 2007, 68, 626–631 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Quim. Nova, 2007, 30, 1167–1170 Search PubMed.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4185 CrossRef CAS.
- C. G. Cannon, Spectrochim. Acta, 1958, 10, 341–368 CrossRef CAS.
- A. Nikolić, B. Jović, S. Csanady and S. Petrović, J. Mol. Struct., 2007, 834–836, 249–252 Search PubMed.
- M. Gorman, J. Chem. Educ., 1957, 34, 304–306 CAS.
- A. J. Michell, Aust. J. Chem., 1975, 28, 335–341 Search PubMed.
- A. N. Egorochkin and S. E. Skobeleva, Russ. Chem. Rev., 1979, 48, 1198–1211 Search PubMed.
- A. Fujii, G. N. Patwari, T. Ebata and N. Mikami, Int. J. Mass Spectrom., 2002, 220, 289–312 Search PubMed.
- F.-X. Coudert, R. Vuilleumier and A. Boutin, ChemPhysChem, 2006, 7, 2464–2467 CrossRef CAS.
- R. K. Thomas, Proc. R. Soc. London, Ser. A, 1971, 325, 133–149 CAS.
- N. L. Pivonka, C. Kaposta, M. Brümmer, G. von Helden, G. Meijer, L. Wöste, D. M. Neumark and K. R. Asmis, J. Chem. Phys., 2003, 118, 5275–5278 CrossRef CAS.
- A. S. N. Murthy and C. N. R. Rao, Appl. Spectrosc. Rev., 1968, 2, 69–191 CrossRef.
- M. Goswami and E. Arunan, Phys. Chem. Chem. Phys., 2009, 11, 8974–8983 RSC.
- J. Yarwood and G. N. Robertson, Nature, 1975, 257, 41–43 Search PubMed.
- M. Banno, K. Ohta, S. Yamaguchi, S. Hirai and K. Tominaga, Acc. Chem. Res., 2009, 42, 1259–1269 CrossRef CAS.
-
(a) H. T. Flakus and B. Hachula, Spectrochim. Acta, Part A, 2011, 79, 1276–1284 Search PubMed;
(b) B. Bandyopadhyay, P. Pandey, P. Banerjee, A. K. Samanta and T. Chakraborty, J. Phys. Chem. A, 2012, 116, 3836–3845 Search PubMed.
- R. Gupta and A. Chandra, J. Mol. Liq., 2012, 165, 1–6 Search PubMed.
- R. Diniz, H. A. De Abreu, W. B. de Almeida, N. G. Fernandes and M. T. C. Sansiviero, Spectrochim. Acta, Part A, 2005, 61, 1747–1757 Search PubMed.
- M. Rozenberg, C. Jung and G. Shoham, Phys. Chem. Chem. Phys., 2003, 5, 1533–1535 RSC.
- M. Rozenberg, A. Loewenschuss and Y. Marcus, Phys. Chem. Chem. Phys., 2000, 2, 2699–2702 RSC.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2012, 116, 4089–4096 Search PubMed.
- B. G. Oliveira and L. F. C. C. Leite, THEOCHEM, 2009, 915, 38–42 Search PubMed.
- Y. Yang, W. Zhang, S. Pei, J. Shao, W. Huang and X. Gao, THEOCHEM, 2005, 732, 33–37 CrossRef CAS.
- Y. Liu, W. Liu, Y. Yang and J. Liu, Int. J. Quantum Chem., 2006, 106, 2122–2128 CrossRef CAS.
- C. L. Braun and S. N. Smirnov, J. Chem. Educ., 1993, 70, 612–614 CrossRef CAS.
- S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 12789–12799 Search PubMed.
- O. Donoso-Tauda, P. Jaque and J. C. Santos, Phys. Chem. Chem. Phys., 2011, 13, 1552–1559 RSC.
- S. Inagaki, H. Murai and T. Takeuchi, Phys. Chem. Chem. Phys., 2012, 14, 2008–2014 RSC.
- X. Lu, H. Shi, J. Chen and D. Ji, Comput. Theor. Chem., 2012, 982, 34–39 Search PubMed.
- B. G. Oliveira and R. C. M. U. Araújo, Can. J. Chem., 2012, 90, 368–375 Search PubMed.
- I. V. Alexandrov, Chem. Phys., 1980, 51, 449–457 Search PubMed.
- Y. Zhao and W. Liang, Chem. Soc. Rev., 2012, 41, 1075–1087 RSC.
- F. A. S. Chipem, A. Mishra and G. Krishnamoorthy, Phys. Chem. Chem. Phys., 2012, 14, 8775–8790 RSC.
- H. R. Leverentz, K. A. Maerzke, S. J. Keasler, J. I. Siepmann and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 7669–7678 RSC.
- N. Gresh, P. Claverie and A. Pullman, Int. J. Quantum Chem., 1986, 29, 101–118 CAS.
-
(a) M. in het Panhuis, P. L. A. Popelier, R. W. Munn and J. G. Ángyán, J. Chem. Phys., 2001, 114, 7951–7961 CrossRef;
(b) I. V. Stasyuk, R. Y. Stetsiv and Y. V. Sizonenko, Condens. Matter Phys., 2002, 5, 685–706 Search PubMed.
- B. G. Oliveira and R. C. M. U. Araújo, Quim. Nova, 2007, 30, 791–796 Search PubMed.
- G. Uyeda, J. C. Williams, M. Roman, T. A. Mattioli and J. P. Allen, Biochemistry, 2010, 49, 1146–1159 CrossRef CAS.
- E. Clot, O. Eisentein and R. H. Crabtree, New J. Chem., 2001, 25, 66–72 RSC.
- A. Koll, S. M. Melikova, A. Karpfen and P. Wolschann, J. Mol. Struct., 2001, 559, 127–145 CrossRef CAS.
- D. J. Nesbitt, Chem. Rev., 1988, 88, 843–870 CrossRef CAS.
- R. C. M. U. Araújo and M. N. Ramos, J. Braz. Chem. Soc., 1998, 9, 499–505 CAS.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Struct. Chem., 2009, 20, 663–670 Search PubMed.
- S. Pinchas, Anal. Chem., 1955, 27, 2–6 CrossRef CAS.
- J. L. Adcock and H. Zhang, J. Org. Chem., 1995, 60, 1999–2002 CrossRef CAS.
- G. Trudeau, J.-M. Dumas, P. Dupuis, M. Guérin and C. Sandorfy, Top. Curr. Chem., 1980, 93, 91–125 CAS.
- P. Hobza, V. Špirko, Z. Havlas, K. Buchhold, B. Reiman, H.-D. Barth and B. Brutschy, Chem. Phys. Lett., 1999, 299, 180–186 CrossRef CAS.
- P. Hobza and Z. Havlas, Chem. Phys. Lett., 1999, 303, 447–452 CrossRef CAS.
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, F. F. Chagas, A. B. Carvalho and M. N. Ramos, J. Mol. Model., 2008, 14, 949–955 Search PubMed.
- D. Blume, M. Lewerenz, F. Huisken and M. Kaloudis, J. Chem. Phys., 1996, 105, 8666–8683 CrossRef CAS.
- I. E. Boldeskul, I. F. Tsymbal, E. V. Ryltsev, Z. Latajka and A. J. Barnes, J. Mol. Struct., 1997, 436–437, 167–171 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, THEOCHEM, 2009, 908, 79–83 CrossRef CAS.
- A. H. Pakiari and Z. Jamshidi, THEOCHEM, 2004, 685, 155–161 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, Orbital: Electron. J. Chem., 2009, 1, 156–166 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, THEOCHEM, 2010, 944, 168–172 CrossRef.
- I. Alkorta, J. Elguero and C. Foces-Foces, Chem. Commun., 1996, 1633–1634 RSC.
- S. N. Delanoye, W. A. Herrebout and B. J. van der Veken, J. Am. Chem. Soc., 2002, 124, 11854–11855 CrossRef CAS.
- B. G. Oliveira and M. N. Ramos, Int. J. Quantum Chem., 2010, 110, 307–316 Search PubMed.
- P. Hobza and Z. Havlas, Theor. Chem. Acc., 2002, 108, 325–334 CrossRef CAS.
- E. Cubero, M. Orozco and F. J. Luque, Chem. Phys. Lett., 1999, 310, 445–450 Search PubMed.
- J. Chocholoušová, V. Špirko and P. Hobza, Phys. Chem. Chem. Phys., 2004, 6, 37–41 RSC.
- J. M. Hermida-Ramón and A. M. Graña, J. Comput. Chem., 2007, 28, 540–546 CrossRef CAS.
- A. Dkhissi, L. Adamowicz and G. Maes, J. Phys. Chem. A, 2000, 104, 5625–5630 CrossRef CAS.
- E. S. Kryachko and T. Zeegers-Huyskens, J. Phys. Chem. A, 2001, 105, 7118–7125 CrossRef CAS.
- J. L. Alonso, S. Antolínez, S. Blanco, A. Lesarri, J. C. López and W. Caminati, J. Am. Chem. Soc., 2004, 126, 3244–3249 CrossRef CAS.
- V. I. Teberekidis and M. P. Sigalas, THEOCHEM, 2007, 803, 29–38 Search PubMed.
- W. B. Jensen, Chem. Rev., 1978, 78, 1–22 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, F. S. Pereira, E. F. Lima, W. L. V. Silva, A. B. Carvalho and M. N. Ramos, Quim. Nova, 2008, 31, 1673–1679 Search PubMed.
- B. G. Oliveira and M. L. A. A. Vasconcellos, Inorg. Chem. Commun., 2009, 12, 1142–1144 Search PubMed.
- N.-N. Wei, C. Hao, Z. Xiu, J. Chen and J. Qiu, J. Comput. Chem., 2010, 31, 2853–2858 Search PubMed.
-
(a) I. Rozas, Phys. Chem. Chem. Phys., 2007, 9, 2782–2790 RSC;
(b) S. G. Olesen and S. Hammerum, J. Phys. Chem. A, 2009, 113, 7940–7944 Search PubMed.
- K. Jordan, Science, 1986, 232, 1559–1560 Search PubMed.
- K. D. Sen, Reviews in Modern Quantum Chemistry: A Celebration of the Contributions of Robert G. Parr, World Scientific, Singapore, 2002 Search PubMed.
- A. S. P. Gomes and C. R. Jacob, Annu. Rep. Prog. Chem., Sect. C, 2012, 108, 222–277 RSC.
- P. Sautet, Quantum Chemistry Methods, in Characterization of Solid Materials and Heterogeneous Catalysts: From Structure to Surface Reactivity, ed. M. Che and J. C. Védrine, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- L. Gagliardi and B. O. Roos, Chem. Soc. Rev., 2007, 36, 893–903 RSC.
- P. Echenique and J. L. Alonso, Mol. Phys., 2007, 105, 3057–3098 CrossRef CAS.
- C. J. Barden and H. F. Schaefer III, Pure Appl. Chem., 2000, 72, 1405–1423 Search PubMed.
- P. Pyykkö and J. F. Stanton, Chem. Rev., 2012, 112, 1–3 Search PubMed.
- A. Ghosh, Coord. Chem. Rev., 2009, 253, 523–525 Search PubMed.
- J. E. H. Koehler, W. Saenger and W. F. van Gunsteren, Eur. Biophys. J., 1988, 16, 153–168.
- W. O. George, B. F. Jones and R. H. Lewis, Philos. Trans. R. Soc. London, Ser. A, 2001, 359, 1611–1629 CrossRef CAS.
- R. L. Snow and J. L. Bills, J. Chem. Educ., 1975, 52, 506–509 Search PubMed.
- V. Sahni, Int. J. Quantum Chem., 1995, 56, 265–283 Search PubMed.
- R. J. Gillespie, Coord. Chem. Rev., 2008, 252, 1315–1327 CrossRef CAS.
- A. M. Simas, V. H. Smith Jr. and A. J. Thakkar, THEOCHEM, 1985, 123, 221–229 Search PubMed.
- S. Scheiner, Annu. Rev. Phys. Chem., 1994, 45, 23–56 CrossRef CAS.
- G. Chałasiński and M. Szcześniak, Chem. Rev., 2000, 100, 4227–4252 CrossRef CAS.
- B. Barbiellini and A. Shukla, Phys. Rev. B: Condens. Matter, 2002, 66, 235101–235105 Search PubMed.
- Y. X. Lu, J. W. Zou, J. C. Fan, W. N. Zhao, Y. J. Jiang and Q. S. Yu, J. Comput. Chem., 2009, 30, 725–732 CrossRef CAS.
- A. Hayashi, M. Shiga and M. Tachikawa, J. Chem. Phys., 2006, 125, 204310–204316 Search PubMed.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS.
- L. Rao, H. Ke, G. Fu, X. Xu and Y. Yan, J. Chem. Theory Comput., 2009, 5, 86–96 CrossRef CAS.
- J. S. Tse, Annu. Rev. Phys. Chem., 2002, 53, 249–290 CrossRef CAS.
- M. F. Carvalho, R. A. Mosquera and R. Rivelino, Chem. Phys. Lett., 2007, 445, 117–124 CrossRef CAS.
- H. Hooshyar, H. Rahemi, M. H. Akhbari Shad and B. Khezri, J. Mol. Model., 2009, 15, 525–536 Search PubMed.
- H. Hu, M. Liu, Z. F. Wang, J. Zhu, D. Wu, H. Ding, Z. Liu and F. Liu, Phys. Rev. Lett., 2012, 109, 055501–055505 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- K. Sharkas, A. Savin, H. J. A. Jensen and J. Toulouse, J. Chem. Phys., 2012, 137, 044104–044113 Search PubMed.
- K. Burke, J. Chem. Phys., 2012, 136, 150901–150909 CrossRef.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS.
- J. Lorenzana, Z.-J. Ying and V. Brosco, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075131–075134 Search PubMed.
- L. Lin and L. Ying, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235144–235153 Search PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 12, 289–320 Search PubMed.
- B. Kaduk, T. Kowalczyk and T. Van Voorhis, Chem. Rev., 2012, 112, 321–370 CrossRef CAS.
- D. C. Young, Semiempirical Methods in Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, John Wiley & Sons, Inc., New York, 2002 Search PubMed.
- M. I. Bernal-Uruchurtu and M. F. Ruiz-López, Chem. Phys. Lett., 2000, 330, 118–124 CrossRef CAS.
- J. J. Dannenberg, THEOCHEM, 1997, 401, 279–286 CrossRef CAS.
- M. Korth, M. Pitoňák, J. Rezáč and P. Hobza, J. Chem. Theory Comput., 2010, 6, 344–352 CrossRef CAS.
- J. Rézać, J. Fanfrlík, D. Salahub and P. Hobza, J. Chem. Theory Comput., 2009, 5, 1749–1760 CrossRef CAS.
- J. Rézáć and P. Hobza, J. Chem. Theory Comput., 2012, 8, 141–151 CrossRef CAS.
- N. L. Allinger, R. A. Kok and M. R. Imam, J. Comput. Chem., 1988, 9, 591–595 CrossRef.
- J. Hermans, Adv. Protein Chem., 2005, 72, 105–119 Search PubMed.
- D. S. Vieira and L. Degreve, Mol. Phys., 2009, 107, 59–69 Search PubMed.
- H. C. Georg, K. Coutinho and S. Canuto, J. Chem. Phys., 2005, 123, 124307–124314 CrossRef.
- V. M. L. Santos, F. G. B. Moreira and R. L. Longo, Chem. Phys. Lett., 2004, 390, 157–161 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho, M. N. Ramos, M. Z. Hernandes and K. R. Cavalcante, THEOCHEM, 2007, 802, 91–97 Search PubMed.
- B. G. Oliveira and M. L. A. A. Vasconcellos, J. Theor. Comput. Chem., 2007, 6, 399–412 Search PubMed.
- M. L. A. A. Vasconcellos, B. G. Oliveira and L. F. C. C. Leite, THEOCHEM, 2008, 860, 13–17 Search PubMed.
- R. Cabot, C. A. Hunter and L. M. Varley, Org. Biomol. Chem., 2010, 8, 1455–1462 RSC.
- H. Wang, L. Wang, S. Shen, W. Zhang, M. Li, L. Du, X. Zheng and D. L. Phillips, J. Chem. Phys., 2012, 136, 124509–124519 Search PubMed.
- A. Aquino, D. Tunega, G. Haberhauer, M. H. Gerzabek and H. Lischka, J. Phys. Chem. A, 2002, 106, 1862–1871 CAS.
- M. A. C. Nascimento, J. Braz. Chem. Soc., 2008, 19, 245–256 CAS.
- S. Hirata, Theor. Chem. Acc., 2012, 131, 1071–1074 Search PubMed.
- E. Schultz, J. Chem. Educ., 2005, 82, 400A–400B Search PubMed.
- P. Geerlings and F. De Proft, Int. J. Mol. Sci., 2002, 3, 276–309 Search PubMed.
- D. E. Ellis and O. Warschkow, Coord. Chem. Rev., 2003, 238–239, 31–53 Search PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon, Oxford, 1990 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2011, 12436–12437 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2011, 12438–12444 Search PubMed.
- R. F. W. Bader, Monatsh. Chem., 2005, 136, 819–854 Search PubMed.
- R. F. W. Bader, P. L. A. Popelier and T. A. Keith, Angew. Chem., Int. Ed. Engl., 1994, 33, 620–631 CrossRef.
- R. F. W. Bader and C. F. Matta, Found. Chem. DOI:10.1007/s10698-012-9153-1.
- R. F. W. Bader, Acc. Chem. Res., 1975, 8, 34–40 CrossRef CAS.
- I. Fernández and G. Frenking, Phys. Chem. Chem. Phys., 2012 10.1039/c2cp41244f.
- P. Needham, Philosophy of Science, 2004, 71, 1038–1047 Search PubMed.
- A. W. Thackray, Isis, 1966, 57, 35–55 Search PubMed.
- R. F. W. Bader and T. T. Nguyen-Dang, Adv. Quantum Chem., 1981, 14, 63–124 CAS.
- R. F. W. Bader, An Introduction to the Electronic Structure of Atoms and Molecules, Clarke, Irwin &
Co. Ltd., Toronto, 1970 Search PubMed.
- M. Goli and S. Shahbazian, Theor. Chem. Acc., 2011, 129, 235–245 CrossRef CAS.
- F. Heidarzadeh and S. Shahbazian, Int. J. Quantum Chem., 2011, 111, 2788–2801 Search PubMed.
- M. Goli and S. Shahbazian, Int. J. Quantum Chem., 2011, 111, 1982–1998 Search PubMed.
- P. A. M. Dirac, Phys. Z. Sowjetunion, 1933, 3, 64–72 Search PubMed.
- R. P. Feynman, Rev. Mod. Phys., 1948, 20, 367–387 CrossRef.
- J. Schwinger, Phys. Rev., 1951, 82, 914–927 Search PubMed.
- R. F. W. Bader, Chem. Phys. Lett., 2006, 426, 226–228 Search PubMed.
- P. Nasertayoob and S. Shahbazian, Int. J. Quantum Chem., 2009, 109, 726–732 CrossRef CAS.
- J. Hernández-Trujillo and R. F. W. Bader, J. Phys. Chem. A, 2000, 104, 1779–1794 CrossRef CAS.
- R. Bryant, P. Griffiths and D. Grossman, Bull. Am. Math. Soc., 2005, 42, 407–412 Search PubMed.
- E. Schrödinger, Ann. Phys., 1926, 79, 734–756 Search PubMed.
- H. Jacobsen, J. Comput. Chem., 2009, 30, 1093–1102 CrossRef.
- R. F. W. Bader, Chem.–Eur. J., 2006, 12, 7769–7772 Search PubMed.
- P. J. MacDougall, M. B. Hall, R. F. W. Bader and J. R. Cheeseman, Can. J. Chem., 1989, 67, 1842–1846 Search PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- R. F. W. Bader, Theor. Chem. Acc., 2001, 105, 276–283 Search PubMed.
- J. A. Platts, Phys. Chem. Chem. Phys., 2005, 7, 3805–3810 RSC.
- R. F. W. Bader, Theor. Chem. Acc., 2002, 107, 381–382 Search PubMed.
- M. Born, Z. Phys., 1926, 37, 863–867 Search PubMed.
- S. Srebrenik and R. F. W. Bader, J. Chem. Phys., 1974, 61, 2536–2540 Search PubMed.
- R. F. W. Bader and P. M. Beddall, J. Chem. Phys., 1972, 56, 3320–3329 CAS.
- R. F. W. Bader and H. J. T. Preston, Int. J. Quantum Chem., 1969, 3, 327–347 CrossRef CAS.
- R. F. W. Bader, T. T. Nguyen-Dang and Y. Tal, Rep. Prog. Phys., 1981, 44, 893–948 CrossRef.
- R. F. W. Bader, A Theory of Molecular Structure in Chemical Applications of Topology and Graph Theory, ed. R. B. King, Elsevier Scientific Publishing, Amsterdam, 1983 Search PubMed.
- P. L. A. Popelier, Atoms in Molecules. An Introduction, Pearson Education, London, 2000 Search PubMed.
- C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Y. A. Abramov, J. Phys. Chem. A, 2011, 115, 12809–12817 Search PubMed.
- C. F. Matta and A. A. Arabi, Future Med. Chem., 2011, 3, 969–994 Search PubMed.
- L. J. Farrugia, C. Evans, D. Lentz and M. Roemer, J. Am. Chem. Soc., 2009, 131, 1251–1268 CrossRef CAS.
- L. J. Farrugia and H. M. Senn, J. Phys. Chem. A, 2010, 114, 13418–13433 Search PubMed.
- I. Love, Comput. Theor. Chem., 2012, 985, 23–29 Search PubMed.
- H. Paulsen, J. A. Wolny and A. X. Trautwein, Monatsh. Chem., 2005, 136, 1107–1118 CrossRef CAS.
-
(a) P. L. A. Popelier, J. Phys. Chem. A, 1999, 103, 2883–2890 CrossRef CAS;
(b) P. L. A. Popelier, Chem. Phys. Lett., 1994, 228, 160–164 CrossRef CAS.
-
(a) S. Hati and D. Datta, J. Comput. Chem., 1992, 13, 912–918 Search PubMed;
(b) R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- G. Gervasio, R. Bianchi and D. Marabello, Chem. Phys. Lett., 2004, 387, 481–484 CrossRef CAS.
- A. Kirfel, T. Lippmann, P. Blaha, K. Schwarz, D. F. Cox, K. M. Rosso and G. V. Gibbs, Phys. Chem. Miner., 2005, 32, 301–313 Search PubMed.
- B. Bankiewicz and and M. Palusiak, Comput. Theor. Chem., 2011, 966, 113–119 Search PubMed.
- B. Bankiewicz, P. Matczak and M. Palusiak, J. Phys. Chem. A, 2012, 116, 452–459 Search PubMed.
- F. Biegler-König, R. F. W. Bader and T. H. Tang, J. Comput. Chem., 1982, 3, 317–328 CrossRef.
- D. Cremer and E. Kraka, J. Am. Chem. Soc., 1985, 107, 3800–3810 CrossRef CAS.
- D. Cremer and E. Kraka, J. Am. Chem. Soc., 1985, 107, 3811–3819 CrossRef CAS.
- R. F. W. Bader, P. J. MacDougall and C. D. H. Lau, J. Am. Chem. Soc., 1984, 106, 1594–1605 CrossRef CAS.
- R. F. W. Bader and G. L. Heard, J. Chem. Phys., 1999, 111, 8789–8798 CrossRef CAS.
- D. J. R. Duarte, M. M. de las Vallejos and N. M. Peruchena, J. Mol. Model., 2010, 16, 737–748 CrossRef CAS.
- V. S. Znamenskiy and M. E. Green, J. Phys. Chem. A, 2004, 108, 6543–6553 Search PubMed.
- L. Claude, S. Mohamed and P. Sébastien, J. Mol. Struct., 2003, 647, 53–64 Search PubMed.
- L. F. Pacios, J. Phys. Chem. A, 2004, 108, 1177–1188 CrossRef CAS.
- R. G. Parr, P. W. Ayers and R. F. Nalewajski, J. Phys. Chem. A, 2005, 109, 3957–3959 CrossRef CAS.
- C. F. Matta and R. F. W. Bader, J. Phys. Chem. A, 2006, 110, 6365–6371 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 2010, 114, 7431–7444 CrossRef CAS.
- C. L. Firme, O. A. C. Antunes and P. M. Esteves, J. Braz. Chem. Soc., 2009, 20, 543–548 Search PubMed.
- E. B. A. Filho, E. Ventura, S. A. do Monte, B. G. Oliveira, C. G. L. Junior, G. B. Rocha and M. L. A. A. Vasconcellos, Chem. Phys. Lett., 2007, 449, 336–340 Search PubMed.
- R. A. Klein, J. Am. Chem. Soc., 2002, 124, 13931–13937 CrossRef CAS.
- S. J. Grabowski, J. Phys. Org. Chem., 2008, 21, 694–702 Search PubMed.
- P. Kolandaivel and V. Nirmala, J. Mol. Struct., 2004, 694, 33–38 Search PubMed.
- C. F. Matta and J. Hernández-Trujillo, J. Phys. Chem. A, 2003, 107, 7496–7504 CrossRef CAS.
- R. F. W. Bader, C. F. Matta and F. Cortés-Guzmán, Organometallics, 2004, 23, 6253–6263 CrossRef CAS.
- N. Castillo, C. F. Matta and R. J. Boyd, Chem. Phys. Lett., 2005, 409, 265–269 CrossRef CAS.
- M. Ziólkowski, S. J. Grabowski and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 6514–6521 CrossRef CAS.
- L. Albrecht and R. J. Boyd, J. Phys. Chem. A, 2012, 116, 3946–3951 Search PubMed.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2011, 115, 12561–12571 Search PubMed.
- M. D. Esrafili, J. Mol. Model., 2012, 18, 2003–2011 Search PubMed.
- I. Cukrowski and C. F. Matta, Chem. Phys. Lett., 2010, 499, 66–69 Search PubMed.
- T. M. Krygowski and S. J. Grabowski, Chem. Phys. Lett., 2004, 389, 51–57 CrossRef CAS.
- R. G. A. Bone and R. F. W. Bader, J. Phys. Chem., 1996, 100, 10892–10911 CrossRef CAS.
- J. Hernández-Trujillo and C. F. Matta, Struct. Chem., 2007, 18, 849–857 CrossRef CAS.
- S. Wojtulewski and S. J. Grabowski, J. Mol. Struct., 2002, 605, 235–240 Search PubMed.
- S. J. Grabowski, J. Phys. Chem. A, 2007, 111, 3387–3393 CrossRef CAS.
- A. F. Silva, J. V. da Silva Jr., R. L. A. Haiduke and R. E. Bruns, J. Phys. Chem. A, 2011, 115, 12572–12581 Search PubMed.
- I. Alkorta, I. Rozas and J. Elguero, J. Phys. Chem. A, 1997, 101, 9457–9463 CrossRef CAS.
- J. E. Del Bene, Chem. Phys. Lett., 1974, 24, 203–207 CrossRef CAS.
- A. R. Voth, P. Khuu, K. Oishi and P. S. Ho, Nat. Chem., 2009, 1, 74–79 CrossRef CAS.
- P. Metrangolo and G. Resnati, Chem.–Eur. J., 2001, 7, 2511–2519 CrossRef.
- O. V. Shishkin, I. V. Omelchenko, A. L. Kalyuzhny and B. V. Paponov, Struct. Chem., 2010, 21, 1005–1011 Search PubMed.
- M. Kumari, P. V. Balaji and R. B. Sunoj, Phys. Chem. Chem. Phys., 2011, 13, 6517–6530 RSC.
- O. V. Shishkin, R. I. Zubatyuk, V. V. Dyakonenko, C. Lepetit and R. Chauvin, Phys. Chem. Chem. Phys., 2011, 13, 6837–6848 RSC.
- R. N. Behera and A. Panda, RSC Adv., 2012, 2, 6948–6956 RSC.
- S. Scheiner, J. Chem. Phys., 2011, 134, 094315–094323 CrossRef.
- G. Sánchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2012, 14, 9880–9889 RSC.
- S. J. Grabowski and J. Leszczynski, Computational Chemistry: Reviews of Current Trends, 2005, 9, 195–235 Search PubMed.
- G. N. Patwari, T. Ebata and N. Mikami, J. Chem. Phys., 2000, 113, 9885–9889 CrossRef.
- S. Gawinkowski, L. Walewski, A. Vdovin, A. Slenczka, S. Rols, M. R. Johnson, B. Lesyng and J. Waluk, Phys. Chem. Chem. Phys., 2012, 14, 5489–5503 RSC.
- G. M. Florio, C. J. Gruenloh, R. C. Quimpo and T. S. Zwier, J. Chem. Phys., 2000, 113, 11143–11153 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, Struct. Chem., 2008, 19, 185–189 Search PubMed.
- E. Diana, M. R. Chierotti, E. M. C. Marchese, G. Croce, M. Milanesio and P. L. Stanghellini, New J. Chem., 2012, 36, 1099–1107 RSC.
- A. Caballero and F. A. Jalón, Chem. Commun., 1998, 1879–1880 RSC.
- B. G. Oliveira, R. C. M. U. Araújo and M. N. Ramos, Struct. Chem., 2008, 19, 665–670 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, J. J. Silva and M. N. Ramos, Struct. Chem., 2010, 21, 221–228 Search PubMed.
- J. L. López, M. Mandado, M. J. G. Moa and R. A. Mosquera, Chem. Phys. Lett., 2006, 422, 558–564 Search PubMed.
- T. N. Guru Row and S. R. Gadre, Proc.–Indian Acad. Sci., Chem. Sci., 1985, 95, L437–L438 Search PubMed.
- B. G. Oliveira, M. C. A. Lima, I. R. Pitta, S. L. Galdino and M. Z. Hernandes, J. Mol. Model., 2010, 16, 119–127 Search PubMed.
- T. C. Dinadayalane and J. Leszczynski, J. Chem. Phys., 2009, 130, 081101–081104 Search PubMed.
- M. Rozenberg, G. Shoham, I. Reva and R. Fausto, Phys. Chem. Chem. Phys., 2005, 7, 2376–2383 RSC.
- S. A. C. McDowell, Chem. Phys. Lett., 2006, 424, 239–242 Search PubMed.
- S. A. C. McDowell, Phys. Chem. Chem. Phys., 2003, 5, 808–811 RSC.
- I. V. Alabugin, M. Manoharan and F. A. Weinhold, J. Phys. Chem. A, 2004, 108, 4720–4730 CrossRef CAS.
- S.-C. Wang, P. K. Sahu and S.-L. Lee, Chem. Phys. Lett., 2005, 406, 143–147 Search PubMed.
- M. Freindorf, E. Kraka and D. Cremer, Int. J. Quantum Chem., 2012, 112, 3174–3187 Search PubMed.
- A. J. Barnes, J. Mol. Struct., 2004, 704, 3–9 CrossRef CAS.
- J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620–4636 CrossRef CAS.
- N. R. Dhumal and S. P. Gejji, Chem. Phys. Lett., 2004, 393, 355–360 Search PubMed.
- Y. Yang, Int. J. Quantum Chem., 2009, 109, 266–274 CrossRef CAS.
- J.-X. Wang, Y. Feng, L. Liu, X.-S. Li and Q.-X. Guo, Chin. J. Chem., 2004, 22, 642–648 Search PubMed.
- W. Zierkiewicz, D. Michalska, Z. Havlas and P. Hobza, ChemPhysChem, 2002, 3, 511–518 CrossRef CAS.
- X. Li, L. Liu and H. B. Schlegel, J. Am. Chem. Soc., 2002, 124, 9639–9647 CrossRef CAS.
- P.-P. Zhou and W.-Y. Qiu, ChemPhysChem, 2009, 10, 1847–1858 CrossRef CAS.
- K. Hermansson, Int. J. Quantum Chem., 1993, 45, 747–758 Search PubMed.
- W. Qian and S. Krimm, J. Phys. Chem. A, 2002, 106, 6628–6636 CrossRef CAS.
- J. S. Murray, M. C. Concha, P. Lane, P. Hobza and P. Politzer, J. Mol. Model., 2008, 14, 699–704 CrossRef CAS.
- S. Scheiner and T. Kar, J. Phys. Chem. A, 2002, 106, 1784–1789 CAS.
- K. Hermansson, J. Phys. Chem. A, 2002, 106, 4695–4702 CrossRef CAS.
- L. Pejov and K. Hermansson, J. Chem. Phys., 2003, 119, 313–324 CrossRef CAS.
- K. Hermansson, G. Gajewski and P. D. Mitev, J. Phys. Chem. A, 2008, 112, 13487–13494 Search PubMed.
- W. Yu, Z. Lin and Z. Huang, ChemPhysChem, 2006, 7, 828–830 Search PubMed.
- A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, Dalton Trans., 2006, 3077–3087 RSC.
- M. Rahimi and P. Nasertayoob, MATCH, 2012, 67, 109–126 Search PubMed.
- V. Tognetti and L. Joubert, J. Phys. Chem. A, 2011, 115, 5505–5515 Search PubMed.
- D. G. Truhlar, Encyclopedia of Physical Science and Technology, 3rd edn, 2004, vol. 9 Search PubMed.
- T. Aoyama, O. Matsuoka and N. Nakagawa, Chem. Phys. Lett., 1979, 67, 508–510 CrossRef CAS.
- C. J. Wu and E. A. Carter, J. Am. Chem. Soc., 1990, 112, 5893–5895 Search PubMed.
- S. A. McDonald, G. L. Johnson, B. W. Keelan and L. Andrews, J. Am. Chem. Soc., 1980, 102, 2892–2896 CrossRef CAS.
- A. M. Sapse and D. C. Jain, Int. J. Quantum Chem., 1988, 33, 69–76 CAS.
- E. Ramos-Cordoba, D. S. Lambrecht and M. Head-Gordon, Faraday Discuss., 2011, 150, 345–362 RSC.
- A. M. Sapse, C. Pinto and D. C. Jain, J. Cluster Sci., 2000, 11, 381–389 Search PubMed.
- J. Rozière, J. M. Williams, E. Grech, Z. Malarski and L. Sobcyzk, J. Chem. Phys., 1980, 72, 6117–6122 CrossRef CAS.
- R. D. Parra and X. C. Zeng, J. Chem. Phys., 1999, 110, 6329–6338 CrossRef CAS.
- L. C. Remer and J. H. Jensen, J. Phys. Chem. A, 2000, 104, 9266–9275 CrossRef CAS.
- B. Michielsen, J. J. J. Dom, B. J. van der Veken, S. Hesse, M. A. Suhm and W. A. Herrebout, Phys. Chem. Chem. Phys., 2012, 14, 6469–6478 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- P. A. Kollman and L. C. Allen, Chem. Rev., 1972, 72, 283–303 CrossRef CAS.
- V. M. Goldschmidt, Geochemische Vertei-lungsgesetze der Elemente. Skrifter Norske Videnskaps, Akad, Oslo, 1926 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, New Yok, 3rd edn, 1960 Search PubMed.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho, E. F. Lima, W. L. V. Silva, M. N. Ramos and A. M. Tavares, THEOCHEM, 2006, 775, 39–45 Search PubMed.
- T. Tsurusawa and S. Iwata, J. Chem. Phys., 2000, 112, 5705–5710 CrossRef CAS.
- P. R. Varadwaj, J. Mol. Model., 2010, 16, 965–974 Search PubMed.
- K. Nakamoto, M. Margoshes and R. E. Rundle, J. Am. Chem. Soc., 1955, 77, 6480–6486 CrossRef CAS.
- R. K. Thomas and H. Thompson, Proc. R. Soc. London, A, 1970, 316, 303–313 Search PubMed.
- Z. Zhou, Y. Qu, A. Fu, B. Du, F. He and H. Gao, Int. J. Quantum Chem., 2002, 89, 550–558 CrossRef CAS.
- M. Krajewska, A. Olbert-Majkut and Z. Mielke, Phys. Chem. Chem. Phys., 2002, 4, 4305–4313 RSC.
- H. Matsuura, H. Yoshida, M. Hieda, S.-Y. Yamanaka, T. Harada, K. Shin-ya and K. Ohno, J. Am. Chem. Soc., 2003, 125, 13910–13911 CrossRef CAS.
- M. Hippler, J. Chem. Phys., 2007, 127, 084306–084316 CrossRef.
- D. Hauchecorne, N. Nagels, B. J. van der Veken and W. A. Herrebout, Phys. Chem. Chem. Phys., 2012, 14, 681–690 RSC.
- A. J. Barnes and S. L. Paulson, Chem. Phys. Lett., 1983, 99, 326–330 Search PubMed.
- M. Mura, X. Sun, F. Silly, H. T. Jonkman, G. A. D. Briggs, M. R. Castell and L. N. Kantorovich, Phys. Rev. B, 2010, 81, 195412–195422 CrossRef.
- J.-Q. Wang, J. Sun, W.-G. Cheng, K. Dong, X.-P. Zhang and S.-J. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 11021–11026 RSC.
- F. Kollipost, R. W. Larsen, A. V. Domanskaya, M. Nörenberg and M. A. Suhm, J. Chem. Phys., 2012, 136, 151101–151104 Search PubMed.
- S. Kolboe and S. Svelle, J. Phys. Chem. A, 2008, 112, 6399–6400 CrossRef CAS.
- B. Kharat, V. Deshmukh and A. Chaudhari, Struct. Chem., 2012, 23, 637–644 Search PubMed.
- Z. Xu, H. Li, C. Wang, T. Wu and S. Han, Chem. Phys., 2004, 394, 405–409 Search PubMed.
- G. Zhang, A. Ji and D. Chen, THEOCHEM, 2008, 853, 89–96 CrossRef CAS.
- M. Lozynski, D. Rusinska-Roszak and H.-G. Mack, J. Phys. Chem. A, 1998, 102, 2899–2903 CrossRef CAS.
- D. J. Anick, J. Chem. Phys., 2003, 119, 12442–12456 CrossRef CAS.
- N. V. Belkova, O. A. Filippov, A. M. Filin, L. N. Teplitskaya, Y. V. Shmyrova, V. V. Gavrilenko, L. M. Golubinskaya, V. I. Bregadze, L. M. Epstein and E. S. Shubina, Eur. J. Inorg. Chem., 2004, 3453–3461 CrossRef CAS.
- O. A. Filippov, A. M. Filin, N. V. Belkova, V. N. Tsupreva, Y. V. Shmyrova, I. B. Sivaev, L. M. Epstein and E. S. Shubina, J. Mol. Struct., 2006, 790, 114–121 Search PubMed.
- E. S. Shubina, E. V. Bakhmutova, A. M. Filin, I. B. Sivaev, L. N. Teplitskaya, A. L. Chistyakov, I. V. Stankevich, V. I. Bakhmutov, V. I. Bregadze and L. M. Epstein, J. Organomet. Chem., 2002, 657, 155–162 CrossRef CAS.
- L. M. Epstein, E. S. Shubina, E. V. Bakhmutova, L. N. Saitkulova, V. I. Bakhmutov, A. L. Chistyakov and I. V. Stankevich, Inorg. Chem., 1998, 37, 3013–3017 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, J. Chem. Phys., 2007, 127, 024306–024311 CrossRef.
- N. V. Belkova, E. S. Shubina and L. M. Epstein, Acc. Chem. Res., 2005, 38, 624–631 CrossRef CAS.
- S. Aime, E. Diana, R. Gobetto, M. Milanesio, E. Valls and D. Viterbo, Organometallics, 2002, 21, 50–57 CrossRef CAS.
- H. Ishikawa, A. Saito, M. Sugiyama and N. Mikami, J. Chem. Phys., 2005, 123, 224309–224313 CrossRef.
- G. N. Patwari, T. Ebata and N. Mikami, J. Phys. Chem. A, 2001, 105, 8642–8645 CrossRef CAS.
- I. I. Grinvald, G. A. Domrachev and I. Y. Kalagaev, Dokl. Phys. Chem., 2012, 444, 93–95 Search PubMed.
- L. Andrews, Chem. Soc. Rev., 2004, 33, 123–132 RSC.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Trends Heterocycl Chem., 2009, 14, 21–28 Search PubMed.
- A. Zabardasti, A. Kakanejadifard, A. A. Hoseini and M. Solimannejad, Dalton Trans., 2010, 39, 5918–5922 RSC.
- A. Zabardasti and M. Arabpour, Struct. Chem., 2012, 23, 473–477 Search PubMed.
- P. C. Singh and G. N. Patwari, Chem. Phys. Lett., 2006, 419, 5–9 CrossRef CAS.
- E. V. Bakhmutova, V. I. Bakhmutov, N. V. Belkova, M. Besora, L. M. Epstein, A. Lledós, G. I. Nikonov, E. S. Shubina, J. Tomàs and E. V. Vorontsov, Chem.–Eur. J., 2004, 10, 661–671 CrossRef CAS.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, J. Mol. Model., 2011, 17, 2847–2862 Search PubMed.
- Y. Feng, S.-W. Zhao, L. Liu, J.-T. Wang, X.-S. Li and Q.-X. Guo, J. Phys. Org. Chem., 2004, 17, 1099–1106 CrossRef CAS.
- Y. Yang and W. Zhang, THEOCHEM, 2007, 814, 113–117 CrossRef CAS.
- S. J. Grabowski, T. L. Robinson and J. Leszczynski, Chem. Phys. Lett., 2004, 386, 44–48 CrossRef CAS.
- E. C. Aguiar, J. B. P. da Silva and M. N. Ramos, Spectrochim. Acta, Part A, 2008, 71, 5–9 Search PubMed.
- J. Florián, J. Leszczynski and B. G. Johnson, J. Mol. Struct., 1995, 349, 421–426 CrossRef CAS.
- B. G. Oliveira, M. L. A. A. Vasconcellos, R. R. Olinda and E. B. A. Filho, Struct. Chem., 2009, 20, 81–90 Search PubMed.
- B. G. Oliveira and M. L. A. A. Vasconcellos, Struct. Chem., 2009, 20, 897–902 Search PubMed.
- M. M. Vallejos and N. M. Peruchena, J. Phys. Chem. A, 2012, 116, 4199–4210 Search PubMed.
- I. Skarmoutsos, D. Dellis, R. P. Matthews, T. Welton and P. A. Hunt, J. Phys. Chem. B, 2012, 116, 4921–4933 CrossRef CAS.
- O. A. Filippov, N. V. Belkova, L. M. Epstein, A. Lledos and E. S. Shubina, ChemPhysChem, 2012, 13, 2677–2687 Search PubMed.
- T. Kar and S. Scheiner, J. Chem. Phys., 2003, 119, 1473–1482 CrossRef CAS.
-
(a) Y. Liu, Y. Yang, K. Jiang, D. Shi and J. Sun, Bull. Chem. Soc. Jpn., 2011, 84, 191–195 Search PubMed;
(b) P. C. Singh and G. N. Patwari, J. Phys. Chem. A, 2007, 11, 3178–3183 Search PubMed;
(c) Q. Z. Li, T. Hu, X. L. An, B. A. Gong and J. B. Cheng, ChemPhysChem, 2008, 9, 1942–1946 Search PubMed;
(d) T. B. Yisgedu, X. Chen, H. K. Lingam, Z. Huang, E. A. Meyers, S. G. Shore and J.-C. Zhao, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, 1–3 Search PubMed;
(e) X.-Y. Hao, Z.-R. Li, D. Wu, Z.-S. Li and C.-C. Sun, J. Chem. Phys., 2003, 118, 10939–10943 Search PubMed;
(f) T. Kimura, N. Koiso, K. Ishiwata, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 8880–8883 CrossRef CAS;
(g) W. Zierkiewicz and P. Hobza, Phys. Chem. Chem. Phys., 2004, 6, 5288–5296 RSC;
(h) I. Alkorta and J. Elguero, Chem. Phys. Lett., 2003, 381, 505–511 Search PubMed;
(i) E. A. Ruben, M. S. Chapman and J. D. Evanseck, J. Phys. Chem. A, 2007, 111, 10804–10814 Search PubMed.
- B. G. Oliveira, Comput. Theor. Chem., 2012, 998, 173–182 Search PubMed.
- W. Mikenda and S. Steinböck, J. Mol. Struct., 1994, 326, 123–130 CrossRef CAS.
- J. G. Ángyán, M. Loos and I. Mayer, J. Phys. Chem., 1994, 98, 5244–5248 CrossRef CAS.
- J. G. Ángyán, G. Jansen, M. Loos, C. Hättig and B. A. Heß, Chem. Phys. Lett., 1994, 219, 267–273 CrossRef CAS.
- D. B. Chesnut, Chem. Phys., 2006, 327, 327–334 Search PubMed.
- V. Tognetti, L. Joubert and C. Adamo, J. Chem. Phys., 2010, 132, 211101–211104 CrossRef.
- C. Foroutan-Nejad and S. Shahbazian, THEOCHEM, 2009, 894, 20–22 Search PubMed.
- M. T. Carroll, C. Chang and R. F. W. Bader, Mol. Phys., 1988, 63, 387–405 CAS.
- M.-F. Fan, Z. Lin, J. E. McGrady and A. M. P. Mingos, J. Chem. Soc., Perkin Trans., 1996, 2, 563–568 Search PubMed.
- P. L. A. Popelier and R. F. W. Bader, Chem. Phys. Lett., 1992, 189, 542–548 CrossRef CAS.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- C. F. Matta, L. Huang and L. Massa, J. Phys. Chem. A, 2011, 115, 12451–12458 Search PubMed.
- F. Pichierri, Chem. Phys. Lett., 2012, 519–520, 83–88 Search PubMed.
- M. Nasiri, M. Shakourian-Fard and A. Fattahi, J. Phys. Org. Chem., 2012, 25, 803–810 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS.
- S. J. Grabowski and J. M. Ugalde, J. Phys. Chem. A, 2010, 114, 7223–7229 Search PubMed.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 11450–11458 CrossRef CAS.
- R. F. W. Bader, Int. J. Quantum Chem., 1994, 49, 299–308 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- B. G. Oliveira, J. Chil. Chem. Soc., 2009, 54, 43–45 Search PubMed.
- C. F. Matta, M. Sichinga and P. W. Ayers, Chem. Phys. Lett., 2011, 514, 379–383 Search PubMed.
- P. L. A. Popelier and F. M. Aicken, J. Am. Chem. Soc., 2003, 125, 1284–1292 Search PubMed.
- R. Parthasarathi, R. Amutha, V. Subramanian, B. U. Nair and T. Ramasami, J. Phys. Chem. A, 2004, 108, 3817–3828 CrossRef CAS.
- J. S. M. Anderson and P. W. Ayers, J. Phys. Chem. A, 2011, 115, 13001–13006 Search PubMed.
- L. D. Site, Phys. Lett. A, 2001, 286, 61–64 Search PubMed.
- J. R. Mohallem, Theor. Chem. Acc., 2002, 107, 372–374 Search PubMed.
- B. G. Oliveira, F. S. Pereira, R. C. M. U. Araújo and M. N. Ramos, Chem. Phys. Lett., 2006, 427, 181–184 CrossRef.
- E. Cubero, M. Orozco, P. Hobza and F. J. Luque, J. Phys. Chem. A, 1999, 103, 6394–6401 CrossRef CAS.
- M. Domagala and S. J. Grabowski, J. Phys. Chem. A, 2005, 109, 5683–5688 CrossRef CAS.
- H. Roohi and S. Bagheri, Int. J. Quantum Chem., 2011, 111, 961–969 Search PubMed.
- P. G. Puranik and V. Kumar, Proc.–Indian Acad. Sci., Sect. A, 1963, 58, 29–37 Search PubMed.
- F. Fuster and S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 10078–10086 CrossRef CAS.
- S. Wojtulewski and S. J. Grabowski, J. Mol. Struct., 2003, 645, 287–294 CrossRef CAS.
- S. J. Grabowski, Chem. Phys. Lett., 2000, 327, 203–208 CrossRef CAS.
- M. J. Calhorda and P. E. M. Lopes, J. Organomet. Chem., 2000, 609, 53–59 CrossRef.
- L. Feng, F.-Q. Bai, Y. Wu and H.-X. Zhang, Mol. Phys., 2011, 109, 645–653 Search PubMed.
- I. Alkorta, K. Zborowski, J. Elguero and M. Solimannejad, J. Phys. Chem. A, 2006, 110, 10279–10286 CrossRef CAS.
- A. Mohajeri, M. Alipour and M. Mousaee, J. Phys. Chem. A, 2011, 115, 4457–4466 CrossRef CAS.
- M. van Meerssche and J. Feneau-Dupont, Introduction à la Cristallographie, in Chimie Structurale, ed. Oyez é et al., Leuven, Bruxelles, Paris, 1976 Search PubMed.
- B. Raghavendra, P. K. Mandal and E. Arunan, Phys. Chem. Chem. Phys., 2006, 8, 5276–5286 RSC.
- Y. V. Zefirov and P. M. Zorky, Russ. Chem. Rev., 1995, 64, 415–428 Search PubMed.
- S. S. Batsanov, J. Chem. Soc., Dalton Trans., 1998, 1541–1546 RSC.
- F. C. Rangel, A. L. B. Montel and K. C. Mundim, Mol. Simul., 2009, 35, 342–348 Search PubMed.
- E. Kemner, I. M. de Schepper and G. J. Kearley, Chem. Commun., 2001, 2466–2467 RSC.
- M. Carroll and R. F. W. Bader, Mol. Phys., 1988, 65, 695–722 CAS.
- F. Cortés-Guzmán and R. F. W. Bader, Chem. Phys. Lett., 2003, 379, 183–192 CrossRef CAS.
- J. Hernández-Trujillo, F. Cortés-Guzmán, D.-C. Fang and R. F. W. Bader, Faraday Discuss., 2007, 135, 79–95 RSC.
- M. García-Revilla, E. Francisco, A. Costales and A. M. Pendás, J. Phys. Chem. A, 2012, 116, 1237–1250 Search PubMed.
- A. M. Pendás, E. Francisco, M. A. Blanco and C. Gatti, Chem.–Eur. J., 2007, 13, 9362–9371 CrossRef CAS.
- R. Runtz, R. F. W. Bader and R. R. Messer, Can. J. Chem., 1977, 55, 3040–3045 Search PubMed.
- P. F. Zou and R. F. W. Bader, Int. J. Quantum Chem., 1992, 43, 677–699 Search PubMed.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, Chem. Phys. Lett., 2006, 422, 334–339 Search PubMed.
- I. Alkorta, J. Elguero and S. J. Grabowski, J. Phys. Chem. A, 2008, 112, 2721–2727 CrossRef CAS.
- M. P. Barnett and J. F. Harrison, Int. J. Quantum Chem., 2003, 95, 659–662 Search PubMed.
- L. C. Allen, J. Am. Chem. Soc., 1975, 97, 6921–6940 Search PubMed.
- Z. Huang, L. Yu, Y. Dai and H. Wang, THEOCHEM, 2010, 960, 98–105 Search PubMed.
- I. Alkorta, I. Rozas and J. Elguero, Struct. Chem., 1998, 9, 243–247 CrossRef CAS.
- S. Wojtulewski and S. J. Grabowski, Chem. Phys. Lett., 2003, 378, 388–394 Search PubMed.
- S. J. Grabowski, THEOCHEM, 2007, 811, 61–67 CrossRef CAS.
- J. M. Hermida-Ramón and R. A. Mosquera, Chem. Phys., 2006, 323, 211–217 Search PubMed.
- F. T. T. Huque and J. A. Platts, Org. Biomol. Chem., 2003, 1, 1419–1424 RSC.
- M. Mandado, A. M. Graña and R. A. Mosquera, Phys. Chem. Chem. Phys., 2004, 6, 4391–4396 RSC.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2004, 108, 5823–5830 CrossRef CAS.
- P. Tarakeshwar and K. S. Kim, J. Mol. Struct., 2002, 615, 227–238 CrossRef CAS.
- P. Tarakeshwar, K. S. Kim and B. Brutschy, J. Chem. Phys., 2000, 112, 1769–1781 CrossRef CAS.
- E. E. Fileti, P. Chaudhuri and S. Canuto, Chem. Phys. Lett., 2004, 400, 494–499 CrossRef CAS.
- H. Szatylowicz, J. Phys. Org. Chem., 2008, 21, 897–914 CrossRef CAS.
- H. Raissi, M. Yoosefian, F. Mollania, F. Farzad, A. R. Nowroozi and D. Loghmaninejad, Comput. Theor. Chem., 2011, 966, 299–305 Search PubMed.
- T.-Y. Lin, K.-C. Tang, S.-H. Yang, J.-Y. Shen, Y.-M. Cheng, H.-A. Pan, Y. Chi and P.-T. Chou, J. Phys. Chem. A, 2012, 116, 4438–4444 Search PubMed.
- Y. Mo, J. Phys. Chem. A, 2012, 116, 5240–5246 Search PubMed.
- S. J. Grabowski, Chem. Phys. Lett., 2001, 338, 361–366 CrossRef CAS.
- S. J. Grabowski, J. Mol. Struct., 2001, 562, 137–143 CrossRef CAS.
- T. Y. Nikolaienko, L. A. Bulavin and D. M. Hovorun, Phys. Chem. Chem. Phys., 2012, 14, 7441–7447 RSC.
- G. Sello, THEOCHEM, 1995, 340, 15–28 Search PubMed.
- C. F. Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef.
- R. F. Nalewajski, J. Phys. Chem., 1985, 89, 2831–2837 CrossRef CAS.
- P. Bultinck, W. Langenaeker, P. Lahorte, F. De Proft, P. Geerlings, M. Waroquier and J. P. Tollenaere, J. Phys. Chem. A, 2002, 106, 7887–7894 CrossRef CAS.
- P. Bultinck, W. Langenaeker, P. Lahorte, F. De Proft, P. Geerlings, C. van Alsenoy and J. P. Tollenaere, J. Phys. Chem. A, 2002, 106, 7895–7901 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- F. Martin and H. Zipse, J. Comput. Chem., 2005, 26, 97–105 CrossRef CAS.
- R. F. W. Bader, W. H. Henneker and P. E. Cade, J. Chem. Phys., 1967, 46, 3341–3363 CrossRef CAS.
- R. F. W. Bader, P. M. Beddall and P. E. Cade, J. Am. Chem. Soc., 1971, 93, 3095–3107 Search PubMed.
- R. F. W. Bader, S. G. Anderson and A. J. Duke, J. Am. Chem. Soc., 1979, 101, 1389–1395 CrossRef CAS.
- R. F. W. Bader, T. T. Nguyen-Dang and Y. Tal, J. Chem. Phys., 1979, 70, 4316–4329 CrossRef CAS.
- R. F. W. Bader and C. F. Matta, J. Phys. Chem. A, 2004, 108, 8385–8394 CrossRef CAS.
- I. S. Bushmarinov, K. A. Lyssenko and M. Y. Antipin, Russ. Chem. Rev., 2009, 78, 283–302 Search PubMed.
- E. Chamorro, P. Fuentealba and A. Savin, J. Comput. Chem., 2003, 24, 496–504 CrossRef CAS.
- R. P. Sagar and M. Hô, J. Mex. Chem. Soc., 2008, 52, 60–66 Search PubMed.
- D. Vanfleteren, D. van Neck, P. Bultinck, P. W. Ayers and M. Waroquier, J. Chem. Phys., 2010, 132, 164111–164120 CrossRef.
- V. G. Tsirelson, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 632–639 CrossRef.
- O. Hassel, Science, 1970, 170, 497–502 CAS.
- H. Ratajczak and W. J. Orville-Thomas, J. Mol. Struct., 1973, 19, 237–245 Search PubMed.
- H. Ratajczak, W. J. Orville-Thomas and C. N. R. Rao, Chem. Phys., 1976, 17, 197–216 CrossRef CAS.
- Z. Latajka, M. M. Szczçśniak, H. Ratajczak and W. J. Orville-Thomas, J. Comput. Chem., 1980, 1, 417–419 Search PubMed.
- C. N. R. Rao, P. C. Dwivedi, H. Ratajczak and W. J. Orville-Thomas, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 955–966 RSC.
- W. T. King and G. B. Mast, J. Phys. Chem., 1976, 80, 2521–2525 CrossRef CAS.
- W. B. Person, B. Zilles, J. D. Rogers and R. G. A. Maia, J. Mol. Struct., 1982, 80, 297–308 CrossRef CAS.
- M. Gussoni, M. N. Ramos, C. Castiglioni and G. Zerbi, Chem. Phys. Lett., 1989, 160, 200–205 Search PubMed.
- V. H. Rusu, M. N. Ramos and J. B. P. da Silva, Int. J. Quantum Chem., 2006, 106, 2811–2817 Search PubMed.
- C. Castiglioni, M. Gussoni and G. Zerbi, J. Chem. Phys., 1984, 80, 3916–3918 Search PubMed.
- P. L. A. Popelier and G. Logothetis, J. Organomet. Chem., 1998, 555, 101–111 CrossRef CAS.
- C. Foroutan-Nejad, J. Phys. Chem. A, 2011, 115, 12555–12560 CrossRef CAS.
- N. H. Werstiuk and W. Sokol, Can. J. Chem., 2008, 86, 737–744 CrossRef CAS.
- M. Goli and S. Shahbazian, Theor. Chem. Acc., 2012, 131, 1208–1226 Search PubMed.
- J. I. Rodríguez, A. M. Köster, P. W. Ayers, A. Santos-Valle, A. Vela and G. Merino, J. Comput. Chem., 2009, 30, 1082–1092 CrossRef CAS.
- E. Kavitha, N. Sundaraganesan and S. Sebastian, Indian J. Pure Appl. Phys., 2010, 48, 20–30 Search PubMed.
- B. G. Oliveira, J. Arg. Chem. Soc., 2007, 95, 59–69 Search PubMed.
- A. Y. Li, J. Chem. Phys., 2007, 126, 154102–154112 CrossRef.
- I. Alkorta, I. Rozas and J. Elguero, Ber. Bunsen-Ges. Phys. Chem., 1998, 102, 429–435 Search PubMed.
- I. Alkorta and J. Elguero, J. Phys. Chem., 1996, 100, 19367–19370 CrossRef CAS.
- R. L. T. Pereira and S. E. Galembeck, J. Am. Chem. Soc., 2003, 125, 15614–15622 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, J. Phys. Chem. A, 1997, 101, 4236–4244 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, Chem. Soc. Rev., 1998, 27, 163–170 RSC.
- M. Esseffar, A. El Firdoussi, W. Bouab, J. L. Abboud, O. Mó and M. Yáñez, J. Phys. Chem. A, 2009, 113, 14711–14717 Search PubMed.
- H.-J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC.
- M. T. Scerba, A. F. DeBlase, S. Bloom, T. Dudding, M. A. Johnson and T. Lectka, J. Phys. Chem. A, 2012, 116, 3556–3560 Search PubMed.
- L. J. Massa, S. Ehrenson, M. Wolfsberg and C. A. Frishberg, Chem. Phys. Lett., 1971, 11, 196–197 Search PubMed.
- L. Radom, Aust. J. Chem., 1974, 27, 231–239 Search PubMed.
- E. W. Della and C. H. Schiesser, J. Chem. Soc., Chem. Commun., 1994, 417–419 RSC.
- Y. Mo, P. V. R. Schleyer, H. Jiao and Z. Lin, Chem. Phys. Lett., 1997, 280, 439–443 Search PubMed.
- A. Klaer and T. Müller, J. Phys. Org. Chem., 2010, 23, 1043–1048 Search PubMed.
- K. Hirao and S. Yamabe, Chem. Phys., 1984, 89, 237–244 CrossRef CAS.
- M. D. Bojin and D. J. Tantillo, J. Phys. Chem. A, 2006, 110, 4810–4816 CrossRef CAS.
- S. J. Grabowski, J. Phys. Org. Chem., 2003, 16, 797–802 CrossRef CAS.
- N. H. Werstiuk, Can. J. Chem., 2010, 88, 1195–1204 Search PubMed.
- W.-Z. Xu, F.-D. Ren, J. Ren, S.-N. Liu, Y. Yue, W.-L. Wang and S.-S. Chen, J. Mol. Model., 2010, 16, 615–627 CrossRef.
- R. F. W. Bader, THEOCHEM, 2010, 943, 2–18 CrossRef CAS.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37–68 CrossRef CAS.
- D. Wu, Z. R. Li, X. Y. Hao, A. F. Jalbout, L. Adamowicz, R. J. Li and C.-C. Sun, J. Chem. Phys., 2004, 120, 1330–1335 Search PubMed.
- B. G. Oliveira and R. C. M. U. Araújo, Advances in Quantum Theory: Hydrogen Bonds and Stacking Interactions on the DNA Structure: A Topological View of Quantum Computing, ed. Ion I. Cotăescu, IneTech, Rijeka, 2012 Search PubMed.
- F. Weinhold and C. R. Landis, Chem. Educ. Res. Pract., 2001, 2, 91–104 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 1–42 Search PubMed.
- S. J. Grabowski, Croat. Chim. Acta, 2009, 82, 185–192 CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 4772–4779 CrossRef CAS.
- S. J. Grabowski, A. Sokalski, E. Dyguda and J. Leszczynski, J. Phys. Chem. B, 2006, 110, 6444–6446 CrossRef CAS.
- S. S. Batsanov, Inorg.
Mater. (Transl. of Neorg. Mater.), 2001, 37, 871–885 Search PubMed.
- S. J. Grabowski, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS.
- Values of the dihydrogen bond energies collected from ref. 388 and 652 were appraised with BSSE and ZPE corrections according to:
(a) S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef;
(b) J. C. Faver, Z. Zheng and K. M. Merz Jr., Phys. Chem. Chem. Phys., 2012, 14, 7795–7799 RSC;
(c) A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2006, 27, 1203–1210 CrossRef CAS;
(d) F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS;
(e) D. A. McQuarrie, Statistical thermodynamics, Harper and Row, New York, 1973 Search PubMed.
- M. Hazewinkel, Poincaré–Hopf theorem, Encyclopedia of mathematics, Springer, 2001 Search PubMed.
- P. Balanarayan and S. R. Gadre, J. Chem. Phys., 2003, 119, 5037–5044 CrossRef CAS.
- D. Roy, P. Balanarayan and S. R. Gadre, J. Chem. Phys., 2008, 129, 174103–174109 Search PubMed.
- R. F. W. Bader and D. A. Legare, Can. J. Chem., 1992, 70, 657–676 CAS.
- N. O. J. Malcolm and P. L. A. Popelier, J. Phys. Chem. A, 2001, 105, 7638–7645 CrossRef CAS.
- A. Richon, A Scrolling History of Computational Chemistry, Network Science Corporation, Washington – DC, 2001 Search PubMed.
- V. van Speybroeck and R. J. Meier, Chem. Inform., 2003, 34 Search PubMed.
- V. van Speybroeck and R. J. Meier, Chem. Soc. Rev., 2003, 32, 151–157 RSC.
- S. D. Peyerimhoff, The Development of Computational Chemistry in Germany. Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2003, vol. 18 Search PubMed.
- D. B. Boyd, The Development of Computational Chemistry in Canada. Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2007, vol. 15 Search PubMed.
- S. J. Smith and B. T. Sutcliffe, The Development of Computational Chemistry in the United Kingdom. Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, vol. 10 Search PubMed.
- J. D. Bolcer and R. B. Hermann, The Development of Computational Chemistry in the United States. Reviews in Computational Chemistry, 5, ed. K. B. Lipkowitz and D. B. Boyd, VCH Publishers, New York, 2007, vol. 5 Search PubMed.
- J. Limtrakul, ScienceAsia, 2007, 33(s1), 33–37 Search PubMed.
- E. Clementi, AIP Conf. Proc., 2012, 1456, 5–54 Search PubMed.
- R. C. Glen, J. Comput.-Aided Mol. Des., 2012, 26, 47–49 Search PubMed.
- L. J. Farrugia, C. Evans and M. Tegel, J. Phys. Chem. A, 2006, 110, 7952–7961 CrossRef CAS.
- R. W. Soukup, Monatsh. Chem., 2005, 136, 803–818 Search PubMed.
- B. S. Sudhindra and J.-H. Fuhrhop, Int. J. Quantum Chem., 1981, 20, 747–753 Search PubMed.
- H.-M. Xiao, J.-S. Li and H.-S. Dong, J. Phys. Org. Chem., 2001, 14, 644–649 Search PubMed.
- E. J. Meijer and M. Sprik, J. Chem. Phys., 1996, 105, 8684–8689 CrossRef CAS.
- L. F. Molnar, X. He, B. Wang and K. M. Merz Jr., J. Chem. Phys., 2009, 131, 065102–065117 CrossRef.
- C. Aleman and D. Zanuy, Chem. Phys. Lett., 2000, 319, 318–326 Search PubMed.
- I. Kaplan, Intermolecular Interactions Physical Picture, Computational Methods and Model Potentials, Wiley Series in Theoretical Chemistry, John Wiley & Sons, 2006 Search PubMed.
- P. V. Komarov and V. G. Plotnikov, Int. J. Quantum Chem., 2012, 112, 3039–3045 Search PubMed.
- R. Roszak, R. W. Góra and S. Roszak, Int. J. Quantum Chem., 2012, 112, 3046–3051 Search PubMed.
- L. Piela, Int. J. Quantum Chem., 2012, 112, 3091–3097 Search PubMed.
- J. B. P. da Silva, B. B. Neto, M. N. Ramos and R. E. Bruns, Chemom. Intell. Lab. Syst., 1998, 44, 187–195 CrossRef CAS.
- R. C. M. U. Araújo, J. B. P. da Silva, B. B. Neto and M. N. Ramos, Chemom. Intell. Lab. Syst., 2002, 62, 37–46 CrossRef CAS.
- H. Wang, Z. Huang, T. Shen and L. Guo, J. Mol. Model., 2012, 18, 3113–3123 Search PubMed.
- Y. Zeng, M. Zhu, X. Li, S. Zheng and L. Meng, J. Comput. Chem., 2012, 33, 1321–1327 Search PubMed.
- J. N. Latosińska, M. Latosińska, M. A. Tomczak, J. Seliger, V. Žagar and J. K. Maurin, Magn. Reson. Chem., 2012, 50, 89–105 Search PubMed.
- I. Yu. Bagryanskaya, E. V. Bartashevich, D. K. Nikulov, Yu. V. Gatilov and A. V. Zibarev, J. Struct. Chem., 2009, 50, 127–136 Search PubMed.
- B. Ośmiałowski, J. Mol. Model., 2012, 18, 1633–1644 Search PubMed.
- A. G. Papadopoulos and M. P. Sigalas, J. Mol. Model., 2011, 17, 1669–1678 Search PubMed.
- H. Li, Y. Lu, Y. Liu, X. Zhu, H. Liu and W. Zhu, Phys. Chem. Chem. Phys., 2012, 14, 9948–9955 RSC.
- I. Cukrowski, K. K. Govender, M. P. Mitoraj and M. Srebro, J. Phys. Chem. A, 2011, 115, 12746–12757 Search PubMed.
- H. Wang, Z. Huang, T. Shen and L. Guo, Struct. Chem., 2012, 23, 1163–1172 Search PubMed.
- Z. Huang, Y. Dai, L. Yu and H. Wang, J. Mol. Model., 2011, 17, 2609–2621 Search PubMed.
- Z. Huang, L. Yu and Y. Dai, Struct. Chem., 2010, 21, 855–862 Search PubMed.
- M. V. Vener, A. V. Manaev, A. N. Egorova and V. G. Tsirelson, J. Phys. Chem. A, 2007, 111, 1155–1162 CrossRef CAS.
- E. R. Davidson and D. Feller, Chem. Rev., 1988, 86, 681–696.
- R. M. Balabin, J. Chem. Phys., 2010, 132, 211103–211107 CrossRef.
- B. G. Oliveira, R. C. M. U. Araújo, A. B. Carvalho and M. N. Ramos, Acta Chim. Slov., 2009, 56, 704–711 CAS.
- H. Roohi, A.-R. Nowroozi and F. Eshghi, Int. J. Quantum Chem., 2010, 110, 1489–1499 Search PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1372 CrossRef CAS.
- A. Krishtal, K. Vanommeslaeghe, A. Olasz, T. Veszprémi, C. van Alsenoy and P. Geerlings, J. Chem. Phys., 2009, 130, 174101–174108 CrossRef.
- Y. C. Park and J. S. Lee, J. Phys. Chem. A, 2006, 110, 5091–5095 CrossRef.
- D. E. Woon, T. H. Dunning and K. A. Peterson, J. Chem. Phys., 1996, 104, 5883–5891 CrossRef CAS.
- C.-S. Wang, Y. Zhang, K. Gao and Z.-Z. Yang, J. Chem. Phys., 2005, 123, 024307–024314 Search PubMed.
- J. Friedrich, J. Chem. Theory Comput., 2012, 8, 1597–1607 Search PubMed.
- M. Solimannejad, V. Ramezani, C. Trujillo, I. Alkorta, G. Sánchez-Sanz and J. Elguero, J. Phys. Chem. A, 2012, 116, 5199–5206 Search PubMed.
- L. González, O. Mó and M. Yáñez, J. Chem. Phys., 1998, 109, 139–150 CrossRef CAS.
- P. R. Varadwaj, I. Cukrowski and H. M. Marques, THEOCHEM, 2009, 915, 20–32 Search PubMed.
- S. R. Peerannawar and S. P. Gejji, Phys. Chem. Chem. Phys., 2012, 14, 8711–8722 RSC.
- C. Matta, J. Comput. Chem., 2010, 31, 1297–1311 Search PubMed.
- K. Autumn, Y. A. Liang, S. T. Hsieh, W. Zesch, W. P. Chan, T. W. Kenny, R. Fearing and R. J. Full, Nature, 2000, 405, 681–684 CrossRef CAS.
- Y. Zeng, J. Hao, S. Zheng and L. Meng, J. Phys. Chem. A, 2011, 115, 11057–11066 Search PubMed.
-
(a) S. V. Aradhya, M. Frei, M. S. Hybertsen and L. Venkataraman, Nat. Mater., 2012, 11, 872–876 Search PubMed;
(b) M. Kamiya, T. Tsuneda and K. Hirao, J. Chem. Phys., 2002, 117, 6010–6015 CrossRef CAS;
(c) J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 5287–5293 RSC;
(d) A. Hesselmann, J. Phys. Chem. A, 2011, 115, 11321–11330 Search PubMed.
- R. Podeszwa and K. Szalewicz, J. Chem. Phys., 2012, 136, 161102–161105 Search PubMed.
- Y. Geng, S.-X. Wu, H.-B. Li, X.-D. Tang, Y. Wu, Z.-M. Su and Y. Liao, J. Mater. Chem., 2011, 21, 15558–15566 RSC.
- S. M. Cybulski and C. E. Seversen, J. Chem. Phys., 2005, 122, 014117–014125 CrossRef.
- R. J. Bartlett, V. F. Lotrich and I. V. Schweigert, J. Chem. Phys., 2005, 123, 062205–062226 CrossRef.
- G.-Z. Zhao and M. Lu, J. Chin. Chem. Soc., 2012, 59, 550–556 Search PubMed.
- Y. J. Dappe, J. Ortega and F. Flores, Phys. Rev. B, 2009, 79, 165409–165418 CrossRef.
- K. Sadeghian, M. Bocola and M. Schütz, ChemPhysChem, 2011, 12, 1251–1254 Search PubMed.
- F. O. Kannemann and A. D. Becke, J. Chem. Theory Comput., 2009, 5, 719–727 CrossRef CAS.
- F. O. Kannemann and A. D. Becke, J. Chem. Theory Comput., 2010, 6, 1081–1088 CrossRef CAS.
- J. Tao, J. P. Perdew and A. Ruzsinszky, Proc. Natl. Acad. Sci. U. S. A., 2012, 3, 18–21 Search PubMed.
- M. Jabłoński and M. Palusiak, J. Phys. Chem. A, 2010, 114, 12498–12505 CrossRef CAS.
- M. Jabłoński and M. Palusiak, J. Phys. Chem. A, 2010, 114, 2240–2244 CrossRef CAS.
- V. Tognetti, L. Joubert, P. Cortona and C. Adamo, J. Phys. Chem. A, 2009, 113, 12322–12327 CrossRef CAS.
- S. A. C. McDowell and H. K. Yarde, Phys. Chem. Chem. Phys., 2012, 14, 6883–6888 RSC.
- Y.-H. Wang, L. Li, Y.-X. Lu and J.-W. Zou, Chin. J. Chem. Phys., 2007, 20, 531–536 Search PubMed.
- Q.-Z. Li, R. Li, P. Guo, H. Li, W.-Z. Li and J.-B. Cheng, Comput. Theor. Chem., 2012, 980, 56–61 Search PubMed.
- Q.-Z. Li, H. Qi, R. Li, X.-F. Liu, W.-Z. Li and J.-B. Cheng, Phys. Chem. Chem. Phys., 2012, 14, 3025–3030 RSC.
- S. Marincean and J. E. Jackson, J. Phys. Chem. A, 2010, 114, 13376–13380 Search PubMed.
- Q.-Z. Li, X. Dong, B. Jing, W.-Z. Li, J.-B. Cheng, B.-A. Gong and Z.-W. Yu, J. Comput. Chem., 2010, 31, 1662–1669 CAS.
- Y. Zhang, A.-Y. Li and L.-J. Cao, Struct. Chem., 2012, 23, 627–636 Search PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS.
- I. Alkorta, G. Sanchez-Sanz and J. Elguero, J. Phys. Chem. A, 2012, 116, 2300–2308 Search PubMed.
- R. Lo, A. Ballabh, A. Singh, P. Dastidar and B. Ganguly, CrystEngComm, 2012, 14, 1833–1841 RSC.
- K. Eskandari and H. Zariny, Chem. Phys. Lett., 2010, 492, 9–13 CrossRef CAS.
- Y. V. Nelyubina, M. Y. Antipin, D. S. Dunin, V. Y. Kotov and K. A. Lyssenko, Chem. Commun., 2010, 46, 5325–5327 RSC.
- C. Pak, H. M. Lee, J. C. Kim, D. Kim and K. S. Kim, Struct. Chem., 2005, 16, 187–202 CrossRef CAS.
- C. L. Perrin and J. B. Nielson, Annu. Rev. Phys. Chem., 1997, 48, 511–544 CrossRef CAS.
- J. E. Del Bene and M. J. T. Jordan, THEOCHEM, 2001, 573, 11–23 CrossRef.
- R. D. Willett, C. J. Gómez-García, B. Twamley, S. Gómez-Coca and E. Ruiz, Inorg. Chem., 2012, 51, 5487–5493 Search PubMed.
- N. A. G. Bandeira and B. Le Guennic, J. Phys. Chem. A, 2012, 116, 3465–3473 Search PubMed.
- Y. H. Mariam and R. N. Musin, J. Phys. Chem. A, 2008, 112, 134–145 CrossRef CAS.
- P. Lenain, M. Mandado, R. A. Mosquera and P. Bultinck, J. Phys. Chem. A, 2008, 112, 7898–7904 CrossRef CAS.
- E. Sánchez-García and G. Jansen, J. Phys. Chem. A, 2012, 116, 5689–5697 Search PubMed.
- J. D. Durrant and J. A. McCammon, J. Mol. Graphics Modell., 2011, 31, 5–9 Search PubMed.
-
(a) S. A. C. McDowell and A. D. Buckingham, Phys. Chem. Chem. Phys., 2011, 13, 14097–14100 RSC;
(b) W. A. Bueno, Quim. Nova, 1992, 15, 328–336 Search PubMed.
- P. Tarakeshwar, H. S. Choi, S. J. Lee, J. Y. Lee, K. S. Kim, T.-K. Ha, J. H. Jang, J. G. Lee and H. Lee, J. Chem. Phys., 1999, 111, 5838–5850 CrossRef CAS.
- F. Weinhold and R. A. Klein, Mol. Phys., 2012, 110, 565–579 Search PubMed.
- M. L. Huggins, Angew. Chem., Int. Ed. Engl., 1971, 10, 147–152 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. A. Jeffrey, Crystallogr. Rev., 2003, 9, 135–176 CrossRef CAS.
- D. Hadži and W. Thompson, The Hydrogen Bonding, Pergamon Press, London, 1959 Search PubMed.
- G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, Freeman, San Francisco, 1960 Search PubMed.
- W. C. Hamilton and J. A. Ibers, Hydrogen Bonding in Solids, W.A. Benjamin, Reading, 1968 Search PubMed.
- L. Sobczyk, Hydrogen Bonding, PWN, Warszawa, 1969 Search PubMed.
- S. N. Vinogradov and R. H. Linnel, The Hydrogen Bond, van Nostrand-Reinhold, New York, 1971 Search PubMed.
- M. D. Joesten and L. J. Schaad, Hydrogen Bonding, Marcel Dekker, New York, 1974 Search PubMed.
- J. C. Speakman, The Hydrogen Bond and other Intermolecular Forces, The Chemical Society Monograph, London, 1975 Search PubMed.
- P. Schuster, G. Zundel and C. Sandorfi, The Hydrogen Bond Theory, North-Holland, 1976 Search PubMed.
- D. Hadzi and S. Bratos, in The Hydrogen Bond. Recent Developments in Theory and Experiments, ed. P. Schuster, G. Zundel and C. Sandorfy, North-Holland, Amsterdam, 1976 Search PubMed.
- N. D. Sokolov, Hydrogen Bonding, Nauka, Moscow, 1981 Search PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer, Berlin, 1991 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- S. Scheiner, Hydrogen Bonding. A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed.
- D. Hadzi, Theoretical Treatments of Hydrogen Bonding, Wiley & Sons, Chichester, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- K. Szalewicz, The hydrogen bond, Encyclopedia of Physical Science and Technology, 3rd edn, 2004 Search PubMed.
- D. M. P. Mingos, Supramolecular Assembly via Hydrogen Bonds I, Springer-Verlag, Berlin Heidelberg, 2004 Search PubMed.
- M.-C. Bellissent-Funel and J. C. Dore, Hydrogen Bond Network, Springer, 1994 Search PubMed.
- Y. Marechal, The Hydrogen Bond and the Water Molecule, Elsevier, Amsterdam, 2007 Search PubMed.
- P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory, Oxford University Press, New York, 2009 Search PubMed.
- K. L. Han and G.-J. Zhao, Hydrogen Bonding and Transfer in the Excited State, Wiley-VHC, 2011 Search PubMed.
- T. Kumagai, Visualization of Hydrogen-Bond Dynamics, Springer, 2012 Search PubMed.
- R. Ludwig, Water: From Hydrogen Bonding to Dynamics and Structure, Wiley-VHC, 2011 Search PubMed.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–167 CrossRef CAS.
- A. D. Buckingham, J. E. Del Bene and S. A. C. McDowell, Chem. Phys. Lett., 2008, 463, 1–10 CrossRef CAS.
- F. Fuster, B. Silvi, S. Berski and Z. Latajka, J. Mol. Struct., 2000, 555, 75–84 Search PubMed.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1–15 CrossRef CAS.
- A. M. Pendás, M. A. Blanco and E. Francisco, J. Chem. Phys., 2006, 125, 184112–184131 CrossRef CAS.
- M. A. Blanco, A. M Pendás and E. Francisco, J. Chem. Theory Comput., 2005, 1, 1096–1109 CrossRef CAS.
- R. F. W. Bader and G. A. Jones, Can. J. Chem., 1961, 39, 1253–1265 Search PubMed.
- C. F. Matta, Quantum Biochemistry: Electronic Structure and Biological Activity, Wiley-VCH, Weinheim, 2009 Search PubMed.
- R. F. W. Bader, C. F. Matta and F. J. Martin, Atoms in Medicinal Chemistry. Quantum Medicinal Chemistry, ed. P. Carloni and F. Alber, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- B. K. Alsberg, N. Marchand-Geneste and R. D. king, Chemom. Intell. Lab. Syst., 2000, 54, 75–91 CrossRef CAS.
- C. F. Matta, J. Comput. Chem., 2003, 24, 453–463 Search PubMed.
- R. F. W. Bader, Adv. Quantum Chem., 2009, 57, 285–318 Search PubMed.
- P. L. A. Popelier, Struct. Bonding, 2005, 115, 1–56 CrossRef CAS.
- C. Gatti, P. J. McDougall and R. F. W. Bader, J. Chem. Phys., 1988, 88, 3792–3804 Search PubMed.
- R. F. W. Bader, Found. Chem., 2011, 13, 11–37 Search PubMed.
- S. J. Grabowski, J. Phys. Org. Chem., 2004, 17, 18–31 CrossRef CAS.
- S. J. Grabowski, Monatsh. Chem., 2002, 133, 1373–1380 CrossRef CAS.
- Q.-Z. Li, B. Jing, R. Li, Z.-B. Liu, W.-Z. Li, F. Luan, J.-B. Cheng, B.-A. Gong and J.-Z. Sun, Phys. Chem. Chem. Phys., 2011, 13, 2266–2271 RSC.
- M. Koné, B. Illien, C. Laurence and J. Graton, J. Phys. Chem. A, 2011, 115, 13975–13985 Search PubMed.
- E. D. Isaacs, A. Shukla, P. M. Platzman, D. R. Hamann, B. Barbiellini and C. A. Tulk, Phys. Rev. Lett., 1999, 82, 600–603 CrossRef CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2005, 109, 4331–4341 CrossRef CAS.
- H. Jacobsen, Dalton Trans., 2010, 39, 5426–5428 RSC.
- X. An, B. Jing and Q. Li, J. Phys. Chem. A, 2010, 114, 6438–6443 CrossRef CAS.
- B. G. Oliveira and R. C. M. U. Araújo, Monatsh. Chem., 2011, 142, 861–873 Search PubMed.
- S. J. Grabowski and P. Lipkowski, J. Phys. Chem. A, 2011, 115, 4765–4773 CrossRef CAS.
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.