Prospects for hydrogen storage in graphene
Valentina
Tozzini
* and
Vittorio
Pellegrini
NEST-Istituto Nanoscienze – CNR and Scuola Normale Superiore, Piazza San Silvestro 12, 56127 Pisa, Italy. E-mail: v.tozzini@sns.it; Fax: +39 050 509 417; Tel: +39 050 509 433
First published on 31st October 2012
Abstract
Hydrogen-based fuel cells are promising solutions for the efficient and clean delivery of electricity. Since hydrogen is an energy carrier, a key step for the development of a reliable hydrogen-based technology requires solving the issue of storage and transport of hydrogen. Several proposals based on the design of advanced materials such as metal hydrides and carbon structures have been made to overcome the limitations of the conventional solution of compressing or liquefying hydrogen in tanks. Nevertheless none of these systems are currently offering the required performances in terms of hydrogen storage capacity and control of adsorption/desorption processes. Therefore the problem of hydrogen storage remains so far unsolved and it continues to represent a significant bottleneck to the advancement and proliferation of fuel cell and hydrogen technologies. Recently, however, several studies on graphene, the one-atom-thick membrane of carbon atoms packed in a honeycomb lattice, have highlighted the potentialities of this material for hydrogen storage and raise new hopes for the development of an efficient solid-state hydrogen storage device. Here we review on-going efforts and studies on functionalized and nanostructured graphene for hydrogen storage and suggest possible developments for efficient storage/release of hydrogen under ambient conditions.
I. Introduction
Hydrogen is currently considered one of the most promising “green” fuels, owing to the fact that its staggering energy content of 142 MJ kg−1 exceeds that of petroleum by a factor of three and that the product of its combustion is water vapour. However, it should be remembered that hydrogen is not an energy source, but a secondary energy carrier. It means that hydrogen must be produced, and exactly the same amount of energy needed in the production process is subsequently released during its use in fuel cells. Consequently the advantage of hydrogen for energy must be carefully considered with respect to other carriers, such as electricity.In light of this, the issue of finding systems and materials for efficient hydrogen storage assumes a primary importance. Compared to electricity (supplied by the network or stored in accumulators), in fact, hydrogen can potentially solve the problem of energy dispersion, because once energy is chemically stored then, in principle, it can be indefinitely conserved and transported. Practically the problem of energy dispersion is not eliminated, but transformed into a problem of matter (hydrogen) efficient confinement.
The purpose of this review paper is to illustrate the state of-the-art of graphene systems for hydrogen storage, and highlight new potentialities and possible future developments. After a brief introduction on the main available storage systems, we shall focus on the prospects of graphene systems and on methods for hydrogen storage under ambient conditions that exploit its specific structural and chemical properties.
I.1 Overview of available hydrogen storage systems
During the last few decades several means of hydrogen storage were considered. It is important to recall that the efficiency of storage is usually measured by two parameters: the gravimetric density, GD, namely the weight percentage of hydrogen stored to the total weight of the system (hydrogen + container), and the volumetric density, VD, that is the stored hydrogen mass per unit volume of the system. Both parameters are important, since for practical application a hydrogen storage device must be both light and compact. Thus the possible storage systems can be evaluated plotting their GD and VD in a Cartesian diagram, where the good ones occupy the upper right corner. This is shown in Fig. 1. Even the current Department of Energy – USA (DoE) targets are expressed in terms of VD and GD (green crosses in Fig. 1): specifically, for on-board hydrogen storage systems for light-duty vehicles, the expected hydrogen gravimetric capacity should reach in 2015 the level of 5.5% and a volumetric capacity of 0.04 kg m−3, which would correspond to a usable energy per mass of 1.8 kW h kg−1.1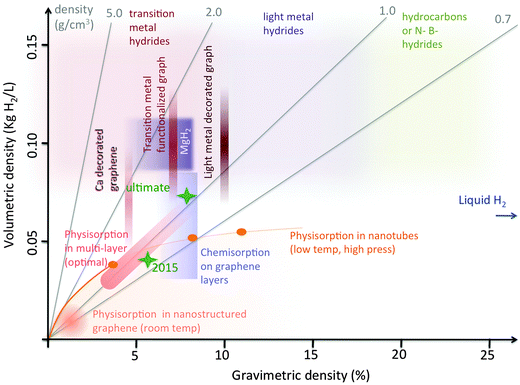 | ||
Fig. 1 Gravimetric vs. volumetric density diagram for several hydrogen storage systems including the graphene-based ones. The orange line represents the optimal relationship for physisorption in nanotubes (the dots correspond to different sizes). The line tends to the value of liquid hydrogen for large sized nanotubes. In general nanostructured physisorption based graphitic systems occupy the area below this line. The oblique shaded strip represents the optimal physisorption within graphene multilayers with spacing nearly double than in graphite (and density nearly one half). Different storage densities in this case correspond to different pressure and temperatures. The vertical dark red strips represent adsorption on decorated or functionalized graphene. These systems have been mostly studied at the level of a single layer, for this reason only the gravimetric density is well defined, while the volumetric density range has been roughly estimated considering variable inter-layer spacing 2–4 times that of graphite. The same criterion has been used to estimate the volumetric density for chemisorption in multilayers (blue rectangle); for this system the gravimetric density has a sharp right edge, corresponding to the maximum loading with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of C and H (∼8%). The storage properties of systems based on material different from graphene (different metal hydrides (including MgH2), hydrocarbons, N- and B-hydrides) are also reported as shaded areas in red, violet and green. The DOE targets (for 2015 and ultimate) are indicated with green stars. The constant density lines are in grey. :1 stoichiometry of C and H (∼8%). The storage properties of systems based on material different from graphene (different metal hydrides (including MgH2), hydrocarbons, N- and B-hydrides) are also reported as shaded areas in red, violet and green. The DOE targets (for 2015 and ultimate) are indicated with green stars. The constant density lines are in grey. | ||
The most obvious storage system is in gaseous form (compressed gas at 30–70 MPa) or as a liquid (at −253 °C) with the advantage of having large GD (between 3% and 10%) and VD (around 30–50 kg m−3). Disadvantages of this storage method are that since it exploits low temperatures and/or high pressures it therefore raises issues related to losses and safety. Another issue to consider is the system's cost. In addition, the VD of liquid hydrogen is not the maximum possible. In fact, it corresponds to the density of liquid hydrogen in molecular form of 0.068 kg l−1, while hydrogen can be further compacted within solid-state systems in the form of hydrides. The storage of atomic hydrogen was indeed realized in several solid systems including transition metal hydrides and light metal hydrides, which display VD between 0.08 and 0.15 kg m−3 (see Fig. 1). The two main drawbacks of these systems are their relatively high weight (low GD) and the presence of high activation barriers for both adsorption and desorption. Another problem is associated with the stability of light hydrides.2,3
Recently several studies have focussed on MgH2,4 which presents many advantages such as its relatively low cost and high weight storage capacity (7.6 wt%). Additionally relatively fast sorption kinetics can be achieved by ball-milling (MgH2 with additives)4,5 or through reaction with small amounts of metal amides.6 Reaction kinetics in 30 s have been also demonstrated.7 Sorption reactions occur in a pressure range of a few bar and a temperature range of 300–370 °C. Further recent significant improvements in terms of storage capacity have been demonstrated by nanostructuring Mg into nanocrystals8 leading to rapid uptake (<30 min at 200 °C), GD < 4% and a volumetric capacity (55 g l−1) greater than that of compressed H2 gas. Finally MgH2 powder compacted with 10 wt% of expanded natural graphite has been shown to lead to magnesium hydride tank displaying enhanced thermal exchanges and improved storage time.9 Finally, sodium alanate, NaAlH4, is also extensively studied as one of the most promising solid-state hydrogen-storage materials.10 In particular when doped with atoms such as Ti, Sc, Ce and Pt they display promising kinetic characteristics.11
Another class of recently considered compounds are the hydrocarbons and N- and B-hydrides12–14 that certainly satisfy the GD and VD requirements, and require chemical reaction to control hydrogen charge/discharge. Graphene, the two-dimensional one-atom-thick crystal composed of carbon atoms arranged in a honeycomb geometry, when considered as a medium for hydrogen storage, falls in this last class, and consequently it inherits some of the advantages. Carbon chemical versatility allows changing from sp2 to sp3 hybridization and efficient binding of hydrogen atoms.15 In addition graphene is stable and robust and, therefore, can be easily transported for long distances. At the same time it is mechanically flexible enabling new charging/discharging strategies under room conditions that exploit the dependence of hydrogen–carbon binding on a local curvature.16 In perspective, assuming that methods for the production of bulk graphene samples will improve with time, graphene's flexibility and unique electronic properties could enable new approaches for hydrogen storage such as the integration of hydrogen-storage modules into flexible and light, all-graphene based devices. It is also possible that integration of graphene into the above-mentioned hydrogen storage materials might offer additional routes for the realization of optimized hybrid tanks.
II. Hydrogen storage in graphene: state of the art
II.1 Physisorption versus chemisorption on single graphene layers
Hydrogen can basically be adsorbed on graphene in two different ways: either by physisorption, i.e. interacting by van der Waals (VdW) forces or by chemisorption, i.e. by forming a chemical bond with the C atoms.Physisorption usually happens with hydrogen in molecular form. The H2 binding energy was theoretically evaluated in the range of 0.01–0.06 eV,16,17 this large spread of values depends on the fact that London dispersion forces are very elusive and difficult to represent. Despite the fact that it is clear that the binding of molecular hydrogen is very weak and requires therefore low temperatures and high pressures to ensure reasonable storage stability. It was shown by a simple empirical argument that under the most favourable conditions (high pressure and low temperature) H2 can form a uniform compact monolayer on the graphene sheet, corresponding to a GD of 3.3%18 (doubled if the two sides are considered). The VD indeed depends on the possibility of compacting graphene sheets in complex structures, which is discussed in the next section.
Chemisorption of molecular hydrogen on graphene presents rather high barriers estimated around ∼1.5 eV,19 because it requires the dissociation of H2 (dissociative adsorption). Conversely, the chemisorption of atomic hydrogen is a rather favourable process: indeed commonly accepted values for H binding energy and chemisorption barriers are ∼0.7 eV and ∼0.3 eV, respectively. These values were extracted from several experimental studies on highly oriented graphite,18,20 and from theoretical studies mainly based on Density Functional Theory (DFT) with model systems mimicking graphite21–24 or graphene.25 More recent scanning tunnelling microscopy (STM) experiments have focussed on atomic-scale imaging of adsorption and clustering phenomena of hydrogen atoms on graphite.26–29 The theoretical studies have shown, in particular, that adsorption of the first H atom locally modifies the graphene structure favouring further H binding, with a collective stabilization effect.30,31 The formation of “dimers” of H on the graphene surface was shown to bring up to ∼1 eV gain in energy with respect to isolated bound H.32,33 Atomic hydrogen absorption on epitaxial graphene on SiC was also investigated by STM showing formation of dimer structures, preferential adsorption of protruding graphene areas and clustering at large hydrogen coverage.34,35
From the point of view of hydrogen storage, the maximum GD reachable in graphene with chemisorption is 8.3% (=1/12), i.e. even larger than the “ultimate” goal of DOE (see Fig. 1). This corresponds to the formation of a completely saturated graphene sheet, with 1:1 C vs. H stoichiometry, namely “graphane”, whose stability was first hypothesized in a DFT-based theoretical study,36 and subsequently studied in experiments.15 The experimental work, in particular, showed one-side hydrogenation of a graphene sheet and its reversibility by thermal annealing. Graphane is a broad band-gap insulator and removal of hydrogen in selected regions can lead to a controllable band-gap opening.37–39 As in the case of physisorption, the VD depends on the possibility of building compact structures with graphene (or graphane) sheets. The energy profiles for the processes of adsorption of hydrogen on graphene are summarized in Fig. 2.
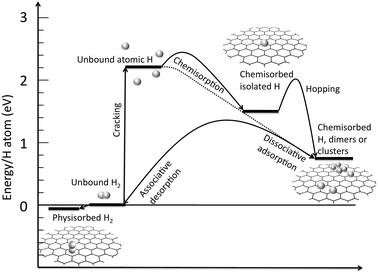 | ||
| Fig. 2 Energy level diagram for the graphene–hydrogen system. The energy is in eV per H atom, i.e. to obtain the values per H2 each energy level and barrier value must be doubled. Values of energy levels are deducted both from experimental and theoretical evaluations with average values taken when different values are available. Barriers are mainly theoretical evaluations. The reference level is the pristine graphene plus unbound molecular hydrogen. | ||
II.2 Hydrogen storage in nano-structured graphene
Being a single graphene layer quasi-2D system, its VD is not well defined, thus in the evaluation of the potentialities of graphene for hydrogen storage one should consider graphene multi-layers, three-dimensional assemblies or nano-structures of graphene. As an example, an interesting prediction regarding the physisorption capabilities of a two-layer graphene system was derived from calculations based on hybrid post-Hartree–Fock/empirical potentials and including quantum treatment for hydrogen.17 It was shown that both the GD and VD depend on the inter-layer separation, with highest values obtained for an interlayer separation of 6–8 Å (the graphite interlayer distance is 3.4 Å). Under these conditions, the physisorption energy is nearly doubled with respect to the monolayer case, reaching values of ∼0.1 eV, because the attractive VdW forces of two layers combine together, inducing a sort of nanopump effect. This is capable of increasing the internal pressure of hydrogen with respect to the external one, reaching a high level of compression. Correspondingly, the GD is increased by ∼30–40% of its single layer value, potentially reaching 8% at high pressure and low temperature, but remaining in the range of 3–4% at room temperature and high pressure. The model was subsequently improved by using more accurate interaction potentials and by exploring a wider range of temperature and pressure.40 The optimal interlayer distance for hydrogen physisorption between the two layers (or equivalently, average diameter for nanoporous systems) was confirmed to be ∼6 Å leading to an “optimal” average density of the storage system roughly one half of that of graphite (see Fig. 1, orange shaded strip). These additional theoretical studies also confirmed that the value of GD of 3% could be reached at room temperature only with high pressures, while at low temperature much higher values can be obtained, even exceeding the compression typical of liquid H2. From the experimental side it was shown that such a layered structure can be realised by using graphene oxide and the interaction between hydroxyl groups and boronic acids.41 This resulted in a sequence of graphene oxide layers connected by benzenediboronic acid pillars displaying an inter-layer separation of 10 Å with a predicted gravimetric capacity of around 6% at 77 K at a pressure of 1 bar. It should be stressed that the natural interlayer distance in graphene oxide of 7 Å is not large enough for molecular hydrogen uptake owing to the presence of the epoxies and hydroxyl groups.VdW interaction self-enhancement can be similarly postulated in any hollow graphene nano-structure. For example, an empirical estimate of the maximal VD vs. GD relation of hydrogen physisorbed on nanotubes can be obtained assuming a level of compression similar to that of liquid H2 and a full occupation of the cavity, and it is represented in Fig. 1 (orange line). Simulations performed both with DFT based methods and empirical force field substantially agree with this line, locating the GD of single walled nanotubes of ∼1 nm and ∼2 nm at ∼4% and 12% respectively.18,42 These values are substantially confirmed by a number of experiments, but correspond to the optimal values reached at high pressures (∼10 Mpa) and low temperatures (∼80 K). The room condition values are much lower. In fact, carbon nanotubes have been extensively considered as hydrogen storage media after the initial report of a GD of 5%.43 Today, however, the best reproducible results yield a GD best value of around 1% at a pressure of 120 bar at room temperature.44
A similar hydrogen-storage capability was theoretically associated with an artificial three-dimensional structure composed of graphene layers placed at an interlayer distance of 12 Å and stabilized by carbon nanotubes inserted perpendicularly to the graphene planes.45Ab initio and Monte Carlo simulations have shown the stability of such pillared graphene structures, demonstrating that GD vs. VD relation lies approximately on the same line as nanotubes. The simulated structures, in particular, display a GD of 8% at low temperature and high pressure, which decreases by an order of magnitude under room conditions, but can be recovered up to 6% at room temperature and ambient pressure after doping the pillared structure with Li cations. From the experimental side, molecular hydrogen adsorption on graphene-like nano-sheets obtained by chemically reducing exfoliated graphite oxide has been studied leading to a molecular hydrogen adsorption capacity of 1.2% at 77 K and a pressure of 10 bar and 0.68% and ambient pressure,46 extending previous controversial experiments on graphitic nanofibers.47 A GD value of 2.7% at 25 bar and room temperature was reported for graphene sheets obtained from graphene oxide after ultrasonic exfoliation in liquid.48
II.3 Improving hydrogen storage by chemical functionalization
Though physisorption within layered or nanostructured graphene can potentially lead to reasonable storage, GD and VD, it appears that large storage capacity values meeting the DoE goals are reached under very unpractical environmental conditions. In order to enhance the hydrogen adsorption under ambient conditions, therefore, various approaches including chemical functionalization of graphene were theoretically proposed. In light of the weak interaction between molecular hydrogen and graphite/graphene, several routes were investigated to enhance both the binding and the gravimetric/volumetric storage capacity.One approach exploits the chemical decoration of graphene with alkali atoms such as Li, Na and K. In the case of Li it was shown that each adsorbed Li on graphene and nanostructured graphene can adsorb up to four H2 molecules amounting to a gravimetric density above 10 wt%.49,50 A change in hydrogen binding energy on K-decorated graphene was also predicted.51 By means of DFT calculations, additionally, it was demonstrated that the combined effect of N-doping of graphene and application of an electric field normal to the sheet could induce dissociative adsorption of H2.52 A different approach is based on the exploitation of the (σ–π/d) Kubas interaction53 to bind the hydrogen on active metal sites on the graphene surface. The advantages for hydrogen storage of this particular chemical interaction, half a way from chemisorption and physisorption, are related to optimization of hydrogen–graphene interaction for room-temperature application. Particularly studied are the cases of chemical functionalization of graphene with different transition metals such as Sc, V and Ti.54–56 Several first-principle and DFT simulations showed the capacity of Ti atoms, for example, to bind up to four hydrogen molecules yielding, if attached to both graphene sides, a nominal gravimetric capacity of above 7%.54,55 It was also predicted that a stable ethylene–titanium complex should be able to attach up to ten hydrogen molecules yielding a further increase of hydrogen storage capacity.57 An interesting experimental result was obtained by decorating graphene obtained by reduction of graphene oxide with Pd atoms. An increase in the GD at 30 bar and at room temperature from 0.6% to 2.5% was reported and associated with a spillover mechanism in which hydrogen molecules undergo dissociative chemisorption on the Pd atoms followed by H migration on the graphene layer.58 The “spillover” effect, therefore, provides a mechanism for hydrogen molecule dissociation into hydrogen atoms. As we shall discuss in the next section this is relevant since atomic hydrogen adsorption/desorption on graphene can be finely controlled offering new strategies for hydrogen storage.
Transition metal elements, however, display large cohesive energies and then prefer to cluster,59 an effect detrimental to the hydrogen storage capacity although strategies to avoid clustering effects, for instance by applying strain to the graphene layer, have been proposed.60 For this reason, decoration with Ca atoms has been also considered.61 In particular recent theoretical studies have proposed Ca-decorated graphene nanoribbons for hydrogen adsorption reaching gravimetric capacities of 5% with negligible clustering of Ca atoms.62 Other approaches to favour the anchoring of transition metals on graphene substrates have been explored. Among them recent theoretical simulations have suggested the potentialities of graphene oxide as a substrate where transition metals such Ti atoms attach to epoxies and hydroxyl groups offering a promising route to gravimetric capacities of around 5%.63
III. Chemical storage in graphene: recent theoretical concepts
The following issues emerge from the previous sections: (i) compared to metal hydrides and other media for hydrogen storage, graphene-based systems have the advantage of being relatively light with good mechanical properties and high surface areas. GD and VD are not excellent but sufficient for the DoE goals. (ii) Within graphene-based systems, the advantages of hydrogen chemisorption with respect to physisorption relay on a much higher stability of the adsorbates, which makes the system very suitable for long time storage or transportation. In addition, at variance with hydrocarbons or N and B based compounds, graphene is a simple covalent crystal with advantages related to robustness, compactness and manageability.There are, however, drawbacks linked to the chemical nature of the interaction (which are, in fact, in common with chemical based storage systems). Both processes of chemisorption and desorption of molecular hydrogen on pristine flat graphene have energetic barriers whose theoretical estimations range in the order of ∼eV. These high barrier values are compatible with recent measurements of dehydrogenation temperatures of graphene or graphite (∼650 K),15,81 thus the problem of catalysis of chemi(de)sorption of molecular hydrogen remains the bottleneck for considering such a system for practical purposes, and any efficient hydrogen storage device must consider ways to overcome, lower, or bypass such energy barriers. However, in contrast to other solid-state systems based on chemisorption, graphene offers the possibility of exploring alternative catalytic strategies exploiting its peculiar mechanical chemical properties.
III.1 Graphene stretching and buckling: effect on H binding
The extended nature of a graphene layer and its large flexibility suggest the possibility of lowering the energy barriers to hydrogen adsorption by means of structural manipulations. We recall that the effect of strain on adsorption of atomic64 and molecular65 hydrogen was recently studied by DFT methods. Both works have indicated that a strain up to ∼10–15% tends to stabilize the adsorbate up to values of ∼1 eV per atom. The impact of strain on the chemisorption/desorption/hopping barrier is less clear. Compression producing an in-plane deformation of the sheet, however, seems to have both the effects of stabilizing the adsorbate and lowering the barrier for associative desorption (or dissociative adsorption).Considering the out of plane deformations, the recent experimental observation that hydrogen clusters are preferentially located on protruding areas of graphene on SiC34,35 is in agreement with the previous observation that atomic H binding energy is increased (and barrier decreased) on fullerenes and nanotubes.66 The empirical explanation of this effect is simple: convex surfaces have the sp2 system distorted towards sp3, which makes the protruding π orbitals more reactive and prone to bind H. The accurate quantification of this effect, however, turns out to be quite complex, involving the interplay between mechanical distortion and change of the electronic structure due to the curvature.67 Indeed the effect of curvature of graphitic nanostructures and in particular carbon nanotubes has been investigated for a long time.68 The chemisorption on the surface of nanotubes and fullerenes was studied in a number of DFT-based theoretical papers in the last decade.69–77 Altogether these works lead to the conclusion that the binding energy of hydrogen on the external surface of nanotubes and fullerenes is increased considerably compared to the flat graphene by values of the order of 1–2 eV per atom (depending on the tube diameter and length, on the chirality, on the coverage and type of decoration). Additionally, in small nanotubes (less than (5,5)) the binding energy is such that even the binding of molecular hydrogen (i.e. via dissociative adsorption) is exothermic. Results about the barriers for adsorption are less clear: the barrier for the adsorption of atomic hydrogen is very small already on flat graphene and disappears for the small convex curvature, while the barrier for dissociative adsorption of H2, thought decreasing with curvature, remains in the range of eV. Thus the process can occur only under specific conditions. For instance, under high pressure the nanotubes can undergo distortion creating more reactive sites. The enhancement of chemical reactivity of a graphene sheet with artificial hemispherical ripples of different radii was subsequently investigated again with DFT.78 The ripples (unstable in the absence of hydrogen) were shown to be stabilized by H adsorption, whose binding energy was found to increase for larger corrugation leading to a stable hydrogen adsorbate with respect to free molecular hydrogen for extreme values of the corrugation.
The above results were rationalized by us by a systematic DFT study of the binding energy of hydrogen as a function of curvature, evaluated on intrinsically rippled graphene structures, stabilized by lateral compression, and displaying a wide range of curvatures.16 We reported a linear increase of the binding energy of atomic H after choosing the value of the local curvature as the independent variable, instead of the average curvature of the ripple (see Fig. 3). The binding energy values for fullerenes and nanotubes lie on the same line. Finally the binding energy of molecular hydrogen can be estimated to follow a similar behaviour, further enhanced by an amount dictated by the cooperative effect of the hydrogen already bound.
![Calculated C–H binding energy versus the local curvature. The local curvature is measured by the puckering distance d of a given C atom with respect to the plane formed by its three first neighbours before H binding (see balls and sticks representation on the X axis). The variation of binding energy with respect to the flat graphene is reported on the left Y axis. The black dots correspond to the binding energy of isolated H evaluated on sampled C sites of the corrugated graphene sheet, shown on the right, with the convex parts in red and the concave ones in blue. The fitting black line is ΔEbind [eV] = −4.449 d [Å]. Binding energy profiles are shown for three sample sites on the convex (red), flat (black), and concave (blue) regions, respectively, as indicated by the arrows on the right (in this case the Y axis reports the absolute binding energy using as a reference system the graphene plus isolated detached atomic H). The arrows on the left indicate the corresponding dots in the main graph. The red squares correspond to literature data of the binding energy on C60 and on nanotubes of different lengths. The structure of graphene sheets hydrogenated on convexities is also indicated by balls and sticks (gray = C, orange = H).](/image/article/2013/CP/c2cp42538f/c2cp42538f-f3.gif) | ||
| Fig. 3 Calculated C–H binding energy versus the local curvature. The local curvature is measured by the puckering distance d of a given C atom with respect to the plane formed by its three first neighbours before H binding (see balls and sticks representation on the X axis). The variation of binding energy with respect to the flat graphene is reported on the left Y axis. The black dots correspond to the binding energy of isolated H evaluated on sampled C sites of the corrugated graphene sheet, shown on the right, with the convex parts in red and the concave ones in blue. The fitting black line is ΔEbind [eV] = −4.449 d [Å]. Binding energy profiles are shown for three sample sites on the convex (red), flat (black), and concave (blue) regions, respectively, as indicated by the arrows on the right (in this case the Y axis reports the absolute binding energy using as a reference system the graphene plus isolated detached atomic H). The arrows on the left indicate the corresponding dots in the main graph. The red squares correspond to literature data of the binding energy on C60 and on nanotubes of different lengths. The structure of graphene sheets hydrogenated on convexities is also indicated by balls and sticks (gray = C, orange = H). | ||
We found that also physisorption of molecular hydrogen depends on the curvature, but in an opposite way: VdW interactions are stronger in the concavities, due to the superposition of attractive basins of different adjacent carbon atoms. However, this effect is detectable only at low temperatures, since the related energies are just a fraction of eV.
III.2 Controlling hydrogen storage with changes of graphene curvature
The results discussed above raise the possibility of exploiting graphene's curvature for implementing practical ways to store/release hydrogen. Similarly the use of the ripples generated by lateral compression was recently proposed to engineer graphane–graphene heterostructures79 displaying finely tunable electronic properties.Concerning hydrogen storage in general, one of the key problems to be solved for practical applications is related to the realization of a chemisorption/desorption mechanism that works under room conditions, without the recourse to extreme temperatures and pressures. Specifically, while it is relatively easy to chemisorb at least atomic hydrogen, the desorption process requires overcoming the associative barrier. To this end we have recently proposed to exploit the control of curvature of graphene to desorb hydrogen.16 The process is schematically depicted in Fig. 4. After corrugation of graphene (e.g. by lateral compression), H atoms are loaded (1 of Fig. 4) and preferentially bind on convexities (2 of Fig. 4). In this configuration hydrogen on graphene can be safely transported, with minimal or no dispersion. When requested, hydrogen can be forced to desorb by inverting the curvature of graphene, i.e. by transforming the convexities into concavities. In this way H atoms will find themselves in the concave part of the corrugated graphene (3 of Fig. 4). Since in this configuration C–H binding is unstable, hydrogen will spontaneously desorb (back to 1). We have explored this process in a Car–Parrinello molecular dynamics simulation in which the curvature inversion is realised by the passage of a transverse acoustic phonon. Hydrogen is seen to desorb and form molecules, the system being able to get around the associative desorption barrier like a sort of “mechanical catalysis” effect (Fig. 5(a)).
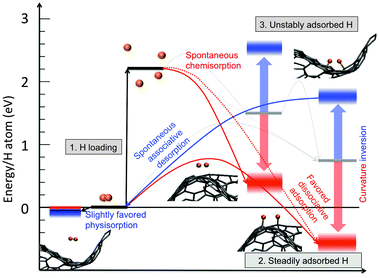 | ||
| Fig. 4 Change of the energy levels due to the local graphene curvature (to compare with Fig. 2). Convexities (red) stabilize the chemisorbed H and slightly destabilize the physisorbed H2. Concavities (blue) act in the opposite way. Convexity causes the complete elimination of chemisorption barriers and the reduction of the dissociative adsorption barrier, concavity eliminates the associative desorption barrier, and favours the physisorption at very low temperatures. Ball–stick representations of bound and physisorbed hydrogen on curved graphene are also reported. Energy reference levels and scales as in Fig. 2. | ||
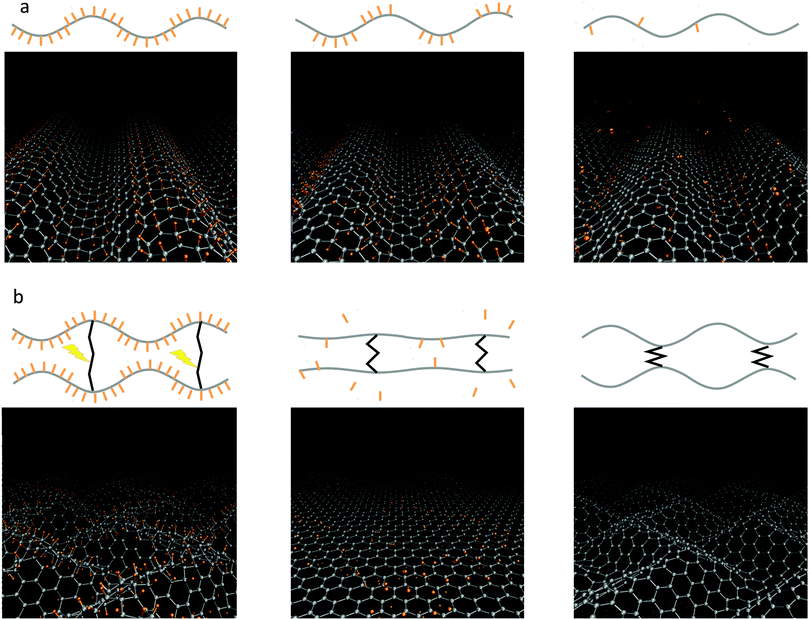 | ||
| Fig. 5 Possible protocols for hydrogen desorption. (a) Travelling wave: the curvature inversion is realized after half a period of a wave of mechanical distortion (an acoustic transverse phonon). This could be obtained using piezoelectric substrates. (b) The inversion is realized directly by changing the size of properly designed intercalants with external stimuli (e.g., optical excitation). The intercalants could function also as spacers. | ||
These findings that combine the large flexibility of graphene layers with large and tuneable C–H binding energy suggest a microscopic working scheme that sets the basis for an hydrogen storage/release device operating at fixed temperature and pressure that exploits the chemisorption as the storage mechanism and can reach estimated gravimetric densities of 8%.
IV. Ideas towards a practical device
The ideal hydrogen storage device should be cheap and robust, store large quantities of hydrogen as set by the DoE1, have a short recharge time, and operate under room conditions. We have briefly reviewed in Section I the different options available so far. Concerning solid-state systems, in particular, current on-board metal hydride storage devices can store up to 7–8% of hydrogen by weight but are costly, and operate under large changes in temperature. An advantage is their relative compactness and the relatively low charging pressure as compared with compressed gas cylinders, which at this time represents a practical solution. Despite the large progress and significant achievements as outlined in Section I.1, none of the solid-state systems developed so far have conclusively addressed all the requirements for commercially suitable hydrogen storage systems. To this respect graphene is an attractive new medium for hydrogen storage because it is readily available also in large quantities (on a ton scale) and potentially at low cost with simple chemical methods.80 However, the quality of the graphene layers produced in bulk quantities remains an issue to be addressed. In principle, as we have seen in Section II, its high surface area combined with the increased adsorption sites after chemical functionalization can lead to high storage capacities. In light of this, graphene can be considered as a promising candidate material for hydrogen storage.A number of approaches based on hydrogen storage in various types of graphene structures and nanostructures are currently studied mostly on theoretical grounds. For those that possess sufficiently large storage capacity, heat must be applied to release hydrogen. In the case of applications on vehicles, this would involve the use of an on-board burner and a heat exchanger.
In an attempt to avoid the requirements of changes in temperatures and pressures, we have proposed in Section III an approach in which hydrogen storage and release can be obtained by means of the control of graphene's curvature. While our simulations suggest the feasibility of this idea and the potentialities for realistic hydrogen-storage applications, there are still a number of practical issues to be solved.
IV.1 Molecular hydrogen chemisorption catalysis
Concerning the loading phase of a possible practical device, our simulations show spontaneous chemisorption of convex surfaces for atomic hydrogen. The experimental demonstration and full characterization of this effect in a corrugated graphene layer is in progress.81 These experiments are currently carried out on a graphene buffer layer grown on SiC that displays a natural corrugation due to interaction with the substrate82 although a factor of ten less than that explored theoretically. In spite of this, the predicted selective chemisorption is observed, monitored by atomically-resolved imaging with a scanning tunnelling microscope in a ultra-high vacuum environment.The energetic data (see Fig. 4) indicate that also chemisorption of molecular hydrogen can be exothermic for large local curvatures. However, in general, at variance with the case of atomic hydrogen, the chemisorption barrier for H2 is likely to remain quite high. In light of this, therefore, if hydrogen is not previously cracked (a process that requires a well-defined amount of energy), additional catalytic mechanisms are necessary.
By analogy with the benzene hydrogenation that can occur, catalysed by transition metals, the functionalization of graphene with Pd has been recently observed to produce chemisorption of H2 by means of a spillover mechanism.58 Besides chemical catalysts, requiring functionalization of graphene often detrimental to its intrinsic properties, other strategies could be considered. Indeed, recent DFT-based calculations have shown that the associative desorption barrier can be decreased or even eliminated by applying an electric field orthogonal to the graphene sheet of appropriate intensity.52 Additionally, such electric fields can create or stabilize graphene's curvature.83 We note that the combined effect of an electric field and an external compression leading to graphene rippling has not been investigated so far, but it is likely to produce cooperative effects and possibly to allow spontaneous adhesion even for molecular hydrogen.
IV.2 Hydrogen desorption
Concerning the desorption phase, in principle the problem is analogous to adsorption, requiring some catalyst to overcome high energetic barriers or the recursion to high temperatures. However the simulation results presented above suggest a possible alternative desorption mechanism that exploits the control of the curvature: hydrogen desorption is seen to spontaneously occur upon curvature inversion. Thus, the problem is moved to finding efficient ways of changing the curvature of graphene sheets.To this respect, several options can be considered. In our simulation the curvature inversion is obtained by a transverse wave. The same effect could be induced by thermally excited transverse phonons. Phonon's proliferation might assist the de-hydrogenation from graphite and graphene and might account for the relatively low temperatures involved, well below the values of barriers predicted by theoretical calculations (usually evaluated at zero temperature). However, a controlled generation of coherent phonons, for example by coupling the system to piezoelectric substrates, should yield an extremely more efficient dehydrogenation. In addition, it has been recently proposed that piezoelectricity could be induced into graphene itself by adsorbing different kind of alkali metals or halides.84 This effect could also be explored to control the curvature inversion.
A different possibility is to functionalize the system with optically active organic molecules such as functionalized azo dyes whose size is controlled by an external radiation at specific wavelength. These could change the distance between layers in specific points, generating corrugation whose concavity could be optically controlled (see Fig. 5(b)).
IV.3 Building the multilayer system and other practical problems
As pointed out in Section II.2, multi-layered graphene should be considered in practically building a medium for hydrogen storage. While macroscopically large epitaxial graphene sheets are currently produced,85 one challenge outlined in Section II.2 is to built and stabilize multi-layers with controllable inter-layer spacing. The already mentioned functionalization with linearly extended organic molecules could serve to that aim, leading to a multi-layer system with tunable volumetric capacity, besides a controllable sheet curvature. Such pillared multi-layers are under consideration by different groups.41 Pillar molecules not only create a tunable inter-layer spacing but also produce a mechanical connection between layers that could propagate the mechanical work delivered by the piezoelectric substrate to all the multilayer system.Finally, one should keep in mind that the curvature enhances the chemical reactivity of graphene in general, particularly to oxygen. Thus the loading phase must be realized in a pure hydrogen atmosphere to avoid loading of other substances. Still in the loading phase, if atomic hydrogen is used, its pressure must be tuned in order to avoid competition with the recombination in molecules. In the desorption phase, on the other hand, the mechanical distortion of the system might produce dissipation of heat that must be kept at minimum. All these practical issues must be carefully considered for the optimization of the device.
V. Conclusions
This review addresses some of the potentialities of graphene and graphene-based structures for hydrogen storage. From the available studies, the majority of them still at a theoretical level, graphene emerges as a promising material for hydrogen storage in terms of GD and VD. The possibility of creating functionalized graphene nanostructures and their peculiar characteristics such as large electrical conduction, robustness, manageability and flexibility opens interesting scenarios for their exploitation in future hydrogen technology. Particularly relevant for hydrogen storage applications is the fact that graphene can be now produced on a large and cost-effective scale by either top-down (such as exfoliation from bulk) or bottom up (atom by atom growth) techniques. Indeed large graphene oxide or graphene flakes can be produced from the exfoliation of pristine graphite. To remark this point we note that, for example, graphene flakes up to several micrometers in sizes can be obtained at high concentration (few mg ml−1) by liquid phase exfoliation86–89 and higher concentration (>5 mg ml−1) can be achieved in ionic liquid.90 However, bulk graphene currently obtained, for example, by liquid phase exfoliation is in the form of aggregates, which limits the surface area available for hydrogen adsorption. Therefore the optimization of graphene production methods will be critical for the successful development of practical hydrogen storage devices.In this perspective paper we have also reported and quantified the tunability of the hydrogen binding energy as a function of graphene local curvature. The large variation of H binding energy makes the chemisorption a favourable process on convex sites and hydrogen release a favourable process on graphene concave sites. On the basis of these predictions we have suggested a multilayer graphene storage system that might lead to a hydrogen device that exploits the controlled change of the local curvature for storing and releasing hydrogen and can operate under room conditions with fast kinetics. The corrugation of graphene flakes and its control combined with the large variety of functionalization approaches and the possibility of bulk production and integration with other materials to form optimized composites raise hopes that graphene can impact the field of hydrogen storage competing or even surpassing the performances of the other solid-state materials explored so far.
Acknowledgements
We thank Ranieri Bizzari, Francesco Bonaccorso, Camilla Coletti, Luigi Crema, Sarah Goler, Stefan Heun, Pasqualantonio Pingue, Vincenzo Piazza and Marco Polini, for useful discussions and suggestions. CINECA supercomputing center resources were obtained by means of INFM-Progetto di Calcolo Parallelo 2009 and Platform “Computation” of IIT (Italian Institute of Technology). Partial support from MIUR through the program “FIRB - Futuro in Ricerca 2010” is also acknowledged.Notes and references
- http://www1.eere.energy.gov/hydrogenandfuelcells/storage/current_technology.html .
- See for example: L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS.
- F. Schüth, B. Bogdanović and M. Felderhoff, Chem. Commun., 2004, 2249 RSC.
- S. Harder, J. Spielmann, J. Intermann and H. Bandmann, Angew. Chem., Int. Ed., 2011, 50, 4156–4160 CrossRef CAS.
- J. Huot, J. F. Pelletier, G. Liang, M. Sutton and R. Schulz, J. Alloys Compd., 2002, 332, 727 CrossRef.
- S. R. Johnson, P. A. Anderson, P. P. Edwards, I. Gameson, J. W. Prendergast, M. Al-Mamouri, D. Book, I. Rex Harris, J. D. Speight and A. Walton, Chem. Commun., 2005, 2823 RSC.
- N. Hanada, T. Ichikawa and H. Fujii, J. Alloys Compd., 2007, 446, 67 CrossRef.
- K.-J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286 CrossRef CAS.
- B. Delhomme, P. de Rango, P. Mart, M. Bacia, B. Zawilski, C. Raufast, S. Miraglia and D. Fruchart, Int. J. Hydrogen Energy, 2012, 37, 9103 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef CAS.
- B. Bogdanovic, M. Felderhoff, A. Pommerin, T. Schuth and N. Spielkamp, Adv. Mater., 2006, 18, 1198 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- K. Müller, K. Stark, B. Müller and W. Arlt, Energy Fuels, 2012, 26, 3691 CrossRef.
- H. W. Li, Y. Yan, S.-i. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185 CrossRef CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS.
- V. Tozzini and V. Pellegrini, J. Phys. Chem. C, 2011, 115, 25523 CAS.
- S. Patchkovskiim, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439 CrossRef.
- A. Zuttel, P. Sudan, Ph. Mauron, T. Kiyobayashi, Ch. Emmenegger and L. Schlapbach, Int. J. Hydrogen Energy, 2002, 27, 203–2012 CrossRef CAS.
- Y. Miura, W. Dino and H. Nakanishi, J. Appl. Phys., 2003, 93, 3395 CrossRef CAS.
- T. Zecho, A. Güttler, X. Sha, B. Jackson and J. Küppers, J. Chem. Phys., 2002, 117, 8486 CrossRef CAS.
- L. Jeloaica and V. Sidis, Chem. Phys. Lett., 1999, 300, 157 CrossRef CAS.
- X. Sha and B. Jackson, Surf. Sci., 2002, 496, 318 CrossRef CAS.
- Y. Ferro, F. Marinelli and A. Allouche, J. Chem. Phys., 2002, 116, 8124 CrossRef CAS.
- N. Rougeau, D. Teillet-Billy and V. Sidis, Chem. Phys. Lett., 2006, 431, 135–138 CrossRef CAS.
- Z. Sljivancanin, E. Rauls, L. Hornekaer, W. Xu, F. Besenbacher and B. Hammer, J. Chem. Phys., 2009, 131, 084706 CrossRef CAS.
- L. Hornekaer, Ž. Šljivančanin, W. Xu, R. Otero, E. Rauls, I. Stensgaard, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2006, 96, 156104 CrossRef CAS.
- L. Hornekaer, E. Rauls, W. Xu, S. Šljivančanin, R. Otero, I. Stensgaard, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2006, 97, 186102 CrossRef CAS.
- A. Andree, M. Le Lay, T. Zecho and J. Küpper, Chem. Phys. Lett., 2006, 425, 99 CrossRef CAS.
- L. Hornekaer, W. Xu, R. Otero, E. Lægsgaard and F. Besenbacher, Chem. Phys. Lett., 2007, 446, 237 CrossRef CAS.
- D. Stojkovic, P. Zhang, P. E. Lammert and V. H. Crespi, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195406 CrossRef.
- S. Casolo, O. M. Løvvik, R. Martinazzo and G. F. Tantardini, J. Chem. Phys., 2009, 130, 054704 CrossRef.
- T. Roman, W. A. Dino, H. Nakanishi, H. Kasai, T. Sugimoto and K. Tange, Carbon, 2007, 45, 203–228 CrossRef.
- Y. Ferro, D. Teillet-Billy, N. Rougeau, V. Sidis, S. Morisset and A. Allouche, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085417 CrossRef.
- R. Balog, B. Jørgensen, J. Wells, E. Lægsgaard, P. Hofmann, F. Besenbacher and L. Hornekær, J. Am. Chem. Soc., 2009, 131, 8744 CrossRef CAS.
- N. P. Guisinger, G. M. Rutter, J. N. Crain, P. N. First and J. A. Stroscio, Nano Lett., 2009, 9, 1462 CrossRef CAS.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- R. Balog, B. Jørgensen, L. Nilsson, M. Andersen, E. Rienks, M. Bianchi, M. Fanetti, E. Lægsgaard, A. Baraldi, S. Lizzit, Z. Sljivancanin, F. Besenbacher, B. Hammer, T. G. Pedersen, P. Hofmann and L. Hornekær, Nat. Mater., 2010, 9, 315 CrossRef CAS.
- V. Tozzini and V. Pellegrini, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 113404 CrossRef.
- P. Sessi, J. R. Guest, M. Bode and N. P. Guisinger, Nano Lett., 2009, 9, 4343 CrossRef CAS.
- I. Cambria, M. J. Lopez and J. A. Alonso, Carbon, 2007, 45, 2649–2658 CrossRef.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902 CrossRef CAS.
- S. M. Lee and Y. H. Lee, Appl. Phys. Lett., 2000, 76, 2877 CrossRef CAS.
- A. C. Dillon, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209 CrossRef CAS.
- G. K. Dimitrakakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 3166 CrossRef CAS.
- G. Srinvas, Y. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Carbon, 2010, 48, 630 CrossRef.
- See for example: V. v. Simonyan and J. K. Johnson, J. Alloys Compd., 2002, 330, 659 CrossRef.
- W. Yuan, B. Li and L. Li, Appl. Surf. Sci., 2011, 257, 10183 CrossRef CAS.
- C. Ataca, E. Aktürk, S. Ciraci and H. Ustunel, Appl. Phys. Lett., 2008, 93, 043123 CrossRef.
- A. Du, Z. Zhu and S. C. Smith, J. Am. Chem. Soc., 2010, 132, 2876 CrossRef CAS.
- A. Tapia, C. Acosta, R. A. Medina-Esquivel and G. Canto, Comput. Mater. Sci., 2011, 50, 2427 CrossRef CAS.
- Z. M. Ao and F. M. Peeters, J. Phys. Chem. C, 2010, 114, 14503 CAS.
- G. J. Kubas, R. R. Ryan, B. I. Swanson, P. J. Vergamini and H. J. Wasserman, J. Am. Chem. Soc., 1984, 106, 451 CrossRef CAS.
- E. Durgun, S. Ciraci and T. Yildirim, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085405 CrossRef.
- Y. Liu, L. Ren, Y. He and H.-P. Cheng, J. Phys.: Condens. Matter, 2010, 22, 445301 CrossRef.
- G. Kim and S.-H. Jhi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085408 CrossRef.
- E. Durgun, S. Ciraci, W. Zhou and T. Yildirim, Phys. Rev. Lett., 2006, 97, 226102 CrossRef CAS.
- V. B. Parambhath, R. Nagar, K. Sethupathi and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 15679 CAS.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582 CrossRef CAS.
- M. Zhou, Y. Lu, C. Zhang and Y. P. Feng, Appl. Phys. Lett., 2010, 97, 103109 CrossRef.
- T. Hussain, B. Pathak, M. Ramzan, T. A. Maark and R. Ahuja, Appl. Phys. Lett., 2012, 100, 183902 CrossRef.
- H. Lee, J. Ihm, M. L. Cohen and S. G. Louie, Nano Lett., 2010, 10, 793 CrossRef CAS.
- L. Wang, K. Lee, Y.-Y. Sun, M. Lucking, Z. Chen, J. J. Zhao and S. B. Zhang, ACS Nano, 2009, 3, 2995 CrossRef CAS.
- M. Yang, A. Nurbawono, C. Zhang, R. Wu, Y. Feng and Ariando, AIP Adv., 2011, 1, 032109 CrossRef.
- H. McKay, D. J. Wales, S. J. Jenkins, J. A. Verges and P. L. de Andres, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 075425 CrossRef.
- R. Ruffieux, O. Gröning, M. Bielmann, P. Mauron, L. Schlapback and P. Gröning, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 245416 CrossRef.
- S. Park, D. Srivastava and K. Cho, Nano Lett., 2003, 3, 1273 CrossRef CAS.
- See for example: H. Cheng, A. C. Cooper, G. P. Pez, M. K. Kostov, P. Piotrowski and S. J. Stuart, J. Phys. Chem. B, 2005, 109, 3780 CrossRef CAS.
- S. P. Change, G. Chen, X. G. Gong and Z.-F. Liu, Phys. Rev. Lett., 2001, 87, 205502 CrossRef.
- J. A. Arellano, L. M. Molina, A. Rubio, M. J. López and J. A. Alonso, J. Chem. Phys., 2002, 117, 2281 CrossRef CAS.
- G. Lu, H. Scudder and N. Kioussis, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 205416 CrossRef.
- K. A. Park, K. Seo and H. H. Lee, J. Phys. Chem. B, 2005, 109, 8967–8972 CrossRef CAS.
- J. T. Tanskanen, M. Linnolahti, A. J. Karttunen and T. A. Pakkanen, Chem. Phys., 2007, 340, 120 CrossRef CAS.
- A. Bilic and J. D. Gale, J. Phys. Chem. C, 2008, 112, 12568 CAS.
- S. Berber and D. Tomanek, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075427 CrossRef.
- A. Kaczamarek, C. Dinadayalane, J. Łukaszewicz and J. Leszczynski, Int. J. Quantum Chem., 2007, 107, 2211 CrossRef.
- G. Ja, J. Li and Y. Zhang, Chem. Phys. Lett., 2006, 418, 40 CrossRef.
- D. W. Boukhvalov and M. I. Katsnelson, J. Phys. Chem. C, 2009, 113, 14176 CAS.
- Z. F. Wang, Y. Zhang and F. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 041403 CrossRef.
- See for example: M. Segal, Nat. Nanotechnol., 2009, 4, 612 CrossRef CAS; Y. Zhu, et al. , Adv. Mater., 2010, 22, 3906 CrossRef.
- S. Goler et al. Submitted. Results are consistent with the theoretical predictions.
- S. Goler, C. Coletti, V. Piazza, P. Pingue, F. Colangelo, V. Pellegrini, K. V. Emtsev, S. Forti, U. Starke, F. Beltram and S. Heun, Carbon, 2013, 51, 249–254 CrossRef CAS.
- Z. Wang, L. Philippe and J. Elias, Phys. Rev. Lett., 2010, 81, 155405 Search PubMed.
- M. T. Ong and E. J. Russel, ACS Nano, 2012, 6, 1387–1394 CrossRef CAS.
- S. Bae, et al. , Nat. Nanotechnol., 2010, 5, 574 CrossRef CAS.
- F. Torrisi, T. Hasan, W. W. Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. Hsieh, S. Jung, F. Bonaccorso, P. J. Paul, D. Chu and A. C. Ferrari, ACS Nano, 2012, 6, 2992 CrossRef CAS.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, X. Tang, C.-H. Tsai and L.-J. Li, Chem. Mater., 2009, 21, 5674 CrossRef CAS.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864 CrossRef CAS.
- V. Alzari, D. Nuvoli, S. Scognamillo, M. Piccinini, E. Gioffredi, G. Malucelli, S. Marceddu, M. Sechi, V. Sanna and A. Mariani, J. Mater. Chem., 2011, 21, 8727 RSC.
- D. Nuvoli, L. Valentini, V. Alzari, S. Scognamillo, S. Bittolo Bon, M. Piccinini, J. Illescas and A. Mariani, J. Mater. Chem., 2011, 21, 3428 RSC.
| This journal is © the Owner Societies 2013 |