Increasing visible-light absorption for photocatalysis with black BiOCl†
Liqun
Ye
a,
Kejian
Deng
b,
Feng
Xu
c,
Lihong
Tian
a,
Tianyou
Peng
a and
Ling
Zan
*a
aCollege of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, China. E-mail: irlab@whu.edu.cn; Fax: 86 27 8737 8727; Tel: 86 27 6875 2919
bKey Laboratory of Catalysis and Materials Science of the State Ethnic Affairs Commission and Ministry of Education, South-Central University for Nationalities, Wuhan, 430074, China
cKey Laboratory for Magnetism and Magnetic Materials of MOE, Lanzhou University, Lanzhou, 730000, China
First published on 11th November 2011
Abstract
Black BiOCl with oxygen vacancies was prepared by UV light irradiation with Ar blowing. The as-prepared black BiOCl sample showed 20 times higher visible light photocatalytic activity than white BiOCl for RhB degradation. The trapping experiment showed that the superoxide radical ( ) and holes (h+) were the main active species in aqueous solution under visible light irradiation.
) and holes (h+) were the main active species in aqueous solution under visible light irradiation.
Semiconductor photocatalysis has attracted increasing attention as a potential environmental technology for wastewater remediation.1 In the past three years, it was found that BiOCl is an excellent semiconductor photocatalyst and exhibits better performance than TiO2 (P25, Degussa) in UV light photocatalytic degradation (PCD) of dyes.2 It is known to have a tetragonal structure with lattice constants of a = 0.3890 nm and c = 0.7890 nm. It has a layered structure which is characterized by [Bi2O2] slabs separated by double slabs of Cl atoms in the tetragonal matlockite structure.3 The layered structure can provide a large enough space to polarize the related atoms and orbitals. The induced dipole can then separate the hole–electron pair efficiently. On the other hand, BiOCl has an indirect-transition band-gap so that the excited electron has to travel a certain k-space distance to be emitted to the valence band (VB) which reduces the recombination probability of the excited electron and the hole. Thus, BiOCl displays high photocatalytic activity under UV light irradiation. But, there are no reports of BiOCl with visible light photocatalytic activity because its band gap is about 3.5 eV,2 except in the form of a heterojunctioned BiOCl photocatalyst.4
It is well known that intrinsic semiconductor photocatalysts with oxygen vacancies can absorb visible-light and display excellent visible-light photocatalytic activity.5,6 However, the oxygen vacancies were generally engendered in difficult processes such as annealing in a reducing atmosphere or doping, which limits the photocatalysts application. In our previous report, we found that the low bond energy and long bond length of the Bi–O bond lead to the production of oxygen vacancies under UV light irradiation and resulted in the formation of black BiOCl.7 Here, black BiOCl was easily prepared by UV light irradiation. By characterizing the black BiOCl with EPR, XPS, HRTEM and DRS techniques, the oxygen vacancies signal was found. Black BiOCl showed 20 times higher photocatalytic activity than white BiOCl for RhB degradation under visible-light irradiation. The photocatalytic mechanism of black BiOCl with oxygen vacancies under visible light irradiation was also investigated.
Fig. 1a shows the XRD pattern of BiOCl samples, which indexes well to the tetragonal structure of BiOCl (JCPDS No: 06-249) and reveals that all the samples are well crystallized. After UV light irradiation, white BiOCl became black and no new peaks appeared in the XRD pattern. It indicates that the UV light irradiation does not result in the appearance of new compounds. Also, the half width and relative intensity of the XRD peaks do not change which implies that the crystal size and orientation have not changed after UV light irradiation.
 | ||
| Fig. 1 (a) XRD pattern of BiOCl samples; (b) EPR spectra of BiOCl samples; (c) HRTEM images of white BiOCl; and (d) black BiOCl. | ||
The EPR technique is effective and is the most common method to detect defects in material science. Here, the EPR technique was applied to detect oxygen vacancies. In order to facilitate the emergence of the EPR signal of BiOCl, all samples were heated in argon at 300 °C for 2 h before testing.7,8Fig. 1b shows the EPR spectra of white and black BiOCl. There is no EPR signal from white BiOCl but, on the contrary, there is a remarkable EPR signal from black BiOCl. The peak at g = 2.001 observed here is a typical signal of oxygen vacancies.9 It indicates that UV irradiation induces the formation of oxygen vacancies. HRTEM images (Fig. 1c, d and Fig. S1†) show that the surfaces of BiOCl became disordered after UV light irradiation, where the disordered outer layer is ∼1 nm in thickness, but on the inside there is good crystallinity. The images again proved the generation of oxygen vacancies.6
The survey XPS spectra of the BiOCl samples are shown in Fig. S2†. According to the survey XPS spectra of white and black BiOCl, except for a carbon contamination signal, the elements present are Bi, O and Cl. This indicates that the BiOCl photocatalyst is pure. In Fig. 2a, the peaks at 165.8 eV and 160.5 eV are indexed as Bi3+ in BiOCl.8,10 The lower binding energies at 163.3 eV and 158.2 eV can be indexed to lower charge Bi ions which are due to the oxygen vacancies present. The same phenomenon has occurred in TiO2 systems, namely, the appearance of oxygen vacancies induced a lower binding energy peak which was due to Ti3+.6,11
 | ||
| Fig. 2 (a) Bi 4f high-resolution XPS spectra of the BiOCl samples; and (b) DRS spectra of the BiOCl samples. | ||
In previous studies on oxygen vacancies in a TiO2 system, researchers proved that oxygen vacancies can form an oxygen vacancy state lying close to the conduction band (CB) of the photocatalyst.5a,6 The photocatalyst with oxygen vacancies can then absorb visible light. The BiOCl with oxygen vacancies is black as shown in the inset of Fig. 2b. Fig. 2b shows the DRS spectra of the BiOCl samples. White BiOCl does not have any absorption in the visible light range. This indicates that no oxygen vacancies existed in white BiOCl before UV irradiation. After UV irradiation, absorption in the visible light range increased. It reveals that the oxygen vacancies emerged after UV light irradiation.
Fig. S3† shows the control experiments for the transformation of white BiOCl to black BiOCl. It can be observed that the oxygen vacancy concentration increased with increasing UV irradiation time (Fig. S3A†). Furthermore, it can be found that the increase in the visible light absorption was very fast over 0.5 h of UV irradiation. After that, the increase became slow. This shows that the generation of oxygen vacancies was focused in the initial 0.5 h. Irradiation wavelength and gas type were also considered as shown in Fig. S3B†. Under UV irradiation, the Ar blowing in the white BiOCl suspension was replaced by air, the absorption intensity decreased under an identical UV irradiation time. This shows that the blowing of inert gas is beneficial in the formation of oxygen vacancies. When UV light was replaced by visible light (λ ≥ 400 nm), the absorption intensity decreased in the visible light range. This indicates that the irradiation wavelength is a dominant influencing factor on the formation of oxygen vacancies.
Fig. 3a displays the PCD results and pseudo first-order reaction kinetics of BiOCl samples. It can be seen that the blank experiment without any catalyst displayed almost no PCD of RhB. The rate constant values of RhB degradation are 0.061, 0.003 and 0.001 min−1 for black BiOCl, white BiOCl and P25, respectively. The results indicate that the black BiOCl with oxygen vacancies displayed good photocatalytic activity under visible light irradiation. Fig. S4† shows the temporal UV-vis absorption spectra during the photocatalytic degradation of RhB on black BiOCl. It shows that the absorption intensity of RhB decreased gradually with visible light irradiation. On the other hand, the maximum peak makes an obvious shift from 554 nm to 500 nm. This can be ascribed to the formation of N-deethylated RhB in the photocatalytic process.12
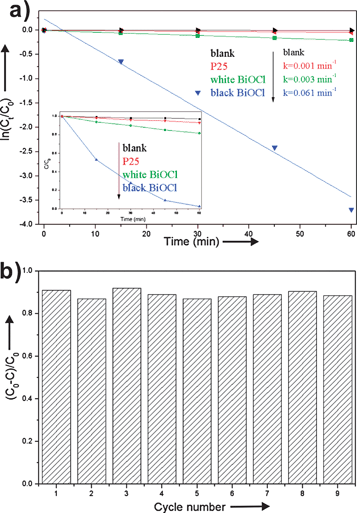 | ||
| Fig. 3 (a) PCD results and pseudo first-order reaction kinetics of BiOCl samples; and photocatalytic stability of black BiOCl. | ||
The black BiOCl exhibits substantial photocatalytic activity and stability under visible light irradiation. Cycled runs of the PCD of RhB under visible light irradiation (λ ≥ 400 nm) over 45 min which were tested using black BiOCl as the photocatalyst, are shown in Fig. 3b. The photocatalytic activity after 9 cycles did not display a decline. Furthermore, the XRD pattern does not change after cycle 9 as shown in Fig. S5†. This indicates that black BiOCl is stable in the oxygen/air and water environments.
The photocatalytic mechanism of white BiOCl as a semiconductor photocatalyst has been studied very few times. For black BiOCl, there has been no reports. Which species is the main active species of the black BiOCl under visible light irradiation? Fig. S6†shows the trapping experiment of active species during the photocatalytic reaction. If hydroxyl radicals act as the main active species in the photocatalytic system for the degradation of RhB, the addition of isopropanol would decrease the degradation kinetics. However, the photocatalytic degradation of RhB was not affected by the addition of 1 mM isopropanol. This is due to the standard redox potential of BiV/BiIII (+1.59 V) being more negative than that of ˙OH/OH− (+1.99 V).13 On the contrary, the photocatalytic degradation of RhB decreased obviously with the addition triethanolamine (TEOA, a quencher of h+)13 and p-benzoquinone (BQ, a quencher of  ).14 Therefore, it can be concluded that
).14 Therefore, it can be concluded that 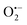 and h+ are the main active species in photocatalysis using black BiOCl with oxygen vacancies in aqueous solution under visible light irradiation.
and h+ are the main active species in photocatalysis using black BiOCl with oxygen vacancies in aqueous solution under visible light irradiation.
To eliminate the sensitization of RhB, a colorless molecular probe, nitroblue tetrazolium (NBT) was chosen to quantify the  concentration and demonstrate the photocatalytic activity of black BiOCl.13,15Fig. 4a and Fig. S7† show the variation in the concentration and UV-vis absorption spectra of NBT in the suspensions of black BiOCl and white BiOCl under visible light irradiation at different times. The decreasing maximum absorbances in the UV-vis spectra of NBT indicates the production of
concentration and demonstrate the photocatalytic activity of black BiOCl.13,15Fig. 4a and Fig. S7† show the variation in the concentration and UV-vis absorption spectra of NBT in the suspensions of black BiOCl and white BiOCl under visible light irradiation at different times. The decreasing maximum absorbances in the UV-vis spectra of NBT indicates the production of  in the black BiOCl suspension. However, there is not any change in UV-vis spectra of NBT in the white BiOCl suspension, which implies no
in the black BiOCl suspension. However, there is not any change in UV-vis spectra of NBT in the white BiOCl suspension, which implies no  production. The result reveals that the photocatalytic activity of black BiOCl and the production of
production. The result reveals that the photocatalytic activity of black BiOCl and the production of 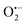 are due to visible light excitation rather than sensitization. According to the reaction equation (Fig. S8†), the production rate of
are due to visible light excitation rather than sensitization. According to the reaction equation (Fig. S8†), the production rate of  during PCD process is four times that of NBT15 and the calculated concentration is shown in Fig. 4b. It was found that the amount of
during PCD process is four times that of NBT15 and the calculated concentration is shown in Fig. 4b. It was found that the amount of  almost linearly increased with the increase of irradiation time. The same phenomenon has been reported in a ZnO system.15 Herein, the yield of
almost linearly increased with the increase of irradiation time. The same phenomenon has been reported in a ZnO system.15 Herein, the yield of  was 5.75 × 10−7 mol g−1 min−1 for black BiOCl.
was 5.75 × 10−7 mol g−1 min−1 for black BiOCl.
 | ||
Fig. 4 (a) Variation of NBT concentration; and (b) variation of  concentration under visible light irradiation (λ ≥ 400 nm). concentration under visible light irradiation (λ ≥ 400 nm). | ||
To understand the function of the oxygen vacancies, PL (photoluminescence) analysis was applied. Fig. 5a shows the PL spectra of the BiOCl. Without UV irradiation, there is only one band at 362 nm (3.43 eV) and because this energy of photoluminescence is nearly equal to the band gap (3.5 eV) of BiOCl, this emission is attributed to the recombination of free electrons from the bottom of the conduction band to the recombination centre at the ground state.16 With 3 h UV irradiation, the bank at 362 nm became weaker, a new strong band at 468 nm (2.65 eV) appeared which relates to the oxygen vacancies.10a,17 This means the separation efficiency of photo-induced electrons and holes was enhanced in the presence of the oxygen vacancies.17 This result is consistent with the previous reports on ZnO18 and TiO2.19Oxygen vacancies can form oxygen vacancy states lying close to the conduction band (CBBiOCl = −1.1 eV) of the photocatalyst.4b,5a The photo-induced electrons on oxygen vacancy states recombine with photo-induced holes at the ground state and cause the new photoluminescence band at 468 nm.
 | ||
| Fig. 5 (a) PL spectra of BiOCl samples; and (b) band structure model and photoreaction process on black BiOCl. | ||
According to previous discussion, the photoreaction process of black BiOCl with oxygen vacancies was proposed. Fig. 5b shows the band structure model and photoreaction process. The electrons are excited up to oxygen vacancy states from the VB under visible light irradiation. Furthermore, the photo-induced electrons on oxygen vacancy states can not recombine easily with photo-induced holes as the oxygen vacancies are active electron traps.20 In general, the life of electrons in electron traps is longer than in the conduction band.21 The electrons in oxygen vacancy states reduce oxygen which is absorbed on the surface of black BiOCl to produce  . Then photocatalytic degradation reactions can be generated by
. Then photocatalytic degradation reactions can be generated by  and h+. So, black BiOCl with oxygen vacancies shows high visible light photocatalytic activity.
and h+. So, black BiOCl with oxygen vacancies shows high visible light photocatalytic activity.
In conclusion, black BiOCl with oxygen vacancies was easily prepared by UV light irradiation and displayed excellent photocatalytic activity under visible light irradiation. It expanded the application range of BiOCl from UV light to the full spectrum. If white BiOCl is used under UV light for few hours, UV-induced oxygen vacancies will appear. Then the as-prepared black BiOCl can be used as a visible light pholocatalyst. This method is much easier than sensitization, heterojunctions and doping to modify BiOCl for visible light application. By trapping the active species during the photocatalytic reaction, superoxide radicals and holes were suggested as the main active species. Under visible light irradiation, electrons were excited to oxygen vacancy states and reacted with O2 to produce  . Then, organic pollutants were degraded by
. Then, organic pollutants were degraded by  and h+.
and h+.
This work was supported by the NSFC and the open foundation of the key laboratory of catalysis and materials science of the State Ethnic Affairs Commission and Ministry of Education, South-Central University for Nationalities. The authors also acknowledge the assistance from the Center for Electron Microscopy, Wuhan University and the Key Laboratory for Magnetism and Magnetic Materials of MOE, Lanzhou University.
Notes and references
- (a) H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS; (b) H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS; (c) M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664–1666 RSC.
- (a) Y. Lei, G. Wang, S. Song, W. Fan and H. Zhang, CrystEngComm, 2009, 11, 1857–1862 RSC; (b) X. Zhang, Z. Ai, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CrossRef CAS; (c) C. Wang, C. Shao, Y. Liu and L. Zhang, Scr. Mater., 2008, 59, 332–335 CrossRef CAS; (d) J. Henle, P. Simon, A. Frenzel, S. Scholz and S. Kaskel, Chem. Mater., 2007, 19, 366–373 CrossRef CAS; (e) M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- (a) K. L. Zhang, C. M. Liu, F. Q. Huang, C. Zheng and W. D. Wang, Appl. Catal., B, 2006, 68, 125–129 CrossRef CAS; (b) J. Geng, W. H. Hou, Y. N. Lv, J. J. Zhu and H. Y. Chen, Inorg. Chem., 2005, 44, 8503–8509 CrossRef CAS.
- (a) L. Zhang, W. Wang, L. Zhou, M. Shang and S. Sun, Appl. Catal., B, 2009, 90, 458–462 CrossRef CAS; (b) S. Y. Chai, Y. J. Kim, M. H. Jung, A. K. Chakraborty, D. Jung and W. I. Lee, J. Catal., 2009, 262, 144–149 CrossRef CAS; (c) X. Changa, G. Yu, J. Huang, Z. Li, S. Zhu, P. Yu, C. Cheng, S. Deng and G. Ji, Catal. Today, 2010, 153, 193–199 CrossRef.
- (a) I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205–212 CrossRef CAS; (b) T. Ihara, M. Miyoshi, Y. Iriyama, O. Matsumoto and S. Sugihara, Appl. Catal., B, 2003, 42, 403–409 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- L. Ye, L. Zan, L. Tian and T. Peng, Chem. Commun., 2011, 47, 6951–6953 RSC.
- (a) H. An, Y. Du, T. Wang, C. Wang, W. Hao and J. Zhang, Rare Met., 2008, 27, 243–250 CrossRef CAS; (b) Y. Wu, M. Xing, J. Zhang and F. Chen, Appl. Catal., B, 2010, 97, 182–189 CrossRef CAS.
- (a) C. Feng, Y. Wang, Z. Jin, J. Zhang, S. Zhang, Z. Wu and Z. Zhang, New J. Chem., 2008, 32, 1038–1047 RSC; (b) D. Pan, G. Xu, L. Lv, Y. Yong, X. Wang, J. Wan and G. Wang, Phys. Rev. Lett., 2006, 89, 082510 Search PubMed.
- (a) W. E. Morgan, W. J. Stec and J. R. V. Wazer, Inorg. Chem., 1973, 12, 953–955 CrossRef CAS; (b) J. M. Song, C. J. Mao, H. L. Niu, Y. H. Shen and S. Y. Zhang, CrystEngComm, 2010, 12, 3875–3881 RSC.
- (a) M. Batzill, E. H. Morales and U. Diebold, Chem. Phys., 2007, 339, 36–43 CrossRef CAS; (b) C. Rath, P. Mohanty, A. C. Pandey and N. C. Mishra, J. Phys. D: Appl. Phys., 2009, 42, 205101 CrossRef; (c) G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H. M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CrossRef CAS.
- (a) H. B. Fu, S. C. Zhang, T. G. Xu, Y. F. Zhu and J. M. Chen, Environ. Sci. Technol., 2008, 42, 2085–2091 CrossRef CAS; (b) L. Ye, C. Yang, L. Tian, L. Zan and T. Peng, Appl. Surf. Sci., 2011, 257, 8072–8077 CrossRef CAS.
- Y. Wang, K. Deng and L. Zhang, J. Phys. Chem. C, 2011, 115, 14300–14308 CAS.
- P. Ji, J. Zhang, F. Chen and M. Anpo, Appl. Catal., B, 2009, 85, 148–154 CrossRef CAS.
- X. Xu, X. Duan, Z. Yi, Z. Zhou, X. Fan and Y. Wang, Catal. Commun., 2010, 12, 169–172 CrossRef CAS.
- S. Cao, C. Guo, Y. Lv, Y. Guo and Q. Liu, Nanotechnology, 2009, 20, 275702 CrossRef.
- Y. Zheng, F. Duan, J. Wu, L. Liu, M. Chen and Y. Xie, J. Mol. Catal. A: Chem., 2009, 303, 9–14 CrossRef CAS.
- Y. Kim and S. Kang, J. Phys. Chem. B, 2010, 114, 7874–7878 CrossRef CAS.
- L. Jing, X. Sun, B. Xin, B. Wang, W. Cai and H. Fu, J. Solid State Chem., 2004, 177, 1213–3382 CrossRef.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS.
- M. Sugawara, Phys. Rev. B: Condens. Matter, 1995, 51, 10743–754 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization. See DOI: 10.1039/c1cp22876e |
| This journal is © the Owner Societies 2012 |