The excited states and vibronic spectroscopy of diphenyldiacetylene and diphenylvinylacetylene†
Joshua A.
Sebree‡
and
Timothy S.
Zwier
*
Department of Chemistry, Purdue University, West Lafayette, IN 47909-2084, USA. E-mail: zwier@purdue.edu
First published on 8th November 2011
Abstract
Laser induced fluorescence (LIF) excitation scans and dispersed fluorescence (DFL) spectra have been recorded for two four-carbon α,ω-diphenyl systems, diphenyldiacetylene (DPDA, ϕ-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC-ϕ) and trans-diphenylvinylacetylene (DPVA, ϕ-CH
C–CC-ϕ) and trans-diphenylvinylacetylene (DPVA, ϕ-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CC-ϕ) as isolated molecules cooled in a supersonic expansion. While these molecules have similar conjugation length, they exhibit strikingly different vibronic spectroscopy and photophysics. The near-UV LIF excitation spectrum of diphenyldiacetylene has its electronic origin at 32
CH–CC-ϕ) as isolated molecules cooled in a supersonic expansion. While these molecules have similar conjugation length, they exhibit strikingly different vibronic spectroscopy and photophysics. The near-UV LIF excitation spectrum of diphenyldiacetylene has its electronic origin at 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 158 cm−1, and a strong progression in the CC stretch (2156 cm−1). All transitions are inherently broad, with widths of ∼30 cm−1 fwhm or greater. The S1 origin DFL spectrum is composed of sharp transitions with Franck–Condon activity mirroring that in the excitation spectrum, and broad emission shifted well to the red ascribable to phosphorescence on the μs timescale. Using ab initio calculations, it is possible to show that DPDA exists as a single, planar conformer with D2h symmetry. In contrast, trans-diphenylvinylacetylene shows intense sharp transitions in both LIF and DFL spectra with an S0–S1 origin of 31183.2 cm−1 and long progressions involving the in-plane fundamentals ν53 (bridge-phenyl bending) and ν51 (bridge-phenyl stretch). A sharp reduction in fluorescence yield in DPVA occurs within 300 cm−1 of the S1 origin. Possible causes for the photophysical processes occurring in the two molecules are discussed.
158 cm−1, and a strong progression in the CC stretch (2156 cm−1). All transitions are inherently broad, with widths of ∼30 cm−1 fwhm or greater. The S1 origin DFL spectrum is composed of sharp transitions with Franck–Condon activity mirroring that in the excitation spectrum, and broad emission shifted well to the red ascribable to phosphorescence on the μs timescale. Using ab initio calculations, it is possible to show that DPDA exists as a single, planar conformer with D2h symmetry. In contrast, trans-diphenylvinylacetylene shows intense sharp transitions in both LIF and DFL spectra with an S0–S1 origin of 31183.2 cm−1 and long progressions involving the in-plane fundamentals ν53 (bridge-phenyl bending) and ν51 (bridge-phenyl stretch). A sharp reduction in fluorescence yield in DPVA occurs within 300 cm−1 of the S1 origin. Possible causes for the photophysical processes occurring in the two molecules are discussed.
I. Introduction
Polyenes (R1-(CHCH)n-R2) and polyynes (R1-(CHCH)n-R2) are simple π-conjugated carbon chains. Polyenes play important roles in nature, most notably in photosynthesis2,3 and vision.4 The electronic character and state ordering of polyenes is very sensitive to the length of the polyene chain, and therefore the electronic spectroscopy and dynamics of the set of polyenes with n = 1–4 have been the subject of numerous previous studies aimed at establishing this ordering and characterizing each of the excited states.5–14
These state ordering effects also apply to phenyl-substituted polyenes. The smallest diphenyl derivative, stilbene (diphenylethene), has been the subject of many spectroscopic investigations and studies of the dynamics of cis-transisomerization.15–25 Based on this work, it is known that the S1 state of trans-stilbene is of 1Bu symmetry with an energy of 32235 cm−1 above the ground state in the gas-phase. S1 levels near the electronic origin have a large relative fluorescence quantum yield (ϕf ∼ 1), that falls precipitously about 1200 cm−1 above the origin.25 By contrast, cis-stilbene undergoes rapid isomerization in its first excited state involving fast internal conversion to the ground state that quenches its fluorescence.
Extension of the bridge by one ethene group gives (1,4)-diphenylbutadiene, which can adopt three isomeric forms differing in the cis-trans character of the two double bonds in the butadiene chain. The excitation spectrum of trans,trans-diphenylbutdiene (DPBD) in the near ultraviolet was recorded by Heimbrook et al.5 They observed a symmetry forbidden 1Ag state at 29652.5 cm−1 that was vibronically coupled to the allowed 1Bu transition at 30800.6 cm−1. Much like stilbene, cis forms of DPBD (cis,trans- and cis,cis-) undergo rapid isomerization upon excitation.12–14
The rigid structure and high conjugation of polyynes, particularly diphenylpolyynes, has been of interest in creating molecular wires26 and building organic dendrimers that have unique nonlinear optical properties.27 An advantage of using diphenylpolyynes is the significant reduction in isomeric forms and reduced chemical reactivity through the capping of their reactive termini.28 The nature of the excited states is determined by coupling of the phenyl rings across the polyacetylenic bridge. In a manner similar to the polyenes, the ordering of the excited states is dependent on the length of the connecting bridge,28 as well as the orientation of the phenyl rings (planar vs. perpendicular).29 Many diphenylpolyynes rapidly interconvert to long-lived triplet states with their own unique properties.30–33
Here again, the first member of the series tolane (diphenylacetylene) has been studied in a supersonic expansion, revealing the presence of two close-lying excited states.34–36 In addition, extensive computational modeling of the phenyl torsional motion has been carried out, including studies of the possible role of πσ* states in the excited state dynamics.35–38
Similar condensed phase studies have been carried out for diphenyldiacetylene (DPDA). A recent study by Thulstrup et al., showed that the absorption features of DPDA arise from a combination of both planar and perpendicular phenyl orientations.29 Until now, there have been no similar studies on longer diphenylpolyynes in the gas phase. This may be due in part to the inherently broadened spectra of many polyyne compounds, even under jet-cooled conditions. For example, both diacetylene (C4H2) and phenyldiacetylene (PhC4H) exhibit broad absorption features under jet-cooled conditions, and no gas-phase fluorescence spectra of either molecule have been collected.39,40
While the pure polyene and polyyne families have been studied extensively, much less is known about enyne bridges. Our recent studies of the spectroscopy and isomerization of trans- and cis-phenylvinylacetylene (PVA) have led to the conclusion that the vinylacetylene substituent produces vibronic spectra similar in many ways to those of styrene and stilbene.41,42 A reversal of the order of substitution to make 1-phenyl-1-butyn-3-ene (PAV) produces a vibronic spectrum with characteristics similar to those of phenylacetylene.43
Capping the end of either PVA or PAV with a second phenyl ring produces diphenylvinylacetylene (DPVA). DPVA has received some attention in solution phase studies because of the possibility of a ring closure mechanism that would lead to 1-phenylnaphthalene.44,45
In this work, the gas-phase laser induced fluorescence (LIF) and dispersed fluorescence (DFL) spectra of diphenyldiacetylene (1,4-diphenyl-1,3-butdiyne, DPDA) and diphenylvinylacetylene (1,4-diphenylbut-1-en-3-yne, DPVA) are presented and compared to the analogous data on DPBD. As we shall see, DPDA has an inherently broadened excitation spectrum in the near-ultraviolet, even under jet-cooled conditions, as anticipated based on other diacetylenic counterparts.39,46 Nevertheless, it has been possible to record both LIF excitation and DFL spectra of DPDA. Surprisingly, sharp features characteristic of emission from the originally excited singlet state are observed in the dispersed emission, complemented by red-shifted emission ascribable to the triplet manifold.
In contrast to DPDA's inherent broadening and weak fluorescence brought about by rapid intersystem crossing to the triplet manifold, DPVA has an extraordinarily strong S0–S1 transition with large oscillator strength and extensive sharp vibronic structure, with strong fluorescence near its electronic origin. At the same time, the spectra show evidence for turn-on of a non-radiative process starting only 300 cm−1 above the S1 origin. Much like stilbene, the spectrum of cis-DPVA was not observed, despite extensive efforts to detect it. In what follows, we present the vibronic spectroscopy of both molecules and compare them with predictions from theory to map out the excited states and propose dynamical pathways involved in the two molecules.
II. Methods
A. Experimental details
The laser induced fluorescence (LIF), ultraviolet holeburning (UVHB), and dispersed fluorescence (DFL) spectra of DPVA and DPDA were carried out in the fluorescence chamber described in detail elsewhere.47 A mass-resolved two-color resonance enhanced two-photon ionization (2C-R2PI) spectrum was collected of DPDA in the time-of-flight spectroscopy chamber also described previously.48 Samples were heated to 90 °C, entrained in helium buffer gas (3 bar) and expanded into vacuum through either a 500 or 750 μm diameter pulsed nozzle.The doubled output of a Nd:YAG-pumped dye laser was used for the S0–S1 excitation (∼0.1 mJ/pulse) in both setups, with 193 nm light from an ArF laser (∼0.1 mJ/pulse) as the second photon for the ionization techniques.
UVHB was used to record conformation-specific spectra of each molecule. To accomplish this, a UV holeburn laser at 10 Hz was spatially overlapped with the probe laser (20 Hz) and fired 200 ns prior in time. The wavelength of the holeburn laser is fixed on a transition from the LIF spectrum while the probe laser is tuned. When the probe laser tunes through a transition that shares the same ground state as the holeburn laser, there is a depletion in the signal. The difference between the shots with and without the holeburn laser was recorded using active baseline subtraction using a gated integrator, resulting in the conformer-specific UV spectrum.
DFL spectra were recorded using a Jobin Yvon 750i monochromator and an intensified charged coupled device camera (Andor DH720) operating in photon counting mode. The resolution of the DFL spectra was around 6–8 cm−1 using entrance slit widths of 50–100 μm.
DPDA was purchased from Sigma-Aldrich and used without further purification. DPVA was synthesized via a published procedure.49 Cuprous iodide was added to a mixture of bis(tripenylphosphine) palladium dichloride and phenylacetylene in ethylamine under a nitrogen atmosphere. β-bromostyrene was added dropwise to the flask and the solution was stirred for six hours at room temperature. The solvent, and unreacted starting material, was removed under reduced pressure and the remaining residue was extracted using a water/hexane extraction. GC-MS showed the final product to be a mixture of trans and cis isomers along with DPDA in a 52:20:28 mixture. The cis isomer could be separated from the other two viacolumn chromatography. Trans-DPVA and DPDA could not be separated.
B. Computational details
The ground state geometries of DPDA and DPVA were optimized using density functional theory (DFT) with a B3LYP50,51 functional and 6-311+G(d,p) basis set using Gaussian 09.52 Excited state calculations were carried out using TDDFT methods at the same level of theory.53 Franck–Condon fitting of prominent vibronic structure observed in the DPVA spectra was carried out using both Gaussian 09 methods and a fitting routine based on the work by Henderson et al.54III. Diphenyldiacetylene
A. Excited state properties
Fig. 1 presents the LIF excitation spectrum of DPDA under jet-cooled conditions. The origin of DPDA was found to be centered at 32158.5 cm−1. The observed transitions are substantially broader than a typical jet-cooled spectrum, with the electronic origin narrowest, but still ∼30 cm−1 fwhm, ten times wider than is typical at jet rotational temperatures of 2–3 K. The observed spectrum is consistent with those taken in solution28,55 and matrix56 studies, with a dominant progression in a mode at 2156 cm−1 ascribable to the totally symmetric CC stretch. The broadness is similar to that seen for the S0–S1 transition in phenyldiacetylene46 and the S0–S2 transition of diacetylene57 and is thus not unexpected, but rather appears as a characteristic property associated with the diacetylenic chain. However, since the spectrum in Fig. 1 was observed via its fluorescence, we have an opportunity to explore the source of this broadening further. Given the intrinsic breadth of the transitions due to DPDA, it is important to verify whether any of this breadth might be due to the presence of more than one ground state level, either from more than one isomer, molecular clusters, or hot bands. To that end, UV holeburning spectroscopy was carried out with the holeburn laser fixed at several frequencies across the broadened origin band. At all holeburn wavelengths, the entire spectrum burned out. An enlargement of one of these UVHB scans in the S0–S1 origin region is presented in Fig. 2b, where it is compared with the LIF excitation spectrum above it (Fig. 2a).
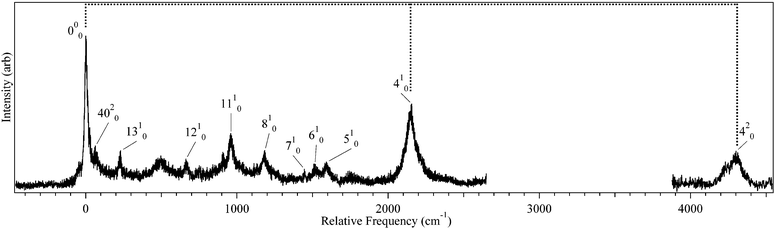 | ||
| Fig. 1
LIF
excitation spectrum of DPDA. The S0-S origin of DPDA is centered at 32158.5 cm−1. Vibronic transitions are based on TDDFT (B3LYP/6-311+G(d,p)) calculations and labeled using Mulliken notation.1 | ||
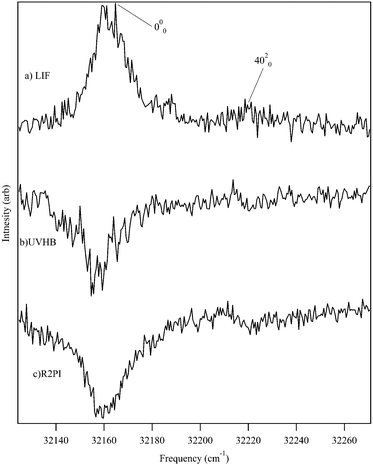 | ||
| Fig. 2 Comparison of the (a) LIF, (b) UVHB, and (c) R2PI spectra of the S0–S1 origin region of DPDA showing that the inherent breadth of transitions in the excitation spectrum is a property of the excited state of DPDA. | ||
As final evidence that the collected spectrum was completely due to DPDA, the mass-resolved 2C-R2PI spectrum was taken in the DPDA monomer mass channel (m/z = 202). Fig. 2c presents the same S0–S1 origin region of DPDA using 193 nm as the ionizing photon, proving that the spectrum is entirely from DPDA monomer. This wavelength was chosen for the second photon because calculations predicted that one-color R2PI would reach very near the ionization threshold. Furthermore, the 193 nm photon also increased the observed signal by allowing for ionization from any nearby dark states to which DPDA may interconvert upon excitation. This was crucial for detection of the R2PI signal from DPDA, as longer wavelengths (>220 nm) were unable to ionize the molecule after a rapid (<1 ns) interconversion process to an unknown state. It is suspected that this state is a long-lived triplet state, similar to what is seen in matrix studies, as a two-color lifetime scan showed the lifetime to be on the order of several microseconds. Nagano and co-workers reported a triplet lifetime of 68 ms for DPDA in methylcyclohexane at 77 K.31
B. Dispersed fluorescence
The dispersed fluorescence spectrum when pumping the S0–S1 origin transition of DPDA is shown in Fig. 3. There are several features of this spectrum that are noteworthy. First, whereas the excited state peaks are inherently broad, the emission to the ground state in the near-resonant region, within 4500 cm−1 of the ground state zero-point level (ZPL), consists of sharp transitions (∼2.5 cm−1) consistent with rotationally cold molecules. Second, the DFL spectrum shows strong Franck–Condon activity to levels over 4500 cm−1 above the S0ZPL, primarily as the result of a strong progression in a mode at 2268 cm−1. Third, the spectrum as a whole exhibits the kind of mirror symmetry one might expect between the excitation spectrum (Fig. 1) and excited state origin DFL spectrum (Fig. 3) if there is minimal vibronic coupling present. Finally, attempts to detect sharp emission bands in this same wavelength region from other vibronic transitions in the excitation spectrum were unsuccessful. Instead, the detected fluorescence was shifted well to the red in a wavelength region where detection by our DFL collection optics was less sensitive. It should also be noted that, relative to DPVA in the following section, DPDA had much lower (>104) relative fluorescence yield.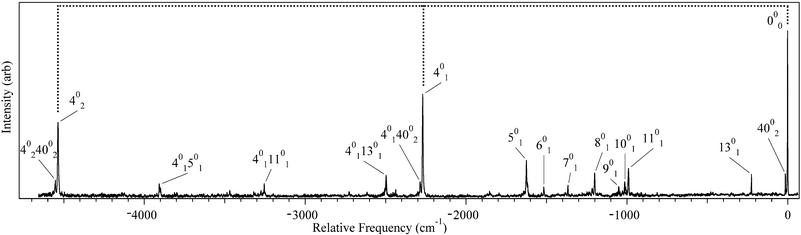 | ||
| Fig. 3 S1 origin DFL spectrum of DPDA. Vibronic assignments are based on DFT (B3LYP/6-311+G(d,p)) calculations and labeled using Mulliken notation.1 See text for further discussion. | ||
Assignment of the vibronic structure of the DPDA spectra was carried out by comparison of the observed transitions to DFT and TDDFT frequency calculations for the ground and excited states, respectively. A summary of select normal modes is shown in Table 1. As can been seen in the Table, the frequencies of most of the modes are not predicted to change significantly upon electronic excitation. The dominant feature in both the DFL and LIF spectra is a progression with spacing of 2268 and 2156 cm−1, respectively (marked by tie lines in Fig. 1 and 3). In diacetylene, a progression of 2105 cm−1 in the absorption spectrum was assigned as the totally symmetric CC stretching mode.39 The calculations predict the equivalent mode in DPDA to have a frequency of 2310 and 2168 cm−1 in the ground and the excited state respectively, in close agreement with experiment. On this basis, we assign the progressions to the ν4CC stretch, whose form is shown in Fig. 4a.
| Mode | Symmetry | S0(DFT) | S0(Exp) | S1(TDDFT) | S1(Exp) | Descriptiona |
|---|---|---|---|---|---|---|
| a Ring modes are giving using Varsanyi notation.58 | ||||||
| 4 | ag | 2310 | 2268 | 2168 | 2156 |
Diacetylene CC stretch |
| 5 | ag | 1635 | 1625 | 1622 | 1592 | Ring deformation 8a |
| 6 | ag | 1529 | 1517 | 1538 | 1514 | Ring deformation (19a) |
| 7 | ag | 1381 | 1366 | 1442 | 1445 | Diacetylenic C–C stretch |
| 8 | ag | 1201 | 1201 | 1199 | 1183 | Ring distortion (9a) |
| 9 | ag | 1051 | 1051 | 1031 | Ring deformation (18a) | |
| 10 | ag | 1018 | 1014 | 1015 | Ring deformation (12) | |
| 11 | ag | 995 | 990 | 974 | 960 | Ring breathing (1) |
| 12 | ag | 677 | 669 | 662 | Ring deformation (6a) | |
| 13 | ag | 230 | 224 | 232 | 231 | Long-axis stretch |
| 40 | ag | 17 | 8 | 31 | 32 | Phenyl torsion |
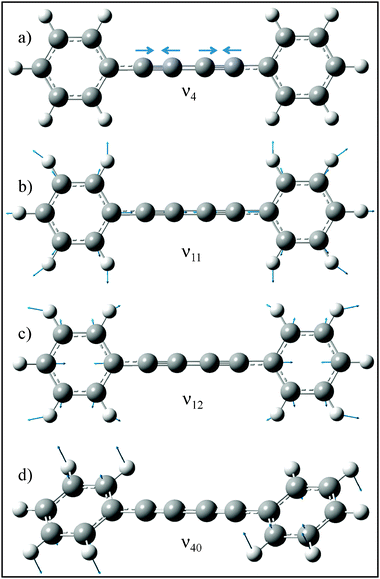 | ||
| Fig. 4 Ground state normal coordinates for modes 4, 11, 12, and 40 calculated at the B3LYP/6-311+G(d,p) level of theory. | ||
DFT and TDDFT calculations predict the geometry of DPDA to retain D2h symmetry upon excitation. If this is the case, then absent vibronic coupling, the fundamentals of only the 13 ag modes will be allowed, while even overtones and combination bands of non-totally symmetric vibrations of the same symmetry could be present. If D2h symmetry is retained, one anticipates these overtones and combination bands to be weak, since no displacement along the non-totally symmetric modes occurs. Indeed, most of the vibronic activity in the spectrum can be assigned to fundamentals of totally symmetric modes via their close correspondence with the frequencies calculated for the ag modes. Furthermore, these modes are symmetric combinations of phenyl ring vibrations on the two rings and are anticipated to carry the Franck–Condon activity associated with a π–π* excitation. Examples of two of these modes associated with ring breathing (ν11, Varsanyi58 mode 1) and in-plane deformation (ν12, Varsanyi58 mode 6a) are shown in Fig. 4b and c.
The lowest frequency transitions observed, 16 and 64 cm−1 in the ground and excited states respectively, are assigned to the 4002 and 4020 transitions involving the phenyl torsion (ν40) about the diacetylenic chain (Fig. 4d). The harmonic calculations predict frequencies for ν40 of 17 and 32 cm−1 (Table 1), a factor of two larger than experiment in S0, but in close agreement in S1. From the experimental changes in frequency of the fundamental upon excitation (from 8 cm−1 to 32 cm−1) one can conclude that the barrier to internal rotation of the phenyl rings is higher in the excited state than the ground state. The implications of this for the electronic states of DPDA will be discussed in more detail below.
In order to assess whether emission at other wavelengths was present in addition to that shown in the DFL spectrum of Fig. 3, the origin region of the spectrum was scanned using different blue cut-off filters placed in front of the LIF excitation photomultiplier tube. Using this series of filters, it was found that light collected in the LIF spectra covered a range over 10000 cm−1 to the red of the origin (See Supplementary Information for data†). Furthermore, this red emission could be detected over 1 μs after the laser pulse. This is to be compared with the short lifetime for sharp fluorescence, with a lifetime much shorter than the laser light pulse (<2 ns vs. 10 ns). The long-lived component could not be directly observed in the time-profile of the emission. Based on the data in hand, we postulate that upon excitation, DPDA undergoes rapid intersystem crossing to a triplet state resulting in lifetime broadening of the singlet-singlet excitation spectrum. A more detailed look at the nature of the intersystem crossing will be taken up in the discussion section below.
IV. Trans-Diphenylvinylacetylene
A. Excited state properties
Fig. 5 shows the LIF and UVHB spectra of trans-DPVA. The LIF spectrum is very intense, with its largest transitions three orders of magnitude greater than those in DPDA. UVHB showed that all the observed transitions belonged to a single conformer showing that no impurities either from the synthesis were present in the sample in detectable amounts. In sharp contrast, all attempts to record the LIF spectrum of the cis-DPVA isomer were unsuccessful, with no detectable fluorescence. Reasons for the lack of cis-DPVA fluorescence will be discussed in more detail below.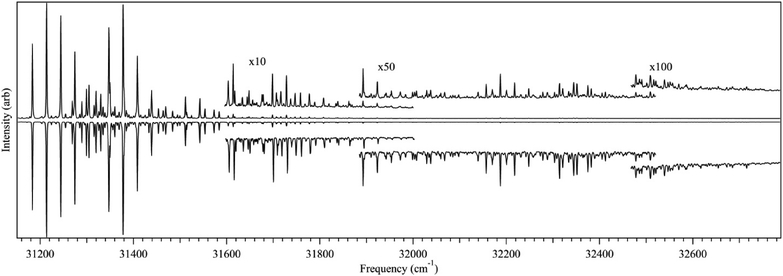 | ||
| Fig. 5 LIF excitation spectrum (top) and UVHB spectrum (bottom) of trans-DPVA. | ||
The S0–S1 origin of trans-DPVA is at 31183.2 cm−1, just under 1000 cm−1 to the red of DPDA. The transitions are sharp, with no hint of the broadening that dominated the spectrum of DPDA. The first 400 cm−1 of trans-DPVA's spectrum is dominated by progressions involving two low-frequency vibrations, of 30.5 cm−1 and 164 cm−1. The spectrum extends more than 1500 cm−1 above the origin; however, the intensity drops sharply about 300 cm−1 above the origin, with the weak transitions near 32500 cm−1 over 1000 times smaller than the origin. Nevertheless, these transitions are still ∼10 times more intense than the fluorescence from DPDA.
B. Dispersed fluorescence
Fig. 6 presents an overview S1 origin DFL spectrum that extends to levels up to 4500 cm−1 above the ground state zero-point level.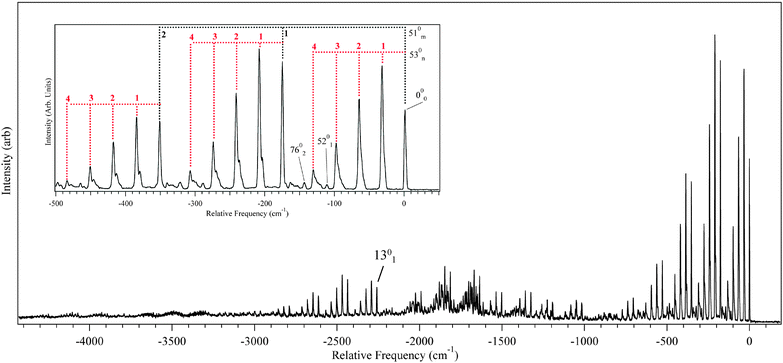 | ||
| Fig. 6 DFL from the origin of DPVA. The majority of the transitions can be assigned as X01510n530m combination bands following the pattern built off the origin (inset). | ||
The assignments shown in the figures are based on a comparison of the experimental transitions with calculated vibrational frequencies in S0 and S1. Table 2 lists the calculated and experimental frequency assignments for select vibrational modes of trans-DPVA, which account for virtually all the observed vibrational structure. Like the excitation spectrum, the DFL spectrum is dominated by progressions involving two vibrational modes with frequencies of 33 cm−1 and 176 cm−1. By comparison with DFT/TDDFT calculations, the modes responsible for these progressions have been assigned to the in-plane fundamentals ν53 (bridge-phenyl bending) and ν51 (bridge-phenyl stretch), respectively, the progressions of which are shown in the inset of Fig. 6. By observing where a ν53/ν51 progression began, it was possible to determine the location of several other vibrational fundamentals. As can be seen in the inset of Fig. 6, the observed transitions exhibit a broadening in higher members. This is postulated to arise due to congestion caused by a 2:1 Fermi resonance between ν78 and ν53 and a 2:2 Fermi resonance between ν77 and ν53.
| Mode | Symmetry | S0 (DFT) | S0 (Exp) | S1 (TDDFT) | S1 (Exp) | D (sim) | D (DFT) |
|---|---|---|---|---|---|---|---|
| 51 | a′ | 179 | 176 | 169 | 164 | 1.77(2) | 1.01 |
| 52 | a′ | 119 | 115 | 114 | 106 | 0.32(6) | 0.20 |
| 53 | a′ | 36 | 33 | 32 | 30.5 | 1.49(2) | 0.90 |
| 76 | a′′ | 78 | 74 | 86 | 66 | ||
| 77 | a′′ | 36 | 31.5 | 41 | 43 | ||
| 78 | a′′ | 28 | 14.5 | 31 | 20.5 |
We have not attempted to assign in detail the extensive vibrational structure between −1000 and −2000 cm−1, since transitions in this region are much weaker and are quite congested. The fundamental located at −2258 cm−1 has been tentatively assigned to the CC stretch (ν13). This is based on the lack of an observable fundamental around −1100 cm−1 and is in good agreement with the predicted value of 2281 cm−1 from the calculations. This mode is similar to ν4 in DPDA both in nature and frequency, but is much less dominant in intensity, indicating a smaller change in CC bond length in DPVA upon electronic excitation than in DPDA.
C. Frank-Condon fitting
DFL spectra of the first several members of the pure 53n0 and 51n0 progressions were recorded, and are shown in Fig. 7 and 8, respectively. A harmonic Franck–Condon analysis was carried out, using FC factors calculated from formulae developed by Henderson et al.54 A global fit for each mode was performed (omitting the scattered light peak) giving displacements of D = 1.77(2) and 1.49(2) for ν53 and ν51 respectively, where D is proportional to the displacement along the given normal mode coordinate.54 Values for D in this range indicate significant geometry change along these normal modes upon excitation. The best-fit intensities are indicated by the markers in the figures. By comparison, the only other observed in-plane fundamental, ν52 (acetylenic wag,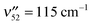 ) showed intensity patterns fit by a displacement D = 0.32(6).
) showed intensity patterns fit by a displacement D = 0.32(6).
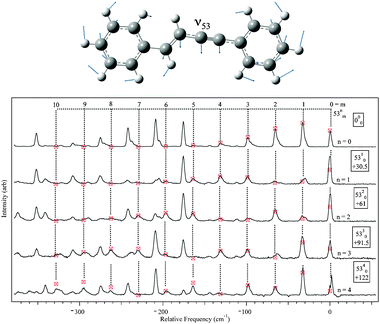 | ||
| Fig. 7 DFL spectra from 53n, n = 0–4 levels with Franck–Condon simulated intensities marked by the markers. | ||
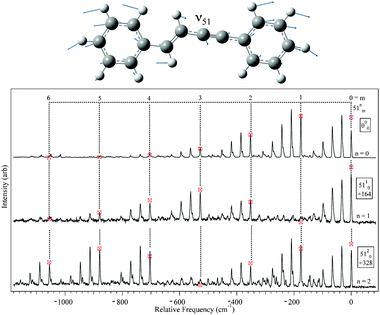 | ||
| Fig. 8 DFL spectra from 51n, n = 0–2 levels with Franck–Condon simulated intensities marked by the markers. | ||
The results of Franck–Condon fitting based on the DFL spectra were used to simulate the LIF excitation spectrum of trans-DPVA, as shown in Fig. 9. For comparison, a first-principles Franck–Condon simulation was carried out based on the optimized DFT/TDDFT geometries, and the calculated normal mode frequencies and forms of the normal modes, using Gaussian09. The result is shown in the bottom trace of Fig. 9. While the resulting spectrum agrees as to which normal modes are important upon excitation (with displacements of 1.01, 0.20 and 0.90 for ν53, ν52 and ν51 respectively), the calculation underestimates the extent of the geometry change.
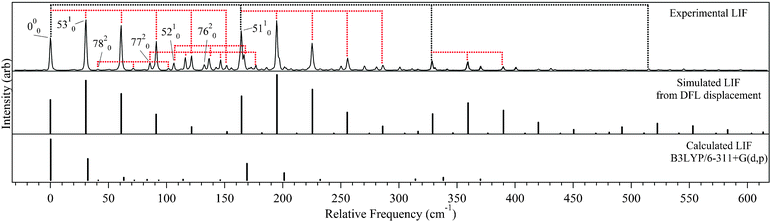 | ||
| Fig. 9 Comparison of the experimental LIF (top) to simulated LIFs from DFL displacement fits (middle) and Gaussian 09 (bottom). Tie lines indicate ν53 and ν51 progressions respectively. | ||
Comparing the experimental LIF excitation spectrum (Fig. 9 top) to that simulated using the S1 origin DFL spectrum (Fig. 9 middle), it is clear that there is a non-radiative process turning on around 300 cm−1 above the S0–S1 origin that decreases the experimental intensities relative to simulation. Several possible sources of non-radiative pathways include excited state trans-cisisomerization (followed by fast quenching of the cis isomer), the interconversion from a ππ* state to a ‘dark’ πσ* state, and ISC to a triplet state. DPVA did not possess detectable red-shifted emission, as was observed in DPDA, so it is less likely that ISC to a triplet state is a factor in the diminished emission. The other options will be examined more closely below.
V. Discussion
A. DPDA
The vibronic spectroscopy of DPDA is intriguing principally because of the inherent breadth of the observed vibronic transitions, even under jet-cooled conditions. In order to better understand why this is so, the excited state potential energy surfaces of DPDA were explored in more detail. As was noted already, the S1 zero-point level of DPDA has limited fluorescence signal, with significant red-shifted emission. Attempts to take DFL spectra from any of the higher vibronic levels were unsuccessful, and only resulted in red-shifted emission that could not be characterized in detail viaDFL. We surmise on this basis that there is some process (intersystem crossing, internal conversion) that is further limiting the fluorescence from these high energy bands.In considering these options, it is worth reflecting first on the photophysics of related molecules. For instance, tolane (Ph–CC–Ph) has sharp transitions upon excitation in the gas phase, but undergoes a similar loss of fluorescence from higher energy transitions.35,36 Subsequent theoretical studies have shown the existence of a state crossing from the initially excited ππ* state to a dark πσ* state of lower energy that accounts for this loss.38,59 The πσ* minimum was found to be planar with C2h symmetry and CC–C angles of 128.9°, giving it a structure much like stilbene. To look at the possible effects of a πσ* state in DPDA, TDDFT (B3LYP/6-311G(d,p)) optimizations on the Au excited state surface were carried out. The CC–C angle was stepped at 10° increments from 180° to 120° keeping a C2h planar symmetry. The results, shown in Fig. 10, predict a state crossing ∼1400 cm−1 above the B1u minimum with the Au minimum being approximately 850 cm−1 higher than the B1u optimized geometry. While this is about half the value of the barrier predicted for tolane, the minimum of the πσ* state in tolane was predicted to be ∼4000 cm−1 lower in energy then the ππ* state.38,59 While the effects of a πσ* state crossing cannot be ruled out, the prediction that the πσ* state is higher in energy then the ππ* state makes this an unlikely scenario for explaining the inherent breadth of the transitions in DPDA.
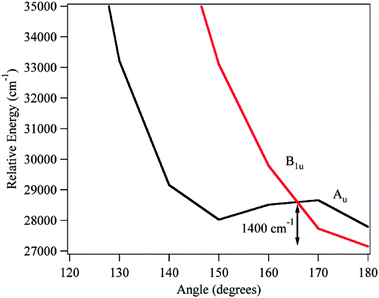 | ||
| Fig. 10 TDDFT (B3LYP/6-311+G(d,p)) electronic states of DPDA optimized on the Au surface. | ||
An additional coordinate of interest in DPDA is the phenyl ring torsion associated with ν40. From the LIF and DFL spectra, it was found that upon electronic excitation, the frequency of ν40 rose from 8 cm−1 to 32 cm−1. This indicates that the barrier to internal rotation is significantly higher in the excited state than in the ground state. Indeed, DFT and TDDFT calculations (B3LYP/6-311+G(d,p)) predict barriers of 88 and 532 cm−1 for the ground and excited states respectively (Fig. 11). In tolane, the ground state barrier was found to be 202 cm−1.36 Furthermore, as Fig. 11 shows, along the phenyl torsional coordinate the dark S2 state crosses the S1 surface only 300 cm−1 above the S1 minimum. Since the S2 state is dark in absorption from S0, interaction with it would lead to a loss of fluorescence. This crossing is low enough in energy to play some role in the loss of sharp emission associated with S1 fluorescence from levels above the So–S1 origin.
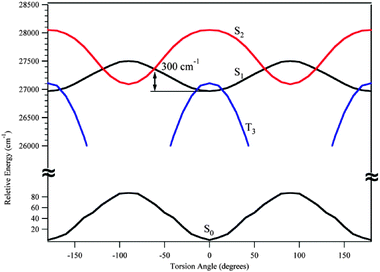 | ||
| Fig. 11 DPDA torsional barriers for the ground state (bottom trace) and excited states (top traces) optimized using DFT and TDDFT methods respectively B3LYP/6-311+G(d,p). Excited state contours S2 and T3 are from the S1 optimized geometry. | ||
Finally, at the S1 optimized geometry, the third triplet state (T3) is found to be only 140 cm−1 above the S1 state with a crossing just 40 cm−1 from the S1 minimum, in very close proximity with the S1 zero-point level. Furthermore, the T3 state has an optimized geometry in which the two rings are perpendicular to one another, with a minimum more than 2800 cm−1 below the S1 minimum. Given that there are three triplet states either below or near the S1 surface, we postulate that upon excitation, DPDA undergoes rapid intersystem crossing to one of the triplet surfaces (probably T3) causing the broadening of the transitions in the LIF excitation spectrum. The fluorescence observed from the S0–S1 origin is due to S1 population that has not crossed to the triplet manifold. At higher energies, it could either be that DPDA is undergoing even faster ISC, or that the combination of ISC and internal conversion to the dark S2 surface has depleted the fluorescing population below detectable limits.
While this scenario is plausible, it is not without its problems. The phenyl torsional coordinate that is present in DPDA is absent in C4H2 and phenyldiacetylene, both of which also show inherent broadening in their singlet-singlet transitions. In fact, much of the vibronic activity in DPDA, particularly the strong progression in the CC stretch, is shared by C4H2 and PDA as well. While the triplet manifold is likely in close proximity in all three molecules, it is unclear whether phenyl torsion plays a direct role in facilitating crossing in DPDA. Second, the lack of any resolved sub-structure in the S0–S1 origin of DPDA indicates strong coupling to a dense manifold of levels. If the broadening were purely due to the short lifetime of the S1 level carrying the oscillator strength in absorption, the 35 cm−1 FWHM of the S0–S1 origin (Fig. 2) would be associated with a 75 fs timescale for the process. Yet, observation of sharp fluorescence from the excited state level (Fig. 3) strongly suggests that the S1 lifetime is considerably longer than this. Given the nsec timescale of our excitation laser, at present we can only place an upper limit to this lifetime of a few nsec. Thus, while triplet states are likely to be the source of the broadening, the exact mechanism is one deserving further exploration both from theory and experiment.
B. DPVA
DPVA is in many ways a close analogue of stilbene. The cis/transisomerization of stilbene has been the subject of many dynamics and photoisomerization studies.16,25,60Cis-stilbene is unbound in the excited state and undergoes rapid isomerization and internal conversion to the ground state, making it impossible to observe viaLIF. The LIF excitation spectrum of trans-stilbene, shows the turn-on of a non-radiative process ∼1200 cm−1 above the S1 origin. Should there be a similar scenario in DPVA, it would explain the lack of an LIF spectrum for cis-DPVA.Fig. 12a shows the calculated barrier for the cis/transisomerization of DPVA. A relaxed scan of the vinyl dihedral angle from 00 to 1800 was carried out on the ground state. At each point on the ground state potential, a TDDFT single-point calculation was used to find the excited state energies by projecting vertically onto the excited state surface. It was found that there is an excited state barrier ∼4500 cm−1 above the trans minimum in the excited state and that cis-DPVA has a well-defined minimum on the excited state surface. We have confirmed the presence of a minimum in the cis configuration by optimization at that geometry. It seems unlikely that the photoisomerization of DPVA is the cause of the drop in fluorescence.
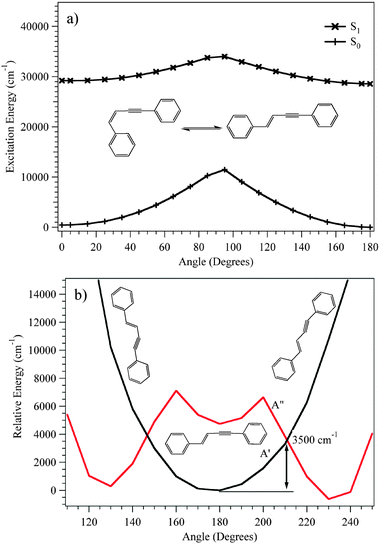 | ||
| Fig. 12 PES showing the energy for (a) the cis/transisomerization of the S0 and S1 states of DPVA optimized on the S0 surface and (b) πσ* crossing of the first A′ and A′′ excited states of trans-DPVA calculated on the first A′′ excited state surface using DFT and TDDFT (B3LYP/6-311+G(d,p)) methods. | ||
As an alternative to isomerization, excited state crossings involving the acetylenic moiety may be responsible for the observed non-radiative turn-on. In previous studies on similar phenylacetylene derivatives, a drop in LIF signal was attributed to interconversion from a ππ* state to a πσ* state.38,42,61 In the case of trans-phenylvinylacetylene, it was found that the πσ* excited state was the lowest excited singlet, with a bent structure with an acetylenic CCH angle of ∼60° and a barrier of ∼2000 cm−1 from the ππ* minimum to the πσ*crossing.61
Fig. 12b shows the calculated relaxed potential energy curve for trans-DPVA along the acetylenic bending coordinate for the first excited A′ and first excited A′′ states. The A′′ minimum was found to be ∼600 cm−1 below that of the A′ state. In addition, the state crossing is predicted to occur 3500 cm−1 above the A′ minimum. Based on these calculations it is hard to state with certainty whether the πσ* state, cis-transisomerization, or a triplet crossing is the most likely source of the non-radiative threshold found experimentally. Once again, a more detailed theoretical exploration of these surfaces would be helpful in finding quantitatively accurate surface crossings and dynamic pathways between them.
As to the lack of fluorescent signal from cis-DPVA, a Franck–Condon analysis with Gaussian09 predicts a large geometry change that would spread intensity out over many higher frequency modes and combination bands. Based on steric effects, cis-DPVA is predicted to be slightly non-planar in the ground state with a vinyl dihedral angle of 1 degree. Upon excitation, the dihedral is predicted to increase to 10 degrees. This same method predicts a change of 6 degrees to 30 degrees for stilbene. If cis-DPVA also undergoes interconversion to a dark state like trans-DPVA, the majority of strongly absorbing transitions may be above the crossing point, resulting in a short-lived excited state with little detectable fluorescence. A future study of cis-DPVAvia direct absorption (e.g., cavity ring-down) would be helpful in seeking to explore the excited state spectroscopy and dynamics of this isomer further.
C. Diphenyl comparison
Table 3 presents a comparison of the properties of DPDA and DPVA with those of trans-diphenylbutadiene (DPBD) and the single-ring series PBD, PVA, and PD. All three molecules in the diphenyl series have very large calculated oscillator strengths for transitions from the ground state to the first allowed excited state (the lower Ag state of DPBD is symmetry forbidden). The calculations consistently underestimate the energy difference between S0 and S1 states relative to experiment; however, relative energies are faithfully reproduced.63 Not surprisingly, conjugative addition of a second phenyl ring lowers the transition energy by 2500–4100 cm−1. In addition, the more unsaturated the C4 chain, the further blue the S0–S1 origin transition is shifted, with trans-DPVA 382 cm−1 and DPDA 1358 cm−1 blue of DPBD.46 This is consistent with what is observed in the corresponding series of C4 phenyl derivatives, where trans-PVA and PDA are 263 and 2916 cm−1 blue of PBD.62,64| Diphenyl a | S1 Symmetry | S1 (Exp) | S1 (Calcb) | Osc. Strengthb | Phenyl equiv.a | S1 (Exp) |
|---|---|---|---|---|---|---|
| a Refers to trans isomers, b Calculated using TDDFT (B3LYP/6-311+G(d,p)) in cm−1, c Refers to S3 in ref. 5, d Ref. 62, e Ref. 41, f Ref. 46. | ||||||
| Diphenylbutadiene c | Bu | 30801 |
26219 |
1.537 | Phenylbutadiene d | 33315 |
| Diphenylvinylacetylene | A′ | 31183 |
26693 |
1.343 | Phenylvinylacetylene e | 33578 |
| Diphenyldiacetylene | B1u | 32159 |
26968 |
0.896 | Phenyldiacetylene f | 36231 |
All three diphenyl molecules in the series undergo rapid internal conversion or intersystem crossing that result in a loss of fluorescence within a few hundred wavenumbers of their origins, though the exact nature of the processes has not been fully determined, and may differ within the series.
One of the most notable differences between DPDA, DPVA, and DPBD is the breadth of their transitions in the near-ultraviolet. DPBD and DPVA have sharp well-defined transitions in both the excitation and dispersed fluorescence spectra, with resolved rotational structure.5 By contrast, the corresponding transitions in DPDA are inherently broad in excitation, with emission divided between sharp transitions ascribable to fluorescence from the singlet state at the S1 origin and long-lived, red-shifted phosphorescence. DPVA possesses no detectable phosphorescence under jet-cooled conditions. Previous studies of DPDB have not commented on the presence or absence of such red-shifted emission.5
Upon closer examination, the molecular orbitals involved in the π–π* transition of all three diphenyls are markedly similar. As seen in Fig. 13, S0–S1 excitation is predicted to occur between the HOMO and LUMO orbitals of each molecule. A cursory inspection of these molecular orbitals indicates that they are nearly one-dimensional particle-in-a-box wave functions with five nodes (HOMO) and six nodes (LUMO) along the long conjugation axis of the molecules. As such, they redistribute the electron density within the box, but have little directionality to the charge redistribution. Not surprisingly, the transition dipole moments point along the ‘a’ inertial axes in all three molecules, as indicated in the Figure.
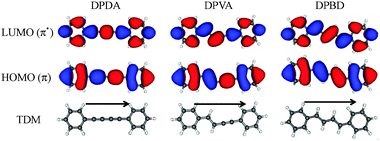 | ||
| Fig. 13 LUMO (top), HOMO (middle) orbitals, and TDM (bottom) predictions for DPDA (left), DPVA (middle), and DPBD (right) calculated using TDDFT B3LYP/6-311+G(d,p) level of theory. | ||
The large TDM's for these transitions suggest a significant charge redistribution between ground and excited states. In seeking to harness such charge redistribution into net transfer of charge along the ‘molecular wire’, one would need to incorporate donor–acceptor character into the two rings that would produce a distinction between the aromatic rings serving as termini. While in DPDA and DPBD, the symmetry of the diyne or diene chains precludes any asymmetry in charge distribution on this basis, in DPVA, the two rings are distinguished by whether they are immediately adjacent to an ethynyl or vinyl substituent. The TDM direction for the S0–S1 transition in trans-DPVA is from the styrene to the phenylacetylene moiety. Thus, molecular wires composed of similar but distinct π sub-units can provide directionality to the charge redistribution even without further distinguishing substitution on the rings. To our knowledge, the incorporation of vinylacetylene sub-units has not been employed in any systematic fashion in creating directionality in molecular wires for molecular electronics applications.
VI. Conclusion
In this study, we have examined the ground and excited state properties of the two non-fused ring PAHs diphenyldiacetylene and trans-diphenylvinylacetylene. It has been observed that DPDA has properties similar to other diacetylene compounds (phenyldiacetylene and diacetylene) with broad absorption features in the excitation spectrum. Unexpectedly, the DFL spectrum show sharp transitions that mirror intensities in excitation, indicating that they arise from singlet emission, with a small but detectable fraction of the population able to fluoresce prior to either undergoing ISC or internal conversion. DPVA, on the other hand, exhibited properties similar to those of phenylvinylacetylene, with a sharp drop in fluorescence most likely due to a surface crossing with a nearby πσ* state.Both of the molecules in this study and the previously studied diphenylbutadiene would benefit from further studies into their absorption profiles viacavity ring-down spectroscopy or UV depletion experiments. It would also be of interest to see if, following ultraviolet photoexcitation, any of the molecules in this series would undergo a ring closure mechanism to form fused-ring PAHs.
Acknowledgements
The authors gratefully acknowledge support from the NASA Planetary Atmospheres program under grant NNX10AB89G for this research.References
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1997–2011.
- Carotenoids in Photosynthesis, ed. A. J. Young and G. Britton, Chapman and Hall, London, 1993 Search PubMed.
- C.-P. Hsu, P. J. Walla, M. Head-Gordon and G. R. Fleming, J. Phys. Chem. B, 2001, 105, 11016–11025 CrossRef CAS.
- Molecular Processes in Vision, ed. E. W. Abrahamson and S. E. Ostroy, Hutchinson Ross Publishing Company, Stroudsburg, 1981 Search PubMed.
- L. A. Heimbrook, B. E. Kohler and T. A. Spiglanin, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 4580–4584 CAS.
- M. T. Allen and D. G. Whitten, Chem. Rev., 1989, 89, 1691–1702 CrossRef CAS.
- G. Orlandi, F. Zerbetto and M. Z. Zgierski, Chem. Rev., 1991, 91, 867–891 CrossRef CAS.
- T. Itoh, Y. Numata and I. Suzuka, Chem. Phys. Lett., 2007, 445, 179–182 CrossRef CAS.
- J. F. Pfanstiel, B. B. Champagne, W. A. Majewsi, D. F. Plusquellic and D. W. Pratt, Science, 1989, 245, 736–738 CrossRef CAS.
- J. H. Pinckard, B. Wille and L. Zechmeister, J. Am. Chem. Soc., 1948, 70, 1938–1944 CrossRef CAS.
- J. Saltiel, T. S. R. Krishna, A. M. Turek and R. J. Clark, Chem. Commun., 2006, 1506 RSC.
- A. Sandoval and L. Zechmeister, J. Am. Chem. Soc., 1947, 69, 553–557 CrossRef CAS.
- J. F. Shepanski, B. W. Keelan and A. H. Zewail, Chem. Phys. Lett., 1983, 103, 9–14 CrossRef CAS.
- L. y. Yang, R. S. H. Liu, K. J. Boarman, N. L. Wendt and J. Liu, J. Am. Chem. Soc., 2005, 127, 2404–2405 CrossRef CAS.
- P. D. Chowdary, T. J. Martinez and M. Gruebele, Chem. Phys. Lett., 2007, 440, 7–11 CrossRef CAS.
- P. M. Felker and A. H. Zewail, J. Phys. Chem., 1985, 89, 5402–5411 CrossRef CAS.
- G. Gershinsky and E. Pollak, J. Chem. Phys., 1997, 107, 812–824 CrossRef CAS.
- S. P. Kwasniewski, L. Claes, J. P. Francois and M. S. Deleuze, J. Chem. Phys., 2003, 118, 7823–7836 CrossRef CAS.
- D. M. Leitner, B. Levine, J. Quenneville, T. J. Martinez and P. G. Wolynes, J. Phys. Chem. A, 2003, 107, 10706–10716 CrossRef CAS.
- J. Quenneville and T. J. Martinez, J. Phys. Chem. A, 2003, 107, 829–837 CrossRef CAS.
- J. Schroeder, T. Steinel and J. Troe, J. Phys. Chem. A, 2002, 106, 5510–5516 CrossRef CAS.
- J. Schroeder, J. Troe and P. Vohringer, Z. Phys. Chem., 1995, 188, 287–306 CrossRef CAS.
- V. C. Vachev, J. H. Frederick, B. A. Brishanin, V. N. Zadkov and N. I. Koroteev, J. Phys. Chem., 1995, 99, 5247–5263 CrossRef CAS.
- R. E. Weston and J. R. Barker, J. Phys. Chem. A, 2006, 110, 7888–7897 CrossRef.
- T. S. Zwier, E. Carrasquillo and D. H. Levy, J. Chem. Phys., 1983, 78, 5493–5505 CrossRef CAS.
- R. W. Wagner and J. S. Lindsey, J. Am. Chem. Soc., 1994, 116, 9759–9760 CrossRef CAS.
- A. Bhaskar, R. Guda, M. M. Haley and Goodson, J. Am. Chem. Soc., 2006, 128, 13972–13973 CrossRef.
- Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, J. Am. Chem. Soc., 2003, 125, 14103–14112 CrossRef CAS.
- P. W. Thulstrup, S. V. Hoffmann, B. K. V. Hansen and J. Spanget-Larsen, Phys. Chem. Chem. Phys., 2011, 13, 16168–16174 RSC.
- Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, J. Chem. Phys., 2001, 114, 1775–1784 CrossRef CAS.
- Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, Chem. Phys. Lett., 1999, 303, 201–208 CrossRef CAS.
- Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, J. Phys. Chem. A, 1998, 102, 5769–5774 CrossRef CAS.
- H. Yoneda, H. Hiura and H. Takahashi, J. Mol. Struct., 1993, 301, 47–56 CrossRef CAS.
- D. R. Borst, S. G. Chou and D. W. Pratt, Chem. Phys. Lett., 2001, 343, 289–295 CrossRef CAS.
- K. Okuyama, M. C. R. Cockett and K. Kimura, J. Chem. Phys., 1992, 97, 1649–1654 CrossRef CAS.
- K. Okuyama, T. Hasegawa, M. Ito and N. Mikami, J. Phys. Chem., 1984, 88, 1711–1716 CrossRef CAS.
- D. Xu and A. L. Cooksy, THEOCHEM, 2007, 815, 119–125 CrossRef CAS.
- M. Z. Zgierski and E. C. Lim, Chem. Phys. Lett., 2004, 387, 352–355 CrossRef CAS.
- R. E. Bandy, C. Lakshminarayan and T. S. Zwier, J. Phys. Chem., 1992, 96, 5337–5343 CrossRef CAS.
- A. G. Robinson, P. R. Winter and T. S. Zwier, J. Phys. Chem. A, 2002, 106, 5789–5796 CrossRef CAS.
- C.-P. Liu, J. J. Newby, C. W. Müller, H. D. Lee and T. S. Zwier, J. Phys. Chem. A, 2008, 112, 9454–9466 CrossRef CAS.
- J. J. Newby, C.-P. Liu, C. W. Müller, W. H. James, E. G. Buchanan, H. D. Lee and T. S. Zwier, J. Phys. Chem. A, 2009, 114, 3190–3198.
- J. A. Sebree, D. F. Plusquellic and T. S. Zwier, J. Mol. Spect., 2011 DOI:10.1016/j.jms.2011.10.001.
- A. H. A. Tinnemans and W. H. Laarhoven, J. Chem. Soc., Perkin Trans. 2, 1976, 1104–1111 RSC.
- A. H. A. Tinnemans and W. H. Laarhoven, Tetrahedron Lett., 1973, 14, 817–820 CrossRef.
- A. G. Robinson, P. R. Winter, C. Ramos and T. S. Zwier, J. Phys. Chem. A, 2000, 104, 10312–10320 CrossRef CAS.
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827–8839 CrossRef CAS.
- R. K. Frost, G. Zavarin and T. S. Zwier, J. Phys. Chem., 1995, 99, 9408–9415 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, PhRvB, 1988, 37, 785 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Wallingford CT, 2009 Search PubMed.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef.
- J. R. Henderson, M. Muramoto and R. A. Willett, J. Chem. Phys., 1964, 41, 580–581 CAS.
- Y. Hirata, T. Okada and T. Nomoto, Chem. Phys. Lett., 1998, 293, 371–377 CrossRef CAS.
- M. Wahadoszamen, T. Hamada, T. Iimori, T. Nakabayashi and N. Ohta, J. Phys. Chem. A, 2007, 111, 9544–9552 CrossRef CAS.
- R. E. Bandy, C. Lakshminarayan and T. S. Zwier, J. Phys. Chem., 1992, 96, 5337–5343 CrossRef CAS.
- G. Varsanyi, Assignments for Vibrational Spectra of 700 Benzene Derivatives, Wiley, New York, 1974 Search PubMed.
- M. Z. Zgierski and E. C. Lim, Chem. Phys. Lett., 2004, 393, 143–149 CrossRef CAS.
- A. Amirav, U. Even and J. Jortner, Mol. Phys., 1983, 49, 899–912 CAS.
- J. J. Newby, C. W. Müller, C.-P. Liu and T. S. Zwier, J. Am. Chem. Soc., 2010, 132, 1611–1620 CrossRef CAS.
- W. J. Buma, B. E. Kohler, J. M. Nuss, T. A. Shaler and K. Song, J. Chem. Phys., 1992, 96, 4860–4868 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS.
- C. P. Liu, J. J. Newby, C. W. Muller, H. D. Lee and T. S. Zwier, J. Phys. Chem. A, 2008, 112, 9454–9466 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cp22857a |
| ‡ NASA Goddard Space Flight Center, Greenbelt, MD 20771, USA. |
| This journal is © the Owner Societies 2012 |