The collaborative role of molecular conformation and energetics in the binding of gas-phase non-covalent polymer/amine complexes†
Justin B.
Renaud
,
Eric
Martineau
,
Gleb G.
Mironov
,
Maxim V.
Berezovski
and
Paul M.
Mayer
*
Chemistry Department, University of Ottawa, 10 Marie Curie, Ottawa, ON K1N 6N5, Canada. E-mail: pmmayer@uottawa.ca; Tel: 1-613-562-5800 ext 6038
First published on 8th November 2011
Abstract
A large series of similar non-covalent complexes were probed using ion mobility spectrometry, molecular mechanics/molecular dynamics (MM/MD), electrospray-tandem mass spectrometry (ESI-MS/MS) and RRKM theory in order to determine the effects of charge state and charge location upon the conformation, the 0 K activation energy (E0) and the entropy of activation (ΔS‡) of the dissociation of these complexes. The non-covalent complexes consisted of poly(methylmethacrylate) oligomers and singly and doubly charged diaminoalkanes of varying length. This allowed for control of the charge separation within the complexes, as well as the size of the complex. A destabilizing effect was observed in complexes containing protons in close proximity, and/or short oligomers. Interestingly, a multiple charge stabilizing effect was observed when charge sites were sufficiently separated and/or when the polymer moiety of the complex was large. ΔS‡ values of doubly charged complexes showed a greater increase with increasing polymer size in comparison to singly charged complexes. This entropic observation is explained by structure, where IMS and MM/MD determined that the charge location was the determining factor of the overall conformation of these complexes and multiple charging resulted in more rigid arrangements. Dissociation of a tightly bound complex is more entropically favorable than a loosely bound complex. Also presented is a MM/MD refinement regime derived from IMS measurements.
1. Introduction
Non-covalent interactions are the foundation of biological activity. Signalling pathways, enzymatic activity and drug action are only some of the multitude of examples that involve non-covalent interactions. Two major aspects that are involved in these interactions are the strength of the interaction and the conformation, though both are highly connected as conformation often dictates the strength and specificity of the contact. Likewise a strong energetic interaction can cause a change in conformation, and in the case of enzymes, leads to a specific change in enzymatic activity.1 Many solution phase methods are capable of measuring the energetics of interactions such as Kinetic Capillary Electrophoresis (KCE), which can measure rate and equilibrium constants that characterize bonding energies.2 Interactions in CE occur over a period of time and complexation happens with various stoichiometries and binding parameters, hence, CE-measured binding values describe the overall palette of interactions and should be considered as “apparent” values, or overall average values. This makes it difficult to understand the role of individual physical states on the overall observable properties. In contrast, mass spectrometry provides a snap shot of the interaction, allowing for investigation of non-bulk properties, such as different charge states and oligomeric species which can lead to better understanding the role each of these has upon the overall observable behaviour of the complex. Measuring energetic properties of non-covalent complexes in the gas phase can be difficult. Probably the most accurate energetic measurements of gas phase non-covalent complexes is through blackbody infrared radiative dissociation (BIRD) where the dissociation rate constant as a function of temperature is recorded, leading to the enthalpy and entropy of activation.3,4,5,6 Using a more conventional instrument, binding energies, as well as proton affinities can be measured using the kinetic method7 (when Δ(ΔS‡) ≈ 0) and the extended kinetic method.8,9 A quick method to assign relative binding energies using variable-energy collision induced dissociation (CID) is accomplished by comparing the point at which 50% of a complex dissociates (E1/2).10 Although fast, great care has to be taken when comparing E1/2 values for different systems because the E1/2 is greatly affected by the number of degrees of freedom (DOF) within the complex,11 as well as its collision cross-section. A previous study that investigated a number of systems where degrees of freedom stability effects were nearly independent of binding affinity showed that the slope of a plot between the stability of the complexes as a function of the number of degrees of freedom produces a linear relationship whose slope can be used to more accurately assign relative binding energies.12 Alternatively, we have demonstrated how the kinetic parameters of zero Kelvin activation energy, E0 and the entropy of activation, ΔS‡, can be extracted from a variable energy CID breakdown diagram using Rice–Ramsperger–Kassel–Marcus (RRKM) theory,13 which inherently takes into account not only the number of DOF, but their different vibrational frequencies.14 Unlike E1/2 which only compares 1 point of a breakdown diagram, this technique uses the entire breakdown diagram to extract the kinetic variables. Specifically, this technique allows for extraction of 3 main variables from a single experimental breakdown diagram: (1) the 0 K activation energy (E0), (2) the entropy of activation (ΔS‡) and (3) α; a linear approximation used to describe the connection between the effective temperature (Teff) and the center-of-mass collision energy (Ecom). Together, these variables can describe a molecular system in a more quantitative fashion.Often, it is necessary to explain energetic observations based on molecular conformations which can be investigated using molecular dynamics (MD). MD is a computational method used for the identification of low energy conformations of molecular systems. When conclusions are being drawn directly from results obtained from MD simulations, it is very important that the conformations obtained accurately coincide with the true molecular structure present in the chemical environment of interest. For gas phase structures, ion-mobility spectrometry (IMS) can be used to measure the molecule’s cross section (Ω), which can be compared with that of the MD-derived structures. With the incorporation of ion-mobility cells within many modern mass spectrometers, its use as an analytical technique is rapidly increasing, and the use of IMS to validate computationally derived structures should also become common practice and lead to a greater confidence level being placed on MD simulation results.
This study aims to connect energetic and entropic data obtained with RRKM theory with the results of conformational analysis, and to identify the links and trends that exist between these 2 properties, by examining a series of more than 50 non-covalent complexes consisting of a synthetic polymer of varying lengths (poly(methylmethacrylate)) with a singly or doubly protonated diaminoalkane substrate.
2. Experimental
2.1 Materials
PMMA (Mw: 1460, Mw/Mn: 1.07) was purchased from Polymer Laboratories (Amherst, MA, USA). PMMA concentrations of 0.1 mg L−1 were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with either diamino(butane, hexane or decane) of concentration 0.1 mg L−1 in methanol. Formic acid was added to give a final solution of 0.1% formic acid. Cross-section calibrants, ubiquitin, myoglobin and lysozyme were purchased from Sigma, (St. Louis).
:1 ratio with either diamino(butane, hexane or decane) of concentration 0.1 mg L−1 in methanol. Formic acid was added to give a final solution of 0.1% formic acid. Cross-section calibrants, ubiquitin, myoglobin and lysozyme were purchased from Sigma, (St. Louis).
2.2 Electrospray ionization mass spectrometry (ESI-MS/MS)
A Micromass Quattro LC (Waters Micromass, Manchester, UK) triple quadrupole mass spectrometer equipped with a Z-spray source was used to carry out the electrospray ionization tandem mass spectrometry (ESI-MS/MS). Sample solutions were introduced via syringe pump at a flow rate of 40 μL min−1. The sample cone voltage was set to 40 V. Collision-induced dissociation (CID) was carried out with argon at a gas pressure of 1 × 10−3 mbar (1 bar = 100 kPa), and laboratory frame collision energies (Elab) ranging from 0 to 100 eV (specific settings are included in figure caption) recorded for 30 s at each collision energy step. The resolution of the first quadrupole was set to unit mass resolution (15 in the Masslynx software) for all experiments. The resolution of the final quadrupole was held constant at 12 to increase the sensitivity of the ESI-MS/MS experiments. All collision energies reported in this paper are center-of-mass (Ecom) energies and derived from Elab using the equation: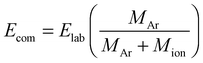 | (1) |
2.3 Extraction of energetic values from breakdown diagrams using RRKM theory
The method used to generate the breakdown curve and fit it to experimental values has been described in detail elsewhere.13 In brief, two main properties were used to generate a theoretical breakdown curve; the unimolecular rate constant, k(E), for the dissociation and the effective internal energy distribution as a function of Ecom.The rate constant was calculated using the RRKM equation:15
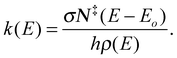 | (2) |
The internal energy distribution as a function of center of mass collision energy was approximated through the use of an effective temperature (Teff) designation in the equation
| Teff = 300 + αEcom | (3) |
The three variables; E0, ΔS‡ and α are altered in an automated manner using an in-house Python/Fortran hybrid program and the score of each fit was assigned based the absolute separation between the Ecom of experimental data points and the Ecom of the point on the theoretical curve with the same % dissociation as experimental (in units of eV) divided by the number of data points in the set. The different combinations of E0, ΔS‡ and α that resulted in a score no less than 0.05% below the best scoring combination were averaged together, and standard deviation taken from this set of Combinations. The listed uncertainty does not take into account the limitations of the model. So, while the precision of the values should be good, the same cannot be guaranteed of their absolute accuracy.
In the interpretation of the value of α, the vibrational heat capacity (Cvib) was calculated using the statistical mechanical equation:
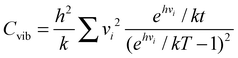 | (4) |
2.4 Ion mobility spectrometry
Experimental cross sections of all complexes were measured following protocol of D. P. Smith et al.19 using myoglobin, ubiquitin and lysozyme tryptic peptides to relate arrival time distributions to molecular cross-sections. A Beckman PA 800 capillary electrophoresis (Beckman Coulter inc., USA) was used to infuse analytes into a Waters Synapt G2 HDMS mass spectrometer (Waters Corp., USA) via the Micromass Capillary Electrophoresis Sprayer (Micromass UK Limited, UK) by applying a constant pressure of 1 psi. A make up solution consisting of a 50:50 methanol:water mixture with 0.1% formic acid, was introduced at a flow rate of 0.7 μL min−1.
2.5 Molecular mechanics/molecular dynamics with simulated annealing (MM/MD-SA)
The AMBER920 program suite using the Generalized AMBER force field (GAFF)21 was used to study the conformations of the complexes. Equilibration and annealing dynamics were performed for 60 different complexes in total; PMMA oligomers (7–16) with diamino(butane/hexane/decane) in both the 1H+ and 2H+ state. Minimized energy conformations of the PMMA/diaminoalkane complex were briefly equilibrated for 20 ps (time step 1 fs) at 300 K before undergoing 2000 cycles of simulated annealing22 (each 18.25 ps total, 0.5 fs time step) starting with drastic heating from 300 K to 800 K over 1.25 ps and equilibrated for 2 ps. This was followed by cooling increments of 100 K over 1 ps with 2 ps of equilibration at each temperature until 300 K. The lowest energy conformation of each simulated annealing cycle was then selected to undergo subsequent rounds of simulated annealing. After each cycle, the low energy conformation was cooled to 0 K over 2 ps, minimized and potential energy measure. Theoretical cross sections of the 50 lowest structures sampled (<6.3 kJ from the lowest structure energy) were measured using the trajectory method in the MOBCAL program.23,24 The 50 lowest energy structures are obtained as .pdb format files from AMBER. In-house software extracted atomic coordinates and their masses, with atomic charges obtained from the topology file used by AMBER. The trajectory method functions through averaging the orientations of a set of coordinates and calculating their interactions with a theoretical helium buffer gas, taking into account long range interactions, scattering and multiple collisions. Uncertainties in these theoretical cross-sections are derived from the standard deviation of the cross-sections obtained for the 50 structures.2.6 IMS validation/MD refinement
In cases where the theoretical cross section did not coincide well with the experimental cross section, a refined MD-SA regime was employed consisting of a second round of MD-SA. Within the 50 lowest energy conformations identified during the first 2000 cycles of MD-SA, the conformation having a theoretical cross section closest to the experimental cross section was equilibrated at 300 K for 400 ps (1 fs time step). This was followed by 1000 cycles of MD-SA using the same regime as the first round and the new lowest energy conformations therein identified. The new conformations were deemed to be a refined structure if: (1) their theoretical cross section is closer to experimental cross section and (2) their MM potential energy are the new global minimum (lower than first round low energy conformations).3. Results and discussion
For brevity, diaminoalkane, 1,4-diaminobutane, 1,6-diaminohexane, 1,10-diaminodecane and dihydrogen terminated poly(methylmethacrylate) will be denoted as DAA, DAB, DAH, DAD and PMMA respectively. A table summarizing all m/z values for the various complexes can be found in supporting information† (Table S2).3.1 IMS MD-SA conformational analysis
Conclusions that are solely based upon MD results can be risky, because the reliability of the computational regime employed can be uncertain and thus MD structures might not coincide well with the actual conformation(s) present. Therefore, IMS was used to obtain experimental cross sections (Ωexp) which were compared to the theoretical cross sections (Ωtheo) obtained from the MD structures using the trajectory method in MOBCAL. Shown in Fig. 1a, Ωtheo obtained from the MD-SA regime of this study agree well with those measured experimentally. When Ωtheo did not coincide with Ωexp, it suggests that the low energy MM structure is most likely not the true conformation. A second round of MD-SA starting from a low energy conformation that was the closest to Ωexp led to structures that not only had an overall lower energy, but also better matched Ωtheo(refined) to Ωexp. For example, following the initial 2000 cycles of MD-SA, the Ωtheo of the complex [PMMA12][DAB + 2H]+2 was overestimated by 18.9%. After MD refinement, the MM potential energy was lowered by 2.9 kJ mol−1 and the refined Ωtheo became only 7.4% larger than Ωexp (Fig. 1b). As an additional point of interest, this PMMA/diaminoalkane system has the potential to be an efficient IMS cross-section calibration standard, as a single injection of a single sample of PMMA spiked with different DAA would yield multiple complexes of different size and charge that have excellent coverage of small to intermediate cross section values.![Theoretical (Ωtheo) vs. experimental (Ωexp) cross section of [PMMAn][DAB + 2H]+2. Dashed diagonal is for Ωtheo = Ωexp. (a) Ωtheo values obtained from 1stround, low energy MM/MD structures using the trajectory method in MOBCAL. (b) Ωtheo obtained from the 2nd MM/MD round (refined).](/image/article/2012/CP/c1cp22936b/c1cp22936b-f1.gif) | ||
| Fig. 1 Theoretical (Ωtheo) vs. experimental (Ωexp) cross section of [PMMAn][DAB + 2H]+2. Dashed diagonal is for Ωtheo = Ωexp. (a) Ωtheo values obtained from 1stround, low energy MM/MD structures using the trajectory method in MOBCAL. (b) Ωtheo obtained from the 2nd MM/MD round (refined). | ||
Due to the strong agreement between Ωexp and Ωtheo, the level of confidence that can be assigned to observations drawn from the MM/MD conformations is increased. The low energy conformers of complexes between PMMA and singly protonated diaminoalkanes show a tendency for the protonated amine to reside predominantly near the center of the PMMA, which adopts a ‘horseshoe’ structure (Fig. 2a,c and e). This allows for the attractive electrostatic forces between the protonated amine and the carbonyl oxygens of the PMMA side-chains to be maximized. When the length of PMMA is small/intermediate (7–12 units), the complexes are fairly rigid as the interaction of singly protonated alkylamine with the center of the PMMA chain being highly favourable. However, as the length of the PMMA increases, the structure becomes less confined and different conformations are observed where the protonated amine is able to interact with different positions of the PMMA chain, not just those from the terminal units. In all cases, the overall conformation was not affected in an evident way by the length of the substrate, meaning complexes containing DAB, DAH or DAD all had very similar conformations.
![Lowest energy conformations obtained using MD-SA for (a) [PMMA8][DAB + 1H]+1, (b) [PMMA8][DAB + 2H]+2, (c) [PMMA8][DAH + 1H]+1, (d) [PMMA8][DAH + 2H]+2, (e) [PMMA8][DAD + 1H]+1 and (f) [PMMA8][DAD + 2H]+2.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f2.gif) | ||
| Fig. 2 Lowest energy conformations obtained using MD-SA for (a) [PMMA8][DAB + 1H]+1, (b) [PMMA8][DAB + 2H]+2, (c) [PMMA8][DAH + 1H]+1, (d) [PMMA8][DAH + 2H]+2, (e) [PMMA8][DAD + 1H]+1 and (f) [PMMA8][DAD + 2H]+2. | ||
Alternatively, the doubly charged complexes, [PMMAn][DAA + 2H]+2 (Fig. 2b,d and f), have an overall structure that is dictated by the DAA, which is protonated on both terminal ends and adopts a linear arrangement that maximizes charge separation. Secondary to the separation of charges, the PMMA adopts a conformation around the DAA in a manner that maximizes interaction between carbonyl oxygens on the PMMA side-chains and the protonated amines. When the DAA is sufficiently short or polymer sufficiently long a ‘horseshoe’ conformation is adopted similar to the corresponding + 1 species occurs, only in this case, one protonated site interacts with the central units of the PMMA oligomer, while the second protonated site interacts with both terminal groups. If the DAA is larger and polymer of short/intermediate length (Fig. 2d,f) a parallel arrangement between the DAA and polymer is seen where each protonation site of the DAA interacts with opposite terminal units of the PMMA (Fig. 2b). The overall trend observed based on the effects of charge state upon conformation is that as the length of the DAA increases, so do the differences between the conformations of singly and doubly protonated species. Similarly, the same observation was verified based on Ωexp measurements. Fig. 3 shows the shortest substrate, DAB, having the smallest effect of charge upon cross section, while the longest, DAD, showed the largest difference. In addition to confirming MM/MD observations, this is also an indirect confirmation that the site of protonation within the complexes is most likely on the amine sites of the DAA.
![Experimental cross sections (Å2) of [PMMA10][DAB + mH]+m, [PMMA10][DAH + mH]+m and [PMMA10][DAD + mH]+m. In black, singly protonated (m = 1), in white, doubly protonated (m = 2).](/image/article/2012/CP/c1cp22936b/c1cp22936b-f3.gif) | ||
| Fig. 3 Experimental cross sections (Å2) of [PMMA10][DAB + mH]+m, [PMMA10][DAH + mH]+m and [PMMA10][DAD + mH]+m. In black, singly protonated (m = 1), in white, doubly protonated (m = 2). | ||
3.2 MS/MS, RRKM energetic analysis
The range of Ecom collision energies used for this study, was relatively small (0.65–1.9 eV) and dissociation of all the [PMMAn][DAA + mH]+m complexes resulted in a single dominant dissociation pathway. Singly charged complexes; [PMMAn][DAA + 1H]+1 dissociated with PMMA leaving as the neutral, and the diamino substrates always retaining the proton (Fig. 4b). The substrates used all have gas phase proton affinities ∼1000 kJ mol−1,25 which indicates that all the PMMA oligomers sampled have gas phase proton affinities of lower values. It should be noted that in cases where the substrate has a lower proton affinity (i.e. glycine, 886.5 kJ mol−1)25 it is possible for the PMMA oligomer to retain the proton after dissociation, and to undergo unique side-chain fragmentation.26 For the doubly charged complexes, [PMMAn][DAA + 2H]+2, all cases sampled resulted in the expected partitioning of charges after dissociation, with one proton being retained by the DAA, and the other leaving with the PMMA oligomer where characteristic PMMA + H+ fragmentation products were observed (Fig. 4a). MS/MS of [PMMAn][DAA + 2H]+2 generates two signals from a single parent ion, therefore the breakdown diagrams were constructed using only the intensity of the DAA + H+ signal relative to that of the parent complex ion. Fitting of the experimental breakdown diagrams results in values for E0, ΔS‡ and α (Fig. 5).![Collision-induced dissociation mass spectra of (a) [PMMA8][DAD + 2H]+2 and (b) [PMMA8][DAD + 1H]+1 obtained at Ecom of 1.26 eV and 0.95 eV respectively. In both cases, only one dissociation pathway exists under these experimental parameters. See experimental procedures for conditions.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f4.gif) | ||
| Fig. 4 Collision-induced dissociation mass spectra of (a) [PMMA8][DAD + 2H]+2 and (b) [PMMA8][DAD + 1H]+1 obtained at Ecom of 1.26 eV and 0.95 eV respectively. In both cases, only one dissociation pathway exists under these experimental parameters. See experimental procedures for conditions. | ||
![Breakdown diagrams of (a) [PMMAn][DAH + 1H]+1 and (b) [PMMAn][DAH + 2H]+2. Experimental measurements obtained from the CID of DAH complexed with PMMA units of 8 mer (□), 10 mer (△), 12 mer (∘) and 15 mer (⋄). Dotted lines obtained by theoretical fitting using RRKM.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f5.gif) | ||
| Fig. 5 Breakdown diagrams of (a) [PMMAn][DAH + 1H]+1 and (b) [PMMAn][DAH + 2H]+2. Experimental measurements obtained from the CID of DAH complexed with PMMA units of 8 mer (□), 10 mer (△), 12 mer (∘) and 15 mer (⋄). Dotted lines obtained by theoretical fitting using RRKM. | ||
Fig. 6 shows that E0 for both singly and doubly protonated complexes increases as the length of the PMMA within the complex increases, which is logical considering an increase of a PMMA unit results in an increase in the overall electrostatic attractive forces between the polymer and the DAA substrate. A table with all values derived in this study is available as supporting information, Tables S3 and S4.† Also observed in the case of the 2+ system is evidence for the PMMA length dependence of E0 reaching an asymptote. If extrapolated further, it could be expected that for much larger PMMA oligomers, an increase in PMMA length would have very little or no effect upon E0.
![Activation energies (E0) of (a) singly protonated complexes; [PMMAn][DAB + 1H]+1 (□), [PMMAn][DAH + 1H]+1 (⋄), [PMMAn][DAD + 1H]+1 (△) and (b) doubly protonated complexes; [PMMAn][DAB + 2H]+2 (□), [PMMAn][DAH + 2H]+2 (⋄), [PMMAn][DAD + 2H]+2 (△). E0 values represent values which resulted in the best fit of experimental breakdown diagrams with theoretical breakdown curve obtained through RRKM.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f6.gif) | ||
| Fig. 6 Activation energies (E0) of (a) singly protonated complexes; [PMMAn][DAB + 1H]+1 (□), [PMMAn][DAH + 1H]+1 (⋄), [PMMAn][DAD + 1H]+1 (△) and (b) doubly protonated complexes; [PMMAn][DAB + 2H]+2 (□), [PMMAn][DAH + 2H]+2 (⋄), [PMMAn][DAD + 2H]+2 (△). E0 values represent values which resulted in the best fit of experimental breakdown diagrams with theoretical breakdown curve obtained through RRKM. | ||
The identity of the DAA substrate does not greatly affect E0 when the complex is singly protonated; [PMMAn][DAA + 1H]+1 (Fig. 6a). In contrast, the opposite is true for the doubly protonated species [PMMAn][DAA + 2H]+2 (Fig. 6b). [PMMAn][DAB + 2H]+2 complexes have the lowest activation energy due to the increase of repulsive forces that are a result of the closer proximity of the charge bearing sites. Likewise, the longest substrate complexes; [PMMAn][DAB + 2H]+2 had much higher E0 values because repulsive forces between charges are minimized. Similar to the conformational analysis, this provides evidence for the site of protonation within the complex to be on the amines.
Previous studies using CID based techniques to probe gas phase binding energies, have showed that often a multiply charged complex that dissociates into 2 charged fragments will have a lower stability in comparison to the singly charged species.12 In this system however, multiple charging of the complex can lead to either a stabilizing or de-stabilizing effect. For [PMMAn][DAB + mH]+m, multiple charging leads to an initial decrease in complex stability until a tipping point when the complex involves PMMA oligomers containing >10 units (Fig. 7a). The E0 of [PMMA8][DAB + 1H]+1 and [PMMA8][DAB + 2H]+2 are 0.85 ± 0.01 eV and 0.63 ± 0.01 eV respectively. As chain length increases, the multiple charge leads to increased stabilization ([PMMA15][DAB + 1H]+1 = 1.43 ± 0.03 eV vs. [PMMA15][DAB + 2H]+2 = 1.77 ± 0.02 eV). Although the charges are in close proximity to each other, there is an increase in the electrostatic attractive forces between the DAA and the polymer and increasing the length of the polymer will eventually lead to attractive electrostatic forces outweighing the repulsive. [PMMAn][DAD + 2H]+2 complexes consistently showed multiple charging leading to complex stabilization vis à vis their singly protonated complement (Fig. 7b).
![Activation energies of (a) [PMMAn][DAB + 1H]+1 (⋄), [PMMAn][DAB + 2H]+2 (△) and (b) [PMMAn][DAD + 1H]+1 (⋄), [PMMAn][DAD + 2H]+2 (△).](/image/article/2012/CP/c1cp22936b/c1cp22936b-f7.gif) | ||
| Fig. 7 Activation energies of (a) [PMMAn][DAB + 1H]+1 (⋄), [PMMAn][DAB + 2H]+2 (△) and (b) [PMMAn][DAD + 1H]+1 (⋄), [PMMAn][DAD + 2H]+2 (△). | ||
The second kinetic value that was altered in order to fit experimental breakdown diagrams was ΔS‡. Altering this predominately changes the slope of the breakdown curve (whereas, E0 is largely responsible for the onset location of the curve). Fig. 8 shows that both singly and doubly protonated, [PMMAn][DAA + mH]+m complexes have an increasing ΔS‡ coinciding with an increase in polymer size. The greatest difference was observed between the singly and doubly charged complexes; the doubly protonated complexes had a steeper slope of increase in comparison with its corresponding singly charged species. The presence of the two charges holds the oligomer chains more rigidly than a single charge site, and so the transition state for dissociation into two singly-charged products is accompanied by a greater relative increase in entropy.
![Entropies of activation (ΔS‡) of (a) [PMMAn][DAB + 1H]+1 (⋄), [PMMAn][DAB + 2H]+2 (△) and (b) [PMMAn][DAD + 1H]+1 (⋄), [PMMAn][DAD + 2H]+2 (△). ΔS‡ values represent values which resulted in the best fit of experimental breakdown diagrams with theoretical breakdown curve obtained through RRKM.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f8.gif) | ||
| Fig. 8 Entropies of activation (ΔS‡) of (a) [PMMAn][DAB + 1H]+1 (⋄), [PMMAn][DAB + 2H]+2 (△) and (b) [PMMAn][DAD + 1H]+1 (⋄), [PMMAn][DAD + 2H]+2 (△). ΔS‡ values represent values which resulted in the best fit of experimental breakdown diagrams with theoretical breakdown curve obtained through RRKM. | ||
The final variable that was necessary to adjust was α, corresponding to the relationship between the effective internal energy distribution, and center-of-mass collision energy. In a previous study that used a similar RRKM protocol to extract energetic data from complexes between the Aβ-40 peptide and drug candidates, the α values were similar for all complexes (∼200 K eV−1) whereas values obtained in this study ranged from 195–458 K eV−1. This is because the overall Aβ-40 peptide complexes were quite similar to each other, whereas, the presently studied complexes cover a much wider range of cross section and degrees of freedom. Unlike E0 and ΔS‡, both singly and doubly protonated complexes show a similar negative slope of α over increasing polymer length (Fig. 9). In essence, α describes the deposition of energy from a collision event into the effective internal energy of the ion, thus the difference in α between a corresponding singly or doubly charged complex of this size should not be significant. The variable α is a parameter that actually accounts for several molecular and instrumental properties, including the number of collision events (collision gas density, ion cross section) and the ability for a unit of collision energy to increase the effective temperature. Using the above properties, a theoretical curve that accurately described the α values of this system was found using the following relation:
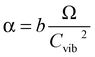 | (5) |
![α values resulting in best fit of experimental breakdown diagrams by theoretical RRKM curves for [PMMAn][DAD + 1H]+1 (△) and [PMMAn][DAD + 2H]+2 (⋄). Under experimental conditions this molecular system can be described by eqn (5) with b = 2.23 × 10−3. Dashed and Solid lines represent theoretical alpha values for [PMMAn][DAD+1H]+1 and [PMMAn][DAD+2H]+2 respectively.](/image/article/2012/CP/c1cp22936b/c1cp22936b-f9.gif) | ||
| Fig. 9 α values resulting in best fit of experimental breakdown diagrams by theoretical RRKM curves for [PMMAn][DAD + 1H]+1 (△) and [PMMAn][DAD + 2H]+2 (⋄). Under experimental conditions this molecular system can be described by eqn (5) with b = 2.23 × 10−3. Dashed and Solid lines represent theoretical alpha values for [PMMAn][DAD+1H]+1 and [PMMAn][DAD+2H]+2 respectively. | ||
4. Conclusions
A major observation, similar to previous reports, is that in these gas phase complexes, electrostatic interactions are the dominant force, and can determine the overall stability and conformation of the complexes. Molecular dynamics is a powerful and widely used tool to investigate molecular structure; however, many classic regimes of MD necessitate the assignment of charge site. In many cases, such as this study, the charge bearing site(s) are apparent, but in other cases involving more complex systems, it could become more difficult. In this example, the location of the charges would greatly affect the overall conformation and energetics, and the location of charges was confirmed on two fronts; through MS/MS and IMS.Acknowledgements
PMM thanks the Natural Sciences and Engineering Research Council of Canada for continuing financial support. JBR thanks the same agency for a graduate scholarship during the tenure of which this work was completed.References
- W. S. Bennett and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4848 CrossRef CAS.
- G. G. Mironov, V. Okhonin, S. I. Gorelsky and M. V. Berezovski, Anal. Chem., 2011, 83, 6 Search PubMed.
- W. D. Price, P. D. Schnier and E. R. Williams, Anal. Chem., 1996, 68, 7 CrossRef.
- R. Daneshfar and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2003, 15, 9 Search PubMed.
- R. C. Dunbar, Mass Spectrom. Rev., 2004, 23, 31 CrossRef.
- P. D. Schnier, W. D. Price, R. A. Jockush and E. R. Williams, J. Am. Chem. Soc., 1996, 118, 11 CrossRef.
- R. G. Cooks and P. S. H. Wong, Acc. Chem. Res., 1998, 31, 7 CrossRef.
- I.-S. Hahn and C. Wesdemiotis, Int. J. Mass Spectrom., 2003, 222, 465 CrossRef CAS.
- P. B. Armentrout, J. Am. Soc. Mass Spectrom., 1999, 11, 8 Search PubMed.
- K. X. Wan, M. L. Gross and T. Shibue, J. Am. Soc. Mass Spectrom., 2000, 11, 7 CrossRef.
- V. Gabelica and E. D. Pauw, J. Mass Spectrom., 2001, 36, 5 CrossRef.
- N. Vinokur and V. Ryzhov, J. Mass Spectrom., 2004, 39, 6 CrossRef.
- P. M. Mayer and E. Martineau, Phys. Chem. Chem. Phys., 2011, 13, 5178 RSC.
- R. A. Marcus and O. K. Rice, J. Phys. Chem., 1951, 55, 14 CrossRef.
- T. Baer and P. M. Mayer, J. Am. Soc. Mass Spectrom., 1996, 8, 12 Search PubMed.
- T. Beyer and D. F. Swinehart, Commun. ACM, 1973, 16, 1 CrossRef.
- J. Casey, A. Alhazmi and P. M. Mayer, Eur. J. Mass Spectrom., 2005, 11, 6 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. T. R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. M. A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian Inc, Wallingford CT, 2004.
- D. P. Smith, T. W. Knapman, I. Campuzano, R. W. Malham, J. T. Berryman, S. E. Radford and A. E. Ashcroft, Eur. J. Mass Spectrom., 2009, 15, 17 CrossRef.
- D. A. Case, T. A. Darden, T. E. C. III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, D. A. Pearlman, M. Crowley, R. C. Walker, W. Zhang, B. Wang, S. Hayik, A. Roitberg, G. Seabra, K. F. Wong, F. Paesani, X. Wu, S. Brozell, V. Tsui, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, P. Beroza, D. H. Mathews, C. Schafmeister, W. S. Ross and P. A. Kollman, University of California, San Francisco, CA, 2006.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. E. Case, J. Comput. Chem., 2004, 25, 17 Search PubMed.
- S. R. Wilson and W. Cui, Biopolymers, 1990, 29, 10 Search PubMed.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 4 CrossRef.
- M. J. Jarrold, online: http://www.indiana.edu/∼nano/software.html.
- E. P. Hunter and S. G. Lias, J. Phys. Chem. Reference Data, 1998, 27, 243 CrossRef.
- A. M. Alhazmi and P. M. Mayer, J. Am. Soc. Mass Spectrom., 2009, 20, 6 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cp22936b |
| This journal is © the Owner Societies 2012 |