Atmospheric hydrocarbon activation by the hydroxyl radical: a simple yet accurate computational protocol for calculating rate coefficients†
Alban S.
Petit
a and
Jeremy N.
Harvey
*b
aLaboratoire de Chimie Théorique, Méthodologies, Modélisation, Université de Montpellier II, Montpellier, France
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, UK. E-mail: jeremy.harvey@bris.ac.uk
First published on 9th November 2011
Abstract
The overall rate coefficient at standard temperature and pressure for the hydrogen abstraction reaction by the hydroxyl radical (HO˙) from common saturated volatile organic compounds (VOCs) is derived theoretically using electronic structure calculations and transition state theory (TST). The computational approach used is based on relatively efficient methods, and hence is applicable to a large number of compounds with only a modest use of computer resources. The key methods used are density functional theory (for the calculation of barrier heights) and simple transition state theory (TST), including a simple correction for tunnelling. All thermally relevant conformers of the reactant and the abstraction TS are included in the study. For all compounds in a test set of thirty-four, the calculated rate coefficient agrees with the experimental value to within better than an order of magnitude, and to within better than a factor of three for all but six cases, so that the accuracy is of predictive utility.
Introduction
Human developments as well as the biomass contribute to release a wide variety of volatile organic compounds (VOCs) into the atmosphere. The tropospheric lifetime of such species is a crucial parameter for establishing the importance of their transport to remote locations and their tendency to accumulate in the atmosphere. The lifetime ranges from a few minutes to several years depending on the compound stability and the degradation pathway.1 The major atmospheric sink of volatile organic compounds is through oxidation processes, which are mainly initiated by species such as HO˙ or NO3˙ radicals or O3. The very reactive HO˙ radical plays an important role in the chemistry occurring in the atmosphere due to its large breadth of action. It can react with H-containing saturated compounds through hydrogen abstraction, add directly to multiple bonds or degrade carbon monoxide. In most cases, the initiation step of oxidation starting from R–H leads to the formation of reactive organic radicals R˙viahydrogen abstraction. These intermediates then rapidly add molecular O2 to form RO2˙ radicals. The subsequent reactions play a crucial role in the O3 budget, the concentration of NOx (x = 1 or 2) and the formation of secondary organic aerosols which are essential to the formation of clouds.2,3 The Earth's albedo,4,5 that is the ratio of reflected to incident solar radiation, is also directly influenced by the size distribution and the altitude of these particles.It is therefore very useful to be able to measure or predict the rate coefficient for H-atom abstraction from VOCs by the HO˙ radical. The branching ratio defining the selectivity for H-atom abstraction from different sites within a given VOC is also important, as isomeric peroxy radicals RO2˙ can lead to different oxidized intermediates. For the majority of anthropogenic and biogenic VOCs present in the troposphere, the reaction with hydroxyl radical is the predominant removal process;1,3 therefore extensive experimental measurements have been carried out. Due to the large variety of relevant compounds it would however be highly challenging to obtain experimental data for all of them. Thus, theoretical prediction of reliable rate coefficients and branching ratios is valuable, and many semi-empirical methods exist, as briefly summarised in the following text. The general details of the hydrogen abstraction reaction by HO˙ are now fairly well-known and understood, from the mechanistic point of view. The transition states for these reactions involve a simple atom transfer with a roughly linear C–H⋯O geometry.6 The barrier heights vary between essentially zero and roughly 40 kJ mol−1. Though these reactions are often simple bimolecular elementary steps, Ravishankara and Smith7,8 have shown the importance of the collisional complex in the entrance channel for the overall dynamics of the reaction, at least for some substrates and under some conditions. Van der Waals interactions, especially hydrogen bonds, are the main stabilizing forces which lead to the existence of such complexes. Stronger interactions of this type tend to lead to longer-lived complexes and hence to a larger impact on the kinetics.
Several methods have been proposed to estimate the abstraction rate coefficients. The most commonly used is the structure–activity relationship (SAR) designed by Atkinson.9 The idea is to divide the reaction of the HO˙ radical with organic compounds into four different pathways: hydrogen atom abstraction, addition on multiple bonds, addition on aromatic rings and reaction with N-, P-, and S- containing groups. For each pathway and each site in the molecule, the estimated rate coefficient takes into account the nature of the neighbouring substituents. The overall rate coefficient is simply the sum of all the individual rates. The latest version of the SAR of Atkinson and Kwok10 contains 89 parameters: 26 group rate coefficients and 63 substituent factors to estimate the overall rate derived from available kinetic databases. For a test set of about 485 molecules, the predicted rate coefficient lies within a factor of 2 with respect to experimental data in 90% of cases. This extremely satisfactory agreement makes this tool very effective for predictions. However, there are some limitations on its use. Substantial deviations occur for halogenated compounds, especially those containing perfluorinated groups, and for ethers. Overall, this method does not perform very well for the chemical classes that are not included in the test set.11
Other approaches for estimating rate coefficients and branching ratios use empirical correlations between measured rate coefficients and some internal physical properties of the compounds under study. For example, the dissociation energy of the C–H bond,12,13 the approximate ionization potential based on the energy of the highest occupied molecular orbital (εHOMO)14 and the NMR chemical shift15 have been used in this way. The linear correlations derived in such models are quite accurate for somewhat small test sets of molecules with a rather limited range of chemical functionalities. For instance, the correlation obtained by Cooper14et al. using εHOMO as a descriptor for hydrofluorocarbons (HFCs) containing one (C1) or two (C2) carbon atoms can predict rate coefficients within a factor of two in 82% of cases. This has been further extended to C3 HFCs and hydrofluoroethers (HFEs). The deviation from experiment showed larger scattering in these further cases. So, extrapolation to a broader range of compounds is not easy, and for some of the models, the data needed are not readily available for larger numbers of compounds.
Electronic structure calculations using high-level ab initio methods such as coupled cluster theory have been applied to reaction of HO˙ with a number of simple VOCs.16,17
Detailed studies of the kinetics based on these calculations and dynamical methods such as variational transition state theory (VTST),18,19RRKM theory,16 and master equation analyses have been carried out. These studies are able to tackle the dynamics of the reaction at a very high level of understanding, including e.g. a qualitative treatment of the quantum mechanical tunneling through semi-classical approaches, such as small-curvature tunneling (SCT),20WKB theory21 or fitting the effective barrier to an Eckart function. Overall, the combination of these statistical methods and a rigorous treatment of the tunneling provides very accurate predictions of the rate coefficients and branching ratios. For instance, Ellingson and Truhlar19 studied the atmospheric oxidation of H2S by HO˙ in order to discover the origin of its unusual temperature-dependence. They predicted a canonical rate coefficient of 4.23 × 10−12 cm3 molecule−1 s−1 at 300 K using VTST/SCT, which is in very good agreement with the recommended rate of 4.5 ± 1.0 cm3 molecule−1 s−1 measured at 298 K. Another example is the reaction between CH3C(O)CH3 and HO˙ for which Caralp16et al. calculated a rate coefficient of 3.30 × 10−14 cm3 molecule−1 s−1, in excellent agreement with the experimental rate of 3.29 ± 0.32 × 10−14 cm3 molecule−1 s−1 at 298 K. However, such calculations are time-consuming and cannot readily be applied to large numbers of VOCs.
The aim of this work is to examine whether simpler methods can be used to predict rate coefficients with reasonable accuracy for large numbers of VOCs. The key technique used is conventional transition state theory (TST)22 in its simplest form. This approach has not been used extensively in the literature to our knowledge, for several reasons among which the most important is perhaps that with some lower-level electronic structure methods very inaccurate results can be obtained. Alternative approaches for related atmospheric processes, e.g.rearrangement of alkyl-oxy radicals, have been reported.23
The input parameters for TST, namely the classical barrier height (ΔE‡) or energy barrier, as well as the rotational constants and harmonic vibrational frequencies of the reactants and the transition state will be derived in this work from density functional theory (DFT) calculations. The choice of latter method is based on the fact that we need a method that can treat electronic correlation at a relatively low computational cost. Quantum mechanical tunneling plays some role in the reaction of HO˙ radicals with VOCs, as the key step is the transfer of a light hydrogen atom.24 This effect will be treated using the simple Wigner25 one-dimensional correction factor. Based on the discussion above of the accuracy of SAR methods, our target is to be able to predict rate coefficients at room temperature to within better than one-half of an order of magnitude i.e. within a factor of three in most cases. A larger error of one order of magnitude (factor of ten) will be acceptable for a small number of the compounds studied. The test set of compounds is composed of 34 atmospherically relevant VOCs belonging to different chemical classes such as alkanes, haloalkanes, ethers, ketones and alcohols. We do not consider unsaturated species in which HO˙ addition to a double bond contributes significantly to the rate coefficient.
The bases of this protocol were established in our group during the study of the atmospheric oxidation of nopinone (6,6-dimethyl-bicyclo[3.3.1]heptan-2-one).26 One part of this work was devoted to the choice of a suitable DFT functional to compute the most accurate possible energy barriers. Three hybrid functionals were tested, namely BH&HLYP,27MPW1K28 and KMLYP,29 which all contain a large amount of Hartree–Fock (exact) exchange of ca. 50%. They were benchmarked against the very accurate ab initio Gaussian-330 (G3) method for a test set of molecules that contain the same bonding pattern as nopinone: methane, ethane, propane, cyclobutane and acetone. The KMLYP functional gave very good agreement for energy barriers with G3 after applying a small additive correction.26 Using the corrected KMLYP potential energy barriers and the corresponding calculated rotational and vibrational parameters for the TS and the reactants, simple TST was used to predict rate coefficients. The ratio of calculated and experimental rate coefficients was found to be good for the test set. E.g. for ethane and propane, the kcalc/kexp ratio was 1.0 and 1.1 respectively. For acetone and methane, the calculated rate was slightly underestimated, with 0.35 and 0.58 kcalc/kexp ratios respectively. For cyclobutane, the rate coefficient was slightly overestimated, by a factor of 1.7. The overall calculated rate coefficient for the atmospheric oxidation of nopinone at room temperature was also in good agreement with experiment: kabs,TST = 6.92 × 10−12 cm3 molecule−1 s−1 compared to kexp = 1.7 × 10−11 cm3 s−1. Moreover, the computed branching ratio corresponding to the first position, corresponding to the tertiary carbon atom adjacent to the carbonyl group, is predicted to be the largest (23.4% of the total rate coefficient at 298 K). This finding is consistent with experimental evidence which showed that products derived from the 1-nopinonyl radical were produced in significant amounts. These encouraging preliminary results suggested that simple computation could indeed predict kinetic data in a reliable way, at least for fairly standard temperature and pressure conditions. The present broader study is aimed at checking that such calculations are indeed a useful addition to experimental methods.
Computational details
All the DFT calculations reported here were performed using the Gaussian 0331 program package, the functional used throughout this study is the KMLYP functional of Kang and Musgrave29 and the standard 6-311+G(d,p) basis set has been used on all atoms. Based on our previous work,26 the barrier heights calculated at this level were subjected to an additive correction of 6.89 kJ mol−1, chosen to reproduce barrier heights calculated using the accurate G3 method.30Full geometry optimization was carried out for reactants and the saddle points. Vibrational frequencies were computed so as to provide the zero-point correction for the electronic energy and to characterise minima and saddle points. Standard DFT integration grids were used throughout; some test calculations with ultrafine grids were carried out, leading to negligible changes in structures and relative energies. Computed vibrational frequencies were also insensitive to the grid size (except for the soft vibrational mode discussed below, where significant changes were observed in some cases). A systematic approach was used to locate all energetically relevant conformers of the reactant species as well as all possible saddle points. Specifically, an attempt was made to locate a saddle point for each structurally distinct C–H bond of each low-energy conformer of the reactant VOC.
Single point G3(MP2)32 energy calculations were performed in one case at the KMLYP-optimized structure of the reactants and lowest TS. This composite method requires MP2 calculations with a special large basis (“GTMP2Large” in Gaussian), these were performed in Gaussian31 using an unrestricted Hartree–Fock reference. It also requires MP2 and QCISD(T) calculations using the 6-31G(d) basis; these were performed using MOLPRO33 and a restricted open-shell reference.
Rate coefficients were calculated from conventional canonical transition state theory,22 calculating the partition function of the activated complex at the saddle point of the potential energy surface. The classical energy barrier for each saddle point was derived from the zero-point energy corrected energy difference between the saddle point and the separated reactants, with the VOC species in the same conformation as in the saddle point. Rotational contributions to the partition function assumed rigid body behaviour. With one exception, the partition function for all vibrational degrees of freedom was derived with the harmonic oscillator (HO) approximation using the unscaled KMLYP frequencies. For some VOCs, the calculated vibrational frequency for one mode of the TS, torsion around the C–H⋯O axis, is very low. This leads to an overestimation for the corresponding contribution to the vibrational partition function. Accordingly, when the computed frequency for this mode is smaller than 40 cm−1, a fixed value of 40 cm−1 is used in the TST calculations. This value, valid only near room temperature, was determined based on calculations for CH2F2. The internal rotation partition function for this mode was computed, and the frequency chosen as the one that yields a vibrational partition function with the same value.
TST requires the counting of the statistical factor for reactants and the activated complex. This factor can be derived in several different ways.22 The approach we have used is the following: the rate coefficient has been computed using no statistical factor for each structurally distinct hydrogen atom in the reactant, and the resulting value has been multiplied by the number of corresponding structurally identical hydrogen atoms, e.g. for methane, one saddle point has been considered, and the rate coefficient has been multiplied by four. This is equivalent to using symmetry factors of 12 for methane and 3 for the activated complex. Additionally, the partition functions for the 2Π HO˙ radical and for the activated complex have been multiplied by factors of four and two, respectively, to take into account the electronic degeneracy.
For each saddle point, we include the one-dimensional Wigner factor25 as an approximate treatment of tunneling. This factor is calculated based on the unscaled magnitude of the imaginary mode at the TS as calculated at the KMLYP level of theory.
The TST procedure described above produces a calculated rate coefficient for abstraction of each structurally distinct C–H bond for a particular conformer of the reactant. We refer to this value as being the conformer-specific rate coefficient. The overall rate coefficient is derived from the weighted sum of these conformer-specific rate coefficients (as explained below), and the branching ratios are obtained by taking the ratio of the rate coefficient summed over all identical sites of the molecule and the overall rate coefficient.
Discussion and results
The aim of this work is to show that relatively simple electronic structure calculations can be used to predict rate coefficients for hydrogen abstraction by the HO˙ radical from C–H bonds of VOCs. To do so, only the separated reactants and the abstraction saddle point need to be located and characterized in the calculations.Many of the VOCs we wish to characterize here are molecules containing multiple C–C single bonds, with rotation around these bonds leading to a range of different conformations. These internal rotations usually exhibit low barriers, so that interconversion between conformers can occur very rapidly at room temperature. Also, in many cases, several low-lying conformers exist within a small energy range above the global minimum, so that at room temperature, several conformers will be present in a given sample. For instance, a molecule of butane is a mixture of 72% anti and 28% gauche at 298 K34. These two conformations interconvert rapidly at 298 K but can be expected to have different physical properties including different rate coefficients for reaction with HO˙. It has been shown previously in computational studies of the reactivity of intermediates in VOC oxidation processes such as β-hydroxyalkoxy radicals35,36 and the 1-butoxy radical37 that including explicitly the effects of several conformers has a significant impact on the overall accuracy of the predicted rate coefficient.
Thus, it became desirable for us to implement this strategy to improve the quality of our results. We assume that the conformer population follows a Boltzmann distribution. For pragmatic reasons, we chose a cut-off value, in terms of Gibbs free energy (G), to restrict the population of conformers to a manageable size. This cut-off is expressed as a difference of Gibbs free energy of 6 kJ mol−1 between any conformer and the most stable one; we find that the weight of a population with a ΔG beyond this limit is small enough to be neglected for the present compounds and target accuracy, though the threshold used may need to be larger at higher temperature or for other compounds. In practice, the use of DFT makes this refinement not demanding in terms of computational resources and real time. In the 34 molecules that constitute our test set, 7 of them have one or more additional conformers lying within 6 kJ mole−1 of the lowest energy conformer. These molecules are 1,2-C2H4Cl2, CH3OC2H5, CH3COCH2(CH3)2, CH3COC3H7, CH3CH(OH)C2H5, (CH3)2C(OH)C2H5 and (CH3)2C(OH)CH(CH3)2. For instance, for 1,2-C2H4Cl2 there are only two possible arrangements corresponding to the gauche and anti conformations, with the latter being more stable.
For all the conformers, all non-equivalent abstraction sites have been taken into account.
The expression for the rate coefficient is as follows:
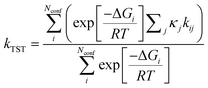
Since we obtain a computed value for kij and kTST, it is straightforward to work out the importance of each channel in the overall rate coefficient, the so-called branching ratios.
All saddle points display similar geometric features (for examples, see Fig. 1): the angle formed by the HO˙ oxygen atom and the C–H breaking bond is almost collinear, with C–H and O–H distances of about 1.2 and 1.3–1.4 Å, respectively. The HOH angle formed by the abstracted H-atom and HO˙ is around 100°, close to that of the product water molecule. A previous study on nopinone25 carried out in our group showed that the PES for rotation around the C–H⋯O axis is flat. As a consequence, the presence of isomeric saddle points might be expected. However, in each case only one preferred orientation around this C–H⋯O axis has been found, so that a unique saddle point is accessible from the reactants for each abstraction site. In other words, for any input orientation of the dihedral angle H–O⋯C–R (θ) the same saddle point is reached upon structural optimization. Nevertheless, this degree of freedom is very floppy and rather different values of θ are obtained for different TSs and different substrates. For molecules such as CH3X (X = F, Cl, Br) or acetone, the dipole–dipole interaction between OH and C–X(O) defines a preferred value for θ where both fragments are eclipsed (θ ≈ 0°). However, somewhat large deviations are observed in the cases of CHCl2Br and CH2F2 where θ is 13.7° and 58°, respectively. In the latter, HO˙ lies in the bisector plan of the F–C–F angle in order to align the dipoles and maximise the interaction. It is noteworthy that the saddle point for methane exhibits the same staggered configuration.
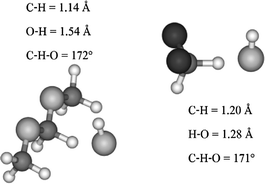 | ||
| Fig. 1 Schematic representation of the abstraction TSs for CH3OCH2OCH3 and CH2F2, computed at the KMLYP/6-311+G(d,p) level of theory and their main geometric features. Entries (12 and 24) of Table 1. | ||
Other theoretical work has been done on the halogenated methane species to predict their reaction rate toward the hydroxyl radical, such as those of Louis38et al. or González-Lafont39et al. They used various high-level ab initio techniques, e.g. MP2/MP4, QCISD along with large basis sets. Our geometries generally agree well with those in these previous studies, though the optimized value of θ varies substantially in some cases, reflecting the floppy nature of this coordinate. Though there is no guarantee that the KMLYP method we use predicts the correct structure in this respect, the impact on calculated energies should be negligible.
Table 1 lists for all the compounds under consideration the relative energy of the lowest hydrogen abstraction TS found, the experimental abstraction rate coefficient, the calculated TST rate coefficient, and their ratio RTST = kTST/kexp. All experimental data are either measured at 300 K or at 297 or 298 K; given the small temperature dependence of the rate coefficients, this should not affect the comparison between experiment and computation. All computational data use T = 300 K. The corrected rate coefficients kcalc will be discussed later.
| Compounds | ΔE‡ (corrected) | k exp | k TST | R TST | k calc | R calc |
|---|---|---|---|---|---|---|
| a The barriers contain the additive correction of 6.89 kJ mol−1 and are ZPE corrected; the numbers shown here correspond to the lowest barrier found for each species. In ref. 40c,h,j: T = 298 K, for ref. 40d: T = 297 K. | ||||||
| CH4 | 22.97 | 6.40 × 10−15 | 1.09 × 10−14 | 1.70 | 1.09 × 10−14 | 1.70 |
| C2H6 | 10.96 | 2.40 × 10−13 | 5.07 × 10−13 | 2.11 | 4.98 × 10−13 | 2.07 |
| C3H8 | 4.65 | 1.10 × 10−12 | 1.70 × 10−12 | 1.55 | 1.60 × 10−12 | 1.46 |
| c-C4H840b | 5.14 | 2.03 × 10−12 | 4.37 × 10−12 | 2.15 | 3.75 × 10−12 | 1.85 |
| c-C6H12 | 2.59 | 8.44 × 10−12 | 1.02 × 10−11 | 1.21 | 7.39 × 10−12 | 0.88 |
| CH3F | 14.42 | 2.07 × 10−14 | 6.71 × 10−14 | 3.24 | 6.70 × 10−14 | 3.24 |
| CH3Cl | 15.06 | 4.36 × 10−14 | 4.35 × 10−14 | 1.00 | 4.35 × 10−14 | 1.00 |
| CH3Br | 16.42 | 2.96 × 10−14 | 2.74 × 10−14 | 0.93 | 2.74 × 10−14 | 0.92 |
| CHF3 | 28.55 | 2.86 × 10−16 | 9.87 × 10−17 | 0.35 | 9.87 × 10−17 | 0.35 |
| CHCl3 | 7.82 | 1.03 × 10−13 | 1.12 × 10−13 | 1.08 | 1.11 × 10−13 | 1.08 |
| CHBr340c | 7.28 | 1.50 × 10−13 | 1.09 × 10−13 | 0.73 | 1.08 × 10−13 | 0.72 |
| CH2F2 | 14.53 | 1.16 × 10−14 | 6.27 × 10−14 | 5.41 | 6.26 × 10−14 | 5.40 |
| CHCl2Br 40d | 7.55 | 6.92 × 10−14 | 1.02 × 10−13 | 1.47 | 1.02 × 10−13 | 1.47 |
| CH3CCl3 | 19.93 | 9.88 × 10−15 | 2.61 × 10−15 | 0.26 | 2.61 × 10−15 | 0.26 |
| 1,1-C2H4Cl240e | 4.92 | 2.97 × 10−13 | 2.90 × 10−13 | 0.98 | 2.87 × 10−13 | 0.96 |
| 1,2-C2H4Cl2 | 9.79 | 1.59 × 10−13 | 1.72 × 10−13 | 1.08 | 1.71 × 10−13 | 1.08 |
| CF3CH2CH340f | 11.81 | 5.42 × 10−14 | 5.51 × 10−14 | 1.02 | 5.50 × 10−14 | 1.01 |
| CF3CHFCH2F 40f | 16.02 | 1.81 × 10−14 | 6.57 × 10−15 | 0.36 | 6.57 × 10−15 | 0.36 |
| CF3CHFCHF240 | 17.54 | 6.40 × 10−15 | 2.68 × 10−15 | 0.42 | 2.66 × 10−15 | 0.42 |
| CHF2CHFCHF2 | 14.16 | 2.02 × 10−14 | 1.20 × 10−14 | 0.59 | 1.20 × 10−14 | 0.59 |
| CH3OCH3 | 2.62 | 2.78 × 10−12 | 3.06 × 10−12 | 1.10 | 2.74 × 10−12 | 0.99 |
| CH3OC2H5 | 2.37 | 5.06 × 10−12 | 2.60 × 10−12 | 0.51 | 2.37 × 10−12 | 0.47 |
| CH3OC4H940g | 1.66 | 3.23 × 10−12 | 2.40 × 10−12 | 0.74 | 2.20 × 10−12 | 0.68 |
| CH3OCH2OCH340h | −7.69 | 4.90 × 10−12 | 5.75 × 10−11 | 11.73 | 1.82 × 10−11 | 3.72 |
| CH3OC(CH3)2OCH340h | 0.50 | 4.70 × 10−12 | 9.13 × 10−12 | 1.94 | 6.80 × 10−12 | 1.45 |
| c-C4H8O240h | −5.09 | 1.26 × 10−11 | 1.13 × 10−11 | 0.89 | 7.93 × 10−12 | 0.63 |
| CH3COCH3 | 9.53 | 1.70 × 10−13 | 1.83 × 10−13 | 1.08 | 1.82 × 10−13 | 1.07 |
| CH3COC2H5 | −0.45 | 1.20 × 10−12 | 2.22 × 10−12 | 1.85 | 2.05 × 10−12 | 1.71 |
| C2H5COC2H5 | −0.74 | 2.06 × 10−12 | 2.20 × 10−12 | 1.07 | 2.04 × 10−12 | 0.99 |
| CH3COC3H7 | −9.82 | 4.56 × 10−12 | 3.54 × 10−11 | 7.76 | 1.52 × 10−11 | 3.34 |
| CH3COCH2CH(CH3)240i | −16.23 | 1.22 × 10−11 | 2.17 × 10−10 | 17.82 | 2.38 × 10−11 | 1.95 |
| CH3CH(OH)C2H540j | −6.22 | 8.77 × 10−12 | 3.01 × 10−11 | 3.43 | 1.41 × 10−11 | 1.61 |
| (CH3)2C(OH)C2H540j | −7.28 | 3.64 × 10−12 | 2.07 × 10−11 | 5.69 | 1.17 × 10−11 | 3.20 |
| (CH3)2C(OH)CH(CH3)240j | −15.34 | 9.08 × 10−12 | 7.72 × 10−11 | 8.50 | 1.98 × 10−11 | 2.18 |
A first point to make is that the RTST values are all reasonably close to 1, meaning that the combination of DFT and simple TST yields results that are fairly accurate, and are able to capture at least most of the variation in the experimental rate coefficients (which span more than five orders of magnitude). This is surprising as several approximations are involved in the TST calculations, each of which could in principle lead to an error of one order of magnitude or more. Clearly, as already emphasized in the study of the atmospheric oxidation of nopinone,26 error cancellation seems to work in our favour.
The neglect of barrier recrossing/variational effects will tend to lead to an overestimate of the rate coefficient, which is presumably compensated for by other errors in this work. In previous work, e.g. on the OH + CH2F2 reaction,39 the overestimate has been shown to be quite large, by a factor of three or more.
One possible candidate for error compensation is the treatment of tunnelling. Unlike in our previous work,26 we do include the simple Wigner correction for tunnelling.25 However, it has been found in some cases e.g. for the reaction of OH with CH3CCl341 that the tunnelling effects can be significantly larger than those predicted by the Wigner formula. In the cited study,41 canonical variational TST along with small-curvature tunnelling (CVT/SCT) approximation42 was used to derive the rate coefficient. At room temperature the ratio of the tunneling-free and SCT rate coefficients, or transmission coefficient, was about 16, whereas the Wigner correction predicts a transmission coefficient of only 4. This suggests that our treatment of tunneling leads to an underestimate of the rate coefficients.
We also considered another reason that might account for larger errors on some calculated rate constants: kinetic complexity. The kTST values calculated so far assume that hydrogen abstraction is the sole, and rate-limiting, step in the reaction. In fact, some of the molecules in this test set can form quite strongly hydrogen-bonded complexes with the hydroxyl radical before the abstraction step. For those where the hydrogen abstraction barrier is low, it is possible that the initial complex-forming step, which we have so far neglected, is rate-limiting. This could account for the large RTST values obtained for some of the species with lower abstraction barriers. A rigorous treatment of such complex kinetics could in principle be performed, by using variational TST to compute the association rate coefficient, locating the hydrogen-bonded complexes, and then using a master equation (ME) microcanonical treatment to compute the net apparent rate coefficient. This type of calculation has indeed been performed previously, e.g. for the reaction of OH with acetone.16 However, it would be too time-consuming for the present purposes.
Instead, we adopted an approximate approach in which the complex, formed with a rate coefficient k1, is assumed not to undergo extensive collisional cooling, and instead either promptly dissociates back to reactants (k−1) or promptly leads to hydrogen abstraction (k2). This assumption is valid only within a low-pressure regime and for relatively weakly bonded complexes, where collisional cooling is less important. It leads us to the following expression for the apparent overall rate coefficient:
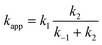
To use this expression, one would in principle need to be able to calculate k1 for each species. Instead, we use experimental data7,8 for the rate constant for quenching of vibrationally excited hydroxyl radical as a good estimate for k1. The experiments show that this is not very dependent on the collider, yielding e.g. values for collision with acetone and nitric acid of (2.67 ± 0.15) × 10−11 and 2.45 × 10−11 cm3 molecule−1 s−1, respectively.7,8 We have used the former value as an approximation for k1 for all species in this work. Note that for smaller values of kTST, kcalc ≈ kTST as one would expect.
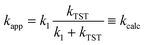
The resulting values of kcalc are shown in Table 1. As can be seen, for all the species where kTST was too large compared to experiment, kcalc is in better agreement with experiment. The ratio of the predicted and the experimental rate coefficients Rcalc is usually very close to one, with the extreme values being 0.26 for CH3CCl3 and 5.40 for CH2F2.
It is possible to obtain very accurate results for those two particular compounds as proven by the work of González-Lafont et al.39 and Liu et al.41 for CH2F2 and CH3CCl3, respectively. They achieved a kpredicted/kexp ratio of about 1.1 in both cases. However, it requires the use of variational transition state theory with interpolated corrections (VTST-IC) along with a robust approximation such as the small or large curvature (S/LCT) or microcanonical optimized multidimensional tunneling (μOMT). Our simple protocol is not as accurate but is much less computationally demanding. It is also more accurate than more simple empirical approaches.
The approach we have used to calculate ro-vibrational partition functions is rather crude. We have not included any rotational–vibrational coupling, and we have used the harmonic oscillator approximation throughout (with one exception). As the reactants and TSs for the larger substrates typically have several low-frequency modes, this approach is bound to lead to errors. While more accurate approaches for soft torsional modes are available,43 we did not wish to use them here, as it would have very considerably complicated our chosen protocol.
The relative success of our approach is based on the fact that many of the soft torsional modes are conserved from reactants to TS, hence any error on partition functions cancels out. The main exception is a very soft torsional motion around the C–H⋯O axis of the TS. This is a transitional mode, corresponding to molecular rotation in the reactants, so error cancellation cannot be expected. Depending on the substrate, the potential energy for torsion of this type can be quite rigid, e.g. if there are hydrogen bonds between the HO˙ fragment and the substrate, or it can be very loose. In the latter case, this type of mode can have extremely low harmonic frequencies, e.g. 16 cm−1 for the CH2F2 saddle point. Such a value leads to a Qvib at 298 K for this mode of 13.45, which can significantly affect the calculated rate coefficient.
The partition function for such modes is likely to be very similar for all TSs where they occur, and be similar to that for the corresponding free rotor. By treating this torsion as an orbiting motion of the hydrogen atom, the moment of inertia can be estimated as 1.37 × 10−47 kg m2, similar to what was obtained by Schwartz et al.44 for related systems (1.46 × 10−47 kg m2). This leads to a free rotor partition function of 5.65 at 298 K, similar to that obtained at the same temperature for a harmonic oscillator with a frequency of 40 cm−1. This explains our chosen protocol: whenever the torsional mode for the OH bond at the TS has a harmonic frequency below 40 cm−1, we describe it instead as a free rotor, which can be effected very simply by assigning to this mode a harmonic frequency of 40 cm−1. This occurs for 5 species (entries 1, 4, 12, 31 and 33 in Table 1), and has the effect of lowering kTST by up to a factor of 2.3. The largest deviation is obtained for CH2F2, with the value for other compounds being much smaller.
Another possible source of error is the accuracy of the computed energy barriers (ΔE‡). We noted that some of the large RTST values correspond to cases where the computed barrier is very low, e.g. for methyl iso-butyl ketoneRTST = 17.82 and ΔE‡ = −16.23 kJ mol−1, i.e. the TS lies 16.23 kJ mol−1 below the energy of the separated reactants. Such a low energy barrier may be related to the tendency of DFT to artificially stabilize saddle points due to self-interaction. This effect is in principle corrected for by using a functional (KMLYP) where self-interaction effects are smaller, and by including an additive correction so as to reproduce more accurately calculated barrier heights for some model compounds. It is nevertheless useful to review the accuracy of the corrected KMLYP approach for some additional compounds, since some of the species considered here are considerably different in reactivity to those considered in our earlier study.26
Accordingly, the energy of the lowest-lying TS for the species giving the lowest barrier, i.e.methyl-isobutyl-ketone, was re-calculated using the accurate G3(MP2) method.32 Including the correction for zero-point energy, this yields a value of −9.54 kJ mol−1—fairly close to the corrected KMLYP value of −16.23 kJ mol−1. While the difference between the two methods is relatively small considering the error bars of the KMLYP and G3(MP2) methods, it is worth noting that it would be large enough to cause a change in kTST by a factor of 15 at 300 K. This large effect arising from a small change in energy barrier highlights the challenge in obtaining accurate rate constants for many different species using ab initio calculations of barrier heights. Also, the barrier heights calculated here for the species CH4–nFn, where n = 0 to 3, were compared to those obtained in a previous G2(CC,MP2) study.45 The results are very similar to one another, with the corrected KMLYP barriers being, respectively, 22.97, 14.42, 14.53 and 28.55 kJ mol−1, vs. 22.18, 17.57, 16.74 and 24.27 kJ mol−1 at the accurate G2 level of theory. Together, this suggests that the corrected KMLYP approach used here to calculate barrier heights yields accurate results for the whole range of species considered.
Fig. 2 represents graphically the correlation we obtained between our results and the experimental values on a logarithmic scale: we plotted log(kcalc) and log(kTST) against log(kexp) and also the residuals (log(kcalc/kexp) and log(kTST/kexp)) against log(kexp). The latter plots show that kTST becomes less accurate for large kexp, but that the accuracy of kcalc remains high even for large values of kexp. The dotted lines represent the boundaries of the region within which the calculated rate constant does not deviate from experiment by more than a factor of three.
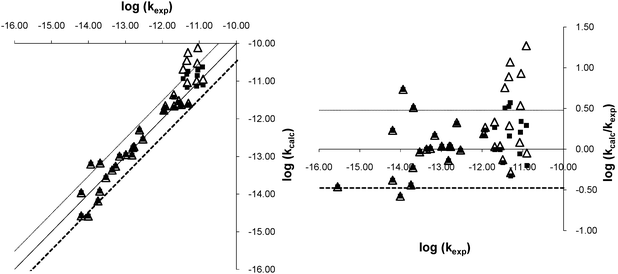 | ||
| Fig. 2 Graphical representation of the agreement between kcalc and kexp on a logarithmic scale. On the left, the plot corresponds to log(kcalc) vs.log(kexp) and on the right, to log(kcalc/kexp) vs.log(kexp). The plain squares correspond to kcalc calculated using the expression for kapp, the triangles to kcalc calculated as kTST. | ||
We note that the prediction of rate coefficient for fluorinated compounds such as CH3F and CF3H appears more challenging than for other halogens. CH3X or CX3H with X = Cl or Br gave good agreement with experimental values as shown by the value of RTST of 1.00 and 0.93 for X = Cl and 1.08 and 0.73 for X = Br. However, when X = F for CH3X and CX3H, larger discrepancies emerge with RTST and Rcalc of 3.24 and 0.35, respectively. Note that concerning CH3F, we obtained a better agreement than the SAR of Atkinson and co-workers did (kSAR/kexp = 5.19).
Several poly-fluorinated compounds such as CF3CH2CH3, CF3CHFCH2F and CF3CHFCHF2 were tested and gave encouraging results, Rcalc = 1.02, 0.36 and 0.42 respectively. For this class of compounds, the SAR of Atkinson and co-workers is known not to perform well.10
The mean unsigned error (MUE) of the logarithm of RTST and Rcalc provides a statistical measure of the overall quality of the transition state theory approach. The MUE is 0.34 for log RTST and 0.25 for log Rcalc, i.e. the rate constants agree with experiment by much better than one order of magnitude on average in both cases. Including the effect of the association step is important, as the MUE on log Rcalc is notably lower.
While the methodology described here is not of the highest accuracy, and is therefore not expected to describe the temperature-dependence of rate constants perfectly, we nevertheless explored this aspect for one compound, methane. Calculation of the rate constant at a range of temperatures close to room temperature, and fitting to an Arrhenius expression led to a calculated activation energy of 20.13 kJ mol−1 and a pre-exponential factor of 3.67 × 10−11 cm3 molecule−1 s−1 at 298 K. This is in fair agreement with the experimental data available, 14.63 (± 0.88) kJ mol−1 and 2.31 × 10−12 cm3 molecule−1 s−1. As can be seen, the calculated activation energy is too large, and also the pre-exponential factor is too large, and these errors cancel out to yield a rate constant that is close to correct.
Conclusions
In this work we present a simple protocol for computing rate coefficients for the reaction between VOCs and the hydroxyl radical. The protocol uses DFT to compute the structure and energy of reactants and transition states, and simple transition state theory to compute rates. We also defined simple yet physically sound corrections for most deficiencies of the methods used, i.e. the presence of pre-reactive complexes in some cases, tunnelling, multi-conformer treatment. This approach is intermediate in complexity between simple empirical or semi-empirical approaches, and high level variational transition state theory approaches using expensive correlated ab initio potential energy surfaces. By being much more computationally affordable than the latter, our approach can readily be applied in a semi-automated way to large number of compounds—in the present study, 34 species were considered. This approach yields good semi-quantitative predictions of rate coefficients, within a factor of three of the experimental value for 29 of the molecules, i.e. more than 80%. The largest deviation from experiment is only by a factor of 5.40. This level of agreement is comparable to that obtained using the SAR of Atkinson and co-workers, and, unlike the latter, performs fairly well for a broad range of compounds, including highly fluorinated species such as CF3CHFCH2F, CF3CHFCHF2 and CHF2CHFCHF2. However, as the method relies to a significant extent on error cancellation, the procedure cannot be expected to yield very high accuracy, especially if one wishes to compute the temperature-dependence of rate constants.References
- R. E. Hester and R. M. Harrison, Volatile Organic Compounds in the Atmosphere, Issues in Environmental Science and Technology, Royal Society of Chemistry, vol. 4, 1995 Search PubMed.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, John Wiley, New York, 1998 Search PubMed.
- V. Ramanathan, J. Geophys. Res., 1987, 92, 4075–4095 CrossRef.
- V. Ramanathan, R. D. Cess, E. F. Harrison, P. Minnis, B. R. Barkstrom, E. Ahmad and D. Hartmann, Science, 1989, 243, 57–63 CAS.
- T. N. Truong and D. G. Truhlar, J. Chem. Phys., 1990, 93, 1761–1769 CrossRef CAS; C. Gonzalez, J. J. W. McDouall and H. B. Schlegel, J. Phys. Chem., 1990, 94, 7467–7471 CrossRef.
- I. W. M. Smith and A. R. Ravishankara, J. Phys. Chem. A, 2002, 106, 4798–4807 CrossRef CAS; D. C. McCabe, S. S. Brown, M. K. Gilles, R. K. Talukdar, I. W. M. Smith and A. R. Ravishankara, J. Phys. Chem. A, 2003, 107, 7762–7769 CrossRef; R. K. Talukdar, T. Gierczak, D. C. McCabe and A. R. Ravishankara, J. Phys. Chem. A, 2003, 107, 5021–5032 CrossRef.
- I. W. M. Smith, J. Chem. Soc., Faraday Trans., 1997, 93, 3741–3750 RSC.
- R. Atkinson, Chem. Rev., 1986, 86, 69–201 CrossRef CAS; R. Atkinson, Int. J. Chem. Kinet., 1987, 19, 799–828 CrossRef; R. Atkinson, Environ. Toxicol. Chem., 1988, 7, 435–442 CrossRef.
- R. Atkinson and E. S. C. Kwok, Atmos. Environ., 1995, 29, 1685–1695 CrossRef.
- A. Sabljic and W. Peijnenburg, Pure Appl. Chem., 2001, 73, 1331–1348 CrossRef CAS.
- See e.g. L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2002, 4, 467–472 RSC.
- J. S. Gaffney and S. Z. Levine, Int. J. Chem. Kinet., 1979, 11, 1197–1209 CrossRef CAS.
- D. L. Cooper, T. P. Cunningham, N. L. Allan and A. McCulloch, Atmos. Environ., 1992, 26A, 1331–1334 CrossRef CAS; D. L. Cooper, T. P. Cunningham, N. L. Allan and A. McCulloch, Atmos. Environ., 1993, 27A, 117–119 CrossRef.
- J. Hodson, Chemosphere, 1988, 17, 2339–2348 CrossRef CAS.
- F. Caralp, W. Forst, E. Henon, A. Bergeat and F. Bohr, Phys. Chem. Chem. Phys., 2006, 8, 1072–1078 RSC.
- L. Masgrau, A. Gonzalez-Lafont and J. M. Lluch, J. Chem. Phys., 2001, 114, 2154–2165 CrossRef CAS; L. Masgrau, A. Gonzalez-Lafont and J. M. Lluch, J. Chem. Phys., 2001, 115, 4515–4526 CrossRef.
- A. Galano, J. R. Alvarez-Idaboy, M. E. Ruiz-Santoyo and A. Vivier-Bunge, J. Phys. Chem. A, 2005, 109, 169–180 CrossRef CAS.
- B. E. Ellingson and D. G. Truhlar, J. Am. Chem. Soc., 2007, 129, 12765–12771 CrossRef CAS.
- R. T. Skodje, D. G. Truhlar and B. C. Garrett, J. Phys. Chem., 1981, 85, 3019–3023 CrossRef CAS.
- B. C. Garrett and D. G. Truhlar, J. Phys. Chem., 1979, 83, 2921–2926 CrossRef CAS.
- K. J. Laidler, Chemical Kinetics, Harper and Row, New York, 3rd edn 1987 Search PubMed.
- L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC.
- Y. P. Liu, D. H. Lu, A. Gonzalez-Lafont, D. G. Truhlar and B. C. Garrett, J. Am. Chem. Soc., 1993, 115, 7806–7817 CrossRef CAS.
- E. Wigner, Z. Phys. Chem., Abt. B, 1932, 19, 203–216 Search PubMed.
- P. J. Lewis, K. A. Bennett and J. N. Harvey, Phys. Chem. Chem. Phys., 2005, 7, 1643–1649 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- C. B. Musgrave and J. K. Kang, J. Chem. Phys., 2001, 115, 11040–11051 CrossRef.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764–7776 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov and J. A. Pople, J. Chem. Phys., 1999, 110, 4703–4709 CrossRef CAS.
- H. J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO, version 2006.1, a package of ab initio programs, Cardiff, UK, 2006 Search PubMed.
- A. Streitwieser, Jr. and C. H. Heathcock, Introduction to organic chemistry, Macmillan, New York, 2nd edn 1981 Search PubMed.
- J. J. Orlando, G. S. Tyndall, M. Bilde, C. Ferronato, T. J. Wallington, L. Vereecken and J. Peeters, J. Phys. Chem. A, 1998, 102, 8116–8123 CrossRef CAS.
- L. Vereecken and J. Peeters, J. Phys. Chem. A, 1999, 103, 1768–1775 CrossRef CAS.
- L. Vereecken and J. Peeters, J. Chem. Phys., 2003, 110, 5159–5170 CrossRef.
- F. Louis, C. A. Gonzalez, R. E. Huie and M. J. Kurylo, J. Phys. Chem. A, 2000, 104, 8773–8778 CrossRef CAS.
- A. Gonzalez-Lafont, J. M. Lluch and J. Espinosa-Garcia, J. Phys. Chem. A, 2001, 105, 10553–10561 CrossRef CAS.
- (a) NIST Chemical Kinetics Database: http://kinetics.nist.gov/kinetics; (b) R. Atkinson, Atmos. Chem. Phys., 2003, 3, 2233–2307 CrossRef CAS; (c) W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb and M. J. Molina, JPL Publ., 1997, 97–4, 1–266 Search PubMed; (d) R. Chiorboli, R. Piazza, M. L. Tosato and V. Carassiti, Coord. Chem. Rev., 1993, 125, 241–250 CrossRef; (e) Z. Jiang, P. H. Taylor and B. Dellinger, J. Phys. Chem., 1992, 96, 8964–8966 CrossRef CAS; (f) B. Rajakumar, R. W. Portmann and J. B. Burkholder, J. Phys. Chem., 2006, 110, 6724–6231 CrossRef CAS; (g) S. Teton, A. Mellouki, G. LeBras and H. Sidebottom, Int. J. Chem. Kinet., 1996, 28, 291–297 CrossRef CAS; (h) E. Porter, J. Wenger, J. Treacy, S. Teton, A. Mellouki, G. LeBras and H. Sidebottom, J. Phys. Chem. A, 1997, 101, 5770–5775 CrossRef CAS; (i) S. Le Calve, D. Hitier, G. Le Bras and A. Mellouki, J. Phys. Chem. A, 1998, 102, 4579–4584 CrossRef CAS; (j) E. Jimenez, B. Lanza, A. Garzon, B. Ballesteros and J. Albaladejo, J. Phys. Chem. A, 2005, 109, 10903–10909 CrossRef CAS.
- J. Y. Liu, Z. S. Li, Z. W. Dai, X. R. Huang and C. C. Sun, J. Phys. Chem. A, 2003, 107, 6231–6235 CrossRef CAS.
- Y. P. Liu, G. C. Lynch, T. N. Truong, D. H. Lu and D. G. Truhlar, J. Am. Chem. Soc., 1993, 105, 2408–2415 CrossRef , and reference therein.
- K. S. Pitzer and W. D. Gwinn, J. Chem. Phys., 1942, 10, 428–440 CrossRef CAS; D. G. Truhlar, J. Comput. Chem., 1991, 12, 266–270 CrossRef; P. Y. Ayala and H. B. Schlegel, J. Chem. Phys., 1998, 108, 2314–2325 CrossRef.
- M. Schwartz, P. Marshall, R. J. Berry, C. J. Ehlers and G. A. Petersson, J. Phys. Chem., 1998, 102, 10074–10081 CrossRef CAS.
- J. Korchowiec, S. Kawahara, K. Matsumura, T. Uchimaru and M. Sugie, J. Phys. Chem. A, 1999, 103, 3548–3553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed data for the case of 2-methyl-2-butanol. See DOI: 10.1039/c1cp21367a |
| This journal is © the Owner Societies 2012 |