Interaction between anions and substituted molecular bowls†
Patxi
García-Novo
a,
Alba
Campo-Cacharrón
a,
Enrique M.
Cabaleiro-Lago
*a and
Jesús
Rodríguez-Otero
b
aDepartamento de Química Física, Facultade de Ciencias, Universidade de Santiago de Compostela, Campus de Lugo. Avda. Alfonso X El Sabio s/n, 27002 Lugo, Galicia, Spain. E-mail: caba.lago@usc.es
bDepartamento de Química Física, Facultade de Química, Universidade de Santiago de Compostela, Avda. das Ciencias, s/n 15782 Santiago de Compostela, Galicia, Spain
First published on 8th November 2011
Abstract
Complexes formed by anions and substituted molecular bowls were studied by means of computational methods. The bowls consisted of corannulene molecules substituted with five or ten F, Cl, or CN groups, whereas Cl−, Br− and BF4− were the anions considered. Substitution with F, Cl and CN hardly affects the geometry of the bowl, but produces an inversion of the molecular electrostatic potential of the bowls, which becomes positive over the two faces of the bowl, therefore interacting favorably with anions. In all cases considered, the most stable complex presents the anion interacting with the concave side of the bowl. The strength of the interaction roughly follows the values of molecular electrostatic potential, being more stable as more positive is the potential. The preference of anions to interact with the concave side of the bowls has its origin in stronger electrostatic and dispersion interactions. Though the solvent produces an important decrease in the stability of the complexes, the results suggest the possibility of employing these substituted buckybowls as anion receptors with a preferential concave complexation, especially for large anions.
1. Introduction
Noncovalent intermolecular interactions are of great importance in modern chemical research, particularly in the area of molecular recognition, supramolecular chemistry, materials science and biochemistry.1–5Klärner et al. showed that for certain molecular tweezers, the electrostatic potential is significantly more negative inside the tweezer than outside, so it could be possible to employ such systems as appropriate receptors for electron-deficient molecules.6 This behavior was not only observed in this kind of molecular tweezers. During the last few years, an interest has aroused with respect to the characteristics of the interactions involving aromatic systems presenting curved surfaces, especially those related to fullerenes, which lead to different properties depending on the face considered.7–11 These molecular bowls are aromatic systems formed by joining six- and five-carbon rings in a similar way as in fullerenes, the five-carbon rings introducing curvature on the delocalized system.7,9 These so-called buckybowls exhibit differences in the electrostatic potential depending on whether the concave or convex face is considered.6,12–14 So, for the simplest of these bowls, corannulene, C20H10, the electrostatic potential is different depending on whether the concave or the convex side of the bowl is considered12–14 and, in fact, cations usually bind corannulene with the convex face.9–13,15–21
Anion receptors are of great interest in different chemical and biological applications.22–24 Recently, the possibility of stabilizing interactions between anions and electron-deficient aromatic systems in the so-called anion⋯π interaction has also aroused interest.22,25,26 The extensive work of Deyá et al. has already shown that the interaction is mainly electrostatic in nature, so one must have an electron deficient aromatic system with positive quadrupole moment, such as hexafluorobenzene or triazine.22,25,27–31 The attractive interaction between this quadrupole and the negative charge of the anion is mainly responsible for the stabilizing interaction, with strengths similar to those observed for cation⋯π interactions.
Most studies in this field have been carried out employing benzene as a model for the aromatic system, so these works have mostly considered hexafluorobenzene, trifluorobenzene or triazine complexes with halogen anions.22,23,25,32 However, there is a lack of studies on the interaction with more complex anions or with more extended aromatic systems. Recently, Hermida-Ramón et al. have performed studies on the interaction of anions with substituted molecular tweezers, showing a significant interaction and suggesting the possibility of complexation of the anion by the tweezers.33–36 It can be expected that for more extended aromatic systems, the contribution of inductive forces will be larger than in benzene derivatives. Also, as indicated by Kim et al.,32 the anion⋯π interaction presents a more dispersive character than the cation⋯π contact, so in complexes formed by large aromatic systems and more complex anions the dispersion contribution could be significant.
In the present work, a computational study of complexes formed by several anions and substituted molecular bowls is presented. The systems studied are shown in Fig. 1. The buckybowls are constructed from corannulene by substituting several hydrogen atoms by the electron-withdrawing chloride, fluoride or cyano groups, pretending to produce an inversion of the molecular electrostatic potential (MEP) thus allowing a stabilising interaction with anions. To the best of our knowledge only the chlorinated derivatives have been already synthesized to date.7 As anions, chloride and bromide were employed as representative of the simplest halogen anions. Fluoride was not considered since it binds to the molecular bowl in an almost covalent way as shown in other anion⋯π interactions.23 Finally, a more structured anion as BF4− was employed to estimate the effect of including larger anions on the dispersive component of the interaction.
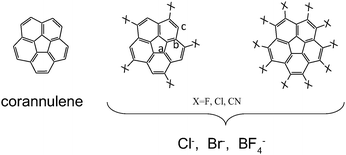 | ||
| Fig. 1 Molecular bowls and anions employed in the present study. | ||
By studying these systems information can be obtained regarding the characteristics of the anion interaction with an extended curved π system. By comparing the different bowls it will be possible to assess the effect produced by changing the molecular electrostatic potential of the bowl on the interaction strength. Also, the minimum energy structures of the complexes will be obtained, giving information about the preference of concave/convex complexation and about the role of dispersion interactions in this kind of systems.
2. Computational details
Clusters formed by substituted corannulenes and the anions indicated in Fig. 1 were computationally studied by using density functional theory and MP2 methods. Starting structures were constructed by placing each of the anions over the different hexagonal or pentagonal faces of the substituted corannulenes, both in the concave and the convex sides. These initial structures were fully optimized employing the BLYP functional corrected by an empirical dispersion term as designed by Grimme (BLYP-D)37,38 together with the aug-cc-pVDZ basis set. Symmetry was only employed for complexes where chloride and bromide anions are located on the symmetry axis of the bowl. Numerical frequency calculations were performed in selected complexes to characterize them as minima in the potential energy surface.The reason for employing a dispersion corrected functional comes from the possibility of dispersion being more important in these systems than in cation⋯π interactions.32 It can be expected that the combination of a large, curved system, together with the presence of anions, will lead to a larger contribution of dispersion which should be almost completely lost with common functionals. BLYP-D was chosen since in previous work it has been shown to give improved results for the interaction in systems containing aromatic units.39,40 Computational effort has been saved by applying the resolution of the identity (RI) approach, the def2-TZVPP basis set being used as auxiliary basis set.41
After locating the stationary points of the potential energy surface of each cluster, the interaction energies were calculated by means of the counterpoise method to avoid basis set superposition error.42–44 Besides BLYP-D, the MP2 method together with the aug-cc-pVDZ basis set was also employed for obtaining interaction energies. MP2 single point calculations were performed at the optimized BLYP-D geometry, applying the resolution of the identity approach with the corresponding auxiliary basis set as implemented in Turbomole.41
It has already been noted that MP2 tends to produce overestimated interaction energies when applied to systems containing aromatic units.4,45–47 Different empirical scalings of the MP2 energies have been proposed to overcome this problem.48–50 In the present work, the parameterization of Hill and Platts (SCSN-MP2) was employed,50 since it was developed employing the same basis set as that used in the present work.
Symmetry Adapted Perturbation Theory calculations (SAPT) were carried out for selected complexes in order to obtain more information about the characteristics of the interaction.51,52 Thus, the interaction energy was decomposed into several contributions such as repulsion, dispersion, induction and electrostatic. In order to include intramonomer correlation effects SAPT(DFT) was used by employing the BLYP functional together with the aug-cc-pVDZ basis set and density fitting to save computation time. Grüning asymptotic correction was applied,53 ionization potentials being obtained at the BLYP/aug-cc-pVDZ level.
Finally, an estimation of the complexation energies in solvents with different dielectric constants has been carried out by employing the COSMO model as implemented in Turbomole.41,54
All calculations were performed with the Orca55 and Turbomole41 programs. SAPT(DFT) calculations were done with Molpro2009.1.56
3. Results
3.1. Substituted molecular bowls
Table 1 lists selected geometrical parameters obtained for the substituted corannulenes at the BLYP-D/aug-cc-pVDZ level of calculation. Among these parameters there are some characteristic distances such as the depth of the bowl measured from the carbon atoms in the rim of the bowl (carbons labelled as c in Fig. 1) to the plane of the pentagonal ring, the distance from these carbon atoms to the center of the pentagonal ring (Rpc) and the diameter of the bowl. Values for corannulene were also included as reference. Comparing with other results in the literature the geometry of corannulene is well reproduced.7,16,18,57 As observed, corannulene is a molecular bowl which presents a depth of 0.94 Å and a diameter of 6.55 Å. Also, carbon atoms present some degree of pyramidalization as indicated by the π-orbital axis vector (POAV) values (the vector which makes equal angles (θσπ) to the three σ-bonds at a conjugated carbon atom, the pyramidalization angle being obtained as θP = (θσπ − 90)).58,59 In any case, it can be observed that deviations from the planarity decrease as the rim of the bowl is approached. The carbons on the pentagonal ring are the most deviated from planarity presenting POAV values of 8.6°.| Coran. | Cl5 | F5 | CN5 | Cl10 | F10 | CN10 | |
|---|---|---|---|---|---|---|---|
| a R pc is the distance from carbon c to the center of the pentagonal ring. | |||||||
| Depth | 0.939 | 0.909 | 0.933 | 0.924 | 0.553 | 0.886 | 0.731 |
| R pc a | 3.409 | 3.404 | 3.399 | 3.413 | 3.412 | 3.397 | 3.420 |
| Diameter | 6.554 | 6.561 | 6.538 | 6.570 | 6.733 | 6.560 | 6.682 |
| POAVa | 8.6 | 8.4 | 8.5 | 8.5 | 5.4 | 8.1 | 6.9 |
| POAVb | 4.2 | 4.0 | 4.4 | 4.0 | 1.8 | 4.3 | 2.7 |
| POAVc | 1.8 | 1.6 | 1.8 | 1.7 | 0.0 | 1.4 | 0.6 |
It can be observed from the data in Table 1 that substitution in five alternating positions hardly affects the geometric characteristics of the bowl. A slight flattening of the bowl is observed as indicated by the values obtained for the bowl depth, which are slightly smaller than for corannulene. The largest deviation corresponds to the chlorinated compound, since this is the most bulky substituent and therefore there is more steric hindrance leading to an opening of the bowl. This effect is also observed for Coran-CN5 whereas for Coran-F5 almost no change is observed. These changes correlate with an increment in the bowl's diameter, though for Coran-F5 there is in fact a small reduction in diameter. POAV values show no significant change.
When the bowls are substituted in the ten available positions of corannulene, more dramatic changes are observed because the substituents are now closer. As expected, the most striking effect corresponds to Coran-Cl10, where the bowl depth is reduced to only 0.55 Å. Therefore, for this species, the bowl is flatter and there will be smaller differences between the concave and convex faces of the bowl. For Coran-CN10 the bowl also flattens, with a depth of 0.73 Å, whereas for Coran-F10 almost no changes are observed though the bowl depth decreases to 0.89 Å. So, any of the decasubstituted bowls is more planar than all the pentasubstituted ones, though the effect is only remarkable with chloride and cyano substituents. Also, POAV values are affected by substitution, with reductions of about 2–3° for a and b carbon atoms.
Therefore, the substituted bowls cannot be expected to introduce a large geometrical effect as compared with the corannulene molecule, with the exception of Coran-Cl10 and maybe Coran-CN10. Though substitution does not significantly alter the geometry of the carbon skeleton, the electronic distribution can be expected to be largely affected by the presence of electron-withdrawing substituents, thus favoring interaction with anions.
Fig. 2 shows the Molecular Electrostatic Potential (MEP) of the substituted bowls together with that for corannulene on a plane which cuts the bowl following the C5 symmetry axis to visualize differences between the concave and convex sides of the bowls. In agreement with results already reported, the corannulene molecule presents negative MEP by both the convex and concave sides, the only positive regions corresponding to the CH groups in the rim of the bowl. However, though both sides of the molecule have negative MEP, the value is more negative in the convex face than in the concave one, so cations interact preferentially with corannulene by the convex side of the bowl.9,12,18 This effect can be easily observed in Fig. 3, where the MEP is represented along the C5 symmetry axis. Corannulene presents negative values, but more negative in the convex side.
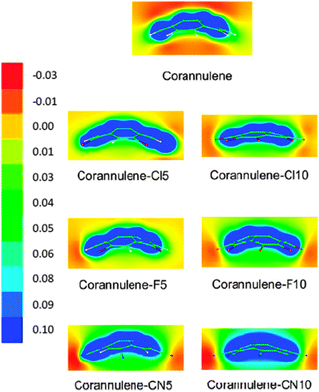 | ||
| Fig. 2 Molecular electrostatic potential of the molecular bowls employed in this study obtained at the BLYP-D/aug-cc-pVDZ level of calculation. The scale is shown in atomic units. | ||
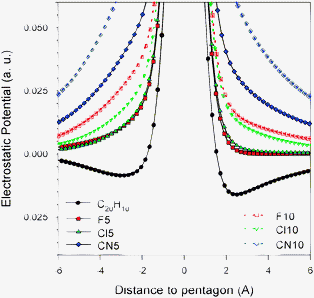 | ||
| Fig. 3 Molecular electrostatic potential of the molecular bowls studied in this work as obtained along the C5 symmetry axis at the BLYP-D/aug-cc-pVDZ level of calculation. Positive distances correspond to the convex face. | ||
When substituents are incorporated the MEP undergoes significant changes. Already for Coran-Cl5 the MEP is much less negative than for corannulene, starting to show positive or almost zero MEP regions. In fact, as observed in Fig. 3, already for Coran-Cl5, the MEP along the symmetry axis always presents positive values, though in the convex side the values are almost zero. Along the axis the MEP is more positive in the concave side of the bowl. An almost identical pattern is obtained for Coran-F5, but the change is larger for Coran-CN5. In this latter case, the MEP is clearly positive in both sides of the bowl, and as observed in Fig. 3 much more positive than either corannulene or any of the other pentasubstituted bowls. Therefore, on a purely electrostatic basis, the Coran-CN5 bowl will interact very favorably with anions, whereas for Coran-F5 and Coran-Cl5, the interaction will be favorable, but not very strong. In decasubstituted bowls the patterns are similar, though in this case the MEPs are clearly positive, and more positive than any of the pentasubstituted bowls with the exception of Coran-CN5. Again, Coran-Cl10 and Coran-F10 behave in a similar manner, though in the concave side the MEP is more positive for the fluorinated compound. For Coran-CN10, the MEP is clearly positive in either side of the bowl, and it will be the most favorable bowl for interacting with anions. This is in agreement with previous observations of Hermida-Ramón et al. in tweezer complexes.34 Therefore, considering only electrostatics the complexation energies with anions should behave as CN10 ≫ CN5 > F10 ≈ Cl10 > F5 ≈ Cl5 ⋙ corannulene.
3.2. Complexes between anions and molecular bowls
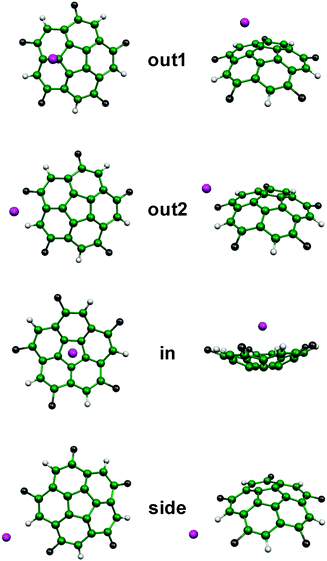 | ||
| Fig. 4 Structural arrangements found after optimization of the complexes formed by substituted bowls and anions. | ||
As shown in Fig. 4, in one of the structures of the convex side (out1) the anion is roughly over one of the carbon atoms of the pentagonal ring. In the other minimum of the convex face (out2) the anion is in a more outer position, interacting with one carbon atom in the rim of the bowl, but also with the hydrogen atom attached to it. As observed in Fig. 4 the difference between structures out2 and side comes from the anion interacting only with the C–H hydrogen in the side structure, establishing a hydrogen bond. Finally, the in structure presents the anion over the symmetry axis and interacting with the concave side of the bowl. The complexes formed with the BF4− anion are not as easily described because the anion is not monoatomic, but the position of the boron atom roughly corresponds to those shown in Fig. 4, with the fluorine atoms contacting with different parts of the bowl. It is also worth noting that for Coran-CN5 no side structures were found, the optimizations going to the out2 minima.
Table 2 lists several values for the distances between the anion and the nearest carbon atom for the complexes shown in Fig. 4. Complexes formed by unsubstituted corannulene and the anions are included as reference, though in this case the out2 structure has not been found. Overall, corannulene substitution results in shorter intermolecular distances to the anions. It can be observed that a chloride anion is located at about 2.8–2.9 Å in the complexes formed by the outer side with Coran-Cl5. The behavior of the fluorinated bowl is similar, though in the out2 structure the chloride anion is located nearer the bowl center, and the nearest carbon atom is of b type. In the complexes formed with Coran-CN5 distances are shorter in the convex face whereas the anion is located at similar distances in the concave side of the bowl. The complexes with bromide anion exhibit a similar behavior, though the distances are larger due to the larger size of the anion. The behavior is more complex for complexes with BF4−, but the location of the anion is similar to that observed in other complexes, though in the out2 structure the anion is located nearer the bowl center, avoiding positions on the edge of the bowl.
| Coran | Cl5 | F5 | CN5 | ||
|---|---|---|---|---|---|
| Cl | side | 3.564 | 3.366 | 3.332 | |
| out1 | 3.034 | 2.806 | 2.872 | 2.555 | |
| out2 | 2.861 | ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif)  ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) |
2.416 | ||
| in | 3.539 | 3.403 | 3.425 | 3.501 | |
| Br | side | 3.751 | 3.626 | 3.568 | |
| out1 | 3.205 | 3.028 | 3.091 | 2.785 | |
| out2 | 3.062 | ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) |
2.704 | ||
| in | 3.678 | 3.539 | 3.560 | 3.501 | |
| BF4 | side | 3.203 | 3.209 | 3.141 | |
| out1 | 3.007 | 2.916 | 2.972 | 2.986 | |
| out2 | ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) |
![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) |
|
||
| in | 2.991 | 2.888 | 2.923 | 2.841 |
Complexation energies were obtained for the clusters studied with the BLYP-D functional and employing the MP2 and the scaled SCSN-MP2 methods (the scaling parameters were developed employing the same basis set as in the present work),49,50 all together with the aug-cc-pVDZ basis set. The results obtained are shown in Table 3, together with values for the complexes formed with corannulene. As expected, complexes formed with corannulene by the convex side are by large the least stable among the complexes studied in this work, in concordance with the MEP data shown in Fig. 2 and 3. In complexes are more stable, but far from the values obtained for any of the complexes formed with substituted corannulenes. Only in the case of the side complexes, where the anions interact with hydrogen atoms on the rim of the bowl, the complexation energies are as large as those found in pentachlorinated of pentafluorinated corannulenes.
| ΔEBLYP-D | ΔEMP2 | ΔESCSN-MP2 | ||
|---|---|---|---|---|
| Coran⋯Cl | side | −56.07 | −60.17 | −60.33 |
| out1 | −9.87 | −8.78 | 0.66 | |
| in | −20.32 | −36.97 | −28.09 | |
| Coran⋯Br | side | −50.52 | −53.15 | −52.76 |
| out1 | −8.46 | −6.93 | 2.54 | |
| in | −21.29 | −36.60 | −27.17 | |
| Coran⋯BF4 | side | −45.81 | −44.11 | −45.74 |
| out1 | −11.04 | −4.26 | 2.33 | |
| in | −37.40 | −27.80 | −21.84 | |
| Cl5⋯Cl | side | −57.3 | −57.8 | −56.5 |
| out1 | −63.7 | −56.7 | −47.6 | |
| out2 | −66.0 | −55.9 | −50.6 | |
| in | −71.8 | −91.0 | −84.5 | |
| Cl5⋯Br | side | −51.0 | −50.1 | −48.1 |
| out1 | −59.9 | −51.7 | −42.8 | |
| out2 | −61.8 | −50.4 | −44.5 | |
| in | −70.3 | −88.0 | −80.5 | |
| Cl5⋯BF4 | side | −49.9 | −45.8 | −45.6 |
| out1 | −55.7 | −46.2 | −42.3 | |
| out2 | −55.0 | −46.8 | −43.2 | |
| in | −78.0 | −67.7 | −63.5 | |
| F5⋯Cl | side | −52.5 | −51.9 | −50.2 |
| out1 | −52.3 | −49.3 | −37.4 | |
| out2 | −58.0 | −51.8 | −44.4 | |
| in | −66.7 | −85.9 | −77.0 | |
| F5⋯Br | side | −45.7 | −44.2 | −42.1 |
| out1 | −49.1 | −44.9 | −33.2 | |
| out2 | −53.9 | −45.6 | −38.2 | |
| in | −65.6 | −82.7 | −73.1 | |
| F5⋯BF4 | side | −41.8 | −37.9 | −38.0 |
| out1 | −48.5 | −41.5 | −34.7 | |
| out2 | −49.1 | −42.4 | −36.8 | |
| in | −74.7 | −64.8 | −59.0 | |
| CN5⋯Cl | out1 | −145.2 | −130.0 | −122.9 |
| out2 | −145.7 | −123.0 | −119.7 | |
| in | −147.1 | −165.0 | −164.2 | |
| CN5⋯Br | out1 | −138.9 | −121.0 | −113.8 |
| out2 | −137.4 | −112.6 | −108.4 | |
| in | −144.5 | −159.5 | −156.7 | |
| CN5⋯BF4 | out1 | −117.3 | −105.3 | −107.0 |
| out2 | −114.0 | −103.0 | −104.5 | |
| in | −144.4 | −131.8 | −133.0 |
Though the actual values differ among the different methods employed, the observed trends are fairly similar in all cases. As expected, the scaled SCSN-MP2 method corrects for a possible overestimation of the MP2 energies leading to less negative complexation energies in most cases (it should be taken into account that the parameterization of the SCSN-MP2 method also tries to include basis enlargement effects).50 The side complexes are the less affected by the empirical scaling whereas larger changes with respect to the original MP2 method are observed for the other structures. On the other hand, the BLYP-D method produces less negative values for the in complexes (with the exception of BF4− complexes) and somewhat more stable complexes for the out1 and out2 structures. In the following the SCSN-MP2 results will be considered though the same conclusions would be reached with the MP2 values. SCSN-MP2 complexation energies of complexes formed with the pentasubstituted corannulenes are shown in Fig. 5.
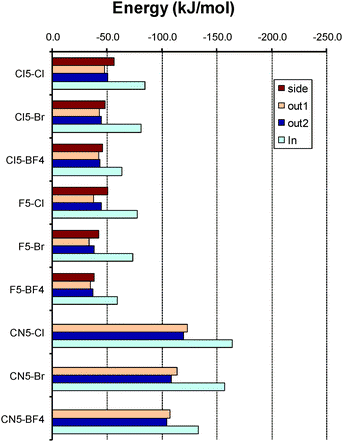 | ||
| Fig. 5 Complexation energy for the complexes with pentasubstituted corannulenes as obtained at the SCSN-MP2/aug-cc-pVDZ level. | ||
Starting with complexes formed with the pentachlorinated corannulene, it can be observed that all anions form quite stable complexes. The two structures considered by the convex face present very similar complexation energies, which are less negative than the side structure at the SCSN-MP2 level (though the contrary happens with BLYP-D). Therefore, these three structures present similar stability, but are less stable than the complex formed by the concave side which is significantly more stable, especially with the SCSN-MP2 method (though less than with the original MP2 method). The complexation energies decrease in the order Cl− > Br− > BF4− following the polarizing power of the anion.
Changing to the pentafluorinated bowl, the results are almost the same as those for the Coran-Cl5 bowl, in correlation with the behavior observed for the MEP. In Coran-CN5 however, the complexation energies dramatically increase leading to complexes stabilized by up to −150 kJ mol−1 (twice more than with other bowls). Taking into account the MEPs in Fig. 2 and 3, this enhancement of the intensity of the interaction should be mainly electrostatic. As observed in Fig. 5 and Table 3, the SCSN-MP2 method predicts much more stable coordination via the concave side of the bowl, with energy differences reaching 20–40 kJ mol−1 with respect to complexes formed by the convex face. Therefore, complexation in substituted bowls is clearly favored on the concave side of the bowl, a behavior quite the opposite to that observed in the complexation of corannulene with cations, which preferentially bind with the convex face.9,12,18
| Cl10 | F10 | CN10 | ||
|---|---|---|---|---|
| Cl | out1 | 2.834 | 2.784 | 2.394 |
| out2 | |
![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) |
|
|
| in | 3.302 | 3.348 | 2.241 | |
| Br | out1 | 3.056 | 3.005 | 2.655 |
| out2 | |
|
|
|
| in | 3.449 | 3.486 | 3.394 | |
| BF4 | out1 | 3.154 | 3.082 | 3.033 |
| out2 | . |
|
|
|
| in | |
|
|
Tables 5 and 6 show the values obtained for the complexation energies of the complexes formed with decasubstituted corannulenes. Comparing the results in Tables 3 and 5 it is clear that decasubstitution leads to more stable clusters as a consequence of more electron-withdrawing groups being included in the molecular bowl (Fig. 6). The stability increases with respect to the pentasubstituted bowls around 20–40 kJ mol−1 for Coran-Cl10 and Coran-F10, whereas for Coran-CN10 the energy changes around 70–90 kJ mol−1. In the case of Coran-Cl10 and Coran-CN10 complexes (with the exception of Coran-CN10⋯BF4−), the smaller energy gains are observed for the in complexes. On the other hand, Coran-F10 complexes show the opposite behavior, the in complexes being the most stabilized by the presence of more electron-withdrawing groups. This behavior could be related to the changes in the curvature of the bowl observed in Coran-Cl10 and Coran-CN10, leading to smaller differences between the concave and convex sides of the bowl.
| ΔEBLYP-D | ΔEMP2 | ΔESCSN-MP2 | ||
|---|---|---|---|---|
| Cl10⋯Cl | out1 | −90.7 | −87.9 | −76.4 |
| out2 | −82.2 | −81.9 | −70.9 | |
| in | −93.2 | −111.2 | −104.4 | |
| Cl10⋯Br | out1 | −86.5 | −81.5 | −70.5 |
| out2 | −77.0 | −74.1 | −63.4 | |
| in | −89.7 | −107.5 | −100.0 | |
| Cl10⋯BF4 | out1 | −83.4 | −77.1 | −69.6 |
| out2 | −79.3 | −72.8 | −67.5 | |
| in | −101.2 | −94.1 | −89.1 | |
| F10⋯Cl | out1 | −89.1 | −90.7 | −73.2 |
| out2 | −89.9 | −88.6 | −74.0 | |
| in | −107.9 | −132.2 | −122.7 | |
| F10⋯Br | out1 | −84.6 | −83.6 | −67.3 |
| out2 | −83.8 | −79.2 | −65.2 | |
| in | −105.3 | −127.0 | −116.9 | |
| F10⋯BF4 | out1 | −82.5 | −78.9 | −70.6 |
| out2 | −80.6 | −76.2 | −69.2 | |
| in | −111.0 | −104.8 | −97.9 | |
| CN10⋯Cl | out1 | −238.1 | −243.2 | −222.1 |
| out2 | −222.1 | −212.1 | −201.9 | |
| in | −229.2 | −247.5 | −249.4 | |
| CN10⋯Br | out1 | −230.7 | −223.6 | −208.3 |
| out2 | −215.3 | −195.7 | −186.3 | |
| in | −223.6 | −239.1 | −239.0 | |
| CN10⋯BF4 | out1 | −190.3 | −184.0 | −186.0 |
| out2 | −183.5 | −177.4 | −179.2 | |
| in | −222.6 | −217.7 | −220.8 |
| CHCl3 | THF | Ethanol | Water | ||
|---|---|---|---|---|---|
| Cl5⋯Br | out1 | 11.3 | 16.9 | 23.6 | 25.3 |
| in | −0.9 | 5.5 | 13.7 | 16.0 | |
| Cl5⋯BF4 | out1 | −11.4 | −7.0 | −1.2 | 0.5 |
| in | −25.1 | −19.4 | −11.4 | −9.0 | |
| F5⋯Br | out1 | 12.6 | 17.2 | 22.5 | 23.9 |
| in | 0.7 | 6.9 | 15.1 | 17.4 | |
| F5⋯BF4 | out1 | −8.4 | −4.5 | 0.7 | 2.2 |
| in | −24.1 | −18.5 | −10.7 | −8.4 | |
| CN5⋯Br | out1 | −11.3 | 3.1 | 23.3 | 29.5 |
| in | −27.8 | −14.5 | 4.5 | 10.4 | |
| CN5⋯BF4 | out1 | −36.0 | −25.8 | −10.6 | −5.8 |
| in | −52.9 | −41.3 | −24.1 | −18.6 | |
| Cl10⋯Br | out1 | 0.7 | 8.6 | 18.8 | 21.7 |
| in | −10.3 | −2.7 | 7.5 | 10.5 | |
| Cl10⋯BF4 | out1 | −24.1 | −17.5 | −8.3 | −5.5 |
| in | −36.7 | −29.4 | −19.2 | −16.1 | |
| F10⋯Br | out1 | 2.5 | 10.7 | 21.6 | 24.8 |
| in | −16.1 | −6.8 | 5.9 | 9.7 | |
| F10⋯BF4 | out1 | −22.5 | −15.7 | −6.0 | −3.0 |
| in | −40.1 | −31.9 | −20.1 | −16.5 | |
| CN10⋯Br | out1 | −55.5 | −32.3 | 2.6 | 14.0 |
| in | −58.7 | −38.0 | −7.0 | 3.1 | |
| CN10⋯BF4 | out1 | −68.3 | −51.0 | −24.3 | −15.5 |
| in | −88.1 | −69.3 | −40.3 | −30.7 |
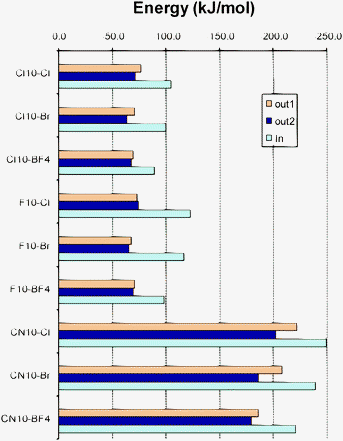 | ||
| Fig. 6 Complexation energy for the complexes with decasubstituted corannulenes as obtained at the SCSN-MP2/aug-cc-pVDZ level. | ||
In any case, the preference for complex formation with the concave side of the bowl is kept, exhibiting values surpassing −100 kJ mol−1 in complexes with Coran-Cl10 or Coran-F10, and reaching more than −220 kJ mol−1 in the case of Coran-CN10. As in the case of pentasubstituted bowls, the interaction loses intensity as the anion becomes less polarizing. Complexes formed with the concave side of Coran-Cl10 are less stable than the corresponding Coran-F10 ones, whereas the opposite was observed in complexes with pentasubstituted bowls.
This is a consequence of Coran-F10 having a significantly more positive MEP than Coran-Cl10 as shown in Fig. 3, though the smaller bowl depth of Coran-Cl10 can also contribute to the weakening of the interaction.
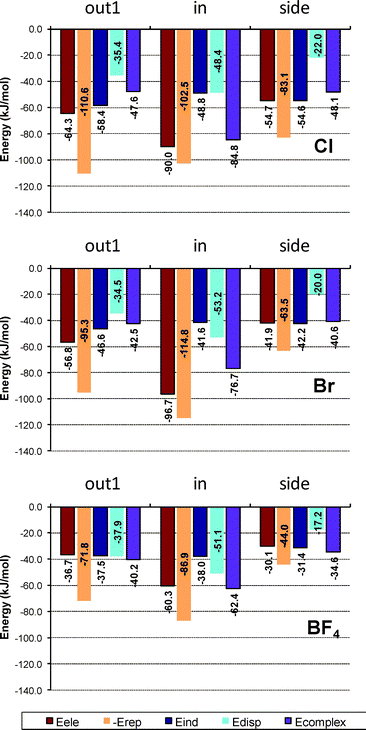 | ||
| Fig. 7 SAPT(DFT) results for complexes formed with Coran-F5. Edisp includes Edisp + Eexch-disp; Eind includes Eind + Eexch-ind + δHF. | ||
It becomes apparent that the stabilization of the complexes with chloride anion comes mainly from the electrostatic term together with the induction contribution, which is of similar magnitude.
The main difference between side and out1 contributions comes from a smaller repulsion term in the side structure, in part compensated by a smaller dispersion contribution than in the out1 structure. The interaction pattern for the in structure is different, presenting the largest electrostatic interaction, whereas induction presents values similar to those observed in other structures. Also, dispersion contribution is significantly larger than for structures formed by the convex side of the bowl. Therefore, the main reason for the in complex to be the most stable is a combination of larger electrostatic and dispersion contributions, which are only partially compensated by a larger repulsion term.
In the case of complexes with bromide, the behavior is similar but, as expected, electrostatic contribution with the convex side of the bowl is smaller, though increases in the concave face. Again, the interaction is controlled by electrostatic plus induction contributions, but dispersion contribution in the in structure is the second most important stabilizing effect. In the case of complexes with boron tetrafluoride, the electrostatic contribution drops significantly, as also does the induction term, as corresponds to a larger, less polarizing anion. However, dispersion contributes significantly in the out1 structure, also becoming the second most stabilizing contribution in the in structure.
The results shown in Fig. 7 allow understanding the effect of the anion on the interaction. In Fig. 8 results are shown for the complexes formed with bromide and all substituted corannulenes, allowing us to determine the effect of bowl substitution on the interaction. As regards pentasubstituted bowls it can be appreciated that Coran-Cl5 and Coran-F5 behave in a very similar way, leading to fairly similar energy contributions, though Coran-Cl5 forms slightly more stable complexes due to greater induction and electrostatic contributions. Coran-CN5 presents a different pattern, with much larger electrostatic contributions, which are mainly responsible for the greater stability of Coran-CN5 complexes. Decasubstituted complexes behave in a very similar way. All complexes present larger electrostatic contributions than their pentasubstituted counterparts, whereas induction only grows significantly in Coran-CN10 complexes.
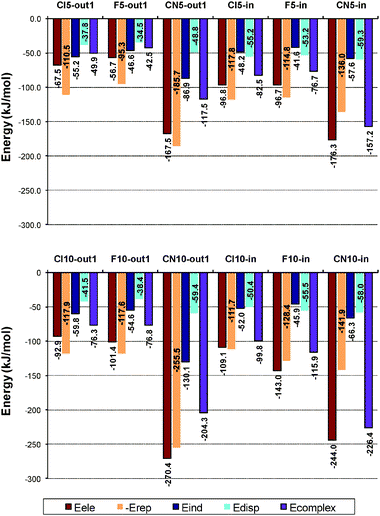 | ||
| Fig. 8 SAPT(DFT) results for complexes formed with bromide. Edisp includes Edisp + Eexch-disp; Eind includes Eind + Eexch-ind + δHF. | ||
Larger differences are observed between Coran-Cl10 and Coran-F10 complexes than in the corresponding pentasubstituted bowls, especially for the in structures as a consequence of the greater planarity of Coran-Cl10. However, though dispersion contribution slightly decreases in Coran-Cl10 complex as a consequence of its smaller curvature, the mainly responsible for the greater stability of the Coran-F10 complex is a significantly larger electrostatic contribution.
In summary, the interaction of anions with substituted molecular bowls is mainly controlled by electrostatics, which roughly determines the intensity of the interaction. Also, larger electrostatic and dispersion interactions favor the formation of complexes with the concave side of the bowl.
Therefore, the results indicate that no stable complex is formed with chloride or bromide anion in water. This behavior is a consequence of the larger desolvation costs of the smaller, more polarizing anions. However, all substituted bowls considered in this work are predicted to form stable complexes with the BF4− anion in water, especially with the concave side of the bowl.
4. Conclusions
The interaction of anions with a series of corannulenes substituted with five and ten electron-withdrawing groups has been studied by means of the BLYP-D and MP2 methods together with the aug-cc-pVDZ basis set. Different structures for the complexes were optimized leading to several geometrical arrangements with the anions located over the concave or the convex faces of the bowls, as well as over the rim of the pentasubstituted corannulenes.Substitution of hydrogen atoms of corannulene by Cl, F or CN groups does not introduce changes on the basic structure of corannulene. Only when ten substituents are included some flattening of the bowl is observed, especially for Coran-Cl10. However, great changes are produced on the MEP of the bowl, which becomes positive, in a similar degree for F and Cl derivatives, but markedly more positive for the CN substituted bowls. Therefore, all substituted bowls present positive MEP regions on the faces of the bowls which allow them to interact favorably with anions via the conjugated system.
Complexes with the anion inside the bowl (concave side) are the most stable, in contrast to cation complexes with corannulene. The complexation energies roughly follow the series of MEPs, the most stable complexes being those formed with corannulenes substituted with CN groups.
A SAPT(DFT) energy decomposition allows recognising the electrostatic contribution as the main stabilizing contribution to complexation. The combination of a larger electrostatic term together with important dispersion contributions results in structures formed with the concave side of the bowl to be the most stable.
Finally, though very stable in the gas phase, complexes are dramatically destabilized by the presence of the solvent. The results obtained show that complexes become much less stable as the dielectric constant increase, to the point that in water only complexes formed with BF4− are stable. In any case, the results point out the possibility of using substituted corannulenes as a means of interacting with anions via the concave side of the bowl, with a preference for large anions where desolvation costs are smaller.
Acknowledgements
The authors thank the financial support from the Ministerio de Ciencia e Innovación (Grant No. CTQ2009-12524,) and Xunta de Galicia (project Incite09209103PR and “Axuda para a Consolidación e Estructuración de unidades de investigación Competitivas do Sistema Universitario de Galicia, 2007/50, cofinanciada polo FEDER 2007-2013”). We are also thankful to the Centro de Supercomputación de Galicia (CESGA) for the use of their computers.References
- P. Hobza and R. Zaradnik, Intermolecular complexes: the role of van der Waals systems in physical chemistry and the biodisciplines, Elsevier, Amsterdam, 1988 Search PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 4120 CrossRef.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CrossRef CAS.
- S. Tsuzuki and T. Uchimaru, Curr. Org. Chem., 2006, 10, 745–762 CrossRef CAS.
- M. L. Waters, Pept. Sci., 2004, 76, 435–445 CrossRef CAS.
- F.-G. Klärner, J. Panitzky, D. Preda and L. T. Scott, J. Mol. Model, 2000, 6, 318–327 CrossRef.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS.
- T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS.
- A. S. Filatov and M. A. Petrukhina, Coord. Chem. Rev., 2010, 254, 2234–2246 CrossRef CAS.
- M. A. Petrukhina and L. T. Scott, Dalton Trans., 2005, 2969–2975 RSC.
- M. A. Petrukhina, Y. Sevryugina, A. Y. Rogachev, E. A. Jackson and L. T. Scott, Organometallics, 2006, 25, 5492–5495 CrossRef CAS.
- J. A. Carrazana-García, J. s. Rodríguez-Otero and E. M. Cabaleiro-Lago, J. Phys. Chem. B, 2011, 115, 2774–2782 CrossRef.
- A. K. Kandalam, B. K. Rao and P. Jena, J. Phys. Chem. A, 2005, 109, 9220–9225 CrossRef CAS.
- L. T. Scott, H. E. Bronstein, D. V. Preda, R. B. M. Ansems, M. S. Bratcher and S. Hagen, Pure Appl. Chem., 1999, 71, 209–220 CrossRef CAS.
- M. V. Frash, A. C. Hopkinson and D. K. Bohme, J. Am. Chem. Soc., 2001, 123, 6687–6695 CrossRef CAS.
- U. D. Priyakumar and G. N. Sastry, Tetrahedron Lett., 2003, 44, 6043–6046 CrossRef CAS.
- U. D. Priyakumar, M. Punnagai, G. P. Krishnamohan and G. N. Sastry, Tetrahedron, 2004, 60, 3037–3043 CrossRef CAS.
- R. C. Dunbar, J. Phys. Chem. A, 2002, 106, 9809–9819 CrossRef CAS.
- T. J. Seiders, K. K. Baldridge, J. M. O'Connor and J. S. Siegel, Chem. Commun., 2004, 950–951 RSC.
- T. J. Seiders, K. K. Baldridge, J. M. O'Connor and J. S. Siegel, J. Am. Chem. Soc., 1997, 119, 4781–4782 CrossRef CAS.
- R. B. M. Ansems and L. T. Scott, J. Phys. Org. Chem., 2004, 17, 819–823 CrossRef CAS.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2007, 129, 48–58 CrossRef CAS.
- P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- A. Frontera, D. Quiñonero and P. M. Deyá, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 440–459 CrossRef CAS.
- H.-J. Schneider and A. Yatsimirski, Principles and Methods in Supramolecular Chemistry, New York, 2000 Search PubMed.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyá, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyá, New J. Chem., 2003, 27, 211–214 RSC.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyá, ChemPhysChem, 2003, 4, 1344–1348 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyá, J. Phys. Chem. A, 2005, 109, 9341–9345 CrossRef CAS.
- J. Rodríguez-Otero, E. M. Cabaleiro-Lago and A. Peña-Gallego, Chem. Phys. Lett., 2008, 452, 49–53 CrossRef.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250–1258 CrossRef CAS.
- J. M. Hermida-Ramón and C. M. Estevez, Chem.–Eur. J., 2007, 13, 4743–4749 CrossRef.
- J. M. Hermida-Ramón, M. Mandado, M. Sanchez-Lozano and C. M. Estevez, Phys. Chem. Chem. Phys., 2010, 12, 164–169 RSC.
- M. Mandado, J. M. Hermida-Ramón and C. M. Estevez, THEOCHEM, 2008, 854, 1–9 CrossRef CAS.
- M. Sanchez-Lozano, N. Otero, J. M. Hermida-Ramón, C. M. Estevez and M. Mandado, J. Phys. Chem. A, 2011, 115, 2016–2025 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- E. M. Cabaleiro-Lago, J. A. Carrazana-Garcia and J. Rodríguez-Otero, J. Chem. Phys., 2009, 130, 234307–234311 CrossRef.
- C. Morgado, M. A. Vincent, I. H. Hillier and X. Shan, Phys. Chem. Chem. Phys., 2007, 9, 448–451 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- G. Chalasinski and M. M. Szczesniak, Chem. Rev., 2000, 100, 4227–4252 CrossRef CAS.
- E. M. Cabaleiro-Lago, J. Rodríguez-Otero and A. Pena-Gallego, Theor. Chem. Acc., 2011, 128, 531–539 CrossRef CAS.
- E. M. Cabaleiro-Lago, J. Rodríguez-Otero and A. Peña-Gallego, J. Phys. Chem. A, 2008, 112, 6344–6350 CrossRef CAS.
- E. M. Cabaleiro-Lago, J. Rodríguez-Otero and A. Peña-Gallego, J. Chem. Phys., 2008, 129, 084305–084308 CrossRef.
- C. D. Sherill, Rev. Comput. Chem., 2009, 26, 1–38 CrossRef.
- R. Distasio Jr. and M. Head-Gordon, Mol. Phys., 2007, 105, 1073–1083 CrossRef.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- J. C. Hill and J. A. Patts, J. Chem. Theor. Comput., 2007, 3, 80–85 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- A. J. Misquitta, R. Podeszwa, B. Jeziorski and K. Szalewicz, J. Chem. Phys., 2005, 123, 214103 CrossRef.
- M. Grüning, O. V. Gritsenko, S. J. A. v. Gisbergen and E. J. Baerends, J. Chem. Phys., 2001, 114, 652–660 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Neese, ORCA. An ab initio, DFT, and Semiempirical Electronic Structure Package, Version 2.6-35, University of Bonn, Germany, 2007 Search PubMed.
- H.-J. Werner, P. J. Knowles, F. R. Manby, M. Schütz, P. Celani, G. Knizia, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Koppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, K. Pfluger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, Molpro version 2009.1, a package of ab initio programs, see http://www.molpro.net, 2009 Search PubMed.
- M. A. Petrukhina, K. W. Andreini, J. Mack and L. T. Scott, J. Org. Chem., 2005, 70, 5713–5716 CrossRef CAS.
- R. C. Haddon, J. Am. Chem. Soc., 1986, 108, 2837–2842 CrossRef CAS.
- R. C. Haddon, J. Phys. Chem., 1987, 91, 3719–3720 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Geometries of the complexes studied and complexation energies for all complexes as modeled with the COSMO method. See DOI: 10.1039/c1cp22823d |
| This journal is © the Owner Societies 2012 |