Application of the dissociative electron transfer theory and its extension to the case of in-cage interactions in the electrochemical reduction of arene sulfonyl chlorides†
Abdelaziz
Houmam
* and
Emad M.
Hamed
Electrochemical Technology Centre, Department of Chemistry, University of Guelph, Guelph, Ontario, N1G 2W1, Canada. E-mail: ahoumam@uoguelph.ca; Fax: +1 (519) 766 1499; Tel: +1 (519) 824 4120 Ext. 56429
First published on 13th September 2011
Abstract
Important aspects of the electrochemical reduction of a series of substituted arene sulfonyl chlorides are investigated. An interesting autocatalytic mechanism is encountered where the starting material is reduced both at the electrode and through homogeneous electron transfer from the resulting sulfinate anion. This is due to the homogenous electron transfer from the two-electron reduction produced anion (arene sulfinate) to the parent arene sulfonyl chloride. As a result, the reduction process and hence the generated final products depend on both the concentration of the substrate and the scan rate. A change is also observed in the reductive cleavage mechanism as a function of the substituent on the phenyl ring of the arene sulfonyl chloride. With 4-cyano and 4-nitrophenyl sulfonyl chlorides a “sticky” dissociative ET mechanism takes place where a concerted ET mechanism leads to the formation of a radical/anion cluster before decomposition. With other substituents (MeO, Me, H, Cl, and F) a “classical” dissociative ET is followed, where the ET and bond cleavage are simultaneous. The dissociative electron transfer theory, as well as its extension to the case of strong in-cage interactions between the produced fragments, along with gas phase chemical quantum calculations results helped us to rationalize both the observed change in the ET mechanism and the occurrence of the “sticky” dissociative ET mechanism. The radical/anion pair interactions have been determined both in solution as well as in the gas phase. The study also shows that despite the low magnitude of in-cage interactions in acetonitrile compared to the gas phase their existence strongly affects the dynamics of the involved reactions. It also shows that, as expected, these interactions are reinforced by the existence of strong electron-withdrawing substituents. The occurrence of an autocatalytic process and the existence of the radical/anion interaction may explain the differences previously observed in the reduction of these compounds in different media.
1. Introduction
Dissociative electron transfer (ET),1–3 where a chemical bond is broken as a result of an ET to an organic or bioorganic molecule, is among the most elementary of all chemical reactions. Its fundamental role in areas including organic synthesis and biological processes is well-known.4–8 To reach a molecular understanding of these processes a rigorous description of the nature and fundamental steps involved in the electron transfer and the subsequent steps is required. Understanding the occurrence of the electron transfer and the factors that control it is therefore necessary.When the dissociative ET follows a stepwise mechanism, where the bond dissociation occurs subsequent to the electron, Hush–Marcus model9–18 of outersphere electron transfer can be applied to the electron transfer step. On the other hand, when the DET is concerted, where the bond breaking and the ET are simultaneous, Savéant's model, which is based on the Morse curve picture of bond breaking, is used.1–3,19–22 For both the stepwise and concerted mechanisms, the electron transfer reaction can be described by a quadratic activation–driving force relationship. For a stepwise ET mechanism, the activation barrier (ΔG≠0) involves only the solvent (λ0) and the inner (λi) reorganization energies (eqn (1)). For a concerted ET mechanism, the bond dissociation energy (DR) of the fragmented bond (R–X) contributes to the activation barrier (eqn (2)).
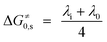 | (1) |
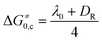 | (2) |
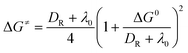 | (3) |
The difference in the reaction free energy between the two ET mechanisms can be expressed by the corresponding standard potentials (eqn (4)).
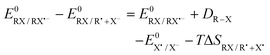 | (4) |
The weaker the bond and the more positive 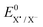 , the more favourable the thermodynamics of the concerted mechanism are.
, the more favourable the thermodynamics of the concerted mechanism are.
The dissociative ET has also been successfully used to describe the formation/dissociation reactions of radical ions.1–3,23–27 More recently, this theory has been extended to describe dissociative ET reactions involving strong interactions between the produced fragments (“sticky” dissociative ET).28–32 Therefore, a new activation free energy––standard free energy quadratic relationship, involving the contribution of the interaction energy in the radical–ion pair (Dp), has been obtained (eqn (5)).28–32 While the accuracy of this model has been demonstrated through its application to heterogeneous28–34 as well as homogeneous35,36 ET reactions, examples undergoing this type of reduction mechanism are very limited. The examples known so far include carbon tetrachloride,28,294-cyanobenzyl chloride,29 haloacetonitriles,30 polychloroacetamides,31benzyl thiocyanates,33 and sulfenyl chlorides.34 Additional experimental examples involving the intermediate formation of such radical–ion pairs (σ––radical ions) would provide more insights into the factors controlling such phenomenon as well as into its consequences on chemical reactions.
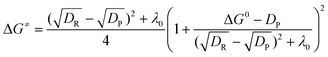 | (5) |
In cyclic voltammetry (CV) the peak characteristics can be used efficiently to obtain accurate mechanistic, kinetic and thermodynamic data by application of the adequate theory. When the single ET product cannot be detected experimentally (high scan rate37,38 and homogeneous catalysis1–3), the transfer coefficient (α), which is directly related to the intrinsic barrier (eqn (6)), can be a sensitive probe of the mechanistic nature of the first electron transfer in dissociative ET processes. Experimentally, the transfer coefficient can readily be determined from the electrochemical peak characteristics (peak width, Ep − Ep/2),39 or the variation of the peak potential, Ep, with the scan rate, v.40 In a concerted mechanism, a value significantly lower than 0.5 is expected, whereas an α value close to or higher than 0.5 is expected in the case of a stepwise mechanism.41–48 The use of the transfer coefficient α value as a tool to ascertain the ET mechanism could however be misleading in situations where the ET at the electrode surface is coupled with chemical reactions that can affect the shape and the position of the cyclic voltammetric signals, and hence extra caution needs to be taken. This was clearly observed with aryl thiocyanates, where the involved autocatalytic mechanism had a dramatic effect on the cyclic voltammograms and hence on the α value.49
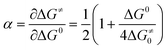 | (6) |
Sulfonyl chlorides and their derivatives are of great importance due to their high reactivity, versatile chemistry and important applications.50–58 Their use in chemistry relies mainly on their facile chemical reduction,50–58 which has been shown to lead to various products through a reaction with a wide range of reagents, and on the readiness to replace the Cl with various substituents.50–58 Despite this wide use of sulfonyl chlorides under reductive conditions, the ET to these compounds and the factors controlling it are still not well understood. A number of electrochemical investigations have been reported in different media but a clear picture of the details of the reduction mechanism is still lacking.59–62 The studies have shown different results in terms of involved mechanisms, number of consumed electrons, and yielded different products.59–62 A more rigorous study would certainly help to understand the reduction mechanism and rationalize the observed differences. Here we are particularly interested in investigating the dynamics of the reduction of a series of arene sulfonyl chlorides through a careful analysis of the involved mechanisms as well as a rigorous determination of the factors controlling its kinetics and thermodynamics. We previously reported the electrochemical reduction of arene sulfenyl chlorides (ArSCl) and showed that their reduction results in the cleavage of the S–Cl bond.34,63 Our investigation showed that the electrochemical reduction of 4-substituted sulfenyl chlorides follows a sticky dissociative ET mechanism involving the formation of a radical/anion (4-substituted phenyl sulfenyl radical/chloride anion) pair.34 This manuscript describes the full investigation of an extended series of 4-substituted arene sulfonyl chlorides 1a–g (Chart 1). Not only is a change of the ET mechanism observed but more interestingly, a clear-cut example of a sticky dissociative ET is encountered. The factors controlling the ET mechanism variation as well as the extend of the in-cage interactions between the reduction fragments are discussed on the basis of the dissociative ET theory and its extension to the case of in-cage interactions (“sticky” dissociative ET).28–34,64 Theoretical calculations help rationalize both the difference in the ET mechanism as well as the cluster formation.
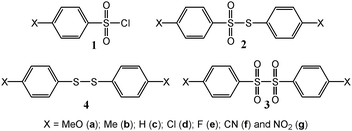 | ||
| Chart 1 | ||
2. Experimental part
2.1 Cyclic voltammetry
Electrochemical measurements were conducted in three electrode glass cells, thermostatted at 25 °C, and under dry nitrogen. The working electrode is a 2 mm diameter glassy carbon electrode (Ekochemie). The electrode was carefully polished and ultrasonically rinsed with ethanol before each run. The reference electrode is a saturated calomel electrode (SCE).The counter electrode was a platinum wire. The electrochemical instrument used is an Autolab PGSTAT30 especially configured to carry out high scan rate CV experiments. A feedback correction was applied to minimize the Ohmic drop between the working and reference electrodes.
2.2 Electrolyses
The electrolyses were carried out in 20 cm3cells with a glassy carbon (Electrosynthesis) rectangular plate working electrode of 8 cm2. The counter electrode was a platinum grid, separated from the cathodic compartment by means of a glass frit. The reference electrode was the same as for CV. The cell was thermostatted at 25 °C, and the solution was kept under a nitrogen stream during the whole electrolysis. The disappearance of the starting material and the formation of the products were followed by in situcyclic voltammetry. The supporting electrolyte, tetramethylammonium hexafluorophosphate, was extracted and chromatographic analyses (HPLC and GCMS) were performed by comparison with authentic samples of the product.2.3 Chemicals
Acetonitrile (Aldrich), the supporting electrolytes, tetramethylammonium and tetrabutylammonium hexafluorophosphate (Fluka, puriss) were used as received.4-Methoxylphenyl sulfonyl chloride (1a), 4-methylphenyl sulfonyl chloride (1b), the phenyl sulfonyl chloride (1c), 4-chlorophenyl sulfonyl chloride (1d), 4-fluorophenyl sulfonyl chloride (1e), 4-cyanophenyl sulfonyl chloride (1f), 4-nitrophenyl sulfonyl chloride (1g), and phenylthiophenylsulfonate (2c) are commercially available (Aldrich) and were used as received. Diphenyl disulfone (3c) was made (see ESI†) through a reaction of the phenyl sulfinate with phenyl sulfonyl chloride (1c). All disulfides 4a–g are commercially available (Aldrich) and were used as received.
2.4 Theoretical calculation
The calculations were performed using the Gaussian 2003 package.65 LUMO orbitals were calculated after a full optimization without imposed symmetry of the conformations using the UHF, B3LYP method with the 6-31G+(d,p) basis set starting from preliminary optimizations performed with semi-empirical methods. We checked that the obtained conformations were real minima by running frequency calculations for the UHF and B3LYP calculations. These calculations could not be performed at the MP2 level because of the too large molecule sizes.3. Results and discussion
3.1 Voltammetric behaviour
The electrochemical reduction of substituted arene sulfonyl chlorides (1a–g) was studied by cyclic voltammetry in acetonitrile, in the presence of tetrabutylammonium hexafluorophosphate (Bu4NPF6 0.1 M) at a glassy carbon electrode. The peak characteristics (peak potential (Ep), peak width (Ep − Ep/2), slope of Ep − log(v) and transfer coefficient (α) values determined from both peak width and Ep − log(v) plots) are summarized in Table 1. Cyclic voltammograms of all studied compounds (1a–g) are reported in Fig. 1.| ArSO2Cl | Substituent | E p1a (V vs.SCE) | n (e-/molecule) | SlopebEp − log(v) | α c | E p −E p/2/V | α d |
|---|---|---|---|---|---|---|---|
| a At v = 200 mV s−1. b In V/unit log(v). c From Ep − log(v) plot. d From peak width. | |||||||
| 1a | MeO | −1.11 | 2 | −0.099 | 0.30 | −0.137 | 0.34 |
| 1b | Me | −1.03 | 2 | −0.106 | 0.28 | −0.147 | 0.32 |
| 1c | H | −1.00 | 2 | −0.100 | 0.30 | −0.142 | 0.33 |
| 1d | Cl | −1.00 | 2 | −0.072 | 0.41 | −0.146 | 0.32 |
| 1e | F | −1.00 | 1.9 | −0.096 | 0.31 | −0.156 | 0.30 |
| 1f | CN | −0.75 | 1.8 | −0.099 | 0.30 | −0.110 | 0.42 |
| 1g | NO2 | −0.40 | 1.8 | −0.071 | 0.42 | −0.111 | 0.42 |
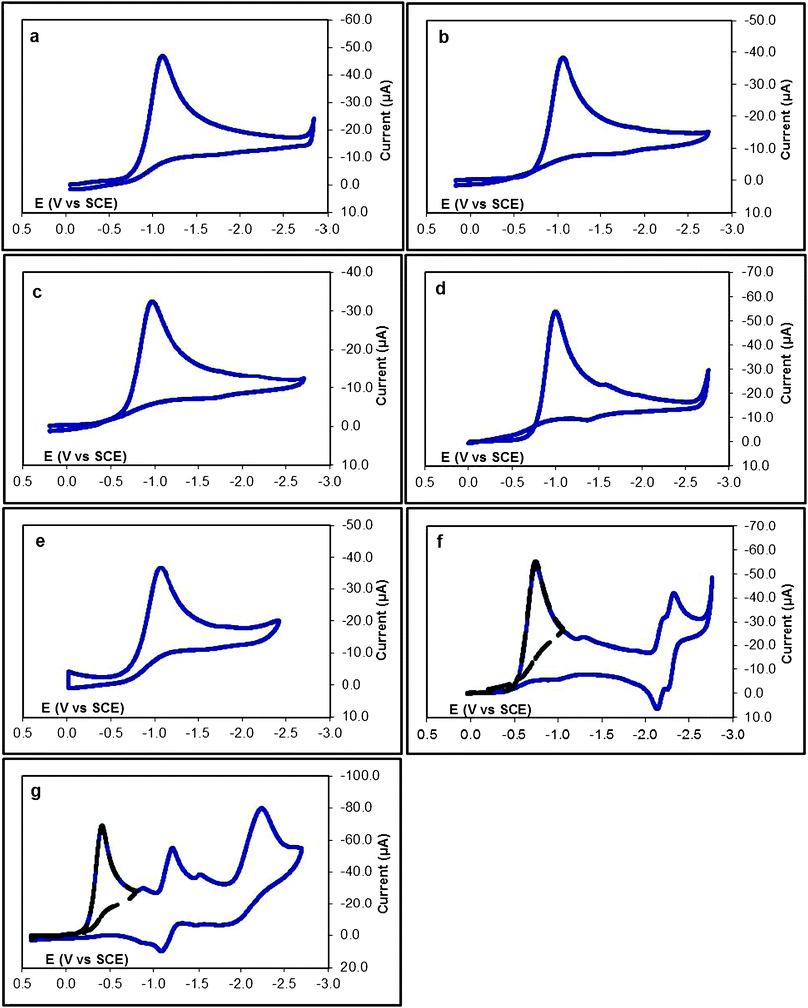 | ||
| Fig. 1 Cyclic voltammetry in CH3CN/Bu4NPF6 (0.1 M) at a glassy carbon electrode at v = 200 mV s−1 of (a) 1a (2.06 mM); (b) 1b (1.52 mM); (c) 1c (1.53 mM); (d) 1d (2.16 mM); (e) 1e (1.20 mM); (f) 1f (2.03 mM); and 1g (1.70 mM). | ||
Compounds 1a–e all show mainly one single reduction peak in cyclic voltammetry. Fig. 1a shows the cyclic voltammogram corresponding to reduction of 4-methoxylphenyl sulfonyl chloride (1a). The reduction peak is observed at a potential Ep = −1.11 V vs.SCE, and corresponds to the irreversible reduction of 1a (Fig. 1a). Its height, measured by reference to the monoelectronic wave of ferrocene corresponds to the consumption of two electrons per molecule. The transfer coefficient values determined from both the first reduction peak width39 and from the Ep − log(v) plot40 correspond to 0.34 and 0.30, respectively, i.e. much lower than 0.5, indicating a reaction kinetically controlled by the electron transfer step.1–3,19–22 Interestingly, trace crossing is observed when the potential scan is inversed. This crossing is not ascribed to the electrode material, as the cyclic voltammogram is reproducible, but corresponds rather to the reduction of a species resulting from the initial reduction and that is easier to reduce than the parent molecule 1a.66–68 A similar crossing was observed in the cyclic voltammetric study of arene thiocyanates.49 For the latter compounds, the trace crossing has been shown to result from an autocatalytic process in which the disulfide, formed through nucleophilic attack of the generated thiolate on the parent thiocyanate, was the intermediate catalyst. A main difference here is that the sulfinate anions generated by the two electron reductive dissociation of the S–Cl bond are not nucleophilic and the autocatalytic process is more likely a “traditional” process involving an electron transfer between this anion and the parent sulfonyl chloride in solution as will be discussed below.66–68 By scanning toward more positive potentials, two irreversible anodic peaks are observed (see ESI†). The first peak located at Ep = 0.43 V vs.SCE corresponds to the oxidation of 4-methoxyphenyl sulfinate, and the second one located at Ep = 1.16 V vs.SCE corresponds to the oxidation of chloride anions, as verified by comparison with the cyclic voltammetry of authentic samples.
Compounds 1f,g show additional reduction peaks. The first peak in all cases corresponds to the irreversible reduction of the initial sulfonyl chloride and the second peak is related to the substituted arene moiety of the sulfinate anion generated at the first peak. For the 4-cyanophenyl sulfonyl chloride (1f) two successive reversible peaks, corresponding to the p-cyanophenyl sulfinate and its protonated form, are observed at E01 = −2.18 V and E02 = −2.30 V vs.SCE. Once the scan rate is increased to 5 V s−1 only one peak is observed corresponding to the reduction of the p-cyanophenyl sulfinate anion (see ESI†). For the 4-nitrophenyl sulfonyl chloride (1g), the second reversible reduction peak is observed at E0 = −1.10 V vs.SCE. For the latter compound a third irreversible reduction peak is observed at Ep = −2.28 V vs.SCE, corresponding to the further reduction of the NO2 group to the NH2 as expected.
Compounds 1a–g show similar characteristics for the first reduction peak as they all present α values between 0.25 and 0.42, much smaller than 0.5, indicating a reaction kinetically controlled by the electron transfer step, and a peak height corresponding to the consumption of two electrons per molecule. Note that due to the autocatalytic nature of the reductive process, shown by the trace crossing in cyclic voltammetry, and its effect on the cyclic voltammetric characteristics, α values were deduced from cyclic voltammograms at low concentrations and relatively higher scan rates (where the trace crossing is totally eliminated). The reason, as we described in detail for the arene thiocyanates,49 is that at low concentration and high scan rates, the experimental electrochemical data correspond to the intrinsic characteristics of the direct reduction of studied sulfonyl chlorides on the electrode, rather than a mixed process involving consumption by both reduction at the electrode and autocatalysis in solution. In the case of 4-nitrophenyl sulfonyl chloride (1g), the peak is narrow and the transfer coefficient obtained from the peak width at 0.2 V s−1 is about 0.56. This value does not provide an indication about the ET mechanism since at this low scan rate, the autocatalytic process is very efficient. The value obtained from the slope of the variation of the peak potential with the scan rate curve is much lower (0.42).
In the framework of a quadratic activation–driving force relationship, which is valid for both the stepwise and the concerted reductive mechanisms, the observed peak characteristics (mainly values much lower than 0.5) mean that the peak potential is more negative than the standard potential of the rate-determining reaction.28–32,35,69–73 This rules out the occurrence of a stepwise mechanism, and tends to indicate the presence rather of a concerted process. The nature of this initial electron transfer will be further investigated below.
3.2 Electrolyses
Electrolyses of compounds 1a–g have been performed in acetonitrile, in the presence of tetramethylammonium hexafluorophosphate (0.1 M) and the results for all studied compounds 1a–g are reported in Table 2. The main product in all cases is the corresponding arene sulfinate (ArSO2−). For most compounds variable quantities of the corresponding thiosulfonates, ArSO2SAr (2a–g), and disulfides ArSSAr (4a–g) are also obtained. The thiosulfonates are usual products in the reduction of these compounds and have been shown to result from chemical reactions in solution and are not formed at the electrode through direct reduction of the parent molecules.59–62 An important result here is that the disulfides result from the reduction of the thiosulfonates as we have demonstrated through electrolysis of the phenylthiophenylsulfonate 2c (see ESI†). The reduction peak of this compound is located at a potential very close to that of the parent sulfonyl chloride (see ESI†) and hence is reduced during the electrolysis, which is set at the reduction potential of the latter compound. Diaryl disulfones, ArSO2SO2Ar (3a–g), have also been detected during the electrolyses but are reduced in situ as indicated by their very low yield at the end of the electrolyses. In a control experiment, diphenyl disulfone (3c) was synthesized and its electrochemistry investigated. Its cyclic voltammogram (see ESI†) shows an irreversible reduction peak at a potential very close to that corresponding to the phenyl sulfonyl chloride (1c). Its electrolysis leads to the phenyl sulfinate quantitatively. The cyano- and nitro-substituted compounds (1f,g) provide only the corresponding aryl sulfinates and no side products, similar to what has been seen for the nitro substituted arene sulfenyl chlorides.34| Electrolysis of ArSO2Cla | ArSO2− (%) | ArSO2SO2Ar (%)b | ArSO2SAr (%) | ArSSAr (%) |
|---|---|---|---|---|
| a In CH3CN + Me4NBF4 (0.1 M) at a glassy carbon electrode. b The electrolyses have been performed at least 3 times and slightly smaller amounts have been obtained in certain cases. | ||||
| 1a (MeO) | 73 | <5 | 18 | 2 |
| 1b (Me) | 60 | <5 | 19 | 9 |
| 1c (H) | 92 | <1 | — | 6 |
| 1d (Cl) | 65 | <8 | 16 | 7 |
| 1e (F) | 90 | <3 | — | 5 |
| 1f (CN) | 99 | — | — | — |
| 1g (NO2) | 99 | — | — | — |
| 2c | 98 | — | — | 98 |
| 3c | 97 | — | — | — |
Based on the electrochemical data, and considering, for now, a two electron reductive dissociation of the S–Cl chemical bond, the reduction mechanism for these compounds can be written as per Scheme 1. The sulfinate anion generated through the two electron reduction process transfers an electron to the parent sulfonyl chloride (reaction B). The generated radical provides the corresponding diaryl disulfone which is immediately reduced to regenerate the sulfinate anion once again (reactions C and D). The radical may also be reduced at the electrode to directly generate the anion (reaction E). These steps are responsible for the observed trace crossing in the cyclic voltammograms.
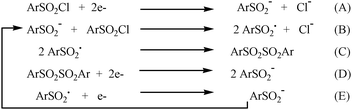 | ||
| Scheme 1 Reduction mechanism of aryl sulfonyl chlorides. | ||
Evidence for the chemical steps (reactions B and C) was provided by a control experiment, in which tetrabutylammonium phenyl sulfinate was added to the phenyl sulfonyl chloride (1c) and the reaction yielded the corresponding disulfone (3c), formed through dimerization of the sulfonyl radical. Evidence for reaction D was provided by cyclic voltammetry of the corresponding disulfone, which showed a reduction peak very close to that of the parent sulfonyl chloride (see ESI†). Electrochemistry of this disulfone has also been previously reported and a similar behavior was observed.74Reduction of the disulfone regenerates the same sulfinate anion. Formation of the sulfinic acid through either protonation of the sulfinate or hydrogen abstraction by the sulfinyl radical is also possible. The autocatalytic process is further confirmed by the cyclic voltammetry data upon changing the concentration and the scan rate. Using a set of dimensionless partial derivative equations, initial and boundary conditions, a main key kinetic dimensionless parameter (λ = RTkC0/Fv) was obtained as in the case of the arene thiocyanates. k is the rate constant of the homogeneous electron transfer step (reaction B).49 This parameter λ represents a measure of the competition between the rate-determining step of the autocatalytic process (reaction B in Scheme 1) and diffusion. Accordingly, the rate of the diffusion process is expected to increase with the sweep rate, while that of the autocatalytic process would increase with an increase of the sulfonyl chloride concentration. An investigation into the effect of the scan rate on the cyclic voltammograms of the studied arene sulfonyl chlorides at two different concentrations shows that this is indeed the case.
The variations of the cyclic voltammograms of the phenyl sulfonyl chloride (1c) in CH3CN/Bu4NPF6 (0.1 M) with the scan rate at two different concentrations (1 mM and 5.9 mM) are shown as a typical example in Fig. 2. For the low concentration (1 mM), the crossing observed at 0.2 V s−1 (Fig. 2a) decreases very quickly as the scan rate is increased to 0.4 V s−1 (Fig. 2b) and is completely eliminated at 1.2 V s−1 (Fig. 2c). Reaction B is rapidly eliminated at these scan rates for this low concentration. For the higher concentration (5.9 mM), the crossing that is more pronounced at 0.2 V s−1 (Fig. 2d) persists when increasing the scan rate to 1.2 V s−1 (Fig. 2e) and disappears only at much higher scan rates (15 V s−1 or higher) as shown in Fig. 2f. These results confirm the occurrence of an autocatalytic process that is more pronounced for high concentrations and low scan rates.
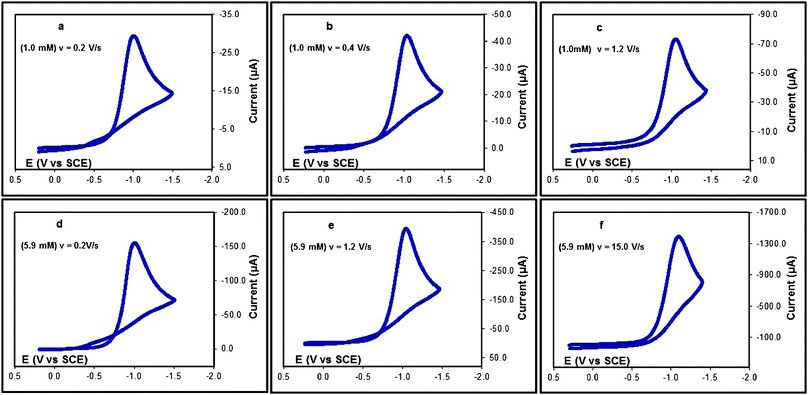 | ||
| Fig. 2 Cyclic voltammetry in CH3CN/Bu4NPF6 (0.1 M) at a glassy carbon electrode 1c at (a) 1 mM, v = 0.2 V s−1; (b) 1 mM, v = 0.4 V s−1; (c) 1 mM, v = 1.2 V s−1; (d) 5.9 mM, v = 0.2 V s−1; (e) 5.9 mM, v = 1.2 V s−1; and (f) 5.9 mM, v = 15 V s−1. | ||
To further confirm the autocatalytic process as described in Scheme 1, control experiments were performed where cyclic voltammetry was done in the presence of a proton donor (pentachlorophenol). The presence of this proton donor results in the total elimination of the trace crossing as expected. Under these conditions, the sulfinate anion is protonated and reaction (B), responsible for the autocatalytic process, is eliminated (see ESI†).
To gain more insights into the nature of the initial ET, a theoretical study at the B3LYP level of the substituted arene sulfenyl chlorides (1a–g) and their corresponding reduced forms has been performed.65,75
3.3 Theoretical study
Fig. 3 shows the LUMOs of compounds 1a–g and the SOMOs of their reduced forms (radical anions or radical/anion pairs).28–34,36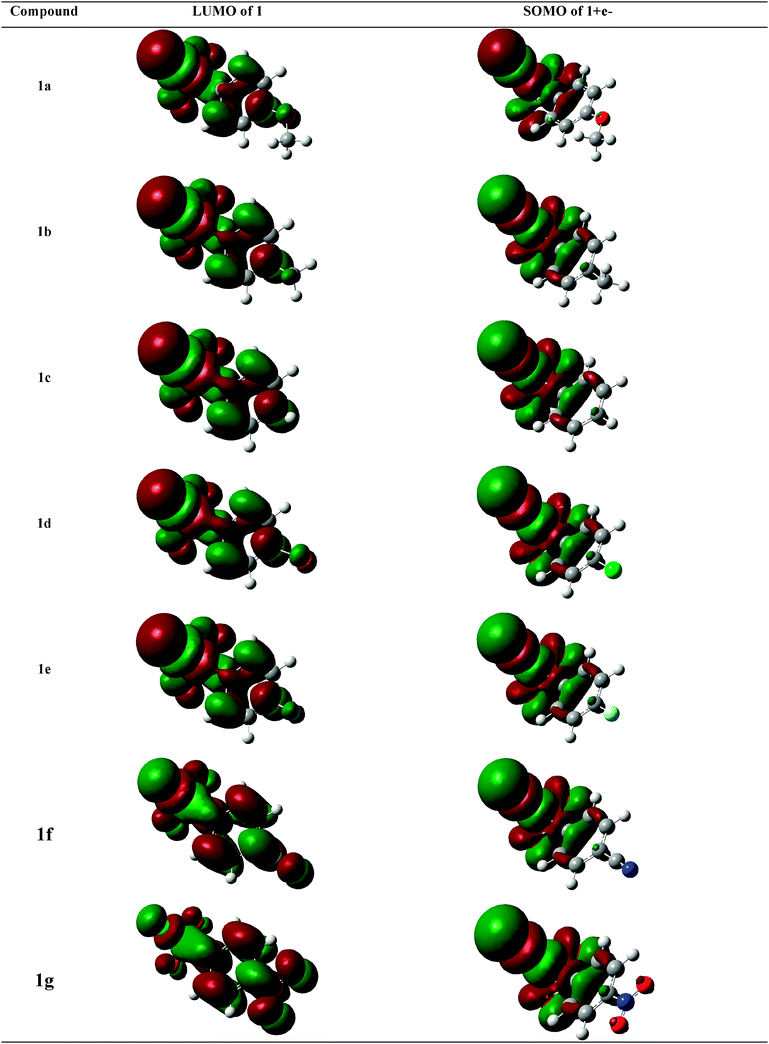 | ||
| Fig. 3 LUMOs and SOMOs of 1a–g and their reduced forms, respectively. | ||
For compounds 1a–e, the LUMOs are more located on the S–Cl group with a lower factor on the aryl moiety, indicating that the extra electron is most likely injected directly into the σ S–Cl bond leading to or concomitant with its dissociation. The SOMOs corresponding to their reduced forms are also more localized on the S–Cl group (σ radical anion or radical/ion pair) and provide thus more support to the injection of the extra electron into the σ S–Cl bond. Note that the reduced forms show large S–Cl distances (2.82 to 2.88 Å), as shown in Table 3, indicating that these reduced forms are radical/ion pairs and not true radical anions. For the cyano and nitro substituted compounds (1f and g), the LUMOs show a higher contribution of the aryl moieties compared to compounds 1a–e. The optimized reduced forms support however the formation of radical/anion pairs, similar to the other compounds, and not radical anions. The SOMOs are indeed located on the sulfonyl chloride group and the S–Cl bond length is 2.82 Å, very similar to the distances found for the other compounds 1a–e, and much larger than what was expected for real radical anions.
| 1a (MeO) | 1b (Me) | 1c (H) | 1d (Cl) | 1e (F) | 1f (CN) | 1g (NO2) | |
|---|---|---|---|---|---|---|---|
| a Bond dissociation energy in kcal mol−1. b Bond length in Å. | |||||||
| D S–Cl a | 50.65 | 50.47 | 50.29 | 50.09 | 50.20 | 49.65 | 49.53 |
| d S–Cl (1)b | 2.14 | 2.13 | 2.13 | 2.13 | 2.13 | 2.12 | 2.12 |
| d S–Cl (1 + e)b | 2.88 | 2.87 | 2.86 | 2.84 | 2.85 | 2.82 | 2.82 |
| ΔdS–Clb | 0.74 | 0.74 | 0.73 | 0.71 | 0.72 | 0.70 | 0.70 |
For all compounds the S–Cl bond distance shows a large increase (0.70 to 0.74 Å) upon injection of the extra electron (Table 3). This confirms that these species are radical/anion pairs and not real radical anions. These results provide therefore more evidence for the non-occurrence of a stepwise mechanism in the initial electron transfer to the arene sulfonyl chlorides as suggested by the electrochemical data. The radical/anion pair formation in the gas phase leads to a question whether the reduction of compounds 1a–g follows a “sticky” dissociative ET mechanism involving the corresponding radical/anion pairs (Scheme 2).
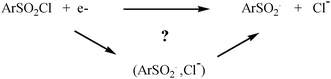 | ||
| Scheme 2 ET mechanism. | ||
To have an idea about the existence and the extent of these radical/anion interactions in the gas phase, potential energy profiles along the S–Cl bond for the XPhSO˙2/Cl− pair for all compounds have been deduced (Fig. 4).76 These energy profiles were calculated at the DFT/B3LYP/6-31G(p,d) level for different values of the S–Cl bond length.65,75 The results confirm the existence of these interactions in the gas phase. The obtained curves have the shape of Morse curves and show a clear energy minimum along the cleaved bond. The deduced interaction energies as well as the values of the bond lengths at the minimum energy (dS–Cl) are reported in Table 4. The results indicate that at least in the gas phase, strong interactions (0.33–0.56 eV) exist indeed between the generated fragments (XPhSO˙2/Cl−). For all compounds the minimum energies are observed at large distances (2.8–2.9 Å), in very good agreement with the ones determined from the earlier optimizations (Table 3), indicating indeed the formation of radical/anion pairs rather than real radical anion intermediates. The 4-cyanophenyl sulfonyl chloride (1f) and 4-nitrophenyl sulfonyl chloride (1g) show, as expected, relatively higher interaction energies. Strong electron-withdrawing groups have indeed been shown to reinforce fragments clustering.28–32
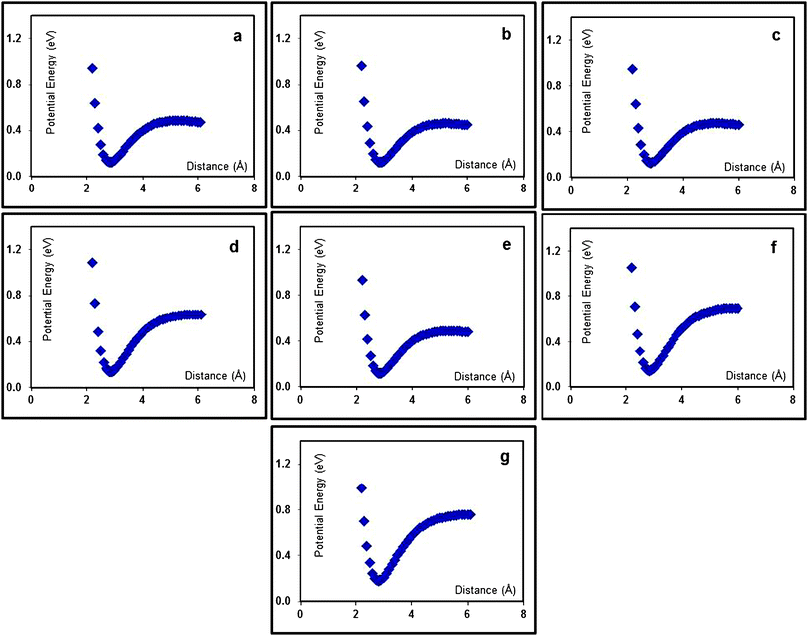 | ||
| Fig. 4 Calculated (B3LYP/6-31G(p,d)) potential energy profiles in the gas phase for the XPhSO2./Cl− pair for (a) 1a; (b) 1b; (c) 1c; (d) 1d; (e) 1e; (f) 1f and (g) 1g. | ||
To clearly ascertain the nature of the first electron transfer and to unravel the extent of the in-cage interactions between the radical/anion fragments upon electrochemical reduction, compounds 1a–g were further investigated through application of both the “classical” dissociative ET model (eqn (3)) and the “sticky” dissociative ET model (eqn (5)). This required comparison of the activation free energy of the reactions, as a function of the driving force, obtained through these two models with the experimental one obtained using convolution analysis77–79 of the cyclic voltammetric data.
To that end a series of parameters were required. The solvent reorganization energies (λ0) were derived from the corresponding radii (a) of the equivalent spheres using eqn (7).64,80
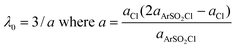 | (7) |
The standard reduction potentials 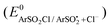 were determined using eqn (8). The values of these parameters are listed in Table 5. The
were determined using eqn (8). The values of these parameters are listed in Table 5. The 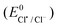 (1.86 V vs.SCE in CH3CN) has been estimated from its known value in water.81,82 The bond dissociation energy (DArSO2–Cl) as well as the bond dissociation entropy (ΔS0) of the S–Cl bond for the aryl sulfonyl chlorides were calculated.83
(1.86 V vs.SCE in CH3CN) has been estimated from its known value in water.81,82 The bond dissociation energy (DArSO2–Cl) as well as the bond dissociation entropy (ΔS0) of the S–Cl bond for the aryl sulfonyl chlorides were calculated.83
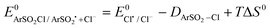 | (8) |
| 1 | Substituent |
ΔS0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a; TΔS0b a; TΔS0b |
|
E
0d |
a Cl e | a ArSO2Cl e | a e | 105Df | Z g | λ 0 h |
|---|---|---|---|---|---|---|---|---|---|---|
a In cal mol−1 K.
b In eV.
c In V vs.SCE.
d
 in V vs.SCE.
e In Å.
f In cm2 s−1.
g cm s−1.
h In eV. in V vs.SCE.
e In Å.
f In cm2 s−1.
g cm s−1.
h In eV.
|
||||||||||
| 1a | MeO | 32.13; 0.405 | 1.86 | 0.079 | 1.81 | 8.48 | 3.23 | 2.9 | 4412 | 0.928 |
| 1b | Me | 32.13; 0.405 | 1.86 | 0.086 | 1.81 | 7.40 | 3.17 | 2.9 | 4593 | 0.946 |
| 1c | H | 28.24; 0.356 | 1.86 | 0.094 | 1.81 | 6.41 | 3.11 | 2.9 | 4772 | 0.964 |
| 1d | Cl | 28.30; 0.357 | 1.86 | 0.103 | 1.81 | 7.03 | 3.15 | 2.9 | 4366 | 0.952 |
| 1e | F | 37.37; 0.472 | 1.86 | 0.098 | 1.81 | 6.81 | 3.14 | 2.9 | 4546 | 0.955 |
| 1f | CN | 33.57; 0.424 | 1.86 | 0.122 | 1.81 | 8.05 | 3.21 | 2.9 | 4467 | 0.934 |
| 1g | NO2 | 32.16; 0.406 | 1.86 | 0.128 | 1.81 | 7.45 | 3.18 | 2.9 | 4260 | 0.943 |
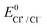
The experimental activation free energy was deduced from the cyclic voltammetric data using eqn (9):
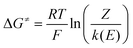 | (9) |
The potential-dependant rate constant of the electron transfer, k(E), was derived from convolution analysis of the cyclic voltammetric data using eqn (10). The convolutive current I was then obtained through convolution of the cyclic voltammetric current (i) according to eqn (11), where Il is the limiting convolutive current Il = FSC0D1/2, with S being the electrode surface area, C0 the substrate concentration and D its diffusion coefficient.
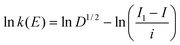 | (10) |
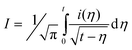 | (11) |
The pre-exponential factor was taken as the collision frequency (Z) and was determined through eqn (12), where M is the molar mass.
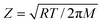 | (12) |
After these parameters were deduced, it was possible to compare the experimental activation free energy–driving force curve to the ones obtained using the “classical” and “sticky” dissociative ET models (eqn (3) and (5)).
For compounds 1a–e a good fitting was observed between the experimental and the “classical” dissociative ET model data. For the two compounds with the cyano (1f) and nitro (1g) substituents the predicted activation energy using the “classical” dissociative ET model was larger than the experimental one. The use of the “sticky” dissociative ET model (eqn (5)), and through tuning of the radical/anion interactions Dp, good fitting of the predicted activation energy to the experimental data was possible (Fig. 5f and g). This showed that for these compounds, the initial electron transfer involves indeed interactions between the radical and the anion fragments (substituted aryl sulfonyl radical/chloride anion). This confirmed the suggestion of the theoretical optimization of their reduced forms, showing a minimum at a large S–Cl distance corresponding to a radical/anion pair. The presence of such interactions decrease indeed the activation energy as has been previously shown.28–32 To get the best fitting the values used for radical/anion interactions were 12 and 10 meV for the 4-cyanophenyl sulfonyl chloride (1f) and 4-nitrophenyl sulfonyl chloride (1g), respectively. For compounds 1a–e, even if the gas phase calculations showed the potential formation of radical/ion interactions, application of the ET theories demonstrated that these interactions are eliminated in acetonitrile for these compounds and that the ET mechanism is a “classical” concerted one (Fig. 5a–e). In Fig. 5a–e, the modeled activation free energy–driving force curves using the “sticky” dissociative ET and a Dp value of 20 meV are also provided for comparison. Another important diagnostic for the ET resides in comparing the peak potential to the standard potential. A large difference is usually observed for “classical” concerted ET processes. For the investigated arene sulfonyl chlorides, the difference is more than 1.1 V for compounds 1a–e, while it is much smaller for compounds 1f and 1g (870 mV and 530 mV, respectively). The relatively smaller differences indicate a reduction of these compounds through intermediate formation of radical/anions pairs, compared to the other ones (1a–e) not involving such interactions.
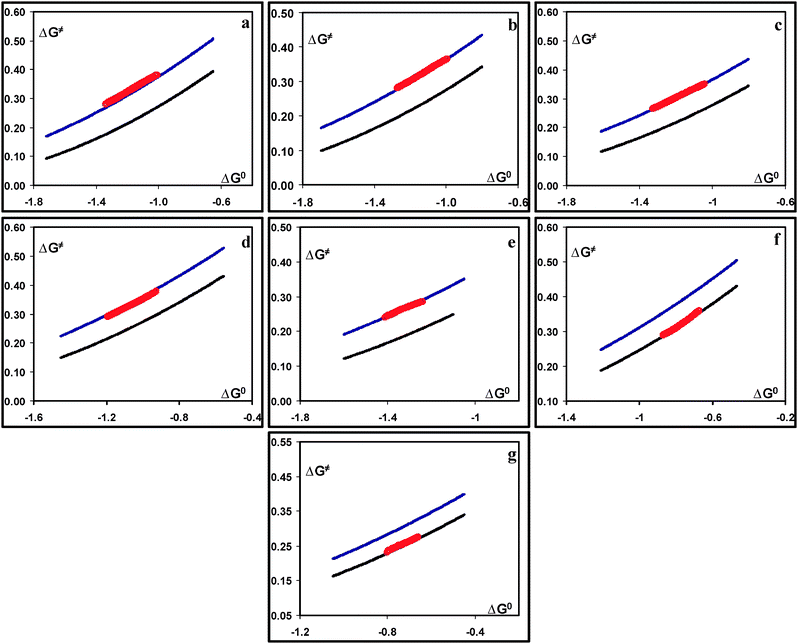 | ||
Fig. 5 Experimental and predicted activation free energy vs. standard free energy plots for (a) 1a; (b) 1b; (c) 1c; (d) 1d; (e) 1e; (f) 1f and (g): 1g. ( ): Predicted using the “classical” dissociative ET Model. ( ): Predicted using the “classical” dissociative ET Model. ( ): Predicted using the “sticky” dissociative ET model. ( ): Predicted using the “sticky” dissociative ET model. ( ): Experimental through convolution analysis. ): Experimental through convolution analysis. | ||
In comparison with the arene sulfenyl chlorides (ArSCl), the sulfonyl chlorides (ArSO2Cl) showed a difference in the ET mechanism. For the former compounds the radical/ion pair formation was observed for most compounds except for the nitro-substituted molecules, which followed a stepwise ET mechanism.34,63 In both series the S–Cl bond is cleaved as a result of the injection of an extra electron yielding the same chloride anion leaving group. The bond dissociation energy of the S–Cl cleaved bond of the studied compounds is only slightly higher than for the arene sulfenyl chlorides. We have recently shown that these electrochemical characteristics made the arene sulfenyl chlorides excellent new precursors for the formation of aromatic self-assembled monolayers (SAMs) on gold surfaces, which are more difficult to obtain than the aliphatic ones.84 This means that sulfonyl chlorides may also be good precursors for the formation of SAMs on metal surfaces. A difference though resides in the reduction potentials in the two series; the sulfonyl chlorides being about 1 V more difficult to reduce than the sulfenyl chlorides. It will be interesting to see how this difference as well as the presence of the oxygen atoms will affect the SAM formation. This is presently under investigation and is of great importance especially that the sulfonyl chlorides are more versatile, more stable and more readily available compared to the sulfenyl chlorides.
4. Conclusions
The reduction of substituted arene sulfonyl chlorides was investigated using cyclic voltammetry, theoretical calculations and convolution analysis. The involved mechanisms were clearly established through application of the “classical” and “sticky” dissociative electron models. For the initial electron transfer, a “sticky” dissociative ET mechanism is encountered with the 4-cyanophenyl sulfonyl chloride (1f) and 4-nitrophenyl sulfonyl chlorides (1g). For these compounds, the electrochemical reduction leads to the intermediate formation of a radical/anion pair, before complete dissociation. The formation of a radical/anion pair was suggested by the electrochemical data. An additional support for the intermediacy of these radical/anion pairs was provided by the gas phase theoretical calculation. Considerable bond elongations (more than 0.7 Å) were indeed observed between the neutral structures and the minima obtained for their reduced forms. For the other compounds (1a–e), a “classical” concerted dissociative ET is followed involving the simultaneous ET and cleavage of the S–Cl chemical bond.For all compounds, the potential energy profile curves of the reduced forms, along the cleaved S–Cl bond, showed the formation of radical/anion pairs through interactions in the gas phase. Moreover, these curves also showed that the interactions are reinforced by strong electron-withdrawing substituents.
Application of the dissociative ET theory and its extension to the case of strong in-cage interactions showed that while these interactions are eliminated through solvation in acetonitrile for compounds 1a–e, they do persist for the sulfonyl chlorides with the strongest electron withdrawing substituents (1f,g). The extent of the in-cage interactions was provided by fitting of the modeled free activation energy–driving force free energy curves using the “sticky” dissociative ET model to the experimental ones. The results showed these interactions to be, as expected, much smaller in acetonitrile compared to the gas phase. These interactions do indeed lower the activation free energy of the electrochemical reaction.
Another important result is that the global mechanism involves the occurrence of an autocatalytic process. Although this process is not as pronounced as that observed for the aryl thiocyanates,49 nevertheless autocatalytic processes are solvent dependent and this, along with the in-cage interactions, which may persist in other solvents even for the other compounds (1a–e), may explain the different products previously reported in various solvents.
Acknowledgements
A. Houmam gratefully acknowledges the Natural Sciences and Engineering Research Council (NSERC), the Canada Foundation for Innovation (CFI), and the Ontario Innovation Trust (OIT) for funding.Notes and references
- J.-M. Savéant, Electron Transfer, Bond Breaking and Bond Formation, in Advances in Physical Organic Chemistry, ed. T. T. Tidwell, Academic Press, New York, 2000, vol. 35, pp. 177–192 Search PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Phys., 2006, 324, 40 CrossRef CAS.
- A. Houmam, Chem. Rev., 2008, 108, 2180 CrossRef CAS.
- J. Grimshaw, Electrochemical reactions and mechanisms in organic chemistry, Elsevier, New York, 2000 Search PubMed.
- S. F. Nelsen, Electron Transfer in Organic Chemistry, in Electron Transfer Chemistry, ed. V. Balzani, Wiley-VCH, New York, 2001, vol. 1, pp. 342–392 Search PubMed.
- H. J. Schäfer, Organic Electrochemistry, in Encyclopedia of Electrochemistry, Wiley-VCH, Weinheim, 2004, vol. 8 Search PubMed.
- S. Torii, Electroorganic Reduction Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- D. G. Peters, Halogenated Organic Compounds, in Organic Electrochemistry, ed. H. Lund and O. Hammerich, Marcel Dekker, New York, 2001, 4th edn, pp. 341–377 Search PubMed.
- R. A. Marcus, J. Chem. Phys., 1955, 24, 4955.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 979 CrossRef CAS.
- N. S. Hush, J. Chem. Phys., 1958, 28, 962 CrossRef CAS.
- N. S. Hush, Trans. Faraday Soc., 1961, 57, 557 RSC.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679 CrossRef CAS.
- R. A. Marcus, Theory and Applications of Electron Transfers at Electrodes and in Solution, in Special Topics in Electrochemistry, ed. P. A. Rock, Elsevier, New York, 1977, pp. 161–179 Search PubMed.
- R. A. Marcus, Faraday Discuss. Chem. Soc., 1982, 74, 77 Search PubMed.
- R. A. Marcus and N. Sutin, Biochem. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- J.-M. Savéant, J. Am. Chem. Soc., 1987, 109, 6788 CrossRef CAS.
- J.-M. Savéant, J. Am. Chem. Soc., 1992, 114, 10595 CrossRef CAS.
- J.-M. Savéant, Acc. Chem. Res., 1993, 26, 455 CrossRef CAS.
- J.-M. Savéant, Dissociative Electron Transfer, in Advances in Electron Transfer Chemistry, ed. P. S. Mariano, JAI Press, New York, 1994, vol. 4, pp. 53–116 Search PubMed.
- J.-M. Savéant, J. Phys. Chem., 1994, 98, 3716 CrossRef CAS.
- D. Laage, I. Burghardt, T. Sommerfield and J. T. Hynes, J. Phys. Chem. A, 2003, 107, 11292 CrossRef CAS.
- I. Burghardt, D. Laage and J. T. Hynes, J. Phys. Chem. A, 2003, 107, 11271 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 106, 16051 CrossRef.
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16834 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2000, 122, 9829 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2001, 123, 11908 CrossRef CAS.
- A. Cardinale, A. A. Isse, A. Gennaro, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2002, 124, 13533 CrossRef CAS.
- C. Costentin, C. Louault, M. Robert and A.-L. Teillout, J. Phys. Chem. A, 2005, 109, 2984 CrossRef CAS.
- J.-M. Savéant, J. Phys. Chem. B, 2001, 105, 8995 CrossRef CAS.
- E. M. Hamed, H. Doai, C. K. McLaughlin and A. Houmam, J. Am. Chem. Soc., 2006, 128, 6595 CrossRef CAS.
- C. Ji, M. Ahmida, M. Chahma and A. Houmam, J. Am. Chem. Soc., 2006, 128, 15423 CrossRef CAS.
- C. Costentin, P. Hapiot, M. Médebielle and J.-M. Savéant, J. Am. Chem. Soc., 2000, 122, 5623 CrossRef.
- A. A. Isse and A. Gennaro, J. Phys. Chem. A, 2004, 108, 4180 CrossRef CAS.
- C. P. Andrieux, P. Hapiot and J.-M. Savéant, Chem. Rev., 1990, 90, 723 CrossRef CAS.
- C. P. Andrieux, P. Hapiot and J.-M. Savéant, J. Phys. Chem., 1988, 92, 5987 CrossRef CAS.
- α = (RT/F)(1.85/Ep/2 − Ep).
- ∂Ep/∂log v = −29.5/α at 20 °C.
- Cases have been encountered where an important internal intrinsic barrier is associated with a stepwise ET and in certain cases a low transfer coefficient42–48.
- M. G. Severin, M. C. Avéralo, F. Maran and E. Vianello, J. Phys. Chem., 1993, 97, 150 CrossRef CAS.
- S. Jakobson, H. Jensen, S. U. Pederson and K. Daasbjerg, J. Phys. Chem. A, 1999, 103, 4141 CrossRef CAS.
- T. B. Christensen and K. Daasbjerg, Acta Chem. Scand., 1997, 51, 307 CrossRef CAS.
- K. Daasbjerg, H. Jensen, R. Benassi, F. Taddei, S. Antonello, A. Gennaro and F. Maran, J. Am. Chem. Soc., 1999, 121, 1750 CrossRef CAS.
- S. Antonello, R. Benassi, G. Gavioli, F. Taddei and F. Maran, J. Am. Chem. Soc., 2002, 124, 7529 CrossRef CAS.
- S. Antonello, K. Daasbjerg, H. Jensen, F. Taddei and F. Maran, J. Am. Chem. Soc., 2003, 125, 14905 CrossRef CAS.
- A. A. Isee, A. Gennaro and F. Maran, Acta Chem. Scand., 1999, 53, 1013 CrossRef.
- A. Houmam, E. M. Hamed and I. E. J. Still, J. Am. Chem. Soc., 2003, 125, 7258 CrossRef CAS.
- D. K. H. Ho, L. Chan, A. Hooper and P. E. Brennan, Tetrahedron Lett., 2011, 52, 820 CrossRef CAS.
- M. Harmata, P. Zheng, C. Huang, M. G. Gomes, W. Ying, K.-O. Ranyanil, G. Balan and N. L. Calkins, J. Org. Chem., 2007, 72, 683 CrossRef CAS.
- S. R. Dubbaka, P. Steunenberg and P. Vogel, Synlett, 2004, 1235 CAS.
- E. E. Boros, I. Kaldor, P. J. Brown and V. L. Styles, Synth. Commun., 2001, 31, 505 CrossRef CAS.
- S. R. Dubbaka and P. Vogel, J. Am. Chem. Soc., 2003, 125, 15292 CrossRef CAS.
- H. Uchiro and S. Kobayashi, Tetrahedron Lett., 1999, 40, 3179 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi and A. Jamalian, Synlett, 2005, 1447 CrossRef CAS.
- N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- K. G. Petrov, Y.-M. Zhang, M. Carter, G. S. Cockerill, S. Dickerson, C. A. Gauthier, Y. Guo, R. A. Mook Jr., D. W. Rusnak, A. L. Walker, E. R. Wood and K. E. Lackey, Bioorg. Med. Chem. Lett., 2006, 16, 4686 CrossRef CAS.
- J.-G. Gourcy, G. Jeminet and J. C. R. Simonet, Acad. Sci. Paris Série C, 1973, 277, 1079 Search PubMed.
- G. Jeminet, J. Simonet and J.-G. Gourcy, Bull. Soc. Chim. Fr., 1974, 5–6, 1102.
- B. Fleszar and P. Sanecki, Roczniki chemii Ann. Soc. Chim. Polonorum, 1977, 51, 605 Search PubMed.
- A. C. D. Angelo, S. M. A. Jorge and N. R. Stradiotto, Ecletica Quim., 2005, 30, 57 Search PubMed.
- C. Ji, J. D. Goddard and A. Houmam, J. Am. Chem. Soc., 2004, 126, 8076 CrossRef CAS.
- C. P. Andrieux, J.-M. Savéant and C. Tardy, J. Am. Chem. Soc., 1998, 120, 4167 CrossRef CAS.
- M. J. Frisch, et al.Gaussian 2003, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- C. P. Andrieux and J.-M. Savéant, Electrochemical Reactions, in Investigation of Rates and Mechanisms of Reactions, Techniques of Chemistry, ed. C. F. Bernasconi, Wiley, New York, 1986, vol. VI/4 edn., part 2, pp. 305–390 Search PubMed.
- C. Amatore, J. Pinson and J.-M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1980, 107, 59 CrossRef CAS.
- C. P. Andrieux, A. Merz and J.-M. Savéant, J. Am. Chem. Soc., 1985, 107, 6097 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2001, 123, 4886 CrossRef CAS.
- C. P. Andrieux, M. Robert, F. D. Saeva and J.-M. Savéant, J. Am. Chem. Soc., 1994, 116, 7864 CrossRef CAS.
- S. Antonello and F. Maran, J. Am. Chem. Soc., 1997, 119, 12595 CrossRef CAS.
- C. P. Andrieux and J.-M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1986, 205, 43 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158 CrossRef CAS.
- B. Persson, J. Electroanal. Chem. Interfacial Electrochem., 1978, 86, 313 CrossRef CAS.
- Optimization of the reduced forms of the studied structures (1a–g + one electron) at each bond length value at the B3LYP/6-31G(p,d) level has been performed using Gaussian 2003 package65.
- What is represented on the plots in Fig. 4 is not the absolute potential energy but rather the difference between the absolute value and a value lower than the minimum energy.
- J. C. Imbeaux and J.-M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1973, 44, 169 CrossRef CAS.
- J.-M. Savéant and D. Tessier, J. Electroanal. Chem. Interfacial Electrochem., 1975, 65, 57 CrossRef CAS.
- J.-M. Savéant, J. Phys. Chem. B, 2002, 106, 9387 CrossRef CAS.
- J.-M. Savéant, J. Am. Chem. Soc., 1992, 114, 10595 CrossRef CAS.
- The
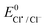 was calculated from the equation
was calculated from the equation 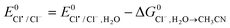 82.
82. - Y. Marcus, Pure Appl. Chem., 1985, 57, 1103 CAS.
- The BDEs have been calculated as the difference between the total molecular energy of the neutral molecules and that of the radicular fragments obtained by homolytic dissociation. The entropies were also deduced as
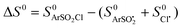 .
. - A. Houmam, H. Muhammad, K. Koczkur, M. Chahma and D. F. Thomas, Chem. Commun., 2011, 47, 7095 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical data, theoretical calculation methodology and data of all studied compounds. See DOI: 10.1039/c1cp22130b |
| This journal is © the Owner Societies 2012 |