XANES and XPS investigations of the local structure and final-state effects in amorphous metal silicates: (ZrO2)x(TiO2)y(SiO2)1−x−y
Michael W.
Gaultois
and
Andrew P.
Grosvenor
*
Department of Chemistry, University of Saskatchewan, Saskatoon, Saskatchewan, Canada S7N 5C9. E-mail: andrew.grosvenor@usask.ca; Fax: +1 (306) 966-4730; Tel: +1 (306) 966-4660
First published on 14th November 2011
Abstract
Amorphous quaternary [(ZrO2)x(TiO2)y(SiO2)1−x−y] and ternary [(ZrO2)x(SiO2)1−x] silicates were synthesized using a sol–gel method and examined viaXPS and XANES. Metal silicates are important industrial materials, though structural characterization is complicated because of their amorphous nature. Hard (Ti K- and Zr K-edge) and soft (Ti L2,3-edge) X-ray XANES spectra suggest the Ti and Zr coordination numbers in the quaternary silicates remain constant as the metal identity or total metal content (x, y, or x + y in the chemical formula) is varied. XPS core-line spectra from the quaternary silicates show large decreases in Ti 2p3/2, Zr 3d5/2, Si 2p3/2, and O 1s binding energies due to increasing final-state relaxation with greater next-nearest neighbour substitution of Si for less-electronegative Ti/Zr, which was confirmed by analysis of the O Auger parameter. These decreases in binding energy occur without any changes in the ground-state energies (e.g., oxidation state) of these atoms, as examined by Ti L2,3-edge, Si L2,3-edge, and O K-edge XANES. Because most spectroscopic investigations are concerned with ground-state properties, knowledge of the contributions from final-state effects is important to understand the spectra from materials of interest.
1. Introduction
Amorphous transition-metal silicates (e.g., (MO2)x(SiO2)1−x; M = Ti, Zr, Hf) have been the subject of many investigations because of their tuneable properties and industrial applications. Many of the desirable properties depend on the metal content, which can be adjusted to control the refractive index, thermal expansion coefficient, hardness, chemical durability, and dielectric constant of the materials.1–10TiO2 is often incorporated into SiO2 to make glasses with a high refractive index and ultra-low thermal expansion, ZrO2 substitution greatly increases the resistance to alkaline solutions, and (HfO2)x(SiO2)1−x has been proposed as a high-k dielectric replacement for SiO2 gate dielectrics in transistors.4–6,11–13 Quaternary ((MO2)x(M′O2)y(SiO2)1−x−y; M, M′ = Ti, Zr, Hf) mixed-metal silicates can also be made, and allow the combination of beneficial properties from each of the ternary metal silicates ((MO2)x(SiO2)1−x). This creates a wealth of chemical possibilities; yet, despite the considerable volume of work on the ternary silicates, the quaternary silicates have received considerably less attention.9,14–18Traditionally, silicates are made via melt syntheses; however, the high temperatures required to melt transition-metal silicates (often >1300 °C) cause phase segregation and crystallization, leading to heterogeneous products.15,17,19 Consequently, low-temperature soft processing techniques, such as sol–gel synthesis, have led the way to making amorphous, homogeneous glasses with high metal content.17,19–21 Solution-based routes have the added benefit of a range of post-synthetic solution processing techniques, such as spin-coating for thin-film applications or fibre drawing and spinning for use in composites or textiles.7,22,23 The lack of long-range order in amorphous materials hinders the use of diffraction-based techniques that are typically used to probe the structure and homogeneity. As a result, amorphous materials are ideal candidates for study by X-ray spectroscopy, which is sensitive to only the first few coordination shells of the element being examined.
In this work, amorphous (ZrO2)x(TiO2)y(SiO2)1−x−y and (ZrO2)x(SiO2)1−x (quaternary and ternary metal silicates, respectively) were synthesized by sol–gel methods and investigated by X-ray absorption near-edge spectroscopy (XANES) and X-ray photoelectron spectroscopy (XPS). This examination has allowed for a study of the influence of substitution on both the local chemical environment of the constituent elements and the overall electronic structure of the material. XPS is routinely used to examine the electronic structure of materials in the solid state, yet many effects remain poorly understood and require further study to understand their role in spectral energy shifts. (The materials examined here are excellent candidates for such a study.) The role played by the next-nearest neighbour (NNN) in causing shifts in XPS binding energies (BE) is one such effect, and is the focus of this study. Knowledge of the many factors that can influence BEs is important to better our understanding of why spectral energies change, which can then be used to aid investigations of materials, such as the silicates discussed here.
2. Experimental
2.1. Synthesis of (ZrO2)x(SiO2)1−x and (ZrO2)x(TiO2)y(SiO2)1−x−y
The amorphous quaternary [(ZrO2)x(TiO2)y(SiO2)1−x−y (x + y = 0.20, 0.30)] and ternary [(ZrO2)x(SiO2)1−x (x = 0.10, 0.20, 0.25, 0.33, 0.50)] silicates were synthesized using a sol–gel method.3,21 (All reactions were performed under a N2(g) atmosphere with constant stirring.) The Si alkoxide was prehydrolyzed by combining tetraethyl orthosilicate (≥99%, Sigma Aldrich), 100% ethanol, and water in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 molar ratio with catalytic amounts of HCl.21 This solution was stirred under reflux conditions at 80 °C for 2 h and then cooled to room temperature. Ethanol was then added to dilute this solution, after which titanium (IV) tert-butoxide (99.95%, Strem) was added drop-wise. (The amount of ethanol added was such that the ethanol:Ti alkoxide molar ratio was 20:1.) After stirring this solution for one hour, ethanol was added and zirconium (IV) tert-butoxide (99%, Strem) was then added drop-wise with stirring (ethanol:Zr alkoxide molar ratio = 20:1). (The alkoxides were added in order of increasing reactivity—Si, Ti, then Zr—to promote the formation of homogenous products.15,24) Once all of the alkoxides were added, water was then added drop-wise with constant stirring to increase the water:alkoxide (Ti, Zr, and Si alkoxide) molar ratio to 2:1. Finally, the mixture was sealed under N2(g) and stirred until a gel formed (7–21 days), after which the material was exposed to the atmosphere for >7 days. (Higher Ti content and/or overall metal content reduced the time required to gel.) Powders were obtained by placing the resulting materials (transparent glasses) under vacuum for >24 h, grinding with a mortar and pestle, and then annealing them in air at 550 °C for 12 h to remove residual organic material. The formation of these materials was confirmed by use of Fourier transform infrared spectroscopy, and the lack of long-range order, even after annealing at temperatures up to 550 °C, was confirmed by powder X-ray diffraction using Cu Kα radiation generated from a Rigaku Rotaflex RU-200 rotating anode X-ray diffractometer.3,9,15
:2:1 molar ratio with catalytic amounts of HCl.21 This solution was stirred under reflux conditions at 80 °C for 2 h and then cooled to room temperature. Ethanol was then added to dilute this solution, after which titanium (IV) tert-butoxide (99.95%, Strem) was added drop-wise. (The amount of ethanol added was such that the ethanol:Ti alkoxide molar ratio was 20:1.) After stirring this solution for one hour, ethanol was added and zirconium (IV) tert-butoxide (99%, Strem) was then added drop-wise with stirring (ethanol:Zr alkoxide molar ratio = 20:1). (The alkoxides were added in order of increasing reactivity—Si, Ti, then Zr—to promote the formation of homogenous products.15,24) Once all of the alkoxides were added, water was then added drop-wise with constant stirring to increase the water:alkoxide (Ti, Zr, and Si alkoxide) molar ratio to 2:1. Finally, the mixture was sealed under N2(g) and stirred until a gel formed (7–21 days), after which the material was exposed to the atmosphere for >7 days. (Higher Ti content and/or overall metal content reduced the time required to gel.) Powders were obtained by placing the resulting materials (transparent glasses) under vacuum for >24 h, grinding with a mortar and pestle, and then annealing them in air at 550 °C for 12 h to remove residual organic material. The formation of these materials was confirmed by use of Fourier transform infrared spectroscopy, and the lack of long-range order, even after annealing at temperatures up to 550 °C, was confirmed by powder X-ray diffraction using Cu Kα radiation generated from a Rigaku Rotaflex RU-200 rotating anode X-ray diffractometer.3,9,15
2.2. XANES
000 eV. Finely ground samples were sandwiched between Kapton tape, and positioned 45° to the X-ray beam. Transmission spectra were measured with N2-filled ionization chambers and partial fluorescence spectra were collected using a Canberra 13-element Ge detector. Comparison with transmission spectra showed no self-absorption effects in the fluorescence spectra presented here. The X-ray energy was increased by 0.15 eV per step through the Ti K absorption edge and 0.3 eV per step through the Zr K-edge. Spectra were calibrated by comparison to spectra collected concurrently in transmission mode from Ti and Zr metal, which have well-known absorption-edge energies (4966 eV and 17998 eV, respectively).25 The Ti K-edge spectrum from anatase (TiO2; 99.6%, Alfa Aesar) was also collected to provide a standard for comparison of pre-edge peak intensities and absorption energies. The precision of the measurements is estimated to be better than ±0.1 eV, as determined by comparison of spectra from samples and standards collected throughout these studies.
2.3. XPS
XPS spectra were collected using a Kratos AXIS 165 spectrometer and a monochromatic Al Kα (1486.7 eV) X-ray source. The resolution of this instrument has been determined to be 0.4 eV and the precision of the measured binding energies (BE) is better than ±0.10 eV.28 Finely ground powders of (ZrO2)x(TiO2)y(SiO2)1−x−y, TiO2 (anatase), SiO2 (99.99%, Alfa Aesar), and monoclinic ZrO2 (99.7%, Alfa Aesar) were pressed into In foil and mounted on an electrically grounded sample holder. High-resolution spectra of the Ti 2p, Si 2p, O 1s, and C 1s core lines were collected using a pass energy of 20 eV, a step size of 0.05 eV, and a sweep time of 180 s. To counter surface charging, a charge neutralizer was used. (Ar+ ion etching was not used to clean the surface of the samples, as testing on related materials ((TiO2)x(SiO2)1−x showed preferential reduction of Ti.29) All spectra were calibrated using the C 1s core-line arising from adventitious C with a fixed value of 284.8 eV. Charge neutralization and calibration using adventitious C has been previously shown to be effective for analyzing these materials.30 A Shirley-type function was applied to remove the background arising from energy loss and spectra were fitted using synthetic peaks having a combined Gaussian (70%) and Lorentzian (30%) line profile.31 The Si 2p3/2 BE was found by fitting the Si 2p core-line envelope with two peaks (Si 2p3/2 and Si 2p1/2) having a 2:1 peak area ratio (2p3/2:2p1/2).
2.4. Electronic structure calculations
To confirm the interpretation of the complicated O K-edge XANES spectra, full-potential linear muffin-tin orbital (FP-LMTO) calculations were performed using LMTART 7 to determine the total and partial (O 2p, Ti 3d, Zr 4d) density of unoccupied states.32–35 Owing to difficulties inherent in modeling amorphous systems, calculations were performed for crystalline systems (ZrSiO4 and TiSiO4); previous studies using this model have accurately reproduced some of the properties (e.g., the dielectric constant) of amorphous Ti and Zr silicates.36–38 (The true electronic structure of these amorphous silicates likely lies somewhere between the two substitutional end-members modeled here.) Self-consistency was achieved by requiring the convergence of the total energy to be smaller than 10−4 eV per unit cell, and the Brillouin zone integration was performed over 1000 k-points (10 × 10 × 10) using the tetrahedron method.39,40 Crystallographic parameters of ZrSiO4 and TiSiO4 used for the calculations were taken from the literature.41,42 Parameters for TiSiO4 were based on a theoretical study, as the structure-type of TiSiO4 is not thermodynamically stable under ambient conditions.423. Results and discussion
3.1. Transition metal K-edge XANES
Transition metal K-edge XANES spectra were collected to examine how the Ti and Zr coordination number (CN) change as a function of the total metal content and Zr:Ti ratio (Fig. 1). Metal K-edge spectra result from 1s → (n − 1)d (quadrupolar, pre-edge) and 1s → np (dipolar, main-edge) transitions, and examination of these spectra from a series of compounds can lead to useful information on changes in CN.43–50 With decreasing CN, the higher energy and more intense, dipolar portion of the spectrum (i.e., the main-edge, above ∼4980 eV for Ti and ∼18010 eV for Zr) decreases in energy and intensity. This change is a result of poorer final-state screening of the core-hole and reduced number of metal–ligand states (e.g., Ti 4p – O 2p or Zr 5p – O 2p in the silicates studied here) available for 1s electrons to be excited to.49–54 The main edge absorption energy is also characteristic of the metal oxidation state, and the Ti K- and Zr K-edge absorption energies for the silicate materials studied here were consistent with an oxidation state of 4+. (This is supported by other investigations of titanium silicates, where multiple techniques have found that Ti has an oxidation state of 4+.55) Examination of the pre-edge region of Ti K-edge XANES spectra (4965–4975 eV) can also lead to important information on how the CN of a Ti centre changes in a series of compounds. As the CN decreases from 6 (and inversion symmetry is lost), Ti 3d states are partially overlapped by 4p states, which lends a dipolar component to the excitation and results in a more intense Ti K-edge pre-edge peak.44,47,48 Although changes in the pre-edge region of Zr K-edge spectra also occur, they are more difficult to follow because the Zr K pre-edge is partially obscured by the main edge. This is a result of the decreased separation between the 4d and 5p states (compared to the Ti 3d and 4p states), the increase in the broadening of the spectra associated with core-hole lifetime effects, and the decrease in instrumental resolution with greater excitation energy.
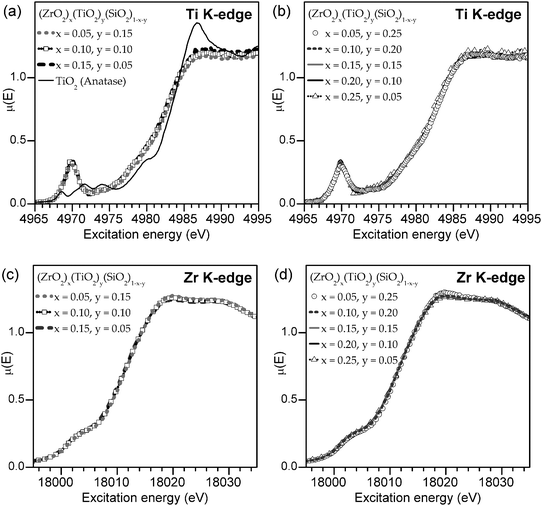 | ||
| Fig. 1 Ti (a, b) and Zr (c, d) K-edge XANES spectra from (ZrO2)x(TiO2)y(SiO2)1−x−y with a fixed total metal content (x + y) of 20 mol% (a, c) and 30 mol% (b, d). The lack of changes in the lineshape is evidence that there is no significant change in either the Zr or Ti coordination number (CN) with increasing metal content. A spectrum from a TiO2 (anatase) standard is shown in (a) as an example of the lineshape expected if the Ti4+ CN was 6. | ||
3.2. Zr 3d5/2, Ti 2p3/2, Si 2p, and O 1s XPS binding energy (BE) shifts
To investigate the changes in electronic structure as the identity and concentration of the metal is changed, high-resolution Zr 3d, Ti 2p, Si 2p, and O 1s core-line XPS spectra were collected. It has been shown that the photoelectron BEs can change as a result of shifts in the ground- and/or final-state energies, though the cause of the shift cannot be determined by XPS alone.62–64 To describe why BEs shift, a version of the charge potential model is shown in eqn (3.1), where ground- and final-state effects have been grouped together in the first and second set of parentheses, respectively.62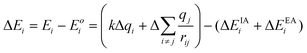 | (3.1) |
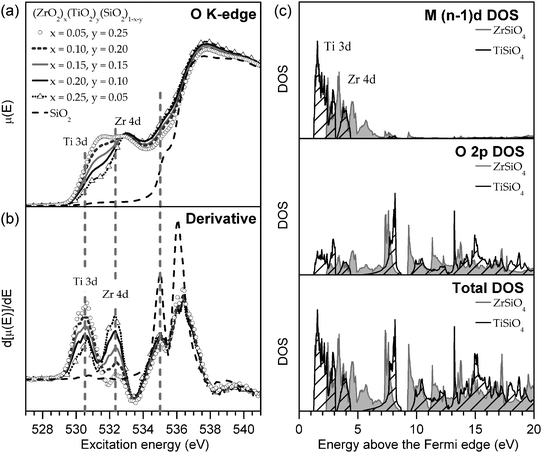
The total binding energy shift, ΔEi, which is the difference between the measured binding energy of the atom of interest, Ei, and a reference binding energy, Eoi, consists of a number of contributions. This shift in BE occurs due to changes at and around the absorbing atom, given by the first and second terms, respectively, in each set of parentheses in eqn (3.1). In the ground state, shifts in BE result from changes in the screening of the nuclear charge of the atom being excited, i. Here, the kΔqi term describes changes in the charge of the atom being excited (Δqi), while the 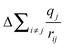 term (often called the Madelung potential) describes changes in the charge of atoms (j) located around atom i at a distance rij.62 (k is a parameter that describes the Coulombic interaction between core and valence electrons.65) In the final state, after a photoelectron has been ejected from atom i and an unscreened core-hole has been produced, the remaining electrons will rearrange to minimize the total energy of the system. In the presence of this newly-created attractive potential, the electrons in the system relax towards the core-hole; shifts in BE owing to final-state relaxation result from changes in the magnitude of relaxation, which depends on the ability of electrons to screen the core-hole produced by removal of a photoelectron. Extra-atomic relaxation of electrons from surrounding atoms, represented by ΔEEAi, is sensitive to changes in the chemical environment, whereas intra-atomic relaxation of electrons localized on the atom of interest is not, so ΔEIAi is often negligible.62
term (often called the Madelung potential) describes changes in the charge of atoms (j) located around atom i at a distance rij.62 (k is a parameter that describes the Coulombic interaction between core and valence electrons.65) In the final state, after a photoelectron has been ejected from atom i and an unscreened core-hole has been produced, the remaining electrons will rearrange to minimize the total energy of the system. In the presence of this newly-created attractive potential, the electrons in the system relax towards the core-hole; shifts in BE owing to final-state relaxation result from changes in the magnitude of relaxation, which depends on the ability of electrons to screen the core-hole produced by removal of a photoelectron. Extra-atomic relaxation of electrons from surrounding atoms, represented by ΔEEAi, is sensitive to changes in the chemical environment, whereas intra-atomic relaxation of electrons localized on the atom of interest is not, so ΔEIAi is often negligible.62
To study how increasing the total metal content affects the electronic structure of these materials, XPS core-line spectra from (ZrO2)x(TiO2)y(SiO2)1−x−y were collected (Fig. 2 and 3). All core-line photoelectron peaks (Zr 3d5/2, Ti 2p3/2, Si 2p, and O 1s) show a significant decrease in BE with increasing total metal content (x + y, mol% ZrO2 + TiO2), but show little to no variation in BE when the total metal content is fixed and the Zr:Ti ratio is varied. The O 1s spectra of the quaternary silicates exhibit a shoulder at low BE that increases in intensity with metal content (Fig. 3b). This low-BE contribution is attributed to the formation of metal-rich domains in the SiO2 matrix, whereas the significantly more intense high-BE peak results from homogeneously mixed transition-metal and Si atoms.66 (This heterogeneity is known to occur in metal silicates at higher metal loadings.3,61,67) Additionally, the O 1s spectrum of the monoclinic ZrO2 (m-ZrO2) standard shows a high-BE shoulder at energies characteristic of surface adsorbates, which is understandable given that the standard was not pre-treated by Ar+ sputtering and/or in vacuoannealing.68
![(a) Zr 3d and (b) Ti 2p3/2XPS core-line spectra of the quaternary silicates and binary oxides. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y. The Zr 3d and Ti 2p BEs decrease with increasing total metal content. (Small variations (±0.1 eV) were observed between samples with identical total metal content, though these variations are near the limits of instrumental precision (±0.1 eV) and lack a monotonic trend.)](/image/article/2012/CP/c1cp22717c/c1cp22717c-f2.gif) | ||
| Fig. 2 (a) Zr 3d and (b) Ti 2p3/2XPS core-line spectra of the quaternary silicates and binary oxides. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y. The Zr 3d and Ti 2p BEs decrease with increasing total metal content. (Small variations (±0.1 eV) were observed between samples with identical total metal content, though these variations are near the limits of instrumental precision (±0.1 eV) and lack a monotonic trend.) | ||
![(a) Si 2p and (b) O 1s XPS core-line spectra of the quaternary silicates and binary oxides. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y. The Si 2p and O 1s binding energies (BE) decrease with increasing total metal content. The O 1s spectra of the quaternary silicates have a shoulder at low BE that increases in intensity with metal content. This peak is the result of metal-rich domains, which are known to exist in metal silicates at high metal loadings.3,61,66,67](/image/article/2012/CP/c1cp22717c/c1cp22717c-f3.gif) | ||
| Fig. 3 (a) Si 2p and (b) O 1s XPS core-line spectra of the quaternary silicates and binary oxides. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y. The Si 2p and O 1s binding energies (BE) decrease with increasing total metal content. The O 1s spectra of the quaternary silicates have a shoulder at low BE that increases in intensity with metal content. This peak is the result of metal-rich domains, which are known to exist in metal silicates at high metal loadings.3,61,66,67 | ||
To make analysis feasible for the many samples investigated, the BE of the photoelectron peak maximum is plotted as a function of total metal content (Fig. 4). BEs from ternary silicates ((ZrO2)x(SiO2)1−x and (TiO2)x(SiO2)1−x) are also plotted to show the close agreement with values obtained from the quaternary silicates at similar total metal content (≤0.1 eV difference when the total metal content is identical).29 As the total metal content increases, the variation of the quaternary BEs at a fixed total metal content increases, though there is no monotonic trend in BE as the Zr:Ti ratio is changed (see Fig. 2 and Fig. 3 for the individual spectra), and the small deviations are at the limits of instrumental precision (±0.1 eV). Such small deviations may be due to minor differences in synthetic conditions or heterogeneity, the latter of which is well known in metal silicates when the metal content is high.3,61,67 As the total metal content (x + y) increases from 0.1 → 1, the Zr 3d5/2, Ti 2p3/2, and O 1s BEs decrease by 1.0, 1.3, and 2.5 eV, respectively. Although a similar compositional range is not accessible for Si, the Si 2p3/2 BE decreases by 0.9 eV from (x + y) = 0 → 0.3.
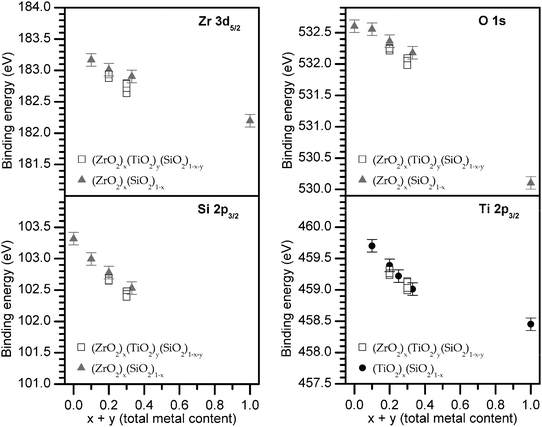 | ||
| Fig. 4 The binding energy (BE) of the photoelectron peak maximum decreases with increasing total metal content (mol% ZrO2 + TiO2). The Ti 2p3/2 BEs from (TiO2)x(SiO2)1−x are from a previous study.29 | ||
It is clear that there are large decreases in BE with increasing total metal content, but the underlying cause of the BE shift is unknown. For example, substitution of Si4+ atoms by more electropositive Ti4+ or Zr4+ atoms (χSi = 1.74, χTi = 1.32, χZr = 1.22) may increase final-state relaxation experienced by electrons in the material, increasing ΔEEAi in eqn (3.1) and decreasing the BE. This substitution could also lead to more electron density at and/or around the absorbing atom in the ground-state, decreasing kΔqi and/or 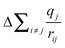 in eqn (3.1) and decreasing the BE.62,63,69,70 Additionally, the incorporation of metal centres into the materials will increase the average bond distances (rSi4+(CN:4) = 0.26 Å, rTi4+(CN:5) = 0.51 Å, rZr4+(CN:8) = 0.84 Å), which could also affect
in eqn (3.1) and decreasing the BE.62,63,69,70 Additionally, the incorporation of metal centres into the materials will increase the average bond distances (rSi4+(CN:4) = 0.26 Å, rTi4+(CN:5) = 0.51 Å, rZr4+(CN:8) = 0.84 Å), which could also affect 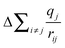 .62,63,71 To separate ground- and final-state effects and identify the major contributors to the observed decrease in BE, complementary techniques, such as XANES, are required.
.62,63,71 To separate ground- and final-state effects and identify the major contributors to the observed decrease in BE, complementary techniques, such as XANES, are required.
3.3. Ti L2,3-, Si L2,3-, and O K-edge XANES
To determine the origin of the BE shifts in the XPS core-line spectra, soft X-ray XANES spectra (Ti L2,3-, Si L2,3-, and O K-edge) were collected and compared to the corresponding XPS spectra (Ti 2p, Si 2p, and O 1s). XANES absorption energies shift for similar reasons as XPS BEs. The important difference between XANES and XPS lies in the final state produced by each method. In XPS, the final state of the system being probed is electron deficient and has an unscreened core-hole (the core electron having been removed from the material), so relaxation effects due to the core-hole are greater than in XANES (where the core electron is promoted to conduction states and can partially screen the core-hole).72–74 Thus, when the ground state is identical and electrons of equal energy (i.e., the same orbital) are probed by both techniques (e.g., Ti 2p3/2XPS and Ti L3-edge XANES), the difference in the final state of analogous XPS and XANES excitations can be exploited to resolve ground- and final-state effects in XPS BE shifts. Additionally, the lineshape of Ti L2,3- and Si L2,3-edge spectra can be used to determine the coordination environment around these elements.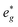 states (see the TiO2 (anatase) spectrum in Fig. 5a).
states (see the TiO2 (anatase) spectrum in Fig. 5a).
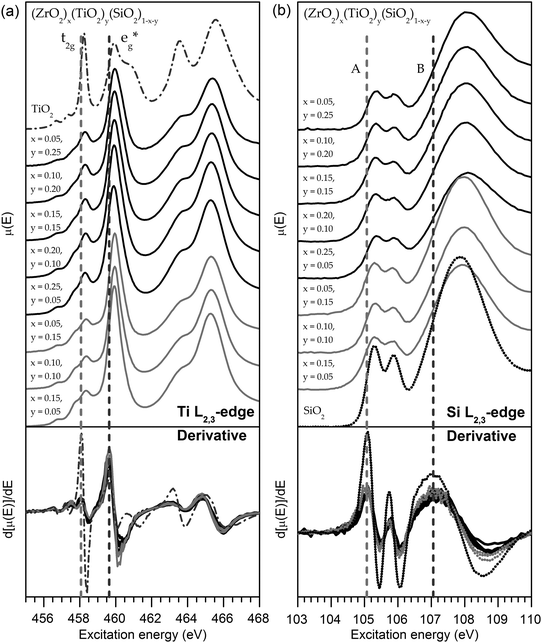 | ||
| Fig. 5 (a) Ti L2,3-edge and (b) Si L2,3-edge XANES spectra of the quaternary silicates. Dashed lines are a guide to indicate the absorption-edge energy, which is taken to be the peak maximum in the first derivative. In both sets of spectra, a <0.1 eV shift in the absorption-edge energy was observed across the series as the total metal content is changed. The lack of substantial shift indicates there is no change in the ground-state energies of these materials. Spectra from TiO2 (anatase) and SiO2 (fused silica) standards are shown as examples of the lineshape expected if the Ti4+ CN was 6 or the Si4+ CN was 4. | ||
Owing to the complex lineshape and numerous spectral features, Ti L2,3-edge spectra are characteristic of the Ti CN, and comparison to crystalline standards with known CNs allows the estimation of the Ti CN.80 The L3-edge spectra show 2 peaks, and the largest distinguishing factor between spectra from Ti having different CNs is the ratio between high- and low-energy peak intensities, which is large when Ti is 5-coordinate (∼3), and small when Ti is either 4- or 6- coordinate (∼1).80 The L2,3-edge spectra collected from the quaternary silicates studied here have lineshapes and absorption-edge energies that are characteristic of 5-coordinate Ti4+ (Fig. 5a), though the presence of other-coordinate species in small amounts cannot be ruled out.80 Further, the lineshape remains largely unchanged with increasing total metal content, suggesting the Ti CN is constant. These findings are consistent with findings from the Ti K-edge spectra, presented earlier (Section 3.1). The static CN is important to isolating the cause of the XPS BE shifts observed earlier (Section 3.2), as changes in CN have been shown to influence BEs.50,81 To separate ground- and final-state effects in the XPS Ti 2p3/2 BE shifts, Ti L3-edge absorption energies were examined to probe the Ti 2p ground-state energies. Ti L3-edge spectra (Fig. 5a) showed no change in the L3-edge absorption energy (<0.1 eV; determined by the maximum in the first derivative shown in Fig. 5a), indicating that ground-state effects are not responsible for the Ti 2p BE shift observed by XPS.
Note here that the Ti K-edge spectra are bulk sensitive, whereas the Ti L2,3-edge spectra are surface sensitive because of the energies and detection methods used.82 Because XPS is surface sensitive, it is important that differences between the surface and the bulk of the material are accounted for, particularly when comparing shifts in XANES absorption energy. Whereas transmission and fluorescence (FLY) measurements rely upon the detection of photons, the signal of a TEY measurement (used for collecting Ti L2,3-edge spectra in this work) is a function of the number of electrons emitted from the sample. Owing to the short inelastic mean free path (IMFP) length of electrons, only electrons emitted near the surface are able to escape from the material.82,83 The IMFP also tends to increase with kinetic energy, and the Ti L2,3-edge is quite low in energy, <500 eV, further enhancing the surface sensitivity.82,83
Comparison of the spectra collected at the Si L2,3-edge reveals only subtle changes in the lineshape (such as a slight broadening of the peaks with increasing metal content) and a <0.1 eV change in absorption energy across all samples (Fig. 5b). The lineshape is similar to SiO2, and confirms that Si has an oxidation state of 4+ and remains in a 4-coordinate, tetrahedral geometry in the materials investigated here.86 This is supported by previous investigations of (TiO2)x(SiO2)1−x using Si K-edge XANES.89 As there are no changes in the Si CN, the decrease in the Si 2p BE cannot be due to CN-induced shifts. Further, the lack of significant changes in the Si L2,3-edge absorption energies (<0.1 eV shift; determined by the maximum in the first derivative shown in Fig. 5b) is testament to the lack of ground-state shifts in the Si 2p BEs with substitution. Previous studies of Ca-, Al-, Mg-, and Fe-containing silicates have shown that Si L2,3-edge absorption energies shift with substitution of the next-nearest neighbour (NNN) cation due to ground-state effects.86 In those instances, ground-state shifts likely arose due to changes in the Madelung potential (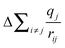 in eqn (3.1)) owing to changes in the NNN charge (qj), but these effects are not present in the Zr- and Ti- silicates studied here, as the oxidation states of Zr4+ and Ti4+ are fixed.
in eqn (3.1)) owing to changes in the NNN charge (qj), but these effects are not present in the Zr- and Ti- silicates studied here, as the oxidation states of Zr4+ and Ti4+ are fixed.
 | ||
| Fig. 6 (a) O K-edge XANES spectra of the quaternary silicates. No shift was observed in the absorption-edge energy (535.0 eV) as Zr replaces Ti, as monitored by (b) the first derivative of the O K-edge spectra. Dashed lines are a guide to indicate the absorption-edge energy, which is taken to be the peak maximum in the first derivative. (c) The metal (n − 1)d and O 2p partial density of states and the total density of states of ZrSiO4 and TiSiO4 were calculated to confirm assignment of the O K-edge spectra. | ||
As with the Ti L2,3- or Si L2,3-edge, any shift in the O K-edge absorption energy would signal that there are changes in ground-state energies with substitution. The O K-edge spectra of the quaternary silicates show a lower-energy feature that becomes more prominent with increasing Ti content (Fig. 6a), owing to the greater number of unoccupied Ti 3d states (Fig. 6c). The Ti 3d states are found at lower energies than the Zr 4d states due to the decreased average distance of 3d electrons to the nucleus (i.e., 3d states are bound more tightly than 4d states). Examination of this feature shows no change in absorption energy (determined by the maximum in the first derivative shown in Fig. 6b) when the metal content is varied. Similar examination of the higher-energy feature in the O K-edge spectra (∼535.0 eV) reveals no shift in absorption energy (Fig. 6a), suggesting that the O ground-state energies are insensitive to changes in the identity of the metal. This was also found to be the case during examination of the Ti L2,3- and Si L2,3-edge spectra (Sections 3.3.1 and 3.3.2).
3.4. Identifying the cause of XPS BE shifts
Previous XPS studies on representative mid- and end-members of the (ZrO2)x(SiO2)1−x series (i.e., SiO2, ZrSiO4, and ZrO2) have also found decreases in core-line BEs with increasing metal content, but the shifts were attributed to increased ground-state charge density at the absorbing atom.30 Core-line BEs and Auger transitions were collected, after which the Auger parameter was used to estimate ΔEEAi and DFT calculations were used to determine the contribution from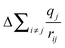 .30 It was found that the calculated increase in the Madelung potential (
.30 It was found that the calculated increase in the Madelung potential (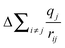 ) was opposed by a measured increase in relaxation of similar magnitude, so it was concluded that the decrease in BEs with increasing metal content was due to increased charge density at the absorbing atom (kΔqi).30 However, XANES absorption energies are also sensitive to changes in charge density in the ground-state, and the spectra presented here show no change in absorption energies with metal content. This suggests that any ground-state contributions, although likely present, oppose each other and do not cause a net change in the ground-state core-level energies.
) was opposed by a measured increase in relaxation of similar magnitude, so it was concluded that the decrease in BEs with increasing metal content was due to increased charge density at the absorbing atom (kΔqi).30 However, XANES absorption energies are also sensitive to changes in charge density in the ground-state, and the spectra presented here show no change in absorption energies with metal content. This suggests that any ground-state contributions, although likely present, oppose each other and do not cause a net change in the ground-state core-level energies.
The XPS core-line spectra, reported in section 3.2, showed a large decrease in the photoelectron BEs of all elements in (ZrO2)x(TiO2)y(SiO2)1−x−y with increasing total metal content. Meanwhile, soft X-ray XANES spectra (Ti L2,3-, Si L2,3- and O K-edge), reported in Section 3.3, showed no changes in absorption energies, implying that ground-state effects are not a major contributor in the XPS BE shifts. This leaves changes in extra-atomic final-state relaxation (ΔEEAi) as the driving cause of these BE shifts. Previous studies on oxygen-deficient perovskite-type materials have shown that a change in CN can change the magnitude of relaxation.81 In the metal silicates studied here, although the average CN of the absorbing atom (from either Si, Zr, or Ti) does not change with increasing total metal content, the average CN of the Si/Zr/Tisite does increase as Si is replaced by metal atoms. (In the silicates examined here, the average CN of Si is 4, Ti is roughly 5, and Zr is >6.) Given the large difference in the average Ti and Zr CNs, a difference between the ternary silicate BEs would be expected if an increase in the average CN of the nearest neighbour or next-nearest neighbour (NNN) site was a major contributor to the BE shifts. However, the core-line BE shifts from (ZrO2)x(SiO2)1−x and (TiO2)x(SiO2)1−x agree within the limits of instrumental precision (±0.1 eV) to those from (ZrO2)x(TiO2)y(SiO2)1−x−y at similar metal loadings (x = 0 → 0.33). The close agreement between the shifts in BE from the ternary and quaternary silicates, which have different average metal CNs, suggests that the average metal CN does not play a major role in the BE shifts observed in the quaternary silicates.29,50
Instead, the BE shifts in the silicates are caused by substitution of Si for Ti or Zr atoms, which are much less electronegative (χSi = 1.74, χTi = 1.32, χZr = 1.22).69,70 The close agreement between ternary BE shifts further supports the conclusion that the electronegativity difference (Δχ) is the primary contributor. As metal atoms replace Si and the average electronegativity of the Si/Zr/Ti site decreases, the electron density becomes less tightly bound. Consequently, electron density from the chemical environment surrounding the absorbing atom can relax to a greater extent around the core-hole produced during an XPS experiment, lowering the final-state energy and leading to a decrease in the BE. Revisiting the charge potential model in eqn (3.1), these shifts resulting from final-state effects can be described as a change in the magnitude of the ΔEEAi term, which describes changes in extra-atomic relaxation.
3.5. The Auger parameter
The electronic structure of a material is sensitive to changes in the chemical environment, and can be probed by many spectroscopic techniques. As was discussed in Sections 3.2 and 3.4, there are numerous factors that influence the BE of a photoelectron peak, making complementary techniques necessary to elucidate the major contributors to a shift in energy. By comparing the shifts in XPS BEs and XANES absorption energies of transitions involving the same core electrons (e.g., the 2p3/2 core-line and the L3-edge), it is argued in this work that the observed decrease in BEs with increasing total metal content is due to greater final-state relaxation. To confirm that final-state effects are responsible for the BE shifts, the O KLL Auger transition was monitored and the Auger parameter, one of the few experimental measures of relaxation, was extracted (Fig. 7 and 8).93,94![O KLL Auger spectra from (a) quaternary and (b) ternary silicates. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y.](/image/article/2012/CP/c1cp22717c/c1cp22717c-f7.gif) | ||
| Fig. 7 O KLL Auger spectra from (a) quaternary and (b) ternary silicates. Dashed lines are a guide to indicate the approximate peak maximum for a set of spectra at a given total metal content, [x + y] in (ZrO2)x(TiO2)y(SiO2)1−x−y. | ||
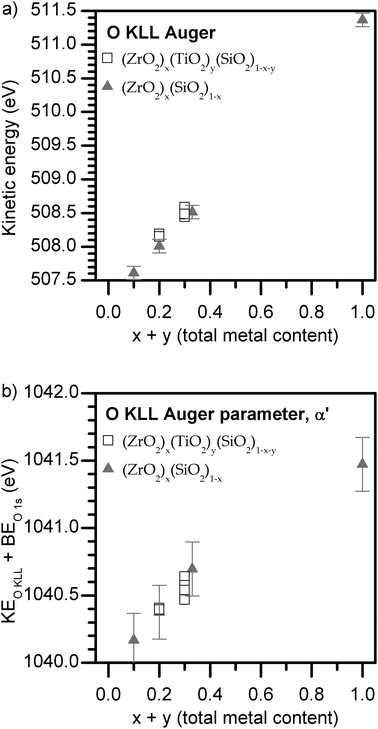 | ||
| Fig. 8 (a) O KLL Auger peak maxima energies and (b) Auger parameters of the quaternary and ternary silicates. The Auger parameter increases with total metal content, confirming that final-state relaxation increases in the same fashion. The precision of α′ is estimated to be twice the BE precision. | ||
After primary photoelectron emission in XPS, the excited atom is left with a core-hole. In the Auger KLL Auger process, this K-shell (1s) core-hole (K+) subsequently decays with relaxation of an L-shell (2p) electron and ejection of an L-shell electron, leaving two holes in the L-shell (L+L+). The kinetic energy (KE) of the Auger electron is approximated by eqn (3.2), where Ei terms represent ground-state energies of core shells, and ΔEEAi terms represent the relaxation energies of core shells. (Where appropriate, ground-state effects have been placed in square brackets to help distinguish them from final-state effects.)
| KEAuger = [Ei(K) − Ei(L) − Ei(L)] − EEAi(K+) + EEAi(L+L+) | (3.2) |
Because the KLL Auger KE depends on the difference in energy between the K and L shells, the Auger KE will increase if the K-shell becomes more stabilized (lower energy), while the Auger KE will decrease if the L-shell becomes more stabilized. (This is why Ei(L) terms have the opposite sign as Ei(K) terms.) Accordingly, a shift in the kinetic energy of the KLL Auger electron is given by eqn (3.3). By analogy, a shift in the BE of a 1s (K-shell) photoelectron examined by XPS is given by eqn (3.4).
| ΔKEAuger = [ΔEi(K) − 2ΔEi(L)] − ΔEEAi(K+) + ΔEEAi(L+L+) | (3.3) |
| ΔBEXps = [ΔEi(K)] − ΔEEAi(K+) | (3.4) |
Provided that only core shells are involved in Auger emission, the shift in ground-state energy of the different core shells is approximately equal (i.e., ΔEi(K) = ΔEi(L) = ΔEi,ground) and the shift in the Auger KE simplifies to eqn (3.5).93
| ΔKEAuger = [−ΔEi,ground] − ΔEEAi(K+) + ΔEEAi(L+L+) | (3.5) |
| Δα′ = ΔKEAuger + ΔBEXPS = 2ΔEEAi | (3.6) |
4. Conclusions
Amorphous quaternary [(ZrO2)x(TiO2)y(SiO2)1−x−y] and ternary [(ZrO2)x(SiO2)1−x] silicates were synthesized using sol–gel methods and examined by XANES and XPS to determine the effect of the next-nearest neighbour (NNN) on XPS BEs of inorganic solids. By varying the composition, Si can be replaced by Zr or Ti, which are both less-electronegative (χSi = 1.74, χTi = 1.32, χZr = 1.22).69,70Zr K-edge, Ti K-edge, Ti L2,3-edge, and Si L2,3-edge XANES spectra confirm that the coordination number (CN) and oxidation state of Zr,4+ Ti4+, and Si4+ do not change with increasing metal content. In the (ZrO2)x(TiO2)y(SiO2)1−x−y series, the substitution of Si for group IV metals (Zr and Ti) with lower electronegativity increases the amount of final-state relaxation experienced by atoms in the material upon excitation, decreasing all core-line binding energies (BE). The observation of significant shifts in the XPS BEs of the quaternary transition-metal silicates without accompanying shifts in analogous XANES absorption energies (e.g., the 2p3/2 core-line and the L3-edge) demonstrates the dominant influence of increasing final-state relaxation with next-nearest neighbour (NNN) substitution in the silicates (Fig. 9). The increase in final-state relaxation with total metal content has been supported through analysis of the Auger parameter. This investigation has provided another example to improve our understanding of the many influences that make analysis of XPS spectra complicated, and has highlighted that large changes in BE can occur without any changes in ground-state energies (e.g., oxidation state).![Plot of XANES absorption energies and XPS BEs for transitions involving the same core electrons (Si 2p). Spectral energies from ternary silicates [(ZrO2)x(SiO2)1−x (◆)] are included to show the close agreement with the quaternary silicates [(ZrO2)x(TiO2)y(SiO2)1−x−y (■)]. The absorption energies remain unchanged while the BEs decrease, indicating that final-state effects are dominant. The height of the symbols represents the precision of the measured energy (±0.1 eV).](/image/article/2012/CP/c1cp22717c/c1cp22717c-f9.gif) | ||
| Fig. 9 Plot of XANES absorption energies and XPS BEs for transitions involving the same core electrons (Si 2p). Spectral energies from ternary silicates [(ZrO2)x(SiO2)1−x (◆)] are included to show the close agreement with the quaternary silicates [(ZrO2)x(TiO2)y(SiO2)1−x−y (■)]. The absorption energies remain unchanged while the BEs decrease, indicating that final-state effects are dominant. The height of the symbols represents the precision of the measured energy (±0.1 eV). | ||
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada through a discovery grant awarded to APG and a Julie Payette research scholarship awarded to MG. The University of Saskatchewan is also thanked for supporting this work through a new-faculty start-up grant awarded to APG. Dr Robert Gordon of Sector 20 at the Advanced Photon Source is thanked for help in carrying out XANES measurements at PNC/XSD-CAT. PNC/XSD facilities at the Advanced Photon Source, and research at these facilities, are supported by the US Department of Energy - Basic Energy Sciences, a Major Resources Support grant from NSERC, the University of Washington, Simon Fraser University and the Advanced Photon Source. Use of the Advanced Photon Source is also supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Mr. Tom Regier, Mr. David Chevrier, Dr. Lucia Zuin, and Mr. Chris Ryan are thanked for help in carrying out XANES measurements using beamlines located at the CLS (11ID-1 and 11ID-2). The CLS is supported by NSERC, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. Dr Dimitre Karpuzov from the Alberta Centre for Surface Engineering and Science, University of Alberta, is thanked for collecting the XPS spectra.References
- M. A. Villegas, A. de Pablos and J. M. Fernandez-Navarro, Glass Technol., 1994, 35, 276–280 CAS.
- G. Mountjoy, D. M. Pickup, R. Anderson, G. W. Wallidge, M. A. Holland, R. J. Newport and M. E. Smith, Phys. Chem. Chem. Phys., 2000, 2, 2455–2460 RSC.
- G. W. Wallidge, R. Anderson, G. Mountjoy, D. M. Pickup, P. Gunawidjaja, R. J. Newport and M. E. Smith, J. Mater. Sci., 2004, 39, 6743–6755 CrossRef CAS.
- K. Kamiya and S. Sakka, J. Mater. Sci., 1980, 15, 2937–2939 CrossRef CAS.
- T. Hayashi, T. Yamada and H. Saito, J. Mater. Sci., 1983, 18, 3137–3142 CrossRef CAS.
- K. Kamiya, S. Sakka and Y. Tatemichi, J. Mater. Sci., 1980, 15, 1765–1771 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol–gel science: the physics and chemistry of sol–gel processing, Academic Press, Boston, 1990 Search PubMed.
- D. Poitras, S. Larouche and L. Martinu, Appl. Opt., 2002, 41, 5249–5255 CrossRef CAS.
- G. Mountjoy, M. A. Holland, G. W. Wallidge, P. Gunawidjaja, M. E. Smith, D. M. Pickup and R. J. Newport, J. Phys. Chem. B, 2003, 107, 7557–7566 CrossRef CAS.
- D. Brassard, D. K. Sarkar, M. A. El Khakani and L. Ouellet, J. Vac. Sci. Technol., A, 2006, 24, 600–605 CAS.
- A. I. Kingon, J. P. Maria and S. K. Streiffer, Nature, 2000, 406, 1032–1038 CrossRef CAS.
- G. D. Wilk, R. M. Wallace and J. M. Anthony, J. Appl. Phys., 2000, 87, 484–492 CrossRef CAS.
- J. Robertson, Rep. Prog. Phys., 2006, 69, 327–396 CrossRef CAS.
- W. Beier, A. A. Goktas and G. H. Frischat, J. Am. Ceram. Soc., 1986, 69, C148–C150 CrossRef.
- U. Wellbrock, W. Beier and G. H. Frischat, J. Non-Cryst. Solids, 1992, 147, 350–355 CrossRef.
- A. Wannagon, N. Mishima, R. Ota, T. Wakasugi and J. Fukunaga, Mater. Sci. Res. Int., 1997, 3, 210–215 CAS.
- A. Wannagon, N. Mishima, T. Wakasugi, R. Ota and J. Fukunaga, J. Ceram. Soc. Jpn., 1997, 105, 940–946 CrossRef CAS.
- P. N. Gunawidjaja, M. A. Holland, G. Mountjoy, D. M. Pickup, R. J. Newport and M. E. Smith, Solid State Nucl. Magn. Reson., 2003, 23, 88–106 CrossRef CAS.
- B. E. Yoldas, J. Mater. Sci., 1979, 14, 1843–1849 CrossRef CAS.
- B. E. Yoldas, J. Mater. Sci., 1977, 12, 1203–1208 CrossRef CAS.
- B. E. Yoldas, J. Non-Cryst. Solids, 1980, 38-9, 81–86 CrossRef.
- M. N. Rahaman, Ceramic processing and sintering, Marcel Dekker, New York, 2003 Search PubMed.
- K. Jiang, A. Zakutayev, J. Stowers, M. D. Anderson, J. Tate, D. H. McIntyre, D. C. Johnson and D. A. Keszler, Solid State Sci., 2009, 11, 1692–1699 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- A. Thompson, D. Attwood, E. Gullikson, M. Howells, K.-J. Kim, J. Kirz, J. Kortright, I. Lindau, P. Pianetta, A. Robinson, J. Scofield, J. Underwood, D. Vaughan, G. Williams and H. Winick, X-Ray data booklet, Lawrence Berkeley National Laboratory, Berkeley, CA, 2009 Search PubMed.
- T. Regier, J. Krochak, T. K. Sham, Y. F. Hu, J. Thompson and R. I. R. Blyth, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 582, 93–95 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- A. P. Grosvenor, R. G. Cavell and A. Mar, J. Solid State Chem., 2007, 180, 2702–2712 CrossRef CAS.
- M. W. Gaultois and A. P. Grosvenor, J. Mater. Chem., 2011, 21, 1829–1836 RSC.
- M. J. Guittet, J. P. Crocombette and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter, 2001, 63, 125117 CrossRef.
- R. L. Martin and D. A. Shirley, J. Chem. Phys., 1976, 64, 3685–3689 CrossRef CAS.
- S. Y. Savrasov and D. Y. Savrasov, Phys. Rev. B: Condens. Matter, 1992, 46, 12181–12195 CrossRef.
- S. Y. Savrasov, Phys. Rev. B: Condens. Matter, 1996, 54, 16470–16486 CrossRef CAS.
- S. Y. Savrasov, Z. Kristallogr., 2005, 220, 555–557 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244–13249 CrossRef.
- A. Sandell, B. Sanyal, L. E. Walle, P. Uvdal and A. Borg, J. Electron Spectrosc. Relat. Phenom., 2011, 183, 107–113 CrossRef CAS.
- G.-M. Rignanese, in Materials Fundamentals of Gate Dielectrics, ed. A. A. Demkov and A. Navrotsky, Springer, Dordrecht, Netherlands, 2005, pp. 249–290 Search PubMed.
- G. M. Rignanese, X. Rocquefelte, X. Gonze and A. Pasquarello, Int. J. Quantum Chem., 2005, 101, 793–801 CrossRef CAS.
- O. Jepsen and O. K. Andersen, Solid State Commun., 1971, 9, 1763–1767 CrossRef.
- O. Jepsen and O. K. Andersen, Phys. Rev. B, 1984, 29, 5965–5965 CrossRef.
- R. J. Finch, J. M. Hanchar, P. W. O. Hoskin and P. C. Burns, Am. Mineral., 2001, 86, 681–689 CAS.
- L. Gracia, A. Beltran and D. Errandonea, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 094105 CrossRef.
- M. Wilke, F. Farges, P. E. Petit, G. E. Brown and F. Martin, Am. Mineral., 2001, 86, 714–730 CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- F. Farges, Am. Mineral., 1997, 82, 36–43 CAS.
- F. Farges, G. E. Brown and J. J. Rehr, Phys. Rev. B: Condens. Matter, 1997, 56, 1809–1819 CrossRef CAS.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- D. Cabaret, A. Bordage, A. Juhin, M. Arfaoui and E. Gaudry, Phys. Chem. Chem. Phys., 2010, 12, 5619–5633 RSC.
- A. P. Grosvenor and J. E. Greedan, J. Phys. Chem. C, 2009, 113, 11366–11372 CAS.
- M. W. Gaultois, J. E. Greedan and A. P. Grosvenor, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 192–195 CrossRef CAS.
- I. S. Elfimov, V. I. Anisimov and G. A. Sawatzky, Phys. Rev. Lett., 1999, 82, 4264–4267 CrossRef CAS.
- C. Weigel, G. Calas, L. Cormier, L. Galoisy and G. S. Henderson, J. Phys.: Condens. Matter, 2008, 20, 135219 CrossRef.
- R. le Toquin, W. Paulus, A. Cousson, C. Prestipino and C. Lamberti, J. Am. Chem. Soc., 2006, 128, 13161–13174 CrossRef CAS.
- G. Berlier, G. Spoto, S. Bordiga, G. Ricchiardi, P. Fisicaro, A. Zecchina, I. Rossetti, E. Selli, L. Forni, E. Giamello and C. Lamberti, J. Catal., 2002, 208, 64–82 CrossRef CAS.
- G. N. Vayssilov, Catal. Rev. Sci. Eng., 1997, 39, 209–251 CAS.
- F. Farges, J. Non-Cryst. Solids, 1996, 204, 53–64 CrossRef CAS.
- F. Farges, G. E. Brown and J. J. Rehr, Geochim. Cosmochim. Acta, 1996, 60, 3023–3038 CrossRef CAS.
- W. B. Kim, S. H. Choi and J. S. Lee, J. Phys. Chem. B, 2000, 104, 8670–8678 CrossRef CAS.
- F. Farges, G. E. Brown, A. Navrotsky, H. Gan and J. J. Rehr, Geochim. Cosmochim. Acta, 1996, 60, 3039–3053 CrossRef CAS.
- D. M. Pickup, G. Mountjoy, G. W. Wallidge, R. J. Newport and M. E. Smith, Phys. Chem. Chem. Phys., 1999, 1, 2527–2533 RSC.
- G. Mountjoy, R. Anderson, R. J. Newport and M. E. Smith, J. Phys.: Condens. Matter, 2000, 12, 3505–3519 CrossRef CAS.
- D. Briggs, Surface analysis of polymers by XPS and static SIMS, Cambridge University Press, Cambridge, U.K., 1998 Search PubMed.
- P. S. Bagus, F. Illas, G. Pacchioni and F. Parmigiani, J. Electron Spectrosc. Relat. Phenom., 1999, 100, 215–236 CrossRef CAS.
- R. J. Cole, D. A. C. Gregory and P. Weightman, Phys. Rev. B: Condens. Matter, 1994, 49, 5657–5661 CrossRef CAS.
- P. A. W. van der Heide, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 79–91 CrossRef CAS.
- A. Y. Stakheev, E. S. Shpiro and J. Apijok, J. Phys. Chem., 1993, 97, 5668–5672 CrossRef CAS.
- R. Anderson, G. Mountjoy, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 1998, 232, 72–79 CrossRef.
- C. M. Pradier, C. Hinnen, K. Jansson, L. Dahl, M. Nygren and A. Flodstrom, J. Mater. Sci., 1998, 33, 3187–3191 CrossRef CAS.
- A. L. Allred and E. G. Rochow, J. Inorg. Nucl. Chem., 1958, 5, 264–268 CrossRef CAS.
- E. J. Little and M. M. Jones, J. Chem. Educ., 1960, 37, 231–233 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. D. Leapman, L. A. Grunes and P. L. Fejes, Phys. Rev. B, 1982, 26, 614–635 CrossRef CAS.
- R. L. Opila, G. D. Wilk, M. A. Alam, R. B. van Dover and B. W. Busch, Appl. Phys. Lett., 2002, 81, 1788–1790 CrossRef CAS.
- A. Nilsson, R. E. Palmer, H. Tillborg, B. Hernnas, R. J. Guest and N. Martensson, Phys. Rev. Lett., 1992, 68, 982–985 CrossRef CAS.
- J. P. Crocombette and F. Jollet, J. Phys.: Condens. Matter, 1994, 6, 10811–10821 CrossRef CAS.
- T. Hoche, P. Ohle, R. Keding, C. Russel, P. A. Van Aken, R. Schneider, H. J. Kleebe, X. Q. Wang, A. J. Jacobson and S. Stemmer, Philos. Mag., 2003, 83, 165–178 CrossRef CAS.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1990, 42, 5459–5468 CrossRef CAS.
- F. De Groot, Coord. Chem. Rev., 2005, 249, 31–63 CrossRef CAS.
- J. C. Fuggle and S. F. Alvarado, Phys. Rev. A: At., Mol., Opt. Phys., 1980, 22, 1615–1624 CrossRef CAS.
- G. S. Henderson, X. Liu and M. E. Fleet, Phys. Chem. Miner., 2002, 29, 32–42 CrossRef CAS.
- M. W. Gaultois and A. P. Grosvenor, J. Phys. Chem. C, 2010, 114, 19822–19829 CAS.
- M. Kasrai, W. N. Lennard, R. W. Brunner, G. M. Bancroft, J. A. Bardwell and K. H. Tan, Appl. Surf. Sci., 1996, 99, 303–312 CrossRef CAS.
- C. J. Powell, A. Jablonski, S. Tanuma and D. R. Penn, J. Electron Spectrosc. Relat. Phenom., 1994, 68, 605–616 CrossRef CAS.
- Y. Iguchi, Sci. Light, 1977, 26, 161–181 CAS.
- D. Li, G. M. Bancroft, M. Kasrai, M. E. Fleet, X. H. Feng and K. H. Tan, Am. Mineral., 1994, 79, 785–788 CAS.
- L. A. J. Garvie and P. R. Buseck, Am. Mineral., 1999, 84, 946–964 CAS.
- J. A. van Bokhoven, T. Nabi, H. Sambe, D. E. Ramaker and D. C. Koningsberger, J. Phys.: Condens. Matter, 2001, 13, 10247–10260 CrossRef CAS.
- D. Li, G. M. Bancroft, M. Kasrai, M. E. Fleet, X. H. Feng, K. H. Tan and B. X. Yang, Solid State Commun., 1993, 87, 613–617 CrossRef CAS.
- G. S. Henderson and M. E. Fleet, J. Non-Cryst. Solids, 1997, 211, 214–221 CrossRef CAS.
- L. A. Grunes, R. D. Leapman, C. N. Wilker, R. Hoffmann and A. B. Kunz, Phys. Rev. B, 1982, 25, 7157–7173 CrossRef CAS.
- F. M. F. de Groot, M. Grioni, J. C. Fuggle, J. Ghijsen, G. A. Sawatzky and H. Petersen, Phys. Rev. B, 1989, 40, 5715–5723 CrossRef CAS.
- H. Kurata, E. Lefevre, C. Colliex and R. Brydson, Phys. Rev. B: Condens. Matter, 1993, 47, 13763–13768 CrossRef CAS.
- C. D. Wagner, Faraday Discuss. Chem. Soc., 1975, 60, 291–300 RSC.
- G. Moretti, J. Electron Spectrosc. Relat. Phenom., 1998, 95, 95–144 CrossRef CAS.
- T. D. Thomas and P. Weightman, Phys. Rev. B, 1986, 33, 5406–5413 CrossRef CAS.
- T. D. Thomas, J. Electron Spectrosc. Relat. Phenom., 1980, 20, 117–125 CrossRef CAS.
- N. D. Lang and A. R. Williams, Phys. Rev. B, 1979, 20, 1369–1376 CrossRef CAS.
- G. Hohlneicher, H. Pulm and H. J. Freund, J. Electron Spectrosc. Relat. Phenom., 1985, 37, 209–224 CrossRef CAS.
- R. J. Cole, P. Weightman and J. A. D. Matthew, J. Electron Spectrosc. Relat. Phenom., 2003, 133, 47–53 CrossRef CAS.
- P. S. Bagus, A. Wieckowski and H. Freund, Chem. Phys. Lett., 2006, 420, 42–46 CrossRef CAS.
- M. Satta and G. Moretti, J. Electron Spectrosc. Relat. Phenom., 2010, 178, 123–127 CrossRef.
| This journal is © the Owner Societies 2012 |