Excited-state proton-relay dynamics of 7-hydroxyquinoline controlled by solvent reorganization in room temperature ionic liquids†
Hyeongtaek
Lim
a,
Hyeok
Jeong
a,
Sun-Young
Park
a,
Jin Yong
Lee
b and
Du-Jeon
Jang
*a
aSchool of Chemistry, Seoul National University, NS60, Seoul 151-742, Korea. E-mail: djjang@snu.ac.kr; Fax: +82-2-875-6624; Tel: +82-2-880-4368
bDepartment of Chemistry, Sungkyunkwan University, Suwon 440-746, Korea
First published on 10th November 2011
Abstract
The excited-state triple proton relay of 7-hydroxyquinoline (7HQ) along a hydrogen-bonded methanol chain in room temperature ionic liquids (RTILs) has been investigated using picosecond time-resolved fluorescence spectroscopy. The rate constant of the proton relay in a methanol-added RTIL is found to be slower by an order of magnitude than that in bulk methanol and to have unity in its kinetic isotope effect. These suggest that the excited-state tautomerization dynamics of 7HQ in methanol-added RTILs is mainly controlled by the solvent reorganization dynamics to form a cyclically hydrogen-bonded complex of 7HQ·(CH3OH)2 upon absorption of a photon due to high viscosity values of RTILs. Because the cyclic complex of 7HQ·(CH3OH)2 at the ground state is unstable in RTILs, the collision-induced slow formation of the cyclic complex should take place upon excitation prior to undergoing subsequent intrinsic proton transfer rapidly.
1. Introduction
Proton transfer plays an important role in a wide variety of chemical and biological phenomena such as water autoionization, fast proton diffusion, acid–base reactions, DNA mutagenesis, enzyme catalysis, and proton pumping through membrane protein channels.1–11 In particular, long-distance proton transport in biological systems is often mediated by hydrogen (H)-bonded chains.7–9 The size, the structure, and the motion of mediating solvent molecules as well as the nature of prototropic groups determine the dynamics of such long-ranged proton transport.9,10 Thus, it is essential to study solvent-mediated proton transfer in detail to understand proton transport in fundamental chemical and biological processes. A number of researchers have investigated the mechanisms of long-ranged proton transport in biological systems using simple molecular models to avoid the structural complexity and the massiveness of the systems. One of the widely used experimental molecular models is 7-hydroxyquinoline (7HQ),2,4,11–29 which is an amphoteric heterocyclic aromatic molecule having both a photoacidic enolic group and a photobasic imino group. For the excited-state proton transfer (ESPT) of 7HQ, protic solvents such as water and alcohols are indispensible because the two prototropic groups of 7HQ are too far from each other to donate or accept a proton directly. On one hand, 7HQ in neat water has been reported to undergo ESPT in a stepwise manner via forming an anionic intermediate species.18,19 On the other hand, in a neat alcohol (ROH), the two-step ESPT mechanism shown in Fig. 1 has been widely discussed.17,24–28 The first step is the solvent reorganization (k1) of normal 7HQ (N) to form a cyclically H-bonded 7HQ·(ROH)2 complex (NCC), and the second step is intrinsic proton transfer (k2) catalyzed by the complexed solvent molecules to produce tautomeric 7HQ (T). In one limit, solvent reorganization can be the rate-determining step so that the observed ESPT rate constant (kPT) of 7HQ becomes k1. In the opposite limit, as equilibrium (K = k1/k−1) between solvent reorganization and solvent randomization (k−1) is rapid relative to k2, kPT is independent of solvent dynamics and is expressed as Kk2. The static role of solvation is reported to be kPT = k2 exp(−ΔG‡/kBT) in the manner of the transition-state theory,30 where ΔG‡ is the free energy of formation for NCC. Solvation to achieve an appropriate configuration is generally believed to be prerequisite to efficient proton tunneling.4,28,30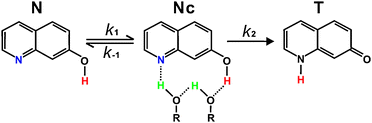 | ||
| Fig. 1 Two-step model for the excited-state proton relay of 7HQ along a hydrogen-bonded alcohol chain. | ||
Varma and co-workers have observed that T* fluorescence generated by the photoexcitation of 7HQ in an alcohol reveals two rise components, concluding that there are two different excited-state tautomerization processes.24 They have suggested that the fast process is associated with the intrinsic proton transfer of NCC formed already at the moment of excitation while the slow process arises from solvent reorganization to form NCC, which subsequently undergoes proton transfer rapidly. Because the simultaneous observation of two different ESPT pathways probably causes difficulties in interpreting experimental results, it is strongly desirable to explore each step separately to understand the alcohol-mediated ESPT of 7HQ fully. To avoid the interference of solvent reorganization in ESPT, we have prepared NCC in the ground state by adding a small amount of an alcohol in a nonpolar aprotic solvent.28 Consequently, we have studied the intrinsic ESPT of NCC directly to show that the ESPT of 7HQ along a H-bonded alcohol chain is initiated by the protonation of the imino group and subsequently completed by the fast deprotonation of the enol group. However, in this study, we will show that the ESPT dynamics of 7HQ in a room temperature ionic liquid (RTIL) having a small amount of methanol is entirely governed by solvent reorganization to form NCC*.
RTILs are molten salts having melting points below room temperature. RTILs have attracted considerable attention due to their remarkable properties such as low volatility, high polarity, high viscosity, easy recycling, high ionic conductivity, miscibility with other solvents, and high selectivity.31–44 They have been widely used as media in organic and inorganic syntheses, separation processes, electrochemical studies, and catalytic processes.34–40 In particular, their negligible vapor pressure makes them suitable solvents for green chemistry. RTILs typically consist of nitrogen- or phosphorus-containing large organic cations and organic or inorganic anions.31 Because the physicochemical properties of RTILs depend on both their cationic and anionic constituents, RTILs with desired properties can be obtained by the proper choice of individual ionic components. The roles of RTILs as media in various photophysical and photochemical processes have also been studied extensively,29,41–43 revealing that the solvent effects of RTILs on photoinduced processes are largely different from those of conventional solvents. However, it is often difficult to interpret experimental results owing to the unique properties of RTILs.43 Thus, a variety of studies on photoinduced processes in RTILs are required to understand the effects and the properties of RTILs thoroughly.
Previously, Bhattacharya and Samanta29 studied the ESPT of 7HQ in methanol-added RTILs of 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim][PF6], Chart 1) and 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide to observe the delayed ESPT. Considering that the ESPT rate became fast as the viscosity of RTILs was lowered with the addition of methanol, they suggested that the high viscosity of RTILs caused difficulty in solvent fluctuations around NCC* which gave rise to the delayed ESPT. However, they could not obtain a clear relationship between the RTIL viscosity and the ESPT rate, indicating that the microviscosity around a 7HQ molecule is quite different from the bulk viscosity. In this study, we investigate the excited-state proton relay of 7HQ along a H-bonded methanol chain in two different RTILs of [bmim][PF6] and 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4], Chart 1) using picosecond time-resolved fluorescence spectroscopy. Our results also show that the excited-state proton-relay dynamics of 7HQ in methanol-added RTILs is remarkably retarded. However, although Bhattacharya and Samanta implicitly assumed that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 cyclic complexes of NCC formed already in the ground state,29 we have found that NCC is unstable in RTILs and that the collision-induced formation of NCC* should occur within the lifetime of N* to produce T*. Accordingly, the excited-state proton-relay dynamics of 7HQ along a methanol chain is entirely controlled by solvent reorganization to form NCC*. The unique properties of RTILs such as high viscosity and ionic characters are considered to exert strong influence on the excited-state tautomerization dynamics of 7HQ molecules in methanol-added RTILs. Because water contacting with the surface of a protein or confined in a cell membrane is known to be much more viscous than bulk water, highly viscous media of RTILs can be good model systems of biological environments. Thus, our results would provide significant information to understand the medium effect on proton transfer dynamics in biological systems.
:2 cyclic complexes of NCC formed already in the ground state,29 we have found that NCC is unstable in RTILs and that the collision-induced formation of NCC* should occur within the lifetime of N* to produce T*. Accordingly, the excited-state proton-relay dynamics of 7HQ along a methanol chain is entirely controlled by solvent reorganization to form NCC*. The unique properties of RTILs such as high viscosity and ionic characters are considered to exert strong influence on the excited-state tautomerization dynamics of 7HQ molecules in methanol-added RTILs. Because water contacting with the surface of a protein or confined in a cell membrane is known to be much more viscous than bulk water, highly viscous media of RTILs can be good model systems of biological environments. Thus, our results would provide significant information to understand the medium effect on proton transfer dynamics in biological systems.
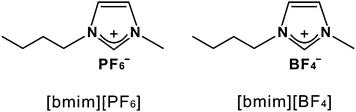 | ||
| Chart 1 Structures of employed room temperature ionic liquids. | ||
2. Experimental methods
RTILs of [bmim][PF6] and [bmim][BF4], purchased from C-TRI, were purified by the following known procedures41 and were kept in a vacuum before use. 7HQ (99%) from Acros, CH3O1H (anhydrous, ≥99.8%) and CH3O2H (isotopic purity ≥99.5%) from Sigma-Aldrich were used as-purchased. The concentrations of 7HQ were kept at 0.1 mM for all of the samples in this study. While absorption spectra were measured with a UV/vis spectrophotometer (Scinco, S-3100), emission spectra were obtained with a home-built fluorometer consisting of a Xe lamp of 75 W (Acton Research, XS432) with a monochromator of 0.15 m (Acton Research, Spectrapro-150) and a photomultiplier tube (Acton Research, PD438) attached to a monochromator of 0.30 m (Acton Research, Spectrapro-300). A mode-locked Nd:YAG laser of 25 ps (Quantel, Pizzicato) and a streak camera of 10 ps (Hamamatsu, C2830) attached to a CCD detector (Princeton Instruments, RTE128H) were employed for the excitation and the detection of fluorescence kinetic profiles, respectively. Samples were excited with 315 nm pulses generated through a Raman shifter filled with methane at 15 atm and pumped by the fourth-harmonic pulses (266 nm) of the laser. Emission wavelengths were selected by combining band-pass filters and cut-off filters. Fluorescence kinetic constants were extracted by fitting kinetic profiles to computer-simulated exponential curves convoluted with instrumental response functions. All of the measurements were carried out at room temperature.3. Results and discussion
The lowest absorption of 7HQ in [bmim][PF6] shifts to the red and loses its vibronic structures with the concentration increase of methanol (Fig. 2). The spectral changes imply that 7HQ molecules associate with methanol molecules by H bonding to produce complexes in RTILs. We have obtained Benesi–Hildebrand plots45 to find out that 7HQ and methanol molecules form 1:2 complexes in [bmim][PF6] with an association constant (Ka) of 0.32 M−2. Similar behaviors were also observed in the absorption of 7HQ in a nonpolar aprotic solvent with the addition of an alcohol.25,27,28 Compared with the Ka value of 7HQ·(CH3OH)2 in n-heptane (9500 M−2),28 the Ka value in [bmim][PF6] is extremely small because the medium is ionic to be highly miscible with methanol. The small Ka value of 0.32 M−2 suggests that 7HQ·(CH3OH) complexes as well as free 7HQ molecules also exist considerably with 7HQ·(CH3OH)2 complexes in RTILs. The most stable structure of 7HQ·(CH3OH)2 in the ground state has been reported to have the cyclic geometry of NCC.26–28 However, our kinetic measurements indicate that the stable structure of 7HQ·(CH3OH)2 in RTILs is not the cyclic structure of NCC (see below). The addition of methanol does not result in perceivable absorption around 410 nm, suggesting that proton-translocated tautomeric molecules in RTILs are not stable to exist at the ground state.
![Absorption and emission spectra, with excitation at 320 nm, of 0.1 mM 7HQ in [bmim][PF6] having methanol concentrations indicated inside. Note that background luminescence originating from neat [bmim][PF6] was subtracted from each of the emission spectra. The absorption spectrum of neat [bmim][PF6] is provided in Fig. S1 of the ESI, while the emission spectra of neat [bmim][PF6] and 7HQ in [bmim][PF6] (before subtraction) are shown in Fig. S2 of the ESI.](/image/article/2012/CP/c1cp22329a/c1cp22329a-f2.gif) | ||
| Fig. 2 Absorption and emission spectra, with excitation at 320 nm, of 0.1 mM 7HQ in [bmim][PF6] having methanol concentrations indicated inside. Note that background luminescence originating from neat [bmim][PF6] was subtracted from each of the emission spectra. The absorption spectrum of neat [bmim][PF6] is provided in Fig. S1 of the ESI†, while the emission spectra of neat [bmim][PF6] and 7HQ in [bmim][PF6] (before subtraction) are shown in Fig. S2 of the ESI.† | ||
Only the UV emission band around 370 nm from N* appears in neat RTILs having no methanol. This implies that 7HQ molecules in neat RTILs cannot produce T* upon excitation although the hydrogen atom bound to the C2 of the imidazolium cation of RTILs is known to participate in H bonding.29,44 With the addition of methanol, the UV emission band shifts to the red while the visible fluorescence band around 530 nm increases in intensity. Because largely Stokes-shifted visible fluorescence around 530 nm is known to arise from T* generated by ESPT, we consider that the ESPT of 7HQ in RTILs is operative in the presence of methanol. In addition, the intensity increase of T* fluorescence with the increment of [CH3OH] suggests that the photoexcitation of 7HQ in methanol-added RTILs produces T*via forming NCC* and subsequently undergoing intrinsic proton transfer (see below). Note that we present only the absorption and emission spectra of 7HQ in [bmim][PF6] because the spectra obtained in [bmim][BF4] show very similar behaviors.
The fluorescence kinetic profiles of samples containing 0.1 mM 7HQ in neat RTILs, monitored at 380 nm, showed biexponential decay times (Fig. 3 and Table 1). For example, in [bmim][PF6], two decay times of 1000 ps (65%) and 3000 ps (35%) were observed at 380 nm. Whereas the fast decay time (k−1f) of 1000 ps is attributed to the relaxation time (k−1rel) of N*, the slow decay time of 3000 ps is considered to arise from the background luminescence decay of the RTIL. The latter is evidenced by the fact that the observed slow decay time in [bmim][PF6] is similar to that in [bmim][BF4] and is almost invariant with methanol addition (Table 1). Furthermore, the slow decay time at 380 nm was also observed in 7HQ-free neat RTILs. On excitation of a sample containing 0.1 mM 7HQ in [bmim][PF6] with 1 M methanol at 315 nm, fluorescence monitored at 380 nm has a biphasic decay profile consisting of 700 ps (60%) and 3000 ps (40%), while tautomer fluorescence recorded at 550 nm rises in 1100 ps and decays in 2400 ps (Table 1). As the concentration of methanol increases, both the fast decay time at 380 nm and the rise time at 550 nm become short in time. Those two time constants are reasonably correlated with each other, implying that the fast decay time at 380 nm (k−1f) is associated with the ESPT time (k−1PT) of N* by the relation kf = krel + kPT. Assuming that the k−1rel value is the fast decay time of fluorescence at 380 nm (1000 ps) in a sample containing 0.1 mM 7HQ in neat [bmim][PF6], we can calculate the k−1PT value to be 2300 ps for a sample with 1 M methanol. However, the k−1PT value cannot be assigned to the intrinsic proton transfer time (k−12) of NCC* to form T*. The intrinsic ESPT time of 62 ps was deduced from the fast decay time at the normal fluorescence of 7HQ in methanol-added n-heptane, and its value remained invariant with changes of methanol concentrations.28 In methanol-added RTILs, both the fast decay time of N* fluorescence and the rise time of T* fluorescence, correlated with the ESPT time, decrease with the concentration of methanol, indicating that the collision-induced formation of NCC* occurs to conduct ESPT within the lifetime of N*. Thus, we suggest that the kPT obtained from this study is mostly determined by the solvent reorganization (k1) of N* to produce NCC*, as described using the two-step ESPT model shown in Fig. 1.
![Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having methanol concentrations of 0 M (triangles), 2 M (squares), and 4 M (circles). Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants.](/image/article/2012/CP/c1cp22329a/c1cp22329a-f3.gif) | ||
| Fig. 3 Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having methanol concentrations of 0 M (triangles), 2 M (squares), and 4 M (circles). Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants. | ||
| RTIL | [CH3OH]/M | [CH3CN]/M | λ em/nm | Rise time/ps | Decay time/ps |
|---|---|---|---|---|---|
| a The viscosity values of [bmim][PF6] and [bmim][BF4] at 20 °C are 312 and 233 cP, respectively,31 while those of methanol and acetonitrile at 25 °C are 0.54 and 0.37 cP, respectively.47 The viscosity value of [bmim][PF6] containing 4 M CH3OH was measured to be 96 cP at room temperature while that of [bmim][PF6] containing 1 M CH3OH and 3 M CH3CN was measured to be 82 cP. b Instant. c Initial amplitude percentage of each component. d The slow component of each kinetic profile at 380 nm is attributed to background luminescence from a neat RTIL. | |||||
| [bmim][PF6] | 0 | 0 | 380 | —b | 1000 (65%)c + 3000 (35%)d |
| 1 | 0 | 380 | — | 700 (60%) + 3000 (40%) | |
| 550 | 1100 | 2400 | |||
| 2 | 0 | 380 | — | 600 (60%) + 3000 (40%) | |
| 550 | 600 | 2400 | |||
| 4 | 0 | 380 | — | 300 (60%) + 3000 (40%) | |
| 550 | 300 | 2500 | |||
| 1 | 1 | 380 | — | 700 (60%) + 3200 (40%) | |
| 550 | 1100 | 2500 | |||
| 1 | 3 | 380 | — | 700 (55%) + 2600 (45%) | |
| 550 | 1100 | 2500 | |||
| [bmim][BF4] | 0 | 0 | 380 | — | 800 (75%) + 3000 (25%) |
| 1 | 0 | 380 | — | 700 (70%) + 3000 (30%) | |
| 550 | 1300 | 3000 | |||
| 2 | 0 | 380 | — | 600 (65%) + 2400 (35%) | |
| 550 | 900 | 2700 | |||
| 4 | 0 | 380 | — | 400 (65%) + 3000 (35%) | |
| 550 | 500 | 2300 |
On one hand, in nonpolar media such as n-alkanes, Nc exists stably in the ground state, so that their intrinsic ESPT can be observed directly without being interfered by solvent reorganization.28 Accordingly, the ESPT time of Nc* in a nonpolar medium is invariant regardless of the concentration of methanol, indicating that the collision-induced formation of Nc* does not take place to conduct ESPT. On the other hand, in the polar media of RTILs having ionic character, Nc is not stable to exist in the ground state, so that upon excitation, the formation of Nc* should occur prior to intrinsic ESPT. As a result, the overall rate constant of ESPT of Nc* in polar RTILs is much smaller than the ESPT rate constant of Nc* in nonpolar n-heptane which has been reported to be 62 ps.28 The rate constant of the ESPT of 7HQ·(alcohol)2 complexes is influenced considerably by the polarity of a medium. We have previously reported that the ESPT energy barrier of 7HQ·(ethanol)2 complexes decreases with the increasing dielectric constant (ε) of a medium; the intrinsic ESPT of 7HQ·(ethanol)2 complexes becomes faster in a more polar medium due to the lower energy barrier of the ESPT.17 Moreover, we have previously shown that the intrinsic ESPT rate of 7HQ·(water)2 complexes is larger in diethyl ether (ε = 4.27 at 20 °C) than in dipropyl ether (ε = 3.38 at 20 °C); ether molecules associate with water molecules of cyclic 7HQ·(water)2 complexes via H bonds, and at the rate-determining step, a more polar diethyl ether molecule helps a water molecule to accept a proton from the enolic group of 7HQ more facilely.14 In this regard, the intrinsic ESPT of Nc* in RTILs is supposed to occur faster than the ESPT in n-heptane (62 ps) because RTILs having ionic character are more polar than n-heptane. However, in our present work, we have not observed the intrinsic ESPT time of Nc* directly in RTILs because solvent reorganization to form Nc* prior to intrinsic ESPT is the rate determining step in the overall ESPT of 7HQ in methanol-added RTILs.
Due to the high miscibility of methanol with RTILs, the collision-induced formation of Nc* would be affected by the diffusion of a methanol molecule in RTILs. Thus, we have estimated the diffusion rate of a methanol molecule in a RTIL simply using the Stokes–Einstein equation, assuming that the hydrodynamic radius of a methanol molecule is 1.5 Å. If a methanol molecule travels the root-mean-square distance of 1 Å to form a H bond with a 7HQ molecule, the diffusion rate of a methanol molecule is deduced to be (340 ps)−1 in [bmim][PF6]. By the relation kf = krel + kPT, the value of kPT is deduced to be (430 ps)−1 in [bmim][PF6] having 4 M methanol. Comparing the diffusion rate of a methanol molecule with the ESPT rate of 7HQ in methanol-added [bmim][PF6], we have found out that the ESPT time of 7HQ is similar to the diffusion time of a methanol molecule. As mentioned above, the collision-induced formation of Nc* by solvent reorganization is the rate-determining step of the ESPT of 7HQ in methanol-added RTILs. However, because of the high miscibility of methanol with the polar media of RTILs having ionic character, methanol molecules have much more chances to associate with RTILs rather than 7HQ. Thus, to form Nc*, methanol molecules should move toward 7HQ molecules, and they are supposed to move slowly by diffusion because of the extremely high viscosity values of RTILs. On the other hand, acid–base reactions are usually treated as a diffusion-controlled reaction.48–52 However, the ESPT of 7HQ in methanol-added RTILs is a proton-relay reaction rather than an acid–base reaction. Thus, although the collision-induced formation of Nc*, which is the rate-determining step of the ESPT of 7HQ in methanol-added RTILs, is relevant closely to the molecular diffusion of methanol, the intrinsic ESPT would not be controlled mainly by proton diffusion.
The fluorescence kinetic profiles of 7HQ in [bmim][PF6] having 1 M CH3O2H further confirm that the deduced ESPT time (k−1PT) is associated with solvent reorganization (Fig. 4). The kinetic isotope effect (KIE) of the ESPT rate constant (kPT) is defined as the ratio of kPT obtained with CH3O1H to kPT with CH3O2H. If intrinsic proton transfervia tunneling, which is isotopically sensitive, dominates the overall ESPT process, the KIE value is expected to be large. The overall proton transfer of 7HQ in neat methanol is reported23 to have the KIE value of 1.4 whereas the KIE value for the ESPT of NCC in n-heptane is 15.2 at room temperature.28 This suggests that while the ESPT rate of 7HQ in neat methanol is largely determined by solvent reorganization, the rate-determining step for the ESPT of NCC in n-heptane is intrinsic proton transfervia tunneling. On excitation of a sample containing 7HQ in [bmim][PF6] with 1 M CH3O2H at 315 nm, the two decay times of 700 ps (60%) and 3000 ps (40%) were measured at 380 nm while the rise time of 1100 ps and the decay time of 2500 ps were observed at 550 nm. Consequently, the KIE value of kPT was obtained to be unity. This indicates that solvent reorganization, which is isotopically insensitive to form NCC*, is the rate-determining step of the overall ESPT in the RTIL. In addition, considering the difference between the KIE values of 1.4 in neat methanol and 1.0 in the RTIL, we suggest that solvent reorganization involving large motion is required for the excited-state tautomerization of 7HQ in methanol-added RTILs.
![Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having 1 M CH3O1H (circles) and CH3O2H (squares). Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants.](/image/article/2012/CP/c1cp22329a/c1cp22329a-f4.gif) | ||
| Fig. 4 Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having 1 M CH3O1H (circles) and CH3O2H (squares). Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants. | ||
Compared with the ESPT time of 200 ps viasolvent reorganization in bulk methanol,24 that of 2300 ps in [bmim][PF6] is remarkably slow. This delayed ESPT can be explained by considering the unique properties of RTILs. As mentioned in the introduction section, Bhattacharya and Samanta29 observed the slow ESPT of 7HQ in methanol-added RTILs and attributed the decreased ESPT rate mainly to the high viscosity values of RTILs. Our experimental results given in Fig. 3 and Table 1 also show that the fast decay time at 380 nm in a RTIL decreases with the concentration increase of methanol. However, the observed ESPT times of N* in both RTILs are nearly the same in spite of the great difference in the viscosity values of [bmim][PF6] (312 cP) and [bmim][BF4] (233 cP). This is in line with the observation of Bhattacharya and Samanta29 that there is no clear relationship between the RTIL viscosity and the ESPT rate. Accordingly, we consider that the high viscosity values of RTILs cannot provide full explanation for the origin of the slow ESPT of N. To have clear understanding, we also measured the fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having both methanol and acetonitrile (Fig. 5 and Table 1). Of note is that the viscosity of acetonitrile is similar to that of methanol while acetonitrile, unlike methanol, cannot transport a proton directly. At methanol and acetonitrile concentrations of 1 and 3 M, respectively, in [bmim][PF6], fluorescence at 380 nm shows the two decay times of 700 ps (60%) and 3000 ps (40%) while tautomer fluorescence at 550 nm rises in 1100 ps and decays in 2500 ps. If the ESPT rate of N* is determined solely by the viscosity, the time constants obtained with 1 M methanol and 3 M acetonitrile should be similar to those values with 4 M methanol and 0 M acetonitrile. We have observed, however, that the time constants are rather analogous to those values with acetonitrile-free 1 M methanol. This supports the idea that another factor also plays a crucial role in the ESPT of N in RTILs although the viscosity has a great influence on the ESPT dynamics. Recall that the collision-induced formation of NCC* is suggested to occur to conduct ESPT within the lifetime of N* on the basis of the fact that both the fast decay time at 380 nm and the rise time at 550 nm are observed to decrease with the concentration increase of methanol in RTILs.
![Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having methanol and acetonitrile concentrations of 1 and 0 M (circles) and 1 and 3 M (squares), respectively. Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants.](/image/article/2012/CP/c1cp22329a/c1cp22329a-f5.gif) | ||
| Fig. 5 Fluorescence kinetic profiles of 0.1 mM 7HQ in [bmim][PF6] having methanol and acetonitrile concentrations of 1 and 0 M (circles) and 1 and 3 M (squares), respectively. Samples were excited at 315 nm and monitored at 380 nm (open) and 550 nm (closed). Solid lines are best-fitted curves to extract kinetic constants. | ||
We suggest that the delayed ESPT of N in RTILs is caused by the slow formation of NCC* from 7HQ, 7HQ·(CH3OH), or noncyclic 7HQ·(CH3OH)2 upon excitation due to the high viscosity of the media. Among 7HQ·(ROH)2 complexes, the cyclically H-bonded complex (NCC), which is the most stable ground-state structure in the gas phase, has been reported to form readily in a nonpolar aprotic solvent with a small amount of an alcohol.27,28 Because a 7HQ molecule goes through ESPTvia a H-bonded alcohol chain consisting of two alcohol molecules, the formation of NCC is essential for the tautomerization of N.17,23–28 The intrinsic ESPT of NCC is known to occur via tunneling assisted by heavy-atom motions to bring about an optimal precursor configuration.28 Thus, if NCC is already formed in the ground state, the rate constant of ESPT in a RTIL should be comparable with that in a nonpolar aprotic solvent. In addition, the KIE value should be larger than unity because NCC species undergoes ESPT directly via tunneling, without going through solvent reorganization, as soon as it is excited. However, our results show that the rate constant of the ESPT of N in RTILs is slower by an order of magnitude than that in bulk methanol and that the KIE value is unity. Thus, we consider that solvent reorganization to produce cyclically H-bonded 7HQ·(CH3OH)2 complexes (NCC*) is the rate-determining step in the ESPT of N in RTILs, while intrinsic proton transfer is the rate-determining step in the ESPT of N in nonpolar aprotic media such as n-alkanes. The formation energy of solute–solvent complexes or solute–solute dimers in the ground state highly depends on media. For example, the association constants of 7HQ·(H2O)2 were reported to be 9.8 M−2 in diethyl ether and 230 M−2 in di-n-propyl ether.14 Similarly, 7-azaindole molecules were reported to form dimers in acetonitrile and n-heptane with the association constants of 32 M−1 and 1800 M−1, respectively.46 In this regard, the formation of NCC is thought to be much more unfavorable in RTILs than in nonpolar aprotic solvents due to the ionic character of RTILs. We have plotted lnkPTversusln[CH3OH] linearly, finding out that kPT increases proportionally to [CH3OH]1.4. 1.4 as the reaction order with respect to the concentration of methanol indicates that the collision-induced formation of NCC* should take place within the lifetime of N* prior to undergoing subsequent intrinsic proton transfer rapidly.
4. Conclusions
The excited-state proton relay of 7HQ along a H-bonded methanol chain in RTILs of [bmim][PF6] and [bmim][BF4] has been observed to be considerably different mechanistically and dynamically from that in nonpolar aprotic solvents. Whereas the intrinsic proton transfer of cyclically H-bonded 7HQ·(CH3OH)2 complexes (NCC) is the rate-determining step in methanol-added nonpolar aprotic solvents, the slow solvent reorganization upon absorption of a photon to yield NCC* determines the ESPT rate of 7HQ in methanol-added RTILs. The rate constant of the proton relay in a methanol-added RTIL is found to be slower by an order of magnitude than that in bulk methanol and to have unity in its kinetic isotope effect. These suggest that the excited-state tautomerization dynamics of 7HQ in methanol-added RTILs is totally controlled by the formation dynamics of NCC* upon absorption of a photon because the viscosity values of RTILs are extremely high. The cyclic complex of 7HQ·(CH3OH)2 in RTILs is unstable at the ground state because RTILs having ionic character are highly miscible with methanol. Thus, the collision-induced slow formation of the cyclic complex should take place upon excitation prior to undergoing subsequent intrinsic proton transfer rapidly.Acknowledgements
This work was financially supported by research grants through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (2011-0001216 and 2011-0001211). H.L. is also thankful to a scholarship of BK21.Notes and references
- P. L. Geissler, C. Dellago, D. Chandler, J. Hutter and M. Parrinello, Science, 2001, 291, 2121–2124 CrossRef CAS.
- C. Tanner, C. Manca and S. Leutwyler, Science, 2003, 302, 1736–1739 CrossRef CAS.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- S.-Y. Park, Y.-S. Lee, O.-H. Kwon and D.-J. Jang, Chem. Commun., 2009, 926–928 RSC.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 964–967 CrossRef CAS.
- A. Kohen, R. Cannio, S. Bartolucci and J. P. Klinman, Nature, 1999, 399, 496–499 CrossRef CAS.
- G. Mathias and D. Marx, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6980–6985 CrossRef CAS.
- M. A. Lill and V. Helms, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2778–2781 CrossRef CAS.
- M. Gutman and E. Nachliel, Annu. Rev. Phys. Chem., 1997, 48, 329–356 CrossRef CAS.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS.
- M. Kasha, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2379–2392 RSC.
- I. García-Ochoa, P. B. Bisht, F. Sánchez, E. Martinez-Atáz, L. Santos, H. B. Triphathi and A. Douhal, J. Phys. Chem. A, 1998, 102, 8871–8880 CrossRef.
- P.-T. Chou, C.-Y. Wei, C.-R. C. Wang, F.-T. Hung and C.-P. Chang, J. Phys. Chem. A, 1999, 103, 1939–1949 CrossRef CAS.
- S.-Y. Park, B. Kim, Y.-S. Lee, O.-H. Kwon and D.-J. Jang, Photochem. Photobiol. Sci., 2009, 8, 1611–1617 CAS.
- S.-Y. Park and D.-J. Jang, J. Am. Chem. Soc., 2010, 132, 297–302 CrossRef CAS.
- H. Lim, S.-Y. Park and D.-J. Jang, J. Phys. Chem. A, 2010, 114, 11432–11435 CrossRef CAS.
- B. Kang, K. C. Ko, S.-Y. Park, D.-J. Jang and J. Y. Lee, Phys. Chem. Chem. Phys., 2011, 13, 6332–6339 RSC.
- S.-I. Lee and D.-J. Jang, J. Phys. Chem., 1995, 99, 7537–7541 CrossRef CAS.
- T.-G. Kim, S.-I. Lee, D.-J. Jang and Y. Kim, J. Phys. Chem., 1995, 99, 12698–12700 CrossRef CAS.
- P. J. Thistlethwaite and P. J. Corkill, Chem. Phys. Lett., 1982, 85, 317–321 CrossRef CAS.
- P. J. Thistlethwaite, Chem. Phys. Lett., 1983, 96, 509–512 CrossRef CAS.
- M. Itoh, T. Adachi and K. Tokumura, J. Am. Chem. Soc., 1983, 105, 4828–4829 CrossRef CAS.
- M. Itoh, T. Adachi and K. Tokumura, J. Am. Chem. Soc., 1984, 106, 850–855 CrossRef CAS.
- J. Konijnenberg, G. B. Ekelmans, A. H. Huizer and C. A. G. O. Varma, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 39–51 RSC.
- T. Nakagawa, S. Kohtani and M. Itoh, J. Am. Chem. Soc., 1995, 117, 7952–7957 CrossRef CAS.
- W.-H. Fang, J. Am. Chem. Soc., 1998, 120, 7568–7576 CrossRef CAS.
- S. Kohtani, A. Tagami and R. Nakagaki, Chem. Phys. Lett., 2000, 316, 88–93 CrossRef CAS.
- O.-H. Kwon, Y.-S. Lee, B. K. Yoo and D.-J. Jang, Angew. Chem., Int. Ed., 2006, 45, 415–419 CrossRef CAS.
- B. Bhattacharya and A. Samanta, J. Phys. Chem. B, 2008, 112, 10101–10106 CrossRef CAS.
- S. Mente and M. Maroncelli, J. Phys. Chem. A, 1998, 102, 3860–3876 CrossRef CAS.
- S. Carda-Broch, A. Berthod and D. W. Armstrong, Anal. Bioanal. Chem., 2003, 375, 191–199 CAS.
- C. Chiappe and D. Pieraccini, J. Phys. Org. Chem., 2005, 18, 275–297 CrossRef CAS.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- M. C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef CAS.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS.
- R. Sheldon, Chem. Commun., 2001, 2399–2407 RSC.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS.
- F. van Rantwijk, R. M. Lau and R. A. Sheldon, Trends Biotechnol., 2003, 21, 131–138 CrossRef CAS.
- A. Paul, P. K. Mandal and A. Samanta, J. Phys. Chem. B, 2005, 109, 9148–9153 CrossRef CAS.
- A. Samanta, J. Phys. Chem. B, 2006, 110, 13704–13716 CrossRef CAS.
- A. Paul and A. Samanta, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2010, 49, 649–661 Search PubMed.
- S. Tsuzuki, H. Tokuda and M. Mikami, Phys. Chem. Chem. Phys., 2007, 9, 4780–4784 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- O.-H. Kwon and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8703–8708 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC, Boca Raton, 78th edn, 1999 Search PubMed.
- E. Pines, B.-Z. Magnes, M. J. Lang and G. R. Fleming, Chem. Phys. Lett., 1997, 281, 413–420 CrossRef CAS.
- L. Genosar, B. Cohen and D. Huppert, J. Phys. Chem. A, 2000, 104, 6689–6698 CrossRef CAS.
- M. Rini, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, Science, 2003, 301, 349–352 CrossRef CAS.
- M. Rini, D. Pines, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, J. Chem. Phys., 2004, 121, 9593–9610 CrossRef CAS.
- K. Adamczyk, M. Premont-Schwarz, D. Pines, E. Pines and E. T. J. Nibbering, Science, 2009, 326, 1690–1694 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectrum of neat [bmim][PF6] and emission spectra of neat [bmim][PF6] and 7HQ in [bmim][PF6] before subtraction. See DOI: 10.1039/c1cp22329a |
| This journal is © the Owner Societies 2012 |