Acetylene and argon adsorption in a supramolecular organic zeolite†
Loredana
Erra‡
a,
Consiglia
Tedesco
*ab,
Valeria R.
Cipolletti§
a,
Liana
Annunziata¶
a,
Carmine
Gaeta
ab,
Michela
Brunelli||
c,
Andrew N.
Fitch
c,
Christina
Knöfel**
d,
Philip L.
Llewellyn
d,
Jerry L.
Atwood
e and
Placido
Neri
ab
aDipartimento di Chimica e Biologia, Università di Salerno, via Ponte don Melillo, I-84084 Fisciano, Italy. E-mail: ctedesco@unisa.it; Fax: +39 089-969603; Tel: +39 089-969586
bNANO_MATES, Research Centre for NANOMAterials and nanoTEchnology at Salerno University, via Ponte don Melillo, I-84084 Fisciano, Italy
cEuropean Synchrotron Radiation Facility, 8 Rue J. Horowitz, 38043 Grenoble, France
dLaboratoire Chimie Provence, UMR 6264, Universités Aix-Marseille I, II & III—CNRS, Centre de Saint Jérôme, 13397 Marseille, France
eDepartment of Chemistry, University of Missouri-Columbia, Columbia, Missouri 65211, USA
First published on 14th November 2011
Abstract
The adsorption properties of a new nanoporous organic zeolite with respect to acetylene and Ar were studied by volumetric adsorption analysis, microcalorimetric experiments, and synchrotron high-resolution X-ray powder diffraction. This allowed us to locate the guest molecules inside the host channels and characterize the host–guest interactions.
Introduction
Microporous materials are characterized by pores smaller than 20 Å, among them are zeolites,1 activated carbons,2 MOF,3–5COF,6,7silica gels,8polymers,9 or organic molecules.10–12 They continuously attract a considerable research interest because of their important applications.The study of the adsorption properties of microporous systems towards different gases is a promising field both from an applicative and a theoretical point of view.13
Valuable physical–chemical information on the host system can be obtained if gases with completely different characteristics are used. For example, Ar is usually used as a neutral probe to understand the surface properties of an adsorptive system and/or to clarify adsorbate densification effects of the gas within the porous framework,14–16 while gases such as methane,5b,8,17–27carbon dioxide5b,5c,26–33 and acetylene34–42 are used with the purpose to study the real applicability of the porous system.43
The difficulties in storage and purification of acetylene have led to considerable attention towards the adsorption properties of new porous materials. Among the technological applications, 80% are devoted to chemical synthesis, the remaining 20% to fuel gas for welding, cutting and coating processes.44 As for the chemical synthesis purity is the main issue, but the delivery of acetylene at high pressures (more than 10 bar) can be accomplished only by dissolving it in acetone. Pure acetylene cannot be safely delivered over 2 bar (explosion limit).45
Many solid adsorbents with micropores display strong host–guest interactions and large uptake of highly reactive acetylene gas at room temperature (several times beyond the safe use limit) but the selectivity over carbon dioxide is still a challenge.28
Among the different kinds of porous systems, those formed by organic molecules are less investigated although their potential applications are being increasingly recognized.10–12 Organic clathrates often tend to collapse by removing the guest molecules and do not retain a permanent porous structure, but there are notable exceptions to this general rule.23–25,36,46,47 Once an organic molecule is found as a suitable molecular scaffold or a building block for porous materials, the opportunity to easily modify it by functionalization would lead to a wide range of structural diversifications, both at the molecular level and at the supramolecular level.10 In this way it results to be crucial to understand the physisorption mechanism at a molecular level by correlating the structural properties with the physical and chemical ones.
Among others, calixarenes have been successfully used as molecular building blocks. Since their discovery as side-products in the Bakelite synthesis, they became a Graal for the synthetic chemists for three main reasons: (i) the size can be varied according to the number of phenyl units forming the calix, (ii) the conformation can easily be controlled, (iii) the versatility of functionalization.48
We have recently designed, prepared and characterized a new crystalline solid based on p-But-calix[4]dihydroquinone 1 revealing permanent porosity (Scheme 1).
![1,2-dimethoxy-p-tert-butylcalix[4]dihydroquinone
1.](/image/article/2012/CP/c1cp22569c/c1cp22569c-s1.gif) | ||
| Scheme 1 1,2-dimethoxy-p-tert-butylcalix[4]dihydroquinone 1. | ||
As shown in Fig. 1, the compound crystallizes in a cubic structure (a = 36.412(4) Å), featuring water channels and large hydrophobic empty cavities (988 Å3), as demonstrated by single crystal synchrotron X-ray diffraction.49,50
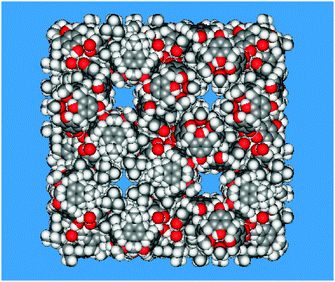 | ||
| Fig. 1 Space-filling model of the unit cell showing the polar open channels (upper left and lower right) and two hydrophobic cages (lower left and upper right) contoured by tert-butyl groups. | ||
The structure acts as a zeolite, in fact the supramolecular framework is preserved after the removal of channel water molecules, as shown by thermogravimetric analysis and X-ray powder diffraction (XRPD) experiments.49
The BET surface area corresponds to 230 m2 g−1 (N2 at 77 K) and the pore distribution plot clearly confirms that the pore diameter of the cubic structure is below 20 Å.51
Previous studies showed that carbon dioxide is absorbed at room temperature inside its nanochannels of the apohost with high selectivity with respect to H2 gas.51 Moreover, also methane molecules can be adsorbed in the nanochannels with a host/guest ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶2, featuring CH–π interactions with the calixarene host-framework.52
∶2, featuring CH–π interactions with the calixarene host-framework.52
The thermal stability of the material, until 493 K in the cubic zeolite-like structure,49 the peculiar packing arrangement with interconnected channels and cavities, the encouraging results on the adsorption of carbon dioxide51 and methane52 led us to explore also the acetylene and Ar adsorption.
Gas sorption analysis and variable temperature high resolution XRPD measurements have been combined to characterize the adsorption properties of this new material and to probe the host–guest interactions by locating the guest molecules inside the host pores. In this way structural and macroscopic properties could be correlated to investigate the physisorption mechanism of this nanoporous molecular material.
Experimental
Crystalline powders of 1 were obtained by following the previously described procedures.49Adsorption studies
Acetylene volumetric sorption analyses were performed using a homemade system.53 A ground sample (0.235 g) was placed in a chamber of a volume of 0.018 L. The sample was evacuated overnight (20 mbar at 333 K) in order to remove water molecules from the crystalline structure.The acetylene adsorption isotherms were carried out at 3 different temperatures: 298 K, 278 K and 203 K. In order to reach the last two temperatures, both the sample chamber and reference chamber were immersed in a Dewar filled with water and ice water in the former case, with dry ice and methanol in the latter case.
The data were collected at different initial pressures between 0.1 and 0.5 bar, until the equilibrium point. The process is almost complete in 5 minutes.
Microcalorimetric analysis was used to understand the interactions of the Ar with the porous framework: a Tian–Calvet apparatus placed upside down in liquid nitrogen connected to a homemade gas dosing system has been used.54
A ground sample (0.148 g) was placed in the measurement chamber after overnight activation under vacuum at 333 K. The Ar sorption isotherms were carried out at 77 K. Data were collected in a pressure range between 0.00 bar and 0.10 bar.
X-Ray powder diffraction
High resolution XRPD (HR-XRPD) measurements were performed at a ID31 ESRF beam line (wavelength 0.80314(4) Å for the acetylene loaded sample and 0.79987(3) Å for the Ar loaded sample) using a rotating glass capillary cell with a gas handling system to allow in situ studies by XRPD.551.0 mm Lindemann capillaries were filled under inert atmosphere with anhydrous powder samples and then mounted on the gas cell. To be sure of the absence of water in the samples, the powder, once placed on the gas cell, was additionally evacuated using a turbomolecular pump (10−5 mbar) and then loaded with the appropriate gas for 30 minutes.
The cell was disconnected from the gas supply and mounted on a goniometer head. A liquid nitrogen cryostat (Oxford Cryostream) was used to perform the measurements under controlled temperature.
To confirm that the guest adsorption does not involve any change in the crystal system and in the space group, considering as a reference structure the result obtained by single crystal synchrotron X-ray diffraction, the HR-XRPD profiles were indexed,56 obtaining always a cubic cell (with values ranging from 36.2130(3) Å at 298 K to 36.2709(3) Å at 173 K in the case of an acetylene loaded sample and from 36.2127(3) Å at 298 K to 36.231(1) Å at 80 K in the case of an Ar loaded sample). Analysis of systematic absences in the HR-XRPD profiles confirmed the space group Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n.
n.
To locate the guest molecules inside the host structure we used the simulated annealing procedures as implemented in the program TOPAS.57
The structures have been refined by a Rietveld method,57 restraining the molecular geometry of the calixarene molecule accordingly to the model obtained by single crystal X-ray diffraction.49 Two overall temperature factors (B) were refined, one for the atoms of the host molecule and the other for the atoms of the guest molecules, restraining the value between 1 and 60. Bond and angle restraints were weighted by 0.05 Å and 5.00°, respectively. Hydrogen atoms were added to the model at their expected positions. The C–H distances were weighted by 0.001 Å.
Six overall parameters for the calixarene molecule (3 translations plus 3 rotations), 5 overall parameters for each acetylene molecule (3 translations plus 2 rotations), 2 isotropic displacement parameters, the site occupancies of the acetylene carbon atoms, the profile and background parameters were refined. In the case of an Ar loaded sample, for the guest atoms just the translations, the isotropic temperature factor and the site occupancies were refined.
Monte Carlo simulation
To confirm the guest positions inside the unit cell the Sorption task available in Materials Studio 4.158 package has been used. The procedure is a canonical Monte Carlo sampling of the search space during which the temperature is gradually decreased. The COMPASS (Condensed-phase Optimized molecular Potentials for Atomistic Simulation Studies) force-field has been applied considering both the electrostatic and the van der Waals terms. During the simulation, sorbate molecules within the framework are randomly rotated and translated. The configuration, resulting from one of these steps, is accepted or rejected according to the selection rules of the Monte Carlo method being used for the simulation.The final positions of the guest molecules as obtained by applying the simulated annealing process implemented in the program TOPAS57 and the Monte Carlo simulation are reported in Tables S1 and S2 (see ESI†), for acetylene and Ar loaded samples, respectively.
Results and discussion
The adsorption measurements have been performed with two different devices for the two gases: while the acetylene adsorption has been studied using a volumetric home-made system53 which provided an immediate understanding of the adsorption phenomenon from a quantitative point of view, the Ar adsorption has been studied using a Tian–Calvet microcalorimeter, particularly powerful to perform an in-depth examination of the surface state of adsorbents.54The acetylene adsorption isotherms at three different temperatures are reported in Fig. 2. In the reported range the isotherms are of first order.
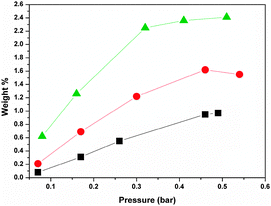 | ||
| Fig. 2 Adsorption isotherm for C2H2 at 298 K (squares), 278 K (circles) and 203 K (triangles). Lines are to guide the eyes. | ||
It is evident that increasing the pressure and lowering the temperature the acetylene uptake is higher, in spite of the limited range of considered values. In particular, a value of 2.4 wt% of acetylene can be adsorbed at 203 K under only 0.5 bar pressure. Under these conditions the acetylene density is 52 times higher than the value of the compression limit for a safe use.
For comparison acetylene adsorption data at 298 K for several microporous crystals of metal–organic and organic compounds are reported in Table 1. The most efficient system is Co2(DHTP), DHTP = 2,5-dihydroxyterephthalate, which adsorbs 198 cm3 g−1 of acetylene at 295 K and 1 bar.42 In our case the adsorption is of 16.9 cm3 g−1 at 0.5 bar and 298 K corresponding to a storage efficiency of 0.97% by weight; this value is comparable to the one reported for the calix[4]arene,38 which is 18 cm3 g−1 at 298 K and 1 bar.
| Compound | BET surface area/m2 g−1 | V p/cm3 g−1 | C2H2 uptake/cm3 g−1 | Ref. |
|---|---|---|---|---|
| a 295 K. b 0.5 bar. pzdc = pyrazine-2,3-dicarboxylate, pyz = pyrazine, etz = 3,5-diethyl-1,2,4-triazole, DHTP = 2,5-dihydroxyterephthalate. | ||||
| 1 | 230 | 0.14 | 17b | This work and 51 |
| Calix[4]arene | 18 | 38 | ||
| Mn(HCOO)2 | 297 | 0.13 | 51 | 35 |
| Cucurbit[6]uril | 210 | 0.13 | 52 | 36 |
| Mg(HCOO)2 | 284 | 0.14 | 66 | 35 |
| Cu[(etz)]n, | 70 | 40 | ||
| Cu2(pzdc)2(pyz) | 0.14 | 42 | 34 | |
| Co2(DHTP) a | 198 | 42 |
Under mild conditions of ambient pressure and temperature our sample shows an efficiency for methane storage of 0.29 wt%, while in the case of CO2 molecules the value was 0.5 wt%.51 It is interesting to note that, at ambient temperature, despite the fact that the pressure used in the case of acetylene is lower, the uptake is higher in comparison with methane and CO2.
The selectivity over CO2 and methane could possibly be understood considering the host–guest interactions, for this reason surface properties and adsorbate densification effects of the gas within the porous framework have been studied by means of Ar adsorption and structural studies have been performed by means of high resolution X-ray powder diffraction.
The Ar adsorption isotherm collected at 77 K as well as the variation of the partial molar enthalpy of adsorption (or differential enthalpy of adsorption) as a function of the relative pressure is reported in Fig. 3.
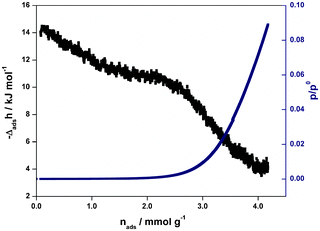 | ||
| Fig. 3 Enthalpy of adsorption and moles of adsorbed gas obtained for Ar in an evacuated sample of 1 at 77 K and up to 0.1 bar. | ||
As shown in Fig. 3 the initial interactions are around −14.5 kJ mol−1, which are in the region of those observed for other quite neutral microporous samples. Indeed, the interactions observed here can be compared to around −13.5 kJ mol−1 observed for Ar adsorption on silicalite14 and −17 kJ mol−1 for Ar adsorption on activated carbon.15
Interestingly, there seems to be three energy domains in this system. The first, up to 1.5 mmol g−1, shows a slight decrease in energy from −14.5 kJ mol−1 to around −11 kJ mol−1 which could be interpreted by an arrangement of the molecules in the polar channels in which the molecular polarizability of argon is affected by the channel polarity. The second regime, up to 2.2 mmol g−1, is almost horizontal at 11 kJ mol−1 which indicates a far more energetically homogeneous domain which could be attributed to the progressive filling of the channels by further Ar molecules. The third energetic domain, from 2.2 mmol g−1 to 4 mmol g−1, corresponds to a large decrease in enthalpy and is the signature of adsorption on the external surface of the solid.
HR-XRPD measurements were performed at 0.5 bar pressure and variable temperature. The corresponding XRPD profiles are reported in Fig. 4a and b for acetylene and Ar loaded samples, respectively. For comparison, measurements have been performed also on an evacuated sample (Fig. S1, ESI†).
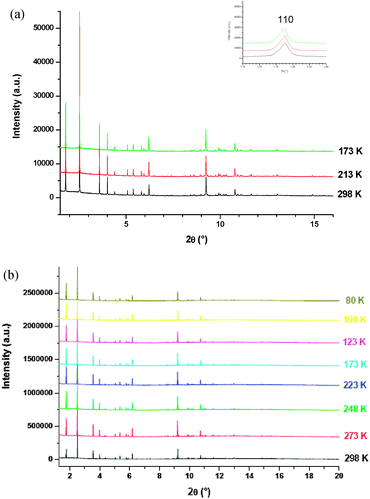 | ||
| Fig. 4 (a) HR-XRPD profiles for the C2H2 loaded sample. In the inset the 2θ range with the first peak, indexed as 110. (b) HR-XRPD profiles for the Ar loaded sample. | ||
Indexing confirmed that the guest adsorption does not involve any change in the crystal system and in the space group. Both for the guest loaded and evacuated samples the cell parameters have been evaluated by Le Bail refinement. In the case of guest loaded samples the cell parameter is always greater than the corresponding evacuated sample at a given temperature (Fig. 5).
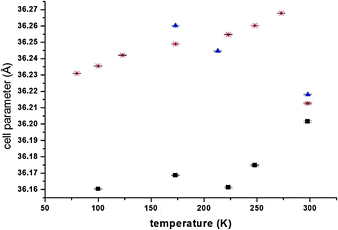 | ||
| Fig. 5 Cell parameters vs. temperature for evacuated (squares), acetylene-loaded (triangles) and Ar loaded samples (stars). Error bars are reported vertically. | ||
While the evacuated sample shows a progressive decrease of the lattice parameter with lowering the temperature, the acetylene loaded sample shows an increase. This indicates that acetylene is able to enter into the porous structure.
For the Ar loaded sample the lattice parameter first shows an increase with lowering the temperature and then a decrease to values that anyway are always greater than the corresponding values for the evacuated sample.
HR-XRPD data collected at 3 different temperatures were used with the aim to locate the acetylene molecules inside the host structure. To locate the acetylene molecules inside the host structure we used the simulated annealing procedures as implemented in the program TOPAS.57 Two different models have been obtained, one for the data at 173 and 213 K and one for the data at 298 K. For the structure at low temperature the asymmetric unit contains two crystallographically independent acetylene molecules located into the channels of the host structure with slightly different occupancy factors (0.43 and 0.52 respectively). The structure obtained using the data at room temperature contains only one crystallographically independent acetylene molecule in the asymmetric unit.
The structures have been refined by the Rietveld method,57 restraining the molecular geometry of the calixarene molecule accordingly to the model obtained by single crystal X-ray diffraction.49 The final results after the refinement are reported in Table 2, the final Rietveld plot for the data collected at 173 K is reported in Fig. 6.
| T/K | R p fitted | w R p fitted | R F 2 | χ 2 | a/Å |
|---|---|---|---|---|---|
| 298 | 0.0546 | 0.0724 | 0.1789 | 3.637 | 36.2180(2) |
| 213 | 0.0494 | 0.061 | 0.1582 | 2.598 | 36.2447(3) |
| 173 | 0.0529 | 0.0649 | 0.1561 | 2.628 | 36.2602(2) |
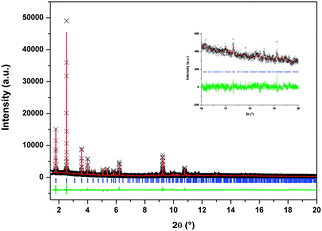 | ||
| Fig. 6 Rietveld refinement results for the acetylene loaded sample at 173 K and 0.5 bar. Observed (black crosses) and calculated X-ray powder diffraction profiles (line above) as well as difference profile (line below) are shown. In the inset the region between 2θ = 16° and 2θ = 20°. | ||
The structure model as obtained from the data at 213 K and 173 K features one acetylene molecule A (magenta in Fig. 7) interacting directly via the CH–π bond59 with one of the methyl groups of the anisole rings of the host framework (HOCH3⋯CC2H2A 2.75 Å, COCH3⋯CC2H2A 3.51 Å). The other non-equivalent acetylene molecule B (yellow in Fig. 7) interacts only with the acetylene A molecule featuring a T shape CH–π bond (CC2H2A⋯CC2H2B distance is 3.06 Å, the CC2H2A ⋯C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC2H2B angle is 127.53°).
CC2H2B angle is 127.53°).
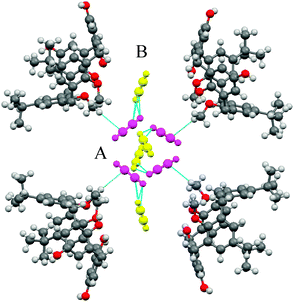 | ||
| Fig. 7 Detailed view of the interactions at 173 K among the acetylene molecules A (magenta), the framework calixarene molecules and the acetylene molecules B (yellow). Only half of the symmetry related acetylene molecules is displayed to account for 0.5 occupancy. | ||
The higher efficiency for acetylene with respect to CO2 and methane can be explained considering that acetylene molecules have the possibility to act both as a H-bond donor as well as a H-bond acceptor interacting both with the host framework and with other acetylene molecules.
There are some similarities between our structure model at low temperatures and that of Cu[(etz)]n at 123 K,40 in that case the guest molecules do not interact with the metal ion but feature the CH–π bond with the host framework and with other C2H2 molecules. This is probably due to similarity in size and shape of the available empty space in the two host frameworks. For both structures also the ratio between the host and guest is 1/1.
The structure model as obtained from the data collected at room temperature shows only one acetylene molecule with partial occupancy (0.70) featuring only CH–π bonds with crystallographically equivalent neighbouring acetylene molecules (CC2H2⋯CC2H2 distances are 2.67 Å and 2.71 Å).
It seems that there are only very weak interactions with the walls of the framework (HOCH3⋯CC2H2 3.24 Å, COCH3⋯CC2H2 3.93 Å). Thus, at room temperature guest–guest interactions prevail against host–guest interactions (Fig. 8).
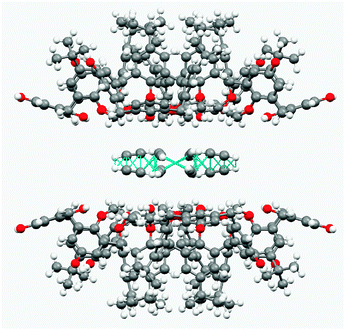 | ||
| Fig. 8 Detailed view of CH–pi interactions at 298 K among acetylene molecules in the host channels. Guest–guest interactions prevail on host–guest interactions. | ||
The host–guest configurational space was also searched by energy based Monte Carlo simulation58 to find the guest preferential sites. The search was performed fixing the host framework, and considering P1 symmetry for the guest molecules. The list of preferential sites was searched to find symmetry related positions along the Pnn space group, obtaining two possible sites really close to the ones obtained by applying the simulated annealing procedure using the HR-XRPD data collected at low temperature. The positions found using the two procedures are listed in Table S1 (see ESI†).
For the Ar loaded sample the HR-XRPD data collected at 80 K and 298 K and 0.5 bar pressure were further analyzed with the aim to locate the guest atoms. The host–guest structure was solved performing the simulated annealing procedure, as implemented in the software package TOPAS.57 Two different models were obtained. For the structure at low temperature the asymmetric unit contains two crystallographically independent Ar atoms located into the channels of the host structure with different occupancy factors, 0.425(7) and 0.27(1).
The structure obtained using the data at room temperature contains only one independent Ar atom in the asymmetric unit. In both cases Ar atoms do not feature any specific interaction with the host.
The apparent close contacts among them can be explained considering the positional disorder as evidenced by occupancy factors less than one. By Rietveld refinement it has been found that the model obtained from the data at 80 K is valid also for the patterns collected up to 273 K.
The final values for the refinement obtained are reported in Table 3, and the final Rietveld plot for the data collected at 80 K is reported in Fig. 9. Ar preferential sites in the host–guest system, found by energy driven Monte Carlo search, match with the one obtained considering HR-XRPD data (Table S2, ESI†).
| T/K | R p fitted | w R p fitted | R F 2 | χ 2 | a/Å |
|---|---|---|---|---|---|
| 298 | 0.0293 | 0.0450 | 1.917 | 0.0293 | 36.2130(3) |
| 273 | 0.0260 | 0.0369 | 5.642 | 0.0263 | 36.2709(3) |
| 248 | 0.0240 | 0.0381 | 5.794 | 0.0240 | 36.2657(2) |
| 223 | 0.0234 | 0.0360 | 5.378 | 0.0234 | 36.2590(3) |
| 173 | 0.026 | 0.0380 | 5.580 | 0.0260 | 36.2547(3) |
| 123 | 0.0218 | 0.0377 | 5.411 | 0.0218 | 36.2501(3) |
| 100 | 0.0216 | 0.0401 | 5.677 | 0.0216 | 36.2436(4) |
| 80 | 0.0189 | 0.0397 | 5.746 | 0.0188 | 36.2381(5) |
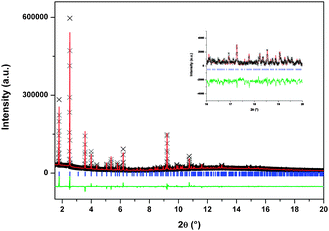 | ||
| Fig. 9 Rietveld refinement results for the Ar loaded sample at 173 K and 0.5 bar. Observed (black crosses) and calculated (line above) X-ray powder diffraction profiles as well as difference profiles (line below) are shown. The region between 2θ = 16° and 2θ = 20° in the inset. | ||
Conclusions
According to the sorption and to the crystallographic analyses we can conclude that our calixarene-based solid behaves as a supramolecular zeolite adsorbing acetylene and Ar inside its nanochannels. Despite the low pressure used the storage efficiency for acetylene results to be higher with respect to methane and carbon dioxide. Structural studies demonstrated that acetylene adsorption is enhanced by the ability of acetylene to act both as a H-bond donor as well as a H-bond acceptor interacting both with the host framework and with other acetylene molecules. Since the hydrophobic cages result to be still unoccupied by any guest molecule, further measurements at higher pressure are underway to assess the possibility that acetylene molecules could occupy also this empty space.Acknowledgements
We acknowledge Dr Mark Johnson (ILL) for useful discussions, Mr Drew Fowler and Mr Jian Tian (University of Missouri) for their experimental support in volumetric adsorption studies, Mr Olivier Grimaldi (ESRF) for technical support in the use of the gas handling cell. We are grateful to ESRF for providing access to ID31 beamline (Proposals CH-2388, CH-2712). Italian MIUR is acknowledged for financial support.Notes and references
- A. Corma, M. J. Diaz-Cabanas, J. L. Jorda, C. Martinez and M. Moliner, Nature, 2006, 443, 842–845 CrossRef CAS.
- B. Daffos, P.-L. Taberna, Y. Gogotsi and P. Simon, Fuel Cells, 2010, 10, 819–824 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS.
- (a) N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902–7904 CrossRef CAS; (b) S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-García, I. Fernández-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664–1672 CrossRef CAS; (c) E. Barea, G. Tagliabue, W.-G. Wang, M. Pérez-Mendoza, L. Mendez-Liñan, F. J. López-Garzon, S. Galli, N. Masciocchi and J. A. R. Navarro, Chem.–Eur. J., 2010, 16, 931–937 CAS.
- (a) C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS; (b) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS.
- W. X. Wang, C. L. Bray, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 11608–11609 CrossRef CAS.
- (a) A. R. Albunia, C. D'Aniello, G. Guerra, D. Gatteschi, M. Mannini and L. Sorace, Chem. Mater., 2009, 21, 4750–4752 CrossRef CAS; (b) For recent reviews on the subject, see G. Guerra and V. Petraccone, Soft Mater., 2011, 9, 105–312 CrossRef.
- P. J. Langley and J. Hulliger, Chem. Soc. Rev., 1999, 28, 279–291 RSC.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS.
- N. B. McKeown, J. Mater. Chem., 2010, 20, 10588–10597 RSC.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS.
- P. Llewellyn, J. P. Coulomb, Y. Grillet, J. Patarin, H. Lauter, H. Reichert and J. Rouquerol, Langmuir, 1993, 9, 1846–1851 CrossRef CAS.
- J. Pikunic, P. Llewellyn, R. Pellenq and K. E. Gubbins, Langmuir, 2005, 21, 4431–4440 CrossRef CAS.
- G. Maurin, P. L. Llewellyn, Th. Poyet and B. Kuchta, Microporous Mesoporous Mater., 2005, 79, 53–59 CrossRef CAS.
- V. C. Menon and S. J. Komarneni, Porous Mater., 1998, 5, 43–58 CrossRef CAS.
- S. Noro, S. Kitagawa, M. Kondo and M. Seki, Angew. Chem., Int. Ed., 2000, 39, 2082–2084 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Watcher, M. O'Keefe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- P. L. Llwellyn, G. Maurin, T. Devic, S. Loera-Serna, N. Rosenbach, C. Serre, S. Bourrelly, P. Horcajada, Y. Filinchuck and G. Férey, J. Am. Chem. Soc., 2008, 130, 12808–12814 CrossRef.
- C. D. Wood, B. Tan, A. Trewin, F. M. Su, J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916–1921 CrossRef CAS.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS.
- A. Comotti, S. Bracco, G. Distefano and P. Sozzani, Chem. Commun., 2009, 284–286 RSC.
- J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS.
- P. K. Thallapally, K. A. Kirby and J. L. Atwood, New J. Chem., 2007, 31, 628–630 RSC.
- (a) W. Li, J. P. Zhang, H. C. Guo and G. Gahungu, J. Phys. Chem. C, 2011, 115, 4935–4942 CrossRef CAS; (b) K. J. Msayib, D. Book, P. M. Budd, N. Chaukura, K. D. M. Harris, M. Helliwell, S. Tedds, A. Walton, J. E. Warren, M. Xu and N. B. McKeown, Angew. Chem., Int. Ed., 2009, 48, 3273–3277 CrossRef CAS.
- B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe, O. M. Yaghi and J. Kim, Nature, 2008, 423, 207–212 CrossRef.
- L. Dobrzanka, G. O Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2006, 128, 698–699 CrossRef.
- J.-R. Li, R. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- S. Surblé, F. Millange, C. Serre, T. Düren, M. Latroche, S. Bourrelly, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2006, 128(46), 14889–14896 CrossRef.
- P. K. Thallapally, J. Tian, M. R. Kishan, C. A. Fernandez, S. Dalgarno, J. McGrail, P. B. Warren and J. L. Atwood, J. Am. Chem. Soc., 2008, 130, 16842–16843 CrossRef CAS.
- K. Nakagawa, D. Tanaka, S. Horike, S. Shimomura, M. Higuchi and S. Kitagawa, Chem. Commun., 2010, 46, 4258–4260 RSC.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS.
- D. G. Samsonenko, H. Kim, Y. Sun, G.-H. Kim, H.-S. Lee and K. Kim, Chem.–Asian J., 2007, 2, 484–488 CrossRef CAS.
- S. Lim, H. Kim, N. Selvapalam, K.-J. Kim, S. J. Cho, G. Seo and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 3352–3355 CrossRef CAS.
- D. Tanaka, M. Higuchi, S. Horike, R. Matsuda, Y. Kinoshita, N. Yanai and S. Kitagawa, Chem.–Asian J., 2008, 3, 1343–1349 CrossRef CAS.
- P. K. Thallapally, L. Dobrzanska, T. R. Gingrich, T. B. Wirsig, L. J. Barbour and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 45, 6506–6509 CrossRef CAS.
- S. Xiang, W. Zhou, J. M. Gallegos, Y. Liu and B. Chen, J. Am. Chem. Soc., 2009, 131, 12415–12419 CrossRef CAS.
- J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS.
- Y. Hu, S.-C. Xiang, W. Zhang, Z. Zhang, L. Wang, J. Bai and B. Chen, Chem. Commun., 2009, 7551–7553 RSC.
- S.-C. Xiang, W. Zhou, Z. Zhang, M. A. Green, Y. Liu and B. Chen, Angew. Chem., Int. Ed., 2010, 49, 4615–4618 CrossRef CAS.
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC.
- R. L. Mayers, The 100 most important chemical compounds, Greenwood Press, 2007, http://www.boc-gases.com Search PubMed.
- S. Budavari, The Merck Index, Merck Research Laboratories, NJ, 12th edn, 1996, p. 16 Search PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS.
- P. K. Thallapally, B. P. McGrail, S. J. Dalgarno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146–150 CrossRef CAS.
- For comprehensive reviews on calixarenes, see: (a) V. Böhmer, Angew. Chem., Int. Ed., 1995, 34, 713–745 CrossRef; (b) A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS; (c) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 1998 Search PubMed; (d) Calixarenes 2001, ed. Z. B. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer, Dordrecht, 2001 Search PubMed; (e) V. Böhmer, in The Chemistry of Phenols, ed. Z. Rappoport, Wiley, Chichester, U.K., 2003, ch. 19 Search PubMed; (f) Calixarenes in the Nanoworld, ed. J. Vicens and J. Harrowfield, Springer, Dordrecht, 2007 Search PubMed.
- C. Tedesco, I. Immediata, L. Gregoli, L. Vitagliano, A. Immirzi and P. Neri, CrystEngComm, 2005, 7, 449–453 RSC.
- For other crystalline forms of this versatile material, see: C. Tedesco, L. Erra, I. Immediata, C. Gaeta, M. Brunelli, M. Merlini, C. Meneghini, P. Pattison and P. Neri, Cryst. Growth Des., 2010, 10, 1527–1533 CAS . For structural studies on a similar distal calix[4]dihydroquinone, see: C. Tedesco, L. Gregoli, I. Immediata, E. Gavuzzo and P. Neri, Cryst. Growth Des., 2008, 8, 3700–3705 Search PubMed.
- P. K. Thallapally, B. P. McGrail, J. L. Atwood, C. Gaeta, C. Tedesco and P. Neri, Chem. Mater., 2007, 19, 3355–3357 CrossRef CAS.
- C. Tedesco, L. Erra, M. Brunelli, V. Cipolletti, C. Gaeta, A. N. Fitch, J. L. Atwood and P. Neri, Chem.–Eur. J., 2010, 16, 2371–2374 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, P. K. Thallapally and T. B. Wirsig, Chem. Commun., 2005, 51–53 RSC.
- P. Llewellyn and G. Maurin, C. R. Chim., 2005, 8, 283–302 CrossRef CAS.
- M. Brunelli and A. N. Fitch, J. Synchrotron Radiat., 2003, 10, 337–339 CrossRef CAS.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- A. Coelho, J. Appl. Crystallogr., 2000, 33, 899–908 CrossRef CAS.
- Sorption Locate Module, Materials Studio 4.3., Accelrys Inc. 2001, 9685 Scranton Road, San Diego, CA92121-3752, USA.
- (a) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC; (b) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The HR-XRPD data for an evacuated sample collected at different temperatures are reported in Fig. S1. The comparison between acetylene and Ar preferential sites by an energy based Monte Carlo simulation method and by diffraction data analysis is reported, respectively, in Tables S1 and S2. The crystallographic information file (cif): C2H2_173K.cif, C2H2_298K.cif, Ar_80K.cif and Ar_298K.cif are reported. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cp22569c |
| ‡ Current address: European Synchrotron Radiation Facility, 8 Rue J. Horowitz, 38043 Grenoble, France. |
| § Current address: Pirelli Tyre SpA Viale Sarca 222, I-20126 Milano, Italy. |
| ¶ Current address: Catalysis and Organometallics, UMR 6226 Sciences Chimiques de Rennes, CNRS–University of Rennes 1, 35042 Rennes Cedex, France. |
| || Current address: Institut Laue-Langevin, 6 Rue J. Horowitz, 38043 Grenoble, France. |
| ** Current address: Fuel Cells and Solid State Chemistry Division, Risø National Laboratory for Sustainable Energy, Technical University of Denmark, DK-4000 Roskilde, Denmark. |
| This journal is © the Owner Societies 2012 |