On the role of thermal activation in selective photochemistry: mechanistic insight into the oxidation of propene on the V4O11−cluster†
Shaohui
Li
ab,
Juri
Demuth
a,
Aldo
Mirabal
a,
Ludger
Wöste
a and
Torsten
Siebert
*a
aInstitut für Experimentalphysik, Freie Universität Berlin, Animallee 14, 14195 Berlin, Germany. E-mail: torsten.siebert@physik.fu-berlin.de
bDepartment of Physics, Shantou University, Shantou 515063, Peoples Republic of China
First published on 9th November 2011
Abstract
An experimental methodology for a mechanistic analysis of gas phase chemical reactions is presented in the context of structure–reactivity relationships of metal oxide clusters relevant to photocatalysis. The spectroscopic approach is demonstrated with the investigation of the photoinduced oxygenation of propene on the V4O11−cluster, where the thermal activation and subsequent photoreaction are deduced with the information from (i) the temperature dependency of the aggregation kinetics in the propene-seeded helium atmosphere of an ion-trap reactor; (ii) the fluence dependency in the yield of different product channels of the photoreaction and (iii) the intensity dependency in the fragmentation of neutral reaction products that are probed via in situ multi-photon ionization. For the thermal reaction, selective hydrogen abstraction from the allylic position of propene accompanied by the linkage to the cluster at the dioxo moiety is postulated as the mechanism in the aggregation of propene on the V4O11−cluster. In accordance with an insightful neutralization–reionization study (Schröder et al., J. Mass. Spectrom., 2010, 301, 84), the subsequent photoinduced reaction is defined by an allylic oxidation in the formation of acrolein from the initial allyloxy radical photoproduct. The relevance of the observed selectivity is discussed in view of the electronic structure and bond motifs offered by high valence oxide systems such as the V4O11−cluster.
Introduction
The distinct electronic properties and structural motifs of vanadium oxides have allowed for their widespread applications in mediating selective and efficient oxidation reactions. The spectrum of applications ranges from the enantio-selective epoxidation of unsaturated hydrocarbons for the synthesis of pharmaceuticals to heterogeneous catalytic processes that supply elementary chemical reagents at an industrial-scale.1–6 The proliferation in utilizing this general reactivity has motivated a systematic study of the structure–reactivity relationship in different classes of vanadium oxide species.1,7–12 In this context, extensive efforts in gas phase studies on the structure of small cluster ions have given a fundamental understanding of the structural motifs in these systems in view of their reactive nature.13–24Within the class of small vanadium oxide cluster ions, the V4O11− system constitutes an exceptional species. Its structure has been shown to deviate from other high valent vanadium oxides that show characteristic peroxo motifs. The DFT analysis given by Sauer and coworkers of the corresponding RPD spectrum acquired by Santambrogio et al. shows a variety of possible peroxo and dioxo structures for this system, but the dioxo motif is determined as the lowest energy species with the best agreement to the experimental data.23 Early collision induced dissociation (CID) experiments by Castleman and coworkers with V4O11− in a xenon atmosphere support the assumption that gas-phase synthesis of this cluster stoichiometry primarily yields a system with a dioxo structure.25 The exceptional reaction channel of atomic oxygen loss together with the localization of spin on a terminal oxygen in the dioxo structure provides the motivation for exploring the possibilities of utilizing this system as a model structure for achieving efficient oxygenation reactions.23 Next to the dioxo-motif, the vanadyl and V–O–V bridge bond motifs also innate to this cluster present a relationship to V4O10− and V2O5 surface structure, which underlines its model nature.1,9,11,13,18
The circumstances described above inspired an initial characterization of the reactivity offered by the V4O11− system in the oxygenation of propene.26 Deviating from the tradition of exploring the thermal reactivity, the work on this system pursued the possibilities opened to photoinduced reactions.27,28 This was partially necessitated by previous reports on the lack of reactivity of small vanadium oxide cluster anions towards hydrocarbons.29 More importantly, this concept was inspired by the general trends seen in different classes of oxidation reactions that were outlined by the groups of Sauer and Schwarz for varying vanadium oxide species and small hydrocarbons.9,13,19,20,30 In this work, specific reactions are clearly associated with the reduction of the vanadium oxide systems giving the occupation of electronic states with high metal d-state character as a consequence of the oxidation process. Photo-activationvia electronic excitation was considered in order to mimic this behavior with ligand-to-metal electron transfer processes, hereby selectively inducing the reactive behavior given for the thermal case.13,27,28 The original photo-reactivity of the V4O11− towards propene showed the formation of an oxygenated hydrocarbon as a consequence of photo-excitation in the [V4O11·C3H6]− complex with femto-second laser pulses at 272 nm.26 Three independent channels were postulated for this reaction due to the capability of the experimental configuration to characterize the charged cluster products as well as neutral reaction participants via in situ multi-photon ionization.
New aspects concerning the mechanism of this reaction have been reported recently in the literature. Particularly noteworthy are the highly insightful neutralization–reionization experiments of the allyl oxide anion given by Schröder et al., which was explicitly analyzed for the interpretation of the reported photoreaction.31 The analysis allowed the unique reaction product in the oxygenation of propene on V4O11− to be unambiguously identified as acrolein. Furthermore, Tian and coworkers report a comprehensive DFT analysis concerning the thermodynamics that govern an oxygenation reaction in the electronic ground state of the reaction complex.24 Importantly, the initial aggregation of propene on the cluster site described in this work will be shown to be highly relevant for understanding the precursor from which the photoreaction initiates. This step involves hydrogen activation and abstraction from the allylic position of propene to the vanadyl moiety of the cluster system (steps 1 and 2 of the “key steps” in the reaction mechanism). The elements of the proposed mechanism explicitly concerning the thermal oxygenation (steps 3 and 4 of the “key steps”) do not apply to the photoreaction at 272 nm (4.56 eV) excitation, which access the unique reactivity characteristic of the respective electronically excited states of the reaction complex. The activity in the literature described above has provided the motivation to revisit this reaction experimentally in order to further establish the findings of these recent works.
Methodology
A tandem-mass spectrometer consisting of a hexadecapole ion trap (1 MHz, home-built) flanked by two quadrupole mass filters (800 kHz, Extrel) coupled to a laser vaporization cluster source constitutes the apparatus utilized for the thermal and photoinduced reactivity studies in this work. Explicitly, synthesis of the metal oxide clusters begins in a standard laser ablation utilizing a 10 Hz frequency-doubled Nd:YAG (Big Sky) at several mJ per pulse, focused with f = 200 mm lens onto the vanadium target. The ablation plasma is carried out in a 2% oxygen-seeded helium gas pulse (oxygen 4.6, Messer-Griesheim; helium 6.0, Linde AG) which is synchronized with a pulsed valve to the 10 Hz laser. The initially formed mixture of cluster species in different charge states is thermalized through expansion of the carrier gas. The mixture is transported to the mass spectrometervia differential pumping and guided through a skimmer on variable potential. Further preparation of the ion beam is carried out in a decapole (1 MHz, Extrel) with thermalization in a helium atmosphere (pHe ≈ 10−4 mbar, helium 6.0, Linde AG) and neutral species are filtered off in the 90° quadrupole deflector that follows. After mass selection of the V4O11−cluster in the first quadrupole filter, a double Einzel lens arrangement couples the ion beam into the hexadecapole ion trap under a constant propene-seeded helium atmosphere (pC3H6 ≤ 10−3 mbar, Propene 2.5, Linde AG; pHe = 2 × 10−2 mbar, Helium 6.0, Linde AG), which is monitored by a baratron (PR400, MKS). The temperature is controlled via a two-stage helium cryostat (CTI-Cryogenics, Head 350 CP; Compressor 8200). The potential of the flanking entrance and exit lenses as well as the bias of the ion trap can be adjusted for cycles of filling, storage and extraction of an ion species in a sequence and at rates that are determined by the application to thermal or photoinduced reactivity. The potentials of the following double Einzel lens arrangement and subsequent 90° quadrupole deflector can be accommodated to guide cationic or anionic species extracted from the ion trap for mass analysis in the second quadrupole filter and detection via a conversion dynode and a channeltron in a pulse counting mode (Burle, model 4873).Specifically, the kinetics in the thermal aggregation of propene on the V4O11−cluster system are measured after mass selection of the V4O11− species and injection at several eV kinetic energy into the ion trap. This is achieved by cycling through a sequence of filling, storage and extraction periods of the ion trap reactor. The potentials on the entrance and exit lenses are switched to allow for the 500 ms filling period and an extraction time of 100 ms. The intermediate storage time is varied from 0 to ≤ 12000 ms in steps of 250 ms after 500 ms has proceeded from the initial injection. For a fixed storage time, the yields in the mass channels of the V4O10− (364 amu), V4O11− (380 amu) and [V4O10(OH)C3H5]− (422 amu) species are determined. A scan of the storage time within this sequence is repeated at 200, 250 and 300 K via control of the helium atmosphere with the helium cryostat for temperature dependent aggregation kinetics.
The study of the photoinduced reactivity of the [V4O10(OH)C3H5]− complex is realized with a femtosecond Ti:Sapphire oscillator and amplifier system. A frequency-doubled Nd:Vanadate laser (Verdi V5, Coherent) at 4.5 W is employed for pumping of a Ti:Sapphire oscillator (Femtosource, Femtolasers) giving laser pulses of 6 nJ centered at ∼805 nm (1.54 eV) with approximately 90 nm (173 meV) bandwidth at 80 MHz repetition rate. Standard chirped pulse amplification (Odin C, Quantronix) is achieved in a multi-pass arrangement at 1 kHz by pumping with a nanosecond frequency-doubled Nd:YLF giving amplified pulses at 807 nm (1.53 eV) and a bandwidth of 36 nm (68.6 meV). The pulses have an energy of 1.4 mJ and durations of sub-40 fs (FWHM). The generation of the third-harmonic from the amplified output is achieved with the standard procedure of sequential frequency doubling (BBO, θ = 29.2°, 0.5 mm) and sum frequency generation of the second harmonic with the residual fundamental (BBO, θ = 55.5°, 0.5 mm). Pulses are obtained with an energy of up to 120 μJ pulse−1, which are centered at 272 nm (4.56 eV) with a bandwidth of approx. 1.0 nm (16.8 meV). The telescope arrangement for the frequency conversion is adjusted so that direct focusing into the ion trap is achieved at approximately f = 700 mm on the axis to the ion trap. The pulse duration of 148 fs (FWHM) of the third harmonic at 272 nm is determined after coupling into the apparatus. This is achieved in the detection of the 3-photon ionization of propene in the ion trap by employing a split mirror configuration with a precision mechanical delay stage for temporal delay of the two sub-pulses obtained from the split mirror reflection.32
The photoinduced reactivity of the [V4O10(OH)C3H5]− complex is characterized at ∼300 K in the helium atmosphere of the ion trap in a continuous irradiation of its contents at 1 kHz repetition rate with the pulses varying from 10 to 100 μJ pulse−1 or a fluence of 8.70 to 80.7 × 10−2 J cm−2 at durations of 148 fs. For the measurement of the mass spectrum of the anionic photoproducts, the filling, storage and extraction cycles are set at 500, 500 and 100 ms, respectively, by applying the appropriate potentials on the exit and entrance lens of the ion trap. For a 500 ms filling and storage time, the reaction kinetics at 300 K for the aggregation of propene with the injected V4O11− have progressed to nearly 70% conversion of V4O11− to the [V4O10(OH)C3H5]− complex. The detection of the neutral photoproducts is achieved by a conversion to the cationic species via in situ multi-photon ionization parallel to photo-excitation of the reaction complex with pulses at 272 nm (4.56 eV) and 20 to 100 μJ pulse−1 or intensities at 1.35 to 6.75 × 1010 W cm−2 with durations of 148 fs. For obtaining the complementary cationic mass spectrum, the filling, storage and extraction cycles are carried out in the same manner, whereby the cationic species generated by ionization of the neutral reaction products are continuously extracted by an exit lens with a potential for containing the anionic species in the ion trap. The extraction cycle is carried out for clearing the ion trap for a new cycle.
Results and discussion
A. The kinetics of propene aggregation on V4O11−
In the initial report on the photoinduced oxygenation reaction of propene, it was assumed that the reaction initiates out of the predefined geometry of the [V4O11·C3H6]− adsorbate complex formed in the collision of propene with the V4O11−cluster ion.26 Considering the general agreement in the literature concerning the virtually inert behavior of small anionic vanadium oxide species towards small hydrocarbons, the complex was assumed to be determined by physisorption, i.e. the chemical nature of the cluster ion and propene is virtually preserved and no covalent linkage is involved in the binding of propene.29 This is in contrast to neutral or cationic vanadium oxide clusters that show a distinct, spontaneous reactivity towards hydrocarbons.9,13,16,19–22,31 In order to gain insight into the validity of this assumption or establish an alternative mechanism for this aggregation process, the conditions of the ion–molecule collisions that determine the nature of this process must be varied. Experimentally, the control of the temperature via the helium bath in the ion trap is the most precise mechanism for varying the collision conditions. Accordingly, Fig. 1 shows the temperature dependent reaction kinetics in the aggregation of propene, which are obtained with the experimental procedure in the Methodology section. Traces of the relative yield in the V4O11−cluster ion and its aggregate complex with propene as well as the V4O10−cluster ion are monitored as a function of the reaction time in the propene-seeded helium atmosphere of the ion trap. Two distinctive features of these measurements are critical for their interpretation: (i) the rate as well as the steady-state level in the relative yield of the V4O10−cluster ion are virtually independent of the temperature and (ii) the relative yield in the consumption and formation of the V4O11−cluster ion and its aggregate complex with propene are highly correlated and show a distinctive increase in the reaction rate for higher temperature.![Aggregation kinetics of propene on the V4O11−cluster system at 200, 250 and 300 K. The experimental data are given in symbols for V4O11− (triangles), [V4O10(OH)C3H5]− (circles) and V4O10− (squares). The fit with the kinetic model (gray lines—red online) given by reactions (1), (2) and (3) is displayed together with the experimental data. For details concerning the kinetic model, see ESI.](/image/article/2012/CP/c1cp22550b/c1cp22550b-f1.gif) | ||
| Fig. 1 Aggregation kinetics of propene on the V4O11−cluster system at 200, 250 and 300 K. The experimental data are given in symbols for V4O11− (triangles), [V4O10(OH)C3H5]− (circles) and V4O10− (squares). The fit with the kinetic model (gray lines—red online) given by reactions (1), (2) and (3) is displayed together with the experimental data. For details concerning the kinetic model, see ESI.† | ||
The observation that the V4O10−cluster ion is generated under thermal conditions requires the discussion of its origin. Two possibilities can be entertained, which are based on the CID work of Castleman and coworkers or the proposed thermal reaction of propene oxidation on V4O11− in the DFT model given by Tian and coworkers.24,25 The latter option can be excluded due to the temperature independent behavior of the V4O10− product and the virtually mono-exponential rise in the relative yield of the aggregate complex. The kinetics of the complex are void of minima or maxima, which would constitute the characteristic behavior in the yield of an intermediate species according to a possible thermal oxygenation reaction. In view of a several eV kinetic energy of the V4O11− upon entering the ion trap reactor, the formation of the V4O10− species can be assigned to CID with the helium bath, analogous to the experiments from the group of Castleman and coworkers. The slow rise in the relative yield of V4O10− which is independent of the rate observed for the other two species is a clear signature of the thermalization of V4O11−via collisions with the helium bath and not the product of an oxygenation reaction. This explains the temperature independent behavior and an important consequence arises for the kinetics of the other reaction species. These must be considered as decoupled from the fully independent rate of the direct CID of V4O11− in the helium atmosphere according to:
| V4O11− → V4O10− + O (kCID) | (1) |
| V4O11− + C3H6 ⇌ [V4O11·C3H6]− (kad/kde) | (2) |
| [V4O11·C3H6]− → [V4O10(OH)C3H5]− (kH) | (3) |
Utilizing the model in (2) and (3) and including the independent rate of (1), the experimental data in Fig. 1 can be fitted for extracting the rate constants. The derivation of the model providing an analytic expression for extracting the rate constants from the data is given in the ESI.† From the model, the rate of complex formation is given by the coupled rates in the equilibrium of the aggregation and the irreversible hydrogen transfer according to kCF = kH [kad/(kde + kH)]. The rates of product formation at 200, 250 and 300 K are given in Table 1. The model is confirmed by numeric integration of the rate equation using these rate constants.
| Temperature/K | Rate constant, kCF/s−1 |
|---|---|
| 200 | 1.98 × 10−1 ± 0.17 × 10−1 |
| 250 | 5.82 × 10−1 ± 0.26 × 10−1 |
| 300 | 7.39 × 10−1 ± 0.27 × 10−1 |
The classical Arrhenius plot with the temperature dependent rate constant kCF is shown in Fig. 2, yielding an activation barrier of Ea = 70.1 ± 17.0 meV (1.61 ± 0.392 kcal mol−1 or 6.75 ± 1.64 kJ mol−1). This experimental value can also be compared to the theoretical value given by Tian and coworkers.24 The result obtained from the kinetics of the propane aggregation on the V4O11−cluster system will play a central role in reevaluating the photoinduced reaction in the following section.
![Arrhenius plot of the rate constant kCF from the model given in the ESI for the formation of the [V4O10(OH)C3H5]− complex with the standard deviation from the model. The values of kCF are determined from the reaction kinetics of propene with V4O11− at 200, 250 and 300 K shown in Fig. 1. The fit to the data (gray line—red online) yields an activation barrier, Ea = 70.1 ± 17.0 meV, for the coupled rates of equilibrium in the aggregation in reaction (2) and the irreversible hydrogen abstraction that follows in reaction (3). The parallel depletion of the V4O10−viaCID in (1) is also considered.](/image/article/2012/CP/c1cp22550b/c1cp22550b-f2.gif) | ||
| Fig. 2 Arrhenius plot of the rate constant kCF from the model given in the ESI† for the formation of the [V4O10(OH)C3H5]− complex with the standard deviation from the model. The values of kCF are determined from the reaction kinetics of propene with V4O11− at 200, 250 and 300 K shown in Fig. 1. The fit to the data (gray line—red online) yields an activation barrier, Ea = 70.1 ± 17.0 meV, for the coupled rates of equilibrium in the aggregation in reaction (2) and the irreversible hydrogen abstraction that follows in reaction (3). The parallel depletion of the V4O10−viaCID in (1) is also considered.† | ||
B. The product channels of the photoreaction
The mechanism of hydrogen abstraction from propene in the initial thermal aggregation on the V4O11−cluster has significant consequences on a photoinduced reaction on this system. From the analysis made in the previous section, the reaction complex that serves as a starting point for a photoinduced reaction is not the [V4O11·C3H6]− adsorbate complex originally postulated based on the previously reported inert behavior of anionic vanadium oxide cluster ions towards small hydrocarbons.26,29 The agreement of the kinetic model with the mechanism of Tian and coworkers for the aggregation process shows the [V4O10(OH)C3H5]− complex to be the precursor for the photochemistry that will be examined in the following.24 From the analysis of Sauer and coworkers, the single occupied molecular orbital in V4O11− with a high radical character on one of the terminal oxygen atoms of the dioxo moiety presents the most logical possibility of a covalent linkage of the resulting allyl radical to the cluster subsequent to the thermally motivated hydrogen abstraction.23 This is further in agreement with the model of propene aggregation given by Tian and coworkers.These considerations define the structure of the aggregate complex and bring the originally reported photochemistry of this system in a new light. Accordingly, the reaction channels resulting from the activation of the [V4O10(OH)C3H5]− complex through electronic excitation with femtosecond pulses at 272 nm (4.56 eV) require further investigation. Panels (a)–(d) in Fig. 3 show the mass spectrum of the anionic cluster species obtained from the ion trap reactor after irradiation, detected with the experimental procedure for the photoreaction described in the Methodology section. As originally reported, UV photo-excitation of this system yields a mass spectrum of the anionic species from the ion trap reactor with a depleted band for the original [V4O10(OH)C3H5]− complex as well as the V4O11−, V4O10− and [V4O8(OH)2]− photoproducts.26 These species were assigned to the corresponding independent reaction channels of photo-dissociation, photo-oxygenation and photo-oxygenation accompanied by multiple hydrogen abstraction. In order to gain further insight into the mechanism with which these three species are formed, the relationship of the individual reaction channels to the initial photo-excitation is investigated. For this, the branching ratio seen in the yield of each photoproduct can be monitored as a function of the fluence of the irradiation that drives the photoinduced reaction, i.e. the number of photons supplied for the reaction is correlated to the yield of each reaction channel. Important for this approach is the consideration that the photo-excitation at 4.56 eV is a one-photon process. In view of the vertical detachment energy given by Sauer and coworkers at 6.66 eV for the V4O11−cluster system, multi-photon excitation at these excitation energies would prepare the reaction complex in the neutral state.23 Since the anionic mass spectrum clearly shows the resulting cluster species from the photoreaction in the anionic charge state, two- or multi-photon excitation can be excluded.
![Yield of the characteristic photoproducts V4O11− (380 amu) and V4O10 (364 amu) and the residual [V4O8(OH)2]− (366 amu) intermediate obtained from the photo-excitation at 272 nm of the [V4O10(OH)C3H5]− complex (422 amu) for varying fluence under the irradiation at (a) 8.70, (b) 34.8, (c) 60.9 and (d) 80.7 × 10−2 J cm−2. For clarity, an overview of the mass spectrum (left) as well as a magnification of the 360–370 amu mass region (right) is given.](/image/article/2012/CP/c1cp22550b/c1cp22550b-f3.gif) | ||
| Fig. 3 Yield of the characteristic photoproducts V4O11− (380 amu) and V4O10 (364 amu) and the residual [V4O8(OH)2]− (366 amu) intermediate obtained from the photo-excitation at 272 nm of the [V4O10(OH)C3H5]− complex (422 amu) for varying fluence under the irradiation at (a) 8.70, (b) 34.8, (c) 60.9 and (d) 80.7 × 10−2 J cm−2. For clarity, an overview of the mass spectrum (left) as well as a magnification of the 360–370 amu mass region (right) is given. | ||
A clear trend can be observed in the yield of the product species shown in Panels (a)–(d) of Fig. 3 for an increasing fluence of 8.70 to 80.7 × 10−2 J cm−2 supplied for the photoreaction. The ratio in the relative yield of the dissociation channel V4O11− and the oxygenation channel V4O10− is nearly inverted, while the ratio of the oxygenation channel given by V4O10− and the reaction channel of oxygenation accompanied by hydrogen abstraction [V4O8(OH)2]− is virtually constant. The latter is significant in view of the mechanism of the reaction. Two possible interpretations can be entertained in view of the behavior of the relative yield of the V4O10− and [V4O8(OH)2]− products as a function of increasing fluence in the photo-excitation. The reaction rates for the processes that initiate these two channels can either be assumed as nearly constant (identical ratio for the rate determining step) or the two cluster species are involved in a single reaction channel. In view of the chemistry involved, the possibility of independent channels with nearly identical ratio in rates is viewed as highly unlikely and discarded in the following interpretation.
By considering the coupled yield of the V4O10− and [V4O8(OH)2]−cluster products seen in the fluence dependency in Panels (a)–(d) of Fig. 3, their participation in the mechanism of the oxygenation reaction can be considered. Recalling the results obtained from kinetic studies summarized in reaction (3) of Section A, the photoreaction initiates from a cluster complex in which hydrogen abstraction has already proceeded in the aggregation of propene on the V4O11−cluster. The resulting allyl radical is assumed to be bound to one of the dioxo oxygen atoms. Keeping this aspect of aggregation in mind, two reaction channels are considered that result from photo-excitation, which are characterized by the position of the bond cleavage in the release of the hydrocarbon from the cluster. These can be summarized by,
| [V4O10(OH)C3H5]− → [V4O10(OH)]− + C3H5 | (4) |
| [V4O10(OH)]− → V4O11− + H | (5) |
| [V4O10(OH)C3H5]− → [V4O8(OH)2]− + C3H4O | (6) |
| [V4O8(OH)2]− → V4O10− + 2 H | (7) |
C. Identification of the neutral reaction products
The analysis of the mechanism underlying the oxidation of propene on the V4O11−cluster given in Sections A and B has been conducted within the classical experimental capabilities available to the mass spectrometry of ion-molecule reactions. With the first report of this photoreaction, a novel experimental approach was introduced into the mechanistic analysis of these types of reactions.26 This was achieved by utilizing high-intensity, femtosecond pulses for the photo-excitation rather than nanosecond or cw sources. The significant intensity innate to ultrashort laser pulses and the enhanced capability to multi-photon excitation and ionization have allowed for the novel capability of detecting and analyzing the neutral participants of the ion–molecule reaction via in situ multi-photon ionization. The complementary nature of the information gained from the detection of the charged anionic cluster products and the ionized neutral photoproducts is significant for the analysis of the mechanism.Importantly, the approach goes beyond obtaining the stoichiometry of the neutral reaction participants via the mass spectrometry of the ionized species. The characteristic fragmentation that accompanies the multi-photon ionization process has opened the possibility of gaining direct insight into the structural isomers that are active in the reaction. This noteworthy capability of acquiring structural information in the context of mass spectrometry could be established in a highly fruitful exchange with Schröder, Schwarz and coworkers, which has been documented in the original publication of the photoreaction and the following analysis of the neutralization–reionization experiments of allyl-oxide anions reported by Schröder et al.26,31 A crucial aspect of interpreting the fragmentation patterns resulting from the ionization is the differentiation of the species generated in the fragmentation of the multi-photon ionization process and products that result from the ion–molecule reaction. While the analysis by Schröder et al. is fully conclusive, this aspect is explored in greater detail by characterizing the intensity dependency of the fragmentation process involved in the photo-ionization of the neutral reaction products.
The procedure for these experiments is described in the Methodology section. The cationic mass spectra of the ionized neutral products obtained from the photoreaction at intensities of 1.35 to 6.75 × 1010 W cm−2 are shown in Panels (a)–(d) of Fig. 4. As previously reported, the cationic mass spectrum shows the fragmentation pattern of a dissociation channel in the region of 38 to 42 amu and an oxygenation product of propene at 53 to 58 amu. The former is correlated with the dissociation channel(4) and the latter to the oxygenation channel(6) in the analysis of the charged cluster products from the photoreaction shown in Section B. In the progression of increasing intensity, the relative yield of the different fragmentation channels of both species varies. From the scattering of different mass channels in Panels (c) and (d), it is evident that strong-field effects that deform the field free electronic potentials of the neutral molecular species begin to disturb the characteristic fragmentation. The focus of the following analysis will therefore proceed primarily from the fragmentation at moderate intensities for the multi-photon ionization in Panels (a) and (b). It is important to note that a comparison to collision induced ionization and fragmentation as performed by Schröder et al. should be made for the most moderate intensity of 1.35 × 1010 W cm−2 shown in Panel (a) of Fig. 4 in view of the distortion that high intensity, ultra-violet femtosecond pulses can impose on the electronic structure of an unsaturated hydrocarbon as well as sequential photo-fragmentation.
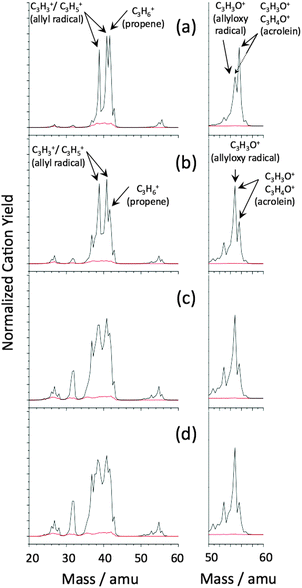 | ||
| Fig. 4 Cationic mass spectrum of the neutral photoproducts of the reaction obtained from in situ multi-photon ionization and fragmentation at (a) 1.35, (b) 2.70, (c) 4.73 and (d) 6.75 × 1010 W cm−2 (black lines) as well as the background ionization without V4O11− in the ion trap (gray lines—red online). The characteristic signals for the allyl radical (39, 41 amu) and propene (42 amu) can be identified. In the magnification of the 50–60 amu mass range, the signals of the allyloxy radical (55 amu) and acrolein (56, 55 amu) generated by the reaction mechanism (8) to (12) provide the capability to identify these products. The intensity dependency of these characteristic signals allows for the superposition of different species to be differentiated. | ||
Examining the relative yield of different fragmentation channels in the mass region of the oxygenated propene species, the pattern obtained directly from the photoreaction given in Panel (a) is in excellent agreement with the reported neutralization–reionization experiments on the allyloxy radical.31 The three fragmentation channels resulting from ionization determined by Schröder et al. can be reviewed for the allyloxy radical,
CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2O˙ → CH2CHCH2O+ (57 amu) + e− CHCH2O˙ → CH2CHCH2O+ (57 amu) + e− | (8) |
| CH2CHCH2O+ → CH2CHCO+ (55 amu) + H2 | (9) |
| CH2CHCH2O˙ → CH2CHCHO + H˙ | (10) |
| CH2CHCHO → CH2CHCHO+ (56 amu) + e− | (11) |
| CH2CHCHO → CH2CHCO+ (55 amu) + H˙ + e− | (12) |
These fragmentation channels can now be considered within the context of the oxygenation reaction of propene on the V4O11−cluster. The mechanism of hydrogen abstraction from the allylic position of propene as a precursor reaction to the photoreaction established in Section A is in accordance with an allylic radical product species. The linkage of the allyl radical on the dioxo site is the first step to oxygenation. Photoinduced ejection of an allyl oxide radical from the V4O11−cluster with further hydrogen loss in the neutral state given by Schröder et al. in reaction (10) can now be seen in the context of the photoinduced reaction of the [V4O10(OH)C3H5]−cluster complex and the detection of the [V4O8(OH)2]− intermediate species. Here, photoinduced cleavage of the dioxo linkage converts the allyl radical to the allyloxy radical. The subsequent hydrogen loss in reaction (10) that yields acrolein is equivalent to the second hydrogen transfer to the cluster in the oxygenation channel given in reaction (6) of Section B. This is confirmed by the anionic mass spectrum in Fig. 3 by the detection of [V4O8(OH)2]− as a photoproduct. This clarifies the full mechanism of the photoinduced oxygenation reaction, from the aggregation of propene as the allyl radical to the ejection of the allyloxy radical and formation of acrolein. It is emphasized that the two hydrogen transfer reactions associated with this channel are documented by the [V4O8(OH)2]− intermediate in the anionic mass spectrum of Fig. 3 and the detection of acrolein in the cationic ionization spectrum of Fig. 4. The agreement of this work with Schröder et al. is noteworthy.26,31
Further attention is now given to the signal at 55 amu in Fig. 4 with respect to the change in the entire fragmentation signature in this region for ionization at higher intensities (Panels (a) to (d)). The ionization mechanism given by Schröder et al., which is reproduced in reactions (8) and (9) for the allyloxy radical, must be considered in this context. Within the photoreaction on the [V4O10(OH)C3H5]−cluster complex, this channel corresponds to ionization of the allyloxy moiety, which is assumed to be in a highly activated state in the process of its dissociation from the cluster due to the substantial energy deposited from photo-excitation of the cluster complex at 272 nm (4.56 eV). For ionization prior to the second hydrogen transfer that produces acrolein, the allyloxy cation is detected after loss of molecular hydrogen at 55 amu. Noteworthy in the progression in Panels (a)–(d) is the rise in the relative yield of precisely this signal from reaction (8) at 55 amu relative to the signals obtained for acrolein (reactions (10) with (11) and (12) at 55 and 56 amu). This confirms the interpretation of Schröder et al. that the fragmentation pattern in the region of the oxygenated product at 55 to 57 amu is primarily composed of the superposition in the ionization and fragmentation channels of the allyloxy radical and acrolein in probing these species from the reaction. Hereby, the two channels can be differentiated due to the selective enhancement of the direct multi-photon ionization of the thermally activated allyloxy radical versusacrolein, which loses thermal energy in the ejection of hydrogen.
An analogous analysis can be made for the dissociation channel given in the photoreaction (4) of Section B. This channel was considered from the anionic mass spectrum in Fig. 3, yielding the V4O11− photoproduct, which would liberate the allyl radical from the [V4O10(OH)C3H5]−cluster complex by cleavage of the C–O linkage at the dioxo moiety. In order to understand the probing of an allyl radical as a photoproduct in this reaction channelvia multi-photon ionization, the intensity dependence shown in Panels (a)–(d) of Fig. 4 must be examined in the region of 38 to 42 amu. For an interpretation of the behavior for increasing intensity, the hydrogen transfer from propene in its initial aggregation to the cluster before the photoreaction must be considered.24 This allows all bands at and below the mass of 41 amu to be considered in their assignment to the allyl cation and its fragments in the probing of the neutral allyl radical. The signal at 42 amu must be assigned to ionized propene. Particularly in the progression to higher intensities from Panels (a) to (b), the bands at a mass of 39 and 41 amu rise in their relative yield in comparison to the signal at 42 amu. Similar to the enhanced ionization efficiency seen for the allyloxy radical for higher intensities, the mechanism of ionization of species ejected from a cluster complex with a substantial energy content from the initial photo-excitation is also enhanced at higher intensities for the allyl radical.
Here it becomes clear that enhanced ionization of the hydrocarbon initially bound to the dioxo unit (allyl or allyloxy radical) originates from the same mechanism of significant energy transfer from the photo-excited cluster system via the covalent linkage. The probing of these two neutral species of the reaction is just a reflection of the general reaction channels of dissociation in reaction (4) and propene oxygenation in reaction (6). In contrast, the steady relative ionization efficiency yielding the signal at 42 amu must be interpreted as the ionization of propene localized on a different region of the cluster, i.e. the photo-ionization captures propene in an association to the V4O11−cluster that has not led or does not lead to hydrogen abstraction and a covalent link to the cluster in the form of an allyl radical. Furthermore, a lack of enhanced ionization at higher intensities in the probing of acrolein at 56 amu can be seen as a result of energy transported out of the photoproduct with the ejection of hydrogen from the initial allyl-oxide radical. In summary, the photo-ionization of products is enhanced from all species that can take up thermal energy from the clustervia the initial covalent linkage at the dioxo group and do not have a mechanism of losing this energy via the kinetic energy involved in ejecting hydrogen (allyl- and allyloxy radical) in the neutral state. This enhanced ionization efficiency is most likely the result of advantageous overlap integrals for photo-ionization from thermally excited vibrational states of these species versus the probability of ionization from the vibrational ground state.
Conclusion
A complete picture of the mechanism underlying the aggregation of propene on the V4O11−cluster system and the subsequent photoinduced oxidation to acrolein from the electronic excitation of the [V4O10(OH)C3H5]− reaction complex has been given. Temperature dependent kinetic measurements of the aggregation process together with the model proposed by Tian and coworkers show the aggregation process to encompass a collision-motivated hydrogen transfer reaction to a vanadyl moiety and the linkage of the resulting allyl radical to the dioxo unit of the V4O11−cluster.23,24 The resulting geometry defines the [V4O10(OH)C3H5]− complex for the photoreaction initiated by electronic excitation of the system at 272 nm (4.56 eV). The fluence dependent detection of the V4O11− and V4O10−cluster products shows two active reaction channels that are characterized by the nature of the cleavage in the linkage of the hydrocarbon to the dioxo moiety. The products of these reaction channels are determined via in situ multi-photon ionization and accompanying fragmentation with subsequent detection of the cationic species. Utilizing the rigorous analysis for this process given by Schröder et al. for interpreting the intensity dependent ionization measurements, the stoichiometry of the product ions can be interpreted and assigned to specific structural isomers.31 For the case of a photoinduced V–O bond cleavage, the allyloxy radical is formed and in the process of ejection from the cluster a second hydrogen transfer can take place in the formation of acrolein. Alternatively, when the link is broken at the C–O bond, the allyl radical is liberated from the cluster complex. The substantial vibrational heating due to a radiationless transition back into the electronic ground state is postulated as the motivation for the cluster products to lose the transferred hydrogen. The detection of the residual [V4O8(OH)2]− intermediate species confirms the double hydrogen transfer in the case of acrolein formation.It is particularly a novel approach of analyzing an ion–molecule reaction by direct detection of the neutral species that has allowed for a significant mechanistic insight into the reaction of propene on the V4O11−cluster system. It completes the picture obtained for the thermal aggregation process and the mechanism in the channels of the photoinduced reaction.24,31 Importantly, the agreement with the model of Schröder et al. confirms the general capability to obtain information on the structural nature of neutral isomers in situ within the context of mass spectrometry.26,29 Beyond the specific information obtained for this reaction, this capability has allowed for a better understanding of the noteworthy reactivity offered by dioxo structures of vanadium oxide species for achieving selective oxidation chemistry. Within the general structural motifs offered by the V4O11−cluster system, it is the cooperative action of the vanadyl moieties in assisting hydrogen abstraction and the precise covalent linkage at the dioxo unit of the resulting radical hydrocarbon species that set the stage for a specific photo-oxygenation.24 Considering the V–O–V bridges that link these cooperatively acting reactive units, a relationship can be drawn to V2O5 surface structures and heterogeneous catalysis as well as the general concept of photo-catalysis for utilizing the unique reactivity innate to electronic excitation.27,28 The question of a possible application of this scheme to methane remains open.
With a last comment to the selectivity of the reaction, it is important to consider that the reaction channels that liberate the allyl and allyloxy radicals are only the result of an enhanced multi-photon ionization innate to the ultrashort laser pulses utilized in the experiment. Should this aspect in the photoreaction be suppressed by utilizing cw or nanosecond sources, it is reasonable to assume that the reaction channel uniquely yielding acrolein could be isolated. This is particularly evident in the formation of the allyloxy radical, where direct ionization of this species circumvents the second hydrogen transfer that leads to the formation of acrolein. Regardless of this circumstance, the oxidation of propene on the V4O11−cluster displays another crucial aspect of achieving selectivity in photoinduced reactions. In the formation of the aggregate complex, it is the thermally motivated hydrogen abstraction and covalent linkage of the hydrocarbon to the cluster system at the dioxo structure that provides a clearly defined starting point for the photo-excitation.24 Without this precursor, the substantial activation of the system by electronic excitation would result in a reactivity that encompasses a variety of geometries defined by the statistics of the collisions. This would lead to an accordingly broad spectrum of photoproducts. It can clearly be seen from this photoreaction that a defined thermal reactivity is a crucial prerequisite for selective photochemistry.
Acknowledgements
The authors gratefully acknowledge the financial support of the Collaborative Research Center (SFB) 546, “Structure, Dynamics and Reactivity of Aggregates of Transition Metal Oxides” of the Deutsche Forschungsgemeinschaft (DFG). The authors highly appreciate the original analysis of the aggregation process by Dr. Michael Walter from the Freiburger Materialforschungszentrum, Albert-Ludwigs-Universität Freiburg, which has significantly facilitated the interpretation of the experimental data. Sincere appreciation is given to Dr. Detlef Schröder from the Academy of Sciences of the Czech Republic and Prof. Dr. Helmut Schwarz from the Technische Universität Berlin for the fruitful discussions and thorough reading of the manuscript.References
- M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Surf. Sci. Rep., 2007, 62, 219 CrossRef CAS.
- W. Zhang and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 286 CrossRef CAS.
- W. Zhang and H. Yamamoto, Angew. Chem., Int. Ed., 2005, 44, 4389 CrossRef CAS.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25 CrossRef CAS.
- G. Ertl, H. Konozinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 1997 Search PubMed.
- C. Batiot and B. Hodnett, Appl. Catal., A, 1995, 137, 170 Search PubMed.
- D. Göbke, Y. Romanyshyn, S. Guimond, J. M. Sturm, H. Kuhlenbeck, J. Döbler, U. Reinhardt, M. V. Ganduglia-Pirovano, J. Sauer and H.-J. Freund, Angew. Chem., Int. Ed., 2009, 48, 3695 CrossRef.
- A. Dinse, S. Khennache, C. H. B. Frank, R. Herbert, S. Wrabetz, R. Schlögl and R. Schomäcker, J. Mol. Catal. A: Chem., 2009, 307, 43 CrossRef CAS.
- N. Dietl, R. F. Höckendorf, M. Schlangen, M. Lerch, M. K. Beyer and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 1430 CrossRef CAS.
- C. Ohde, M. Brandt, C. Limberg, J. Döbler, B. Ziemer and J. Sauer, Dalton Trans., 2008, 326 RSC.
- M.-J. Cheng, K. Chenoweth, J. Oxgaard, A. van Duin and W. A. Goddard, III, J. Phys. Chem. A, 2007, 111, 5115 CAS.
- J. Sauer and J. Döbler, Dalton Trans., 2005, 3116 Search PubMed.
- N. Dietl, M. Engeser and H. Schwarz, Chem.–Eur. J., 2010, 16, 4452 CrossRef CAS.
- J. Roithová and D. Schröder, Chem. Rev., 2010, 110, 1170 CrossRef.
- C. Ohde, C. Limberg, D. Schmidt, M. Enders, S. Demeshko and C. Knispel, Z. Anorg. Allg. Chem., 2010, 636, 2315 CrossRef CAS.
- T. Wende, J. Döbler, L. Jiang, P. Claes, E. Janssens, P. Lievensc, G. Meijer, K. R. Asmis and J. Sauer, Int. J. Mass Spectrom., 2010, 297, 102 CrossRef CAS.
- M. Zboray, A. T. Bell and E. Iglesia, J. Phys. Chem. C, 2009, 113, 12380 CAS.
- K. R. Asmis and J. Sauer, Mass Spectrom. Rev., 2007, 26, 542 CrossRef CAS.
- S. Feyel, D. Schröder and H. Schwarz, J. Phys. Chem. A, 2006, 110, 2647 CrossRef CAS.
- S. Feyel, J. Döbler, D. Schröder, J. Sauer and H. Schwarz, Angew. Chem., Int. Ed., 2006, 45, 4681 CrossRef CAS.
- D. R. Justes, R. Mitrić, N. A. Moore, V. Bonačić-Kouteck Bonačić-Koutecky and A. W. Castleman Jr., J. Am. Chem. Soc., 2003, 125, 6289 CrossRef CAS.
- R. C. Bell and A. W. Castleman Jr., J. Phys. Chem. A, 2002, 106, 9893 CrossRef CAS.
- G. Santambrogio, M. Brummer, L. Wöste, J. Döbler, M. Sierka, J. Sauer, G. Meijer and K. R. Asmis, Phys. Chem. Chem. Phys., 2008, 10, 3992 RSC.
- H.-B. Li, S. X. Tian and J. Yang, J. Phys. Chem. A, 2010, 114, 6542 CrossRef CAS.
- R. C. Bell, K. A. Zemski, D. R. Justes and A. W. Castleman Jr., J. Chem. Phys., 2001, 114, 798 CrossRef CAS.
- S. Li, A. Mirabal, J. Demuth, L. Wöste and T. Siebert, J. Am. Chem. Soc., 2008, 130, 16832 CrossRef CAS.
- H. Garcia, J. Manuel Lopez-Nieto, E. Palomares, H. D. Roth and B. Solsona, J. Mater. Chem., 2006, 16, 216 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- K. A. Zemski, D. R. Justes and A. W. Castleman Jr., J. Phys. Chem. A, 2001, 105, 10237 CrossRef CAS.
- X. Rozanska and J. Sauer, Int. J. Quantum Chem., 2008, 108, 2223 CrossRef CAS.
- D. Schröder, H. Schwarz and J. Roithová, J. Mass Spectrom., 2010, 301, 84 Search PubMed.
- I. Z. Kozma, P. Baum, U. Schmidhammer, S. Lochbrunner and E. Riedle, Rev. Sci. Instrum., 2005, 75, 2323 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: A description of the kinetic model for the aggregation of propene on the V4O11−cluster is given based on the mechanism according to reactions (1)–(3). This includes the procedure for deriving the analytical solutions for the corresponding rate constants and their application in fitting the experimental data. Furthermore, tables for the temperature dependency of all the rate constants involved in the aggregation process are given. See DOI: 10.1039/c1cp22550b |
| This journal is © the Owner Societies 2012 |