A computational study of organic polyradicals stabilized by chromium atoms
Ka-Un
Lao
,
Pei-Kang
Tsou
,
Timm
Lankau
and
Chin-Hui
Yu
*
Department of Chemistry, National Tsing Hua University, 101 KuangFu Road Sec. 2, HsinChu, 30013, Taiwan. E-mail: chyu@oxygen.chem.nthu.edu.tw; Fax: +886-3-5721534; Tel: +886-3-5162080
First published on 9th November 2011
Abstract
Density functional theory has been used to investigate the properties of organic high spin molecules. The M05/cc-pVDZ calculations predict a septet ground state for the 2,3,6,7,10,11-hexahydro-1,4,5,8,9,12-hexaoxocoronene-2,3,6,7,10,11-hexayl radical (coronene-6O). The computations show further that the formation of intermolecular carbon–carbon bonds yields a singlet ground state for the dimer rather than a possible tridectet state as expected from the monomer's multiplicity. A benzene molecule placed between coronene-6O molecules leads to the desired high-spin cluster, but the overall stability of the cluster is low. A chromium atom inserted between two peripheral C6 rings of coronene-6O yields a sandwich structure with the expected tridectet ground state and a binding energy which is 15 times larger than the corresponding tridectet dimer stabilized by a benzene molecule. The presented DFT calculations suggest that a chromium atom can effectively link organic polyradicals to larger magnetic units.
1. Introduction
The addition of magnetic atoms and the incorporation of high spin radicals into non-magnetic molecules are two common methods to produce organic magnetic materials.1–4 The observation of a macroscopic magnetic moment requires the non-destructive coupling of the individual magnetic moments.1–5Previous studies of organic ferromagnets showed that the presence of a diamagnetic molecule, such as a polycyclic aromatic hydrocarbon (PAH), between pairs of ion-radicals generates an effective ferromagnetic coupling among the radicals.6 Nevertheless, the π–π interactions between ion-radicals and PAHs are not strong enough to support a stable magnetic superstructure. Hence, novel methods to stabilize organic ferromagnets are demanded. Organometallic sandwich compounds may be an answer.
Condensation experiments in the gas phase7 suggest that the maximum size of these sandwich stacks is limited to four or five metal atoms. The observed limit in stack size is linked to continuous decrease of the ionization potential with the increase in stack size, which renders long sandwich polymers chemically unstable.7,8 A chromium triple-decker Cr2(C6H6)3 was not observed in the aforementioned laser evaporation experiments,7,9 but its mesitylene analogue Cr2[C6H3(CH3)3]3 has been successfully synthesized10 and characterized.11 The laser evaporation experiments show that the ionization potential of multi-decker sandwich compounds decreases rapidly with the stack size and Lamanna registered, in agreement with this result, an extreme oxygen sensitivity of Cr2[C6H3(CH3)3]3 solutions.10 Jiang et al.12 circumvented the problem of decreasing chemical stability altogether by putting a spacer between the arene units. Looking at the vast amount of bis(arene)chromium compounds13,14 synthesized so far and the even larger number of possibilities to assemble these units into larger structures, this work restricts itself to bis(arene)chromium compounds as possible building blocks in magnetic supermolecules and neglects the details of actual assembly.
The remainder of this paper is organized in three parts. The computational setup section covers computational details such as the evaluation of different density functional theory (DFT) methods for the description of open shell electronic states and the choice of basis sets. The design of the computational experiments as well as links to older one-electron methods will also be discussed in this section. The following results and discussion part is divided into three sub-sections. The first one investigates the possibilities of injecting spin into PAHs by increasing the size of the electronic π-system with oxygen atoms. The second part focuses on the benzene molecule as a link6 between the high-spin arene molecules and the third subsection reports the results for a chromium atom as a linker. Finally, the conclusions from this work are summarized in section 4.
2. Computational details
2.1 Model setup
Coronene (1, Scheme 1) is one of the simplest PAHs and a common model for graphene, but its dimer is still small enough to do all electron calculations to analyze the interactions between layers. Recently, Vergés et al.15 suggested that high-spin organic molecules may be obtained from diamagnetic coronene molecules by the selective hydrogenation of peripheral carbon atoms. The addition of hydrogen atoms effectively reduces the size of the π-system while simultaneously unbalancing it. Herein, we follow the opposite approach by adding oxygen atoms to the edges of the coronene molecule and thereby enlarge the size of the π-system.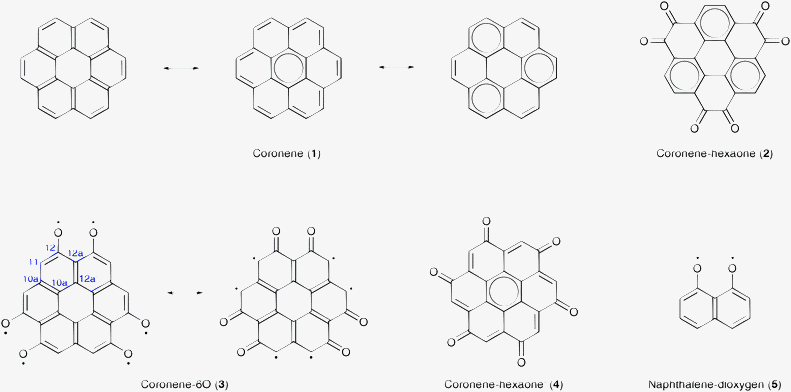 | ||
| Scheme 1 Coronene, coronene-hexaone, and coronene-hexaone′ molecules have singlet ground states with D6h, D3h, and C6h symmetry, respectively. Coronene-6O and coronene-6O′ molecules with D3h symmetry have septet ground states. The naphthalene dioxygen molecule with C2v symmetry has a triplet ground state. The numbers in the aromatic ring of molecule 3 are used to distinguish the different position of carbon atoms as defined in IUPAC. | ||
Hexamethoxycoronene and coronene-1,2,5,6,9,10-hexaone (2, coronene-hexaone) have been synthesized for their possible application in organic semiconductors.16 The oxygen atoms in compound 2 have been rearranged to obtain the coronene-1,4,5,8,9,12-hexaylhexakisoxyl radical (3, coronene-6O), which carries one unpaired electron on each oxygen atom, and coronene-1,3,5,7,9,11-hexone (4, coronene-hexone′) as possible candidates for high-spin organic molecules. In a first approximation, the oxygen atoms in compounds 2, 3 and 4 can be treated in the same way as carbon atoms which results in the same molecular graphs for the oxygenated coronene molecules as those of alternate hydrocarbons with 30 π-electrons (Ne = 30, Ne is the number of electrons). The carbon atoms in alternate hydrocarbons can be separated into two disjoint subsets A and B, where each atom in A has a direct neighbor in B, but none in A, and vice versa. Compounds 2 and 4 are expected to have the same properties as balanced alternate hydrocarbons as both subsets are equally strong (NA = NB, NX is the number of atoms/sites in the subset X). Compound 3 has to be considered as unbalanced, because the first subset contains 6 more atomic sites than the second (NA − NB = 6). Basic Hückel calculations predict a septet ground state for compound 3 and a singlet for compounds 2 and 4, in agreement with the relative sizes of the subsets.17
Another route for the prediction of the ground state multiplicity of alternant hydrocarbons is based on Lieb's second theorem and has been chosen by Vergés et al.15 Lieb's second theorem18 derived from the analysis of the Hubbard Hamiltonian states that the spin S of a unit cell in a solid with a finite bipartite lattice with an even number of crystallographic sites |Λ| = |NA| + |NB| is proportional to the difference in lattice sites S = ½(NB − NA) (Lieb's equation) for a half-filled band (Ne = |Λ|) and a repulsive on-site energy. The bipartite condition is more a character of the hopping matrix than a physical necessity and hence Vergés et al.15 were able to apply Lieb's equation to the analysis of PAHs after dividing the carbon atoms into two disjoint subsets. Lieb's equation predicts a singlet ground state for compounds 2 and 4 and a septet for compound 3 in agreement with the Hückel calculations. This agreement is hardly surprising as the Hückel and Hubbard Hamiltonians are essentially nearest-neighbor interaction Hamiltonians, one from a molecular view and the other from solid-state theory.
The Hückel calculations predict six non-bonding singly occupied molecular orbitals (SOMOs) for compound 3. The easiest way to achieve intramolecular ferromagnetic coupling among these electrons is distance. Once two coronene-6O molecules are so far apart that the overlap between the SOMOs can be neglected, Hund's rule predicts electrons with parallel spin in each of the SOMOs. The total spin would then be an extensive property of the system scaling with the number of coronene-6O units and a long-range magnetic order can be expected. The main objective of this study is therefore to find a spacer, which keeps two coronene-6O molecules sufficiently apart to support ferromagnetic coupling while simultaneously forming chemical bonds to the coronene-6O units strong enough to stabilize an extended superstructure.
Compound 3 is expected to be a highly reactive polyradical. However, it was chosen as a model to study the stacking of organic polyradicals for two reasons. Firstly, the oxygenation offers a theoretically elegant way to extend a π-system to unbalance it. Secondly, the multiplicity of the ground state can be tuned easily by changing the number or position of peripheral oxygen atoms.
2.2 Selecting a DFT method
The energy differences between the lowest singlet and triplet states of methylene and naphthalene-1,8-diylbisoxyl diradical (5, naphthalene-dioxygen) were used to choose a suitable density functional for the present study. The current benchmark values derived from McKellar's experimental data19–21 show that the triplet state of methylene is 0.408 eV lower in energy than the singlet state.22 No experimental data are available for compound 5. Hence, the reference value for the singlet–triplet gap in compound 5 was obtained from CASSCF(12,12) calculations using Dunning's cc-pVDZ basis set.23 These CASSCF(12,12)/cc-pVDZ calculations show that the triplet ground state should be 0.250 eV lower in energy than the lowest lying singlet state.DFT has been widely used in calculations of the properties of chromium sandwich complexes.24–30 However, DFT calculations also have a reputation for describing transition metals and dispersive interactions poorly.31 The M05 functional describes simultaneously dispersive interactions and the chemistry of transition metals correctly32–36 and was therefore chosen for this study. Table 1 lists the results for singlet–triplet gap in methylene and compound 5 calculated with various basis sets. The results indicate that the combination of the M05 functional with the cc-pVDZ basis set reproduces the reference data for the singlet–triplet gap reasonably well: 0.435 eV for methylene and 0.198 eV for compound 5. Hence, the cc-pVDZ basis set was chosen for the non-metal atoms.
| Methods | ΔES–T for methylene | ΔES–T for naphthalene dioxygen |
|---|---|---|
| a Experimental data, ref. 19–21. b CASSCF(12,12)/cc-pVDZ. | ||
| M05/6-31G(d) | 0.547 | 0.463 |
| M05/6-31+G(d) | 0.508 | 0.383 |
| M05/6-31G(d,p) | 0.540 | 0.463 |
| M05/6-31+G(d,p) | 0.503 | 0.382 |
| M05/6-31++G(d,p) | 0.502 | 0.382 |
| M05/6-311G(d) | 0.419 | 0.127 |
| M05/6-311+G(d) | 0.412 | 0.071 |
| M05/6-311G(d,p) | 0.433 | 0.135 |
| M05/6-311+G(d,p) | 0.428 | 0.080 |
| M05/6-311++G(d,p) | 0.430 | 0.080 |
| M05/cc-pvdz | 0.435 | 0.198 |
| Reference data | 0.408a | 0.250b |
The model chemistry for the chromium atom was chosen on the basis of M05 calculations for bis(1,3-i-Pr-indenyl)CrII which has a quintet ground state.37 The non-metal atoms were described by the aforementioned cc-pVDZ basis and Wachters' primitive all-electron valence double zeta basis or the LANL2TZ triple zeta quality basis set, both augmented with one set of f functions.38–40 The corresponding relativistic effective core potentials were used with the LANL2TZ basis set. Both sets of calculations produce very similar results and correctly identify the quintet state as the ground state. The next state is the triplet state, 0.30 eV above the ground state in the LANL2TZ(f) calculation and 0.32 eV in the calculation using Wachter's basis set. Wachters(f) basis set has been successfully used to study the properties of metal sandwich compounds41 and therefore will be used for the presented study.
Geometry optimizations were done routinely with symmetry constraints and all DFT calculations for open-shell high spin states and closed-shell singlet states were done using the spin-unrestricted and spin-restricted formalism, respectively. Unrestricted Kohn–Sham (UKS) singlet determinant calculations have been widely used to study the spin properties of open shell systems, because UKS results do not suffer from spin contaminations as much as unrestricted Hartree–Fock calculations.32,42–49 In our calculations, the expectation values of the S2 operator, S(S + 1), showed that spin contamination is less than one percent.
A natural bond orbital (NBO) analysis50,51 was done to probe the chemical bonds in the systems. All the DFT calculations were done with program suite Gaussian 0952 while the CASSCF calculations were done with program suite MOLPRO 2008.1.53
2.3 Verification of the chosen method
A key element of this study is the correct identification of ground state multiplicities and hence the chosen computational procedure was tested on a set of chromium compounds with well-known ground state multiplicities: The inverted sandwich complex (μ-η6:η6-C6H6) [CrI(DDP)]2 (DDPH is 2-{(2,6-diisopropylphenyl)amino}-4-{(2,6-diisopropylphenyl)imino}pent-2-ene) abbreviated as BCr features a septet ground state.54 A quintet ground state has been reported for bis(1,3-i-Pr-indenyl)CrII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 and CrIICp*Tp (Cp* is pentamethylcyclopentadienyl and Tp is hydrotris(pyrazolyl)borate).55 The CrIICp256,57 complex has a triplet ground state and Cr(C6H6)214 a singlet. The geometries of BCr, bis(1,3-i-Pr-indenyl)CrII, CrIICp*Tp, CrIICp2, and Cr(C6H6)2 were optimized with various numbers of unpaired electrons ranging from singlet to undectet spin states under the constraint of frozen D2, C2h, Cs, D5d, and D6h symmetries, respectively. The different spin states and corresponding optimized ground state geometries for the aforementioned five systems are summarized in Fig. 1 and Table 2. The computational prediction of the ground state spin multiplicities of the five complexes completely agrees with the experimental results and the chosen combination of functional and basis sets are adequate for the molecules studied in this work.
37 and CrIICp*Tp (Cp* is pentamethylcyclopentadienyl and Tp is hydrotris(pyrazolyl)borate).55 The CrIICp256,57 complex has a triplet ground state and Cr(C6H6)214 a singlet. The geometries of BCr, bis(1,3-i-Pr-indenyl)CrII, CrIICp*Tp, CrIICp2, and Cr(C6H6)2 were optimized with various numbers of unpaired electrons ranging from singlet to undectet spin states under the constraint of frozen D2, C2h, Cs, D5d, and D6h symmetries, respectively. The different spin states and corresponding optimized ground state geometries for the aforementioned five systems are summarized in Fig. 1 and Table 2. The computational prediction of the ground state spin multiplicities of the five complexes completely agrees with the experimental results and the chosen combination of functional and basis sets are adequate for the molecules studied in this work.
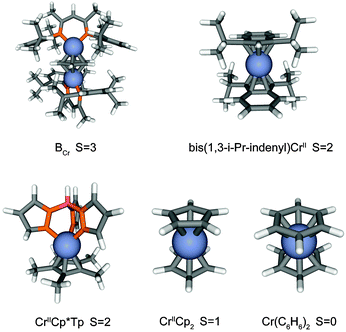 | ||
| Fig. 1 Optimized geometries of BCr, bis(1,3-i-Pr-indenyl)CrII, CrIICp*Tp, CrIICp2, and Cr(C6H6)2 in the septet, quintet, quintet, triplet, singlet spin state under D2, C2h, Cs, D5d, and D6h symmetry, respectively. Carbon atoms are shown in grey, hydrogen atoms in white, nitrogen atoms in orange, boron atom in pink, and chromium atoms in blue. | ||
| Spin states | BCr (D2) | bis(1,3-i-Pr-indenyl)CrII (C2h) | CrIICp*Tp (Cs) | CrIICp2 (D5d) | Cr(C6H6)2 (D6h) |
|---|---|---|---|---|---|
| a The relative state energies (in eV) of five systems are relative to their corresponding spin ground state which is septet spin state for BCr, quintet spin state for bis(1,3-i-Pr-indenyl)CrII and CrIICp*Tp, triplet spin state for CrIICp2, and singlet spin state for Cr(C6H6)2. | |||||
| Undectet | 2.264 | 8.595 | 10.861 | 12.726 | 12.189 |
| Nonet | 1.286 | 4.860 | 6.651 | 8.405 | 8.053 |
| Septet | 0.000 | 2.717 | 2.622 | 3.989 | 2.990 |
| Quintet | 1.996 | 0.000 | 0.000 | 0.374 | 2.088 |
| Triplet | 2.762 | 0.318 | 0.610 | 0.000 | 1.020 |
| Singlet | 3.252 | 1.684 | 1.910 | 1.364 | 0.000 |
3. Results and discussion
3.1 Coronene-6O dimer
Table 3 summarizes the DFT results for compounds 2 to 4. Compound 3 features a septet state while compounds 2 and 4 have singlet ground states as predicted by the initial Hückel calculations. However, while coronene is a flat molecule, the geometry of coronene-6O is curled as the distance between neighboring oxygen atom increases. The lowest energy was observed for the conformer with C3 symmetry with a C1 conformer (Fig. 2) energetically close by (0.013 eV). A planar coronene-6O geometry is a high-order stationary point on the coronene-6O potential energy surface and 0.047 eV higher in energy. Another located stationary point (3 imaginary frequencies) has C2v symmetry and is almost planar. This point is 0.046 eV higher in energy than the global minimum with C3 symmetry. The observed energy differences are small and therefore likely to be method dependent. However, these calculations suggest that the energy of a coronene-6O conformer is controlled by the average distance between oxygen atoms in neighboring carbonyl groups (C3: 2.719 Å, C1: 2.718 Å, C2v: 2.691 Å, D3h: 2.694 Å).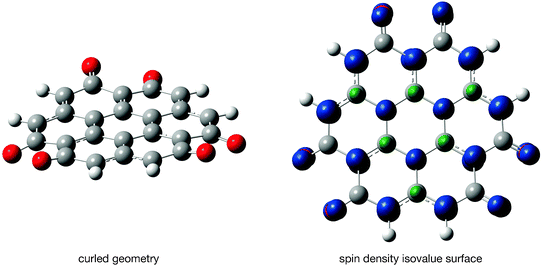 | ||
| Fig. 2 Compound 3 (septet state, C1 symmetry) and its spin density (isovalue: 0.02 a.u.). Carbon atoms are shown in grey, hydrogen atoms in white, and oxygen atoms in red. Blue lobes indicate a positive spin density and green ones a negative. | ||
| Spin states | Compound 2 | Compound 3 | Compound 4 |
|---|---|---|---|
| a The relative state energies (in eV) of three systems are relative to their corresponding spin ground state which is a septet spin state for compound 3, singlet spin state for compound 2 and 4. | |||
| Septet | 5.154 | 0.000 | 3.367 |
| Quintet | 3.476 | 1.122 | 2.638 |
| Triplet | 1.695 | 1.164 | 1.752 |
| Singlet | 0.000 | 2.101 | 0.000 |
The CC bond lengths in coronene-6O with C3 symmetry range between 1.400 and 1.481 Å, while a slightly larger variation was observed in the C2v conformer (1.398 to 1.485 Å). The lower value can be compared with a CC bond in benzene (1.395 Å) while the upper value is similar to a single bond in 1,4-benzoquinone (1.486 Å) and thereby longer than a single bond in 1,3,5-hexatriene (1.448 Å) calculated on the same computational level. The variation in CC bond lengths suggest a breakdown of the aromaticity of the coronene molecule upon oxygenation. Further evidence for this breakdown is given by the calculated 13C NMR shifts calculated using the GIAO method as implemented in Gaussian09 at the current computational level. The calculated 13C NMR shifts are 134 ppm for benzene and 133.5 ± 0.2 ppm (outer ring) and 127.6 ± 0.1 ppm (inner ring) for coronene relative to TMS. These values are in fair agreement with experimental data (128.36, 127.4 (average value), 122.53 ppm58) and are used as internal reference values. The same computational method applied to the C2v conformer of cornene-6O suggests chemical shifts ranging between −12.4 ppm (average value, carbon atom 12a, Scheme 1) to 219.8 ppm (average value, carbon atom 12). A smaller range of values has been observed in the C1 conformer with values starting at 34.5 ppm and ending at 211.8 ppm. In summary, the bond length and NMR data suggest the π-electron system in cornene-6O has to be considered as highly delocalized, rather than aromatic.
Fig. 2 also shows the total spin densities of the septet ground state. Large, positive spin densities are observed on the atom-subset including the oxygen atoms as predicted by the Hückel calculation and small, negative spin densities are observed on the alternate carbon atoms. The even smaller and negative spin densities on the carbonyl carbon atoms are smaller than the isovalue chosen for the plot and therefore not visible in Fig. 2. Negative spin densities are caused by electron-electron correlation effects59 and therefore cannot be predicted with a Hückel model. The pattern in the spin density and the increased OO distance in the ground state geometry suggest that Coulomb repulsion and the Pauli exclusion principle control the energy and shape of compound 3.
The spin densities on the peripheral oxygen and hydrogen atoms are 0.372 and −0.029 electron/Bohr3 in compound 3 and those on the carbon atoms 12, 12a, 12a1, 10a1, 10a and 11 (Scheme 1) are −0.174, 0.564, −0.350, 0.519, −0.301, and 0.616 electron/Bohr3 which suggests that the unpaired electrons are located on the peripheral carbon atoms adjacent to a hydrogen atom (carbon atom 11). A more suitable Lewis structure for compound 3 is that of the 2,3,6,7,10,11-hexahydro-1,4,5,8,9,12-hexaoxocoronene-2,3,6,7,10,11-hexayl radical (Scheme 1). This assignment is further supported by the observation that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length is 1.23 Å in compound 3, which indicates a carbon-oxygen double bond rather than a single bond.
O bond length is 1.23 Å in compound 3, which indicates a carbon-oxygen double bond rather than a single bond.
McConnell's model60 suggests that ferromagnetic spin coupling through space between pancaked aromatic molecules is supported by intermolecular interactions between atoms carrying differently signed spin densities. Regions of positive and negative spin density are aligned in the stacked dimer of compound 3 (Fig. 3, cluster 6) with eclipsed oxygen and hydrogen atoms (OH stacking mode); thus cluster 6 is expected to have a tridectet ground state. Electronic structure calculations with various numbers of unpaired electrons in compound 6 in the OH stacking mode (D3d symmetry) have verified the assumption of a tridectet ground state. This open-shell ground state is 3.778 eV lower in energy than the lowest singlet state. The interlayer distance is 3.60 ± 0.06 Å and the bonding energy (BE) calculated from the ground state energies is 0.457 eV.
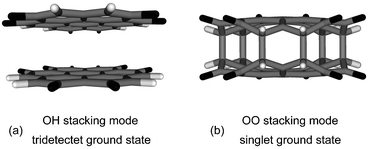 | ||
| Fig. 3 (a) The optimized geometry of compound 6 shows tridectet spin ground state in the OH stacking mode under D3d symmetry. (b) The optimized geometry of compound 6 shows singlet spin ground state in the OO stacking mode under D3h symmetry. Carbon atoms are shown in grey, hydrogen atoms in white, and oxygen atoms in black. | ||
The second possibility to construct a face-to-face dimer is the alignment of oxygen atoms with oxygen atoms (OO stacking mode, Fig. 3) which was unfavorable to form a high spin ground state as indicated by McConnell.60Fig. 4 shows the potential energy curves of the tridectet and singlet state of compound 6 changing from OH stacking mode (Fig. 3(a)) to OO stacking mode (60° of torsional angle) (Fig. 3(b)). The potential energy curves are generated by optimizing the structure in the corresponding spin states with a fixed torsional angle. For illustrative purposes only, a single singlet calculation was done at 60° (OO stacking mode) with the value of the interplanar distance between the coronene-6O molecules fixed at the optimized value from the previous step to prevent the formation of the CC bonds. In Fig. 4, the energy of the tridectet and singlet state increases by 0.519 eV and 1.333 eV moving from the OH (0° torsional angle in Fig. 4) to the OO stacking mode (60° torsional angle in Fig. 4), respectively. Note that the energy of the singlet state (OO stacking mode) drops sharply by 2.423 eV below that of the tridectet state at 60° as C–C bonds form between the two layers (Fig. 3). Each bond is 1.63 Å long and a bonding energy of 2.631 eV for the OO stacked dimer suggests an energy release of approximately 0.4 eV for each pair of combined carbon radicals (Scheme 1). The formation of the single bonds in compound 6 suggests that dimerization as a result of spin pairing is likely to prevent the formation of paramagnetic clusters by simply stacking compound 3.
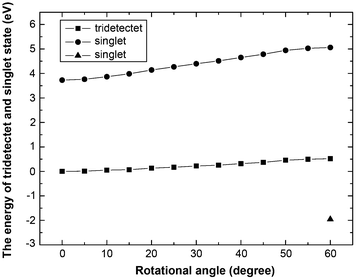 | ||
| Fig. 4 Relative energies of tridectet and singlet state of compound 6 with respect to the torsional angle between two compound 3. The squares and circles represent tridectet and singlet spin states of compound 6. The triangle symbolizes the singlet spin state of compound 6 accompanied with forming six C–C single bonds between two compound 3. All the energies are relative to its tridectet state of OH stacking mode (0 degree for torsional angle). | ||
3.2 A benzene molecule as a spacer
It was shown that diamagnetic molecules inserted as spacers between radical pairs can lead to ferromagnetic materials.6 The benzene molecule is the simplest diamagnetic molecule that supports π–π stacking. In order to prevent the formation of C–C bonds, a benzene molecule is inserted into a coronene-6O dimer in the OH and OO stacking modes. Fig. 5 shows the two orientations of the benzene molecule used in these calculations. All calculations were done with planar coronene-6O molecules to reduce the computational costs as initial calculations showed that the relaxation of the coronene-6O molecules does not affect the benzene–coronene-6O interactions. The tridectet high spin state is found to be the ground state for all four clusters with an optimized interlayer distances ranging between 7.36 and 7.46 Å. The BEs of benzene with the two coronene-6O molecules vary between 0.309 and 0.343 eV with a clear preference for the OH stacking mode (0.341 and 0.343 eV) as to be expected from McConnell's model.60 However, all four bonding energies are considerably lower than the BE of two coronene-6O molecules in OH stacking mode (0.457 eV) suggesting a general weakening of the interactions between the coronene-6O molecules. Hence, benzene as link might not prevent the formation of coronene-6O dimers and the subsequent formation of CC bonds.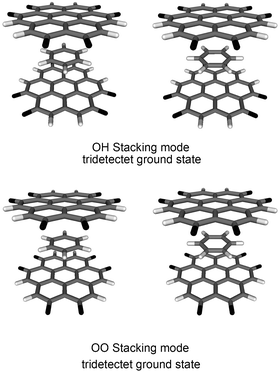 | ||
| Fig. 5 Four tridectet spin ground state optimized geometries of a benzene molecule inserted into a coronene-6O′ dimer with two different positions to separate the two coronene-6O′ molecules in the OH and OO stacking mode. For the above four structures, the optimized interlayer distance of two coronene-6O′ molecules and the binding energies between benzene and two coronene-6O′ molecules are around 7.5 Å and 0.3 eV, respectively. Carbon atoms are shown in grey, hydrogen atoms in white, and oxygen atoms in black. | ||
3.3 A chromium atom as a spacer
Chromium atoms can link two benzene rings.7,61 The coronene molecule offers a wide variety of binding sites for the chromium atom as shown in Fig. 6. The energies of the Cr(coronene)2 sandwich molecules with the chromium atom at different positions and for different spin states are given in Table 4. The most stable Cr(coronene)2 cluster has a singlet ground state and the chromium atom links the center of two peripheral rings (position 2). This result agrees with the observation that metal atoms preferentially binds to the terminal, least anellated benzene ring in polycylic arenes.57 The BE between the chromium atom and two coronene molecules is 1.584 eV which is lower than the BE of 2.914 eV observed for a chromium atom between two benzene rings. The extension of the aromatic system going from benzene to coronene decreases the BE between the chromium atom and the aromatic rings which is likely to be caused by repulsive forces between the parts of coronene molecules without a direct bond to the chromium atom as indicated by the open shape of Cr(coronene)2 in Fig. 6. The singlet ground state suggests that the two coronene molecules create a strong ligand field that quenches the spin on the chromium atom as observed for benzene. Such a spin quenching is not observed in the central position 1 as Cr(coronene)2 with centered chromium atom has a septet ground state.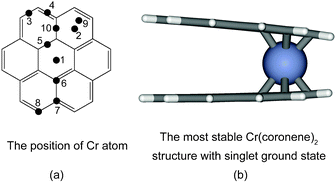 | ||
| Fig. 6 (a) Various positions of chromium atom between the coronene dimer. (b) The most stable structure of Cr(coronene)2 with singlet ground state when chromium is at position 2. Carbon atoms are shown in grey, hydrogen atoms in white, oxygen atoms in black, and chromium atoms in blue. (The numbers used to indicate the positions of the Cr atom above the coronene molecule differ from IUPAC nomenclature.) | ||
| Position | Singlet | Triplet | Quintet | Septet |
|---|---|---|---|---|
| a Relative energies to the singlet spin state of position 2 (in eV). | ||||
| 1 | 1.705 | 2.299 | 1.018 | 0.902 |
| 2 | 0.000 | 0.074 | Moves to site 9 | Moves to site 3 |
| 3 | Moves to site 9 | 1.092 | 0.204 | 0.363 |
| 4 | Moves to site 2 | Moves to site 2 | Moves to site 8 | 0.722 |
| 5 | Moves to site 2 | 2.794 | 0.836 | Moves to site 1 |
| 6 | Moves to site 1 | 2.737 | 0.780 | Moves to site 10 |
| 7 | 4.356 | 2.021 | 0.412 | Moves to site 10 |
| 8 | 4.096 | Moves to site 2 | 0.256 | Moves to site 3 |
| 9 | 1.714 | Moves to site 2 | 0.275 | Moves to site 3 |
| 10 | Moves to site 7 | Moves to site 7 | Moves to site 7 | 0.772 |
The Cr(coronene-6O)2 cluster with the chromium atom linking two peripheral benzene rings (tridectet ground state) is 1.835 eV more stable than the corresponding cluster with a central chromium atom (septet ground state) as to be expected from the calculations on Cr(coronene)2. Hence, all calculations for Cr(coronene-6O)2 (compound 7) in the OH and OO stacking mode have been done with the chromium atom in the center between two peripheral rings (position 2). Electronic structure calculations with various numbers of unpaired electrons (Table 5) show that the Cr(coronene-6O)2 sandwich has a tridectet ground state in the OH stacking conformation as shown in Fig. 7. However it should be noted that the energy difference to the next stable state (septet, OH stacking mode) is just 0.055 eV and thereby close to the error margin of DFT calculations. The energy difference between the tridectet ground state and the next singlet state is about 1 eV in the OH stacking mode and the data in Table 5 hence suggest that the high spin state of coronene-6O does not collapse upon stacking. The data in Table 5 suggest further that the OH stacking mode is preferred to the OO mode.
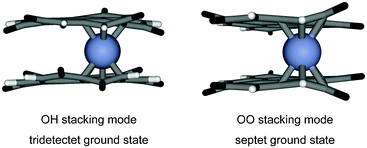 | ||
| Fig. 7 (a) The optimized geometry of compound 7 features tridectet spin ground state in the OH stacking mode. (b) The optimized geometry of compound 7 features septet spin ground state in the OO stacking mode. Carbon atoms are shown in grey, hydrogen atoms in white, oxygen atoms in black, and chromium atoms in blue. | ||
| Spin states | OH stacking mode | OO stacking mode |
|---|---|---|
| a Relative energies to the tridectet spin state in the OH stacking mode (in eV). | ||
| Singlet | 0.998 | 3.337 |
| Triplet | 1.176 | 2.695 |
| Quintet | 1.040 | 1.326 |
| Septet | 0.055 | 0.300 |
| Nonet | 0.666 | 1.618 |
| Undectet | 0.908 | 1.174 |
| Tridectet | 0.000 | 0.550 |
| Quindectet | 0.606 | 0.979 |
| Sepdectet | 2.506 | 2.694 |
| Nondectet | 4.885 | 4.861 |
The BE between the chromium atom and the two coronene-6O molecules is 4.506 eV (tridectet ground state, OH stacking mode) and 4 eV larger than the BE between two coronene-6O molecules (compound 6, tridectet state, OH stacking) as well as 4.2 eV larger than the BE between a benzene molecule and two coronene-6O molecules (section 3.2). Most importantly, the observed BE is significantly larger than the BE of 2.631 eV for the coronene-6O dimmer with C–C bonds. The chromium atom between two peripheral C6 rings is likely to prevent spin quenching by bond formation efficiently. Also, the BE in compound 7 is significantly larger than that for Cr(coronene)2 (1.584 eV) and Cr(benzene)2 (2.914 eV).
The coronene rings in compound 7 show severe buckling with an average CC distance between layers of 3.60 Å and a standard deviation of 0.15 Å. This buckling originates from the C6-rings closest to the chromium atom: Five of the six atoms are on average 2.23 ± 0.12 Å away from the linking chromium atom. The sixth carbon atom is 2.51 Å away from the chromium atom, which suggests an η5 coordination of the C6-rings. The Wiberg bond indices (WBIs) for the individual Cr–C bonds support this assignment. The WBIs for the five short bonds are on average 3.4 times larger than the WBI for the long Cr–C bond. A closer look reveals that the WBIs for the Cr–C bonds are larger for carbon atoms at the edge of the coronene-6O molecule than for those at the center. The Cr–C bonds to the edge are also shorter which indicates the chromium atom has moved out of its center position. The strongest bond in terms of bond length and WBI can be observed for those carbon atoms which carry an unpaired electron (carbon atom 11, Scheme 1).
A similarly bent benzene ring has been reported for the Bis(benzene)chromium (BBC) radical62,63 obtained from addition of organic radicals to BBC. BBC is an efficient organometallic spin scavenger62 and both theory and experiment assign the extra spin to the chromium atom. Fig. 8 shows a plot of the spin density in compound 7. The spin densities on the bonding carbon atoms vanished and the spin density on the chromium atom drastically increased, similar to the observations for the BBC radical. The spin densities per atom (Mulliken: 3.07, NBO: 2.84) suggest three unpaired electrons resting on the chromium atom. The spin transfers from the coronene-6O dimer to the chromium atom happens via the chromiumdz2, dx2−y2 and dxy orbitals (Fig. 9), which form σ and δ bonds in BBC.64 The σ-bonding dz2 orbital is part of the HOMO in BBC and Cr(coronene)2. The δ-orbitals follow directly and the three orbitals lie in the energetic region of SOMOs in coronene-6O. The spin density analysis shows further that each coronene-6O molecule in compound 7 carries 4.5 unpaired electrons (Mulliken: 4.47, NBO: 4.58) which indicates that at least one spin-carrying electron can tunnel between the coronene-6O molecules via the chromium atom.
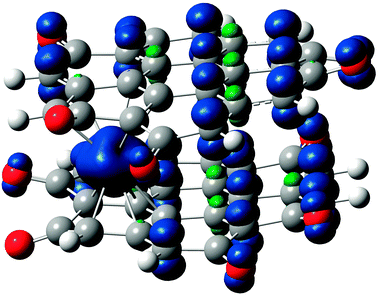 | ||
| Fig. 8 Spin density in Cr(coronene-6O)2 (compound 7, tridectet state, OH stacking). Isovalue for the surface plot set to 0.02 a.u. | ||
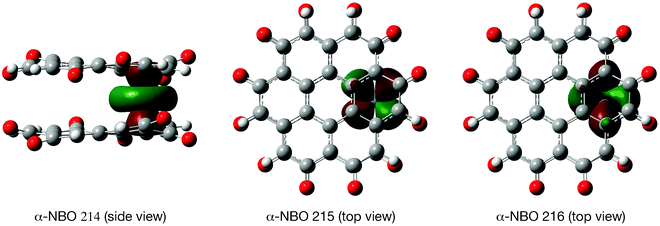 | ||
| Fig. 9 Natural bonding orbitals (isovalue: 0.02 a.u.) for the spin transfer from the coronene-6O sandwich to the chromium atom in Cr(coronene-6O)2 (tridectet state, OH stacking). | ||
Fig. 10 shows the spin density for the septet state and OO stacking. A septet state implies three unpaired electrons per coronene-6O molecule, but Fig. 10 indicates that there are five unpaired electrons per coronene-6O molecule. Two of these electrons have opposing spin and cancel from the counting scheme. The spin density, chromium–carbon bond distances and the Wiberg indices suggest a bonding scheme as shown in Scheme 2: The largest bond distance (2.535 Å) is observed for the carbonyl carbon atom and the low Wiberg index (0.069) suggests a weak or non-existing bond. The shortest (2.187 Å) and strongest (WBI: 0.544) bond is observed between chromium and the carbon atom, which originally carries the spin (carbon atom 11, Scheme 1), while the bond to the carbon atom between the carbonyl groups (carbon atom 12a) are slightly longer (2.229 Å), but still strong (WBI: 0.349). These two bonds pull the chromium atom out of its central position towards the edge of the coronene-6O system. The bond distances to the remaining three carbon atoms vary between 2.33 and 2.37 Å with low Wiberg indices (0.153, 0.153, 0.219). The bond with a WBI 0.219 is included in the set of resonant structures in Scheme 2, but the observed bond lengths clearly indicates the dominance of the bond between the chromium atom and the originally spin carrying carbon atom.
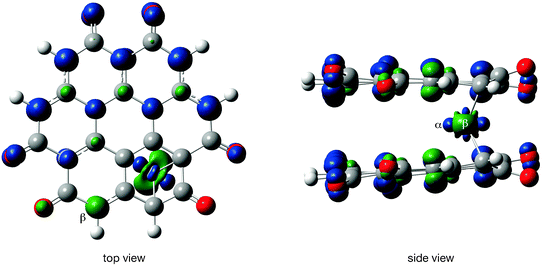 | ||
| Fig. 10 Spin density in Cr(coronene-6O)2 (compound 7, septet state, OO stacking). Isovalue for the surface plot set to 0.02 a.u. | ||
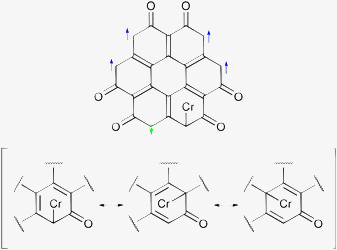 | ||
| Scheme 2 Lewis structures for Cr(coronene-6O)2 with OO stacking in the septet state. | ||
The overall spin density on the chromium atom is small and negative (Mulliken: −0.58, NBO: −0.50), which indicates an electron tunneling process between the coronene-6O molecules and the chromium atom. The surplus of β-spin electrons on the chromium can be explained by the vicinity of the β-electron radicals to the chromium atom. The resonance system in Scheme 2 readily extends to the β-spin carrying chromium atom while the α-spin carrying atom on the other side is separated from the chromium atom by an additional carbonyl group. The non-spherical symmetry of the spin density on the chromium atom shown in Fig. 10 can be explained by different orbitals involved in the tunneling process for the α- and β-electrons. The highest α-spin population can be observed in the chromiumdz2 orbital (NBO: 0.75) while β-electron density can be observed predominantly in the dxy and the dx2−y2 orbitals (NBO: 0.70 and 0.72).
The geometry and spin density of compound 7 in the OH stacking mode and septet state are shown in Fig. 11. The two coronene-6O planes are rotated relative to each other by 30° with the rotational axis running through the chromium atom. The rotation disturbs the preferred matching alignment of spin densities with opposite sign which was visible in the general energetic preference for the OH stacking mode over the OO one so far. Furthermore, the spin density plot shows a nearly spherical β-spin density on the chromium atom. The NBO spin density value suggests that approximately four β-spin and one α-spin electron are localized on the chromium atom, which indicates an overall ferrimagnetic coupling in the system. Three unpaired β-electrons on the chromium atom imply 4.5 unpaired α-electrons per coronene-6O molecule to create a septet state. The number of unpaired electrons is the same as in the tridectet state (vide infra). Moreover, the charge on the chromium atom is always about +1 (NBO charges, OH stacking, septet: 1.04 |e|, OH stacking tridectet: 0.81 |e|, OO stacking septet: 0.73 |e|) and within the DFT framework similar energies have to be expected for the septet and tridectet state in the OH stacking mode.
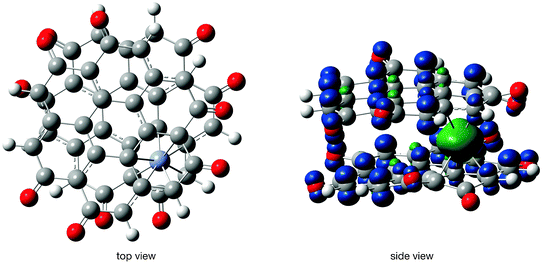 | ||
| Fig. 11 Geometry and spin density in Cr(coronene-6O)2 (compound 7, septet state, OH stacking). Isovalue for the surface plot set to 0.02 a.u. | ||
The calculations on the OO stacked cluster show that a chromium atom can link two coronene-6O molecules while preserving the number of unpaired electrons. The OH stacking misaligns the spin carrying carbon atoms and thereby prevents spin quenching by the formation of carbon–carbon bonds. Also, the chromium atom as angle point for rotation prevents their realignment by a simple rotation (Fig. 11). The side view on the Cr(coronene-6O)2 cluster (OO stacking, septet state, Fig. 10) suggests that the same repulsive forces which pry the Cr(coronene)2 cluster open (Fig. 6) work in the coronene-6O sandwich. A possible candidate for this interaction is the Coulomb repulsion between the extended π-systems. They keep the rings sufficiently away form each other that the spin carrying orbitals on the carbon atoms stay approximately degenerate. As Hund's rule has been applied to this situation, parallel spins are to be expected for these radicals and bond formation is avoided. An external force may overpower this separating force and compel the carbon atoms to interact. The result of such a force will lift the approximate degeneracy of the radical orbitals. Spin pairing and consequently the formation of C–C bonds become likely. Such a force is not part of our model; hence CC bond formation is not observed with a chromium atom as linker.
4. Conclusions
The coronene-6O molecule has a septet ground state as predicted by simple Hückel calculations and Lieb's equation. The unpaired electrons rest on the hydrogen carrying carbon atoms and spin quenching by C–C bond formation prevents the generation of a molecular magnet by stacking coronene-6O units. A benzene molecule between two coronene-6O molecules prevents the formation of C–C bonds and keeps the radicals so far apart that the spins couple ferromagnetically. Thus, a tridectet state is predicted by DFT calculations for (C6H6)(coronene-6O)2, but the Van der Waals interactions between the π-systems are weak and it is likely that such a complex will quickly decompose while C–C bonds from between the coronene-6O units.A chromium atom between two peripheral C6 rings of the coronene-6O molecules forms a stable link, which prevents spin quenching by direct C–C bond formation. The ligand field in the related compounds Cr(C6H6)2 and Cr(cronene)2 is strong enough to form low-spin complexes, which suggests that the chromium atom might not interfere with electron spin on coronene-6O. Since the chromium d orbitals are in the energetic range of the coronene-6O SOMOs, spin transfers between the coronene-6O molecules and the chromium atom in the sandwich complex are therefore inevitable. The DFT calculations predict a tridectet ground state for Cr(coronene-6O)2 as already predicted by the simplified model; however the careful analysis of the DFT results revealed spin transfer between the units and the possibility of spin tunneling between the coronene-6O molecules via the chromium atom. Nonetheless, the high multiplicity of Cr(coronene-6O)2 suggests a very large spin-only magnetic moment for the cluster which indicates a further route to organic magnets via organometallic sandwich compounds.
Acknowledgements
This work is supported by the National Science Council of the Republic of China under Grant NSC 97-2113-M-007-006-MY3. We thank the National Center of High-Performance Computing of the Republic of China for their support of this work.References
- J. S. Miller, Inorg. Chem., 2000, 39, 4392 CrossRef CAS.
- J. S. Miller and A. J. Epstein, Chem. Rev., 1988, 88, 201 CrossRef CAS.
- J. S. Miller and A. J. Epstein, Angew. Chem., Int. Ed. Engl., 1994, 33, 385 CrossRef.
- J. S. Miller and A. J. Epstein, MRS Bull., 2000, 25, 21 CrossRef CAS.
- L. V. Slipchenko, T. E. Munsch, P. G. Wenthold and A. I. Krylov, Angew. Chem., Int. Ed., 2004, 43, 742 CrossRef CAS.
- A. Ivanova, M. Baumgarten, S. Karabunarliev and N. Tyutyulkov, Phys. Chem. Chem. Phys., 2002, 4, 4795 RSC.
- T. Kurikawa, H. Takeda, M. Hirano, K. Judai, T. Arita, S. Nagao, A. Nakajima and K. Kaya, Organometallics, 1999, 18, 1430 CrossRef CAS.
- Yasuike and S. Yabushita, J. Phys. Chem. A, 1999, 103, 4533 CrossRef CAS.
- R. Pandey, B. K. Rao, P. Jena and M. A. Blanco, J. Am. Chem. Soc., 2001, 123, 3799 CrossRef CAS.
- W. M. Lamanna, J. Am. Chem. Soc., 1986, 108, 2096 CrossRef CAS.
- W. M. Lamanna, W. B. Gleason and D. Britton, Organometallics, 1987, 6, 1583 CrossRef CAS.
- J. Jiang, J. R. Smith, Yi Luo, H. Grennberg and H. Ottoson, J. Phys. Chem. C, 2011, 115, 785 CAS.
- D. Seyferth, Organometallics, 2002, 21, 1520 CrossRef CAS.
- D. Seyferth, Organometallics, 2002, 21, 2800 CrossRef CAS.
- J. A. Verges, G. Chiappe, E. Louis, L. Pastor-Abia and E. SanFabian, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 094403 CrossRef.
- R. Rieger, M. Kastler, V. Enkelmann and K. Mullen, Chem.–Eur. J., 2008, 14, 6322 CrossRef CAS.
- J. Reinhold, Quantentheorie der Moleküle, Teubner Studienbücher Chemie, B. G. Teubner, Stuttgart 1994, p. 172 Search PubMed.
- E. H. Lieb, Phys. Rev. Lett., 1989, 62, 1201–1204 CrossRef . Errata: ibid, 1989, 62, 1927.
- P. Jensen and P. R. Bunker, J. Chem. Phys., 1988, 89, 1327 CrossRef CAS.
- E. R. Davidson, D. Feller and P. Phillips, Chem. Phys. Lett., 1980, 76, 416 CrossRef CAS.
- N. C. Handy, Y. Yamaguchi and H. F. Schaefer, J. Chem. Phys., 1986, 84, 4481 CrossRef CAS.
- H. F. Bettinger, P. v. R. Schleyer, P. R. Schreiner and H. F. I. Schaefer, Encyclopedia of Computational Chemistry, 1998, 183 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- Y. I. Kurokawa, Y. Nakao and S. Sakaki, J. Phys. Chem. A, 2010, 114, 1191 CrossRef CAS.
- M. Benard, M.-M. Rohmer, X. Lopez and K. H. Theopold, Angew. Chem., Int. Ed., 2008, 47, 5597 CrossRef CAS.
- M. R. Philpott and Y. Kawazoe, Chem. Phys., 2007, 342, 223 CrossRef CAS.
- M. R. Philpott and Y. Kawazoe, J. Phys. Chem. A, 2008, 112, 2034 CrossRef CAS.
- D. T. Moore, J. Oomens, J. R. Eyler, G. Helden, G. Meijer and R. C. Dunbar, J. Am. Chem. Soc., 2005, 127, 7243 CrossRef CAS.
- T. J. Brunker, J. C. Green and D. O'Hare, Inorg. Chem., 2003, 42, 4366 CrossRef CAS.
- R. Sahnoun and C. Mijoule, J. Phys. Chem. A, 2001, 105, 6176 CrossRef CAS.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, 2nd edn, 2002 Search PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757 RSC.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Phys., 2005, 123, 161103 CrossRef.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 1095 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- J. S. Overby, T. P. Hanusa, S. P. Sellers and G. T. Yee, Organometallics, 1999, 18, 3561 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- C. W. Bauschlicher, S. R. Langhoff and L. A. Barnes, J. Chem. Phys., 1989, 91, 2399 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS.
- J. Zhou, W. Wang and K. Fan, Chem. Phys. Lett., 2006, 424, 247 CrossRef CAS.
- A. J. Boone, C. H. Chang, S. N. Greene, T. Herz and N. G. J. Richards, Coord. Chem. Rev., 2003, 238, 291 CrossRef.
- J. Han, Chem. Phys., 2003, 286, 181 CrossRef CAS.
- J. Han and J. Morales, THEOCHEM, 2005, 756, 55 CrossRef CAS.
- D. Harris, G. H. Loew and A. Komornicki, J. Phys. Chem. A, 1997, 101, 3959 CrossRef CAS.
- D. Rio, R. Sarangi, E. Chufan, K. Karlin, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 11969 CrossRef.
- C. Rong, S. Lian, D. Yin, B. Shen, A. Zhong, L. Bartolotti and S. Liu, J. Chem. Phys., 2006, 125, 174102 CrossRef.
- W. Zierkiewicz and T. Privalov, Dalton Trans., 2006, 1867 RSC.
- R. J. Deeth and N. Fey, J. Comput. Chem., 2004, 25, 1840 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2009, http://www.chem.wisc.edu/∼nbo5 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.2), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby and M. Schutz, MOLPRO, version 2008.1, a package of ab initio programs Search PubMed.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Angew. Chem., Int. Ed., 2007, 46, 6692 CrossRef CAS.
- T. J. Brunker, J. C. Green and D. O'Hare, Inorg. Chem., 2002, 41, 1701 CrossRef CAS.
- F. Z. Engelmann, Naturforsch. B: Anorg. Chem. Org. Chem., 1953, 8B, 775 CAS.
- C. Elschenbroich and A. Salzer, Organometallics: A Concise Introduction, 2nd edn, 1992, p. 345 Search PubMed.
- National Institute of Advanced Industrial Science and Technology, Japan, 17.08.2011, http://riodb01.ibase.aist.go.jp/sdbs/.
- H. M. McConnell, J. Chem. Phys., 1985, 28, 1188 CrossRef.
- H. M. McConnell, J. Chem. Phys., 1963, 39, 1910 CrossRef CAS.
- H. Xiang, J. Yang, J. G. Hou and Q. Zhu, J. Am. Chem. Soc., 2006, 128, 2310 CrossRef CAS.
- E. Samuel, D. Caurant, D. Gourier, Ch. Elschenbroich and K. Agbaria, J. Am. Chem. Soc., 1998, 120, 8088 CrossRef CAS.
- A. Perrier, D. Gourier, L. Joubert and C. Adamo, Phys. Chem. Chem. Phys., 2003, 5, 1337 RSC.
- V. M. Rayón and G. Frenking, Organometallics, 2003, 22, 3304 CrossRef.
| This journal is © the Owner Societies 2012 |