Coumarin derivatives for dye sensitized solar cells: a TD-DFT study†
Rocío
Sánchez-de-Armas
*,
Miguel Ángel
San Miguel
,
Jaime
Oviedo
and
Javier Fdez.
Sanz
Department of Physical Chemistry, University of Seville, Spain. E-mail: rociosa@us.es
First published on 11th November 2011
Abstract
Time dependent density functional theory (TD-DFT) calculations have been carried out to study the electronic structure and the optical properties of five coumarin based dyes: C343, NKX-2311, NKX-2586, NKX-2753 and NKX-2593. We have found out that the position and width of the first band in the electronic absorption spectra, the absorption threshold and the LUMO energy with respect to the conduction band edge are key parameters in order to establish some criteria that allow evaluating the efficiency of coumarin derivatives as sensitizers in Dye Sensitized Solar Cells (DSSC). Those criteria predict the efficiency ordering for the coumarin series in good agreement with the experimental evidence. Presumably, they might be used in the design of new efficient organic based DSSC.
1. Introduction
Dye Sensitized Solar Cells (DSSC)1–3 based on organic dyes adsorbed on nanocrystalline TiO2 electrodes have attracted considerable attention in recent years because of their high incident solar light-to-electricity conversion efficiency and low cost of production.4–7 The photochemical properties of different organic sensitizers have extensively been investigated in an attempt to design dyes with maximal visible light absorption coupled to long-lived excited states. However, major effort is still needed in both developing new sensitizers and finding optimal working conditions to improve the photon-to-current conversion efficiencies.The electronic and optical properties of coumarin based dyes make them one of the most promising classes of organic sensitizers and they have been studied systematically by Hara and Arakawa.8–14 They are considered type-I dyes, in which electron injection occurs through an indirect mechanism: there is a photoexcitation of the dye to an excited state, followed by the electron injection from this state to the semiconductor conduction band.
The first coumarin based molecule studied as a sensitizer for DSSC was C343 (Fig. 1a). Ultrafast electron-injection times of 20–200 fs from C343 into the conduction band of TiO2 have been observed.15,16 However, the conversion efficiency of DSSCs using C343 is much lower than the efficiencies of DSSCs based on Ru complex photosensitizers,17 because of the narrow absorption area that the C343 dye electronic spectrum presents in the visible region.
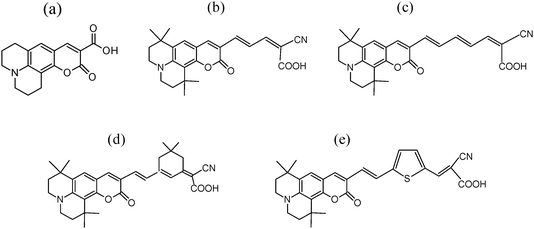 | ||
| Fig. 1 Coumarin dye structures: (a) C343, (b) NKX-2311, (c) NKX-2586, (d) NKX-2753 and (e) NKX-2593. | ||
The usual way to improve the DSSC performance of a given dye, i.e., to widen the absorption in the visible region, consists of a functionalization by either adding chromophore groups or expanding the π system. In this context, it has been found that introducing a methine unit (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) connecting both the cyano (–CN) and carboxyl (–COOH) groups into the coumarin framework expands the π conjugation of the dye resulting in a red shift and an absorption widening in the visible region, desirable for sunlight harvesting. A conversion efficiency of 5.6%, rather higher than that obtained using C343, was reached in 2001 with a cell based on NKX-2311 (Fig. 1b).8 The slow charge recombination, on the order of micro to milliseconds, between NKX-2311 cations and injected electrons into the TiO2 conduction band resulted in efficient charge separation for this system.9 Moreover, the structure of NKX-2311, with a carboxyl group directly connected to the –CHCH– unit, is advantageous for effective electron injection from the dye into the conduction band of TiO2. These dyes are donor–acceptor π-conjugated (D–π–A) dyes possessing both electron-donating (D) and electron-accepting (A) groups linked by π-conjugated bridges. In these dyes, a coumarin skeleton contains an amino group acting as the donor component and cyanoacrylic acid as the acceptor component.
CH–) connecting both the cyano (–CN) and carboxyl (–COOH) groups into the coumarin framework expands the π conjugation of the dye resulting in a red shift and an absorption widening in the visible region, desirable for sunlight harvesting. A conversion efficiency of 5.6%, rather higher than that obtained using C343, was reached in 2001 with a cell based on NKX-2311 (Fig. 1b).8 The slow charge recombination, on the order of micro to milliseconds, between NKX-2311 cations and injected electrons into the TiO2 conduction band resulted in efficient charge separation for this system.9 Moreover, the structure of NKX-2311, with a carboxyl group directly connected to the –CHCH– unit, is advantageous for effective electron injection from the dye into the conduction band of TiO2. These dyes are donor–acceptor π-conjugated (D–π–A) dyes possessing both electron-donating (D) and electron-accepting (A) groups linked by π-conjugated bridges. In these dyes, a coumarin skeleton contains an amino group acting as the donor component and cyanoacrylic acid as the acceptor component.
The absorption maximum of coumarin dyes gradually red-shifts with size expansion of their conjugate system (by increasing the number of methine units) and this shift has been attributed to decreases in the oxidation potentials caused by destabilization of HOMO orbitals. The introduction of a cyano group, with a strong electron-accepting ability, produces an increase in the dye reduction potential due to a stabilization of the LUMO orbital.10 Furthermore, the expansion of the π conjugated system by introducing one or more thiophene rings into the methine chain of NKX-2311 has also been tested. Thiophene simultaneously extended π-conjugation and improved the dye stability relative to other dyes with a long methine chain unit.11,12 This modification does not affect the absorption properties of dyes in solution, but improves the performance of coumarin based DSSCs. Thus, efficiencies up to 7.7% have been reached in cells based on coumarin dyes containing two thiophene units.18
A drawback related to these coumarin dyes is that they are liable to experience π-stacked aggregation on TiO2 surfaces. In particular, dyes containing three methine units (NKX-2586) are very prone to show this effect. This type of aggregation would be advantageous for light harvesting, because it induces a widening in the UV/Vis absorption spectrum. However, it usually leads to inefficient electron injection because of intermolecular energy transfer processes between the dyes, thus resulting in low cell efficiency.
A strategy to prevent dye aggregation is adding coadsorbates, like deoxycholic acid (DCA) and 4-tert-butylpyridine (TBP).13 Although these additives improve the electron injection, they significantly decrease dye adsorption limiting unavoidably the cell performance. Recently, a new coumarin dye (NKX-2753) has been synthesized to reduce dye aggregation while keeping the adsorbed dye amount.14 This molecule has a side ring linked to the alkene chain that prevents dye aggregation, ensuring simultaneously a high surface dye concentration and efficient photon-to-electron conversion. Thus, the use of NKX-2753 has allowed obtaining efficiencies up to 6.7%.
Theoretical methods are a powerful tool for molecular design, and conclusions drawn from calculations are valuable guidelines for synthesis of new efficient dyes. Although coumarin based dyes have been extensively studied from experimental methods in recent years, there are not many theoretical studies about them.19–27 In a recent work, Time-Dependent Density Functional Theory (TD-DFT/B3LYP) has been used to simulate the electronic absorption spectra of free coumarin dyes.24 They obtained excitation energies that agree well with experimental data for small molecules but those energies were underestimated for larger coumarin dyes. Moreover, the inclusion of methanol solvation effect via a PCM model tends to reduce the excitation energies, due to a stabilization of the LUMO orbital. In subsequent works excitation energies that agree better with experiments have been obtained employing a long-range-corrected (LC) exchange–correlation functional.25,26 Furthermore, TD-DFT calculations have been used to establish a relationship between the electronic structure of free coumarin dyes and their efficiency as DSSC sensitizers.27 Thus, the location of the first absorption band and the energy of the HOMO orbital were used as parameters to evaluate the electrochemical efficiency of coumarin dyes. It is worth noting that up to date, all previous theoretical works are limited to the study of electronic structure and optical properties of free dyes and none of them tackles the influence of the adsorption process on the electronic absorption spectra of these dyes.
Recently, we have studied the structural and electronic properties of some organic dyes that have been used as sensitizers in DSSC, both free and supported on several TiO2 clusters with different sizes.28–30 From these works it is shown that the dye adsorption process on the semiconductor produces a reorganization of the electronic states and this fact introduces important changes in the electronic absorption spectra. Moreover, the electronic coupling between the semiconductor conduction band and the dye molecule strongly influences the electron injection mechanism. Therefore, to theoretically determine the convenience of using these dyes as DSSC sensitizers, it is necessary to include the semiconductor effect explicitly in the model. In addition, the cluster size should be large enough to well reproduce the electronic structure of adsorbed dyes.
In this work we have studied the electronic structure and the optical properties of five coumarin based dyes: C343, NKX-2311, NKX-2586, NKX-2753 and NKX-2593 (Fig. 1). First, we consider the geometry of the organic dyes in the electronic ground state as well as the geometries of the five dyes adsorbed on a TiO2 cluster. Second, we explore the optical response of the different structures using TD-DFT. These calculations are performed in a complementary way to simulate the electronic spectra through the conventional mode and the real time propagation within the adiabatic approximation with a basis of localized orbitals. In a previous work we checked that both TD-DFT implementations reach very similar results provided the same exchange–correlation functional even if the employed basis sets are not exactly the same.28
The aim of this work is to understand the physical–chemical aspects that govern the light absorption process in the coumarin based series to find the main parameters that affect the efficiency of these dyes as DSSC sensitizers. Thus, we intend to establish some criteria to evaluate the relative efficiency of coumarin dyes. The use of these criteria could be applied to predict the efficiency of other dyes and presumably to design new more efficient sensitizers, reducing economical cost and synthesis effort.
2. Computational methods
2.1 Technical details
Two complementary sets of calculations have been carried out for each model described in Section 2.2.(1) DFT geometry optimizations have been done by using the PBE generalized gradient approach functional31 together with norm-conserving pseudopotentials32 in the fully non-local Kleinman–Bylander33 form and an auxiliary real-space grid equivalent to a plane-wave cut-off of 130 Ry. A non-standard DZP basis set of Natural Atomic Orbitals (NAOs) constructed from the eigenstates of the atomic pseudopotential was used.34–36 The optical response was then computed using real time TD-DFT (time domain) simulations within the SIESTA implementation.37–39 We consider the system in a finite electric field of 0.1 V Å−1 as an initial state.28 Each system was allowed to evolve for 36 fs with a time-step of 1.5 × 10−3 fs. The damping factor used in the Fourier transformation was 0.1 eV.
(2) Additionally, linear response (LR) frequency domain calculations were performed in the gas phase using the PBE functional and the 6-31G(d,p) basis set. The optical spectra were simulated by conventional TD-DFT (frequency domain) using Gaussian03.40 Over 400 singlet transitions were needed in selected calculations. Before the spectrum calculation, vibrational frequency calculations were done to confirm the stability of all the optimized geometries. With the aim of comparing our results with previous published data, several calculations were repeated using the B3LYP exchange–correlation functional and a discussion of these results is included in the ESI.†
There are some advantages associated with the real-time TD-DFT formulation: all the possible excitations are generated at the same time, so the spectrum is obtained in a wide range of energy; the implementation is relatively simple and although the computational cost is not cheap for small systems, it becomes competitive with traditional methods as the system size increases. However, its main disadvantage is that it is not possible to characterize the nature of the different transitions because the optical response is obtained from the analysis of the electronic dipole moment, where the information from the wavefunctions is integrated. For this reason the combination with conventional TD-DFT is interesting. In previous work28,29 we performed real-time calculations with clusters as large as (TiO2)38 and demonstrated that (TiO2)9 is large enough to reproduce adequately the electronic absorption spectra of the dye–TiO2 systems. Since the (TiO2)9 cluster is affordable from frequency domain TD-DFT calculations, a full analysis of the spectra is possible.
Although it is known that TD-DFT can significantly underestimate the energies of long-range charge transfer states,41 that is not the case with the present calculation. The molecules considered in this paper are not large enough to present a long-range intramolecular charge transfer and therefore, for these molecules the photoexcited state shows a moderate charge transfer (see ESI†). Moreover, it is important to stress that the aim of this work is to establish some criteria to evaluate the relative efficiency of dyes. The established criteria are general, not depending on technical settings.
2.2 Models
The computational methodology has been applied to: free dyes (C343, NKX-2311, NKX-2586, NKX-2753, NKX-2593) and each dye adsorbed onto a (TiO2)9 cluster. The starting geometry for this cluster was taken from the literature.42,43 It was obtained via geometry optimization of the originally spherical shape resulting in a compact structure with only 4-fold coordinated Ti-atoms and with one terminal Ti–O bond. In a previous work,28 we have demonstrated that this cluster size is large enough to reproduce adequately the electronic absorption spectra of some dye–TiO2 systems. Furthermore, this cluster has a critical size which is affordable from both TD-DFT approaches allowing the comparison between them.For C343 there is only one possible configuration, while for the other dyes two different conformational isomers exist. The configuration at the single bond between the coumarin moiety and the methine chain can have either an s-cis or s-trans arrangement. From our calculations we conclude that for the smaller coumarin dyes, such as NKX-2311, the s-cis isomer is slightly favoured, due to steric repulsion between the CO group of the coumarin and the C–N group of the cyanoacrylic acid in the s-trans isomer. In contrast, for the molecules with larger methine chains, such as NKX-2586, the repulsion becomes less important, and the s-trans isomer becomes slightly more stable than the s-cis isomer. These results agree well with previous theoretical works.6,19,25 Nevertheless, energy differences between both isomers are always insignificant (less than 0.03 eV). Moreover, we have checked out that for all dyes the calculated electronic absorption spectra of both isomers are almost identical (differences in the absorption maximum position are less than 0.08 eV).
These dyes also present two different conformations with respect to the nitrogen atom of the unsaturated rings, syn and anti, which have been reported to be almost isoenergetic.19 In this work we have selected the syn conformer which has also been used in previous theoretical work.24,25 Therefore, it has been suggested from ab initio calculations that the absorption spectra of the syn and anti isomers are almost indistinguishable for a coumarin dye.19
Coumarin dyes adsorb on the TiO2 surface through the carboxylic group, and several adsorption configurations are possible, including molecular or dissociative adsorption processes. FTIR spectra for these systems indicate the presence of carboxylate ion after adsorption, which indicates that the deprotonation of the COOH group takes place on the TiO2 surface.14 The formed carboxylate group may coordinate to the surface in several forms. One of them involves the attachment of one dye oxygen atom to one surface titanium atom, leading to a monodentate structure. A second possibility is the formation of a bidentate structure which could be a bidentate bridging (bb) structure with both dye oxygen atoms bound to two surface titanium atoms or a bidentate structure with those two dye oxygens bound to the same surface titanium atom, in a chelating configuration (bc). All previous works agree that monodentate adsorption is not favoured for this kind of dye. Nevertheless, there is some controversy about what bidentate adsorption mode is the most favourable. We carried out geometry optimization of the dyes adsorbed on a (TiO2)9 cluster in both bidentate configurations (bb and bc) (see ESI†). In order to keep the system electroneutrality, the dissociated hydrogen atom was placed on three different oxygen cluster atoms. From this study we concluded that the most stable adsorption mode is, in general, the bidentate chelating configuration (bc), with the two dye oxygens bound to the same surface titanium atom and with the dissociated hydrogen bound to an oxygen in the centre of the cluster. Nevertheless, for some dyes such as NKX-2311, differences between the two bidentate configurations are small (less than 0.15 eV) suggesting that both structures could exist in real systems.
In spite of the energetic differences, the absorption maximum position in the spectra is almost not affected by the adsorption mode (differences less than 0.07 eV). Nevertheless, for the bidentate bridging structure (bb) a new band at lower energy appears in the simulated spectrum, which is not present in the experimental spectrum (see ESI†). This might be considered an indirect evidence to rule out the existence of this adsorption mode, or at least to discard that the bb structure is a main structure.
Considering the relative stabilities of the different isomers and adsorption modes and their (slight) influence on the electronic absorption spectra, we have chosen the s-cis isomer and the bidentate chelating (bc) adsorption mode (with the dissociated hydrogen bound to an oxygen in the centre of the cluster) as a representative model for our study on the electronic structure and the absorption spectrum. Thus, all the results presented in the next section are referred to this model.
3. Results
3.1 Free dyes
The simulated absorption spectra from real-time TD-DFT calculations for free coumarin dyes are shown in Fig. 2. The spectra show a similar profile for all dyes; they present a main intense band with a small shoulder at higher energies. C343 spectrum presents the main peak at 2.96 eV, in excellent agreement with the experimental value (2.81 eV).17 For the rest of dyes the main peak appears red-shifted in the spectrum compared with C343 following the sequence (Table 1): C343 (2.96 eV), NKX-2311 (2.30 eV), NKX-2586 (2.10 eV), NKX-2753 (2.07 eV) and NKX-2593 (1.96 eV). The most intense contribution to the main band is an excitation from the HOMO to the LUMO orbital (Table 1). HOMOs are preferentially localized on the coumarin moiety, which acts as an electron donor, while LUMOs have a large contribution from cyanoacrylic acid and polimethine/thiophene chain (Fig. 3).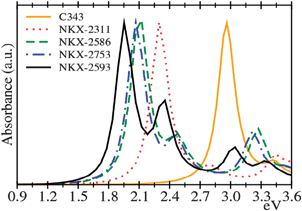
| Dye | Exp./eV | η (%) | TDDFT/eV | Oscillator strength | Wave function |
|---|---|---|---|---|---|
| C343 | 2.81 (MeOH) | 0.9 | 2.96 | 0.3616 | H → L (0.59), H − 2 → L + 1 (−0.14), H − 2 → L (−0.12) |
| NKX-2311 | 2.46 (MeOH) | 5.2 | 2.35 | 1.0538 | H → L (0.56), H − 1 → L (0.14) |
| NKX-2586 | 2.45 (MeOH) | 3.5 | 2.10 | 1.2318 | H → L (0.53), H − 1 → L (0.24), H → L + 1 (−0.14) |
| 2.44 (EtOH) | |||||
| NKX-2753 | 2.52 (EtOH) | 6.7 | 2.07 | 1.0679 | H → L (0.54), H − 1 → L (0.24), H → L + 1 (−0.14) |
| NKX-2593 | 2.43 (EtOH) | 7.2 | 1.96 | 0.9762 | H → L (0.53), H − 1 → L (0.25), H → L + 1 (−0.20) |
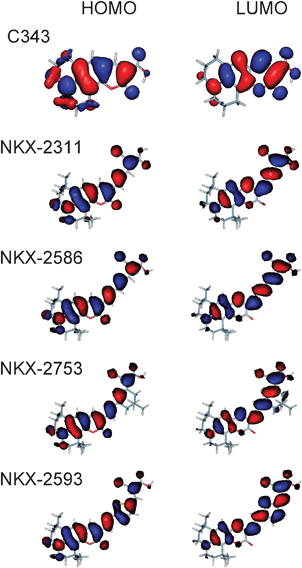 | ||
| Fig. 3 Occupied (left, HOMO) and virtual (right, LUMO) molecular orbitals responsible for the first band in the electronic absorption spectrum of free dyes. The isodensity value used is 0.015. | ||
In the optimized dye geometries, cyanoacrylic acid is coplanar with the polimethine/thiophene chain, indicating strong conjugation across the π orbitals of the CC double bonds. On extending the polimethine chain, the π electron system increases and this fact produces a destabilization of HOMO orbitals (Fig. 4) and a red-shift of the first band. The π electron systems of NKX-2586 and NKX-2753 have the same extension and their HOMO and LUMO orbitals are very similar, which are the reasons why the electronic absorption spectra are almost identical for these two dyes. On the other hand, the introduction of a –CN group (a strong electron acceptor) produces a LUMO stabilization and consequently an important red shift of the first band in the spectra. In this scenario, the C343 main peak is then considerably blue-shifted with respect to the other dyes showing almost no absorption in the visible region.
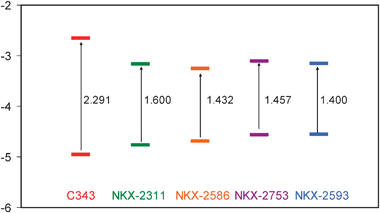 | ||
| Fig. 4 Frontier orbital energies for the different free dyes (eV). Extending the π electron system produces a destabilization of HOMO orbitals while introducing a –CN group produces a LUMO stabilization. | ||
The position (related to the gap between HOMO and LUMO levels) and the width of the first band in the spectrum are the two first parameters that can be related to the dye efficiency, since the absorption shift to lower energies favors the light harvesting process. Nevertheless, in a previous work,28,29 we have shown that dye adsorption on the semiconductor leads to a redistribution of energy levels. For this reason, to theoretically analyze the behavior and efficiency of the dyes as DSSC sensitizers, the semiconductor must be included explicitly in the model. We have to point out that up to date all theoretical works on coumarin dyes are limited to the study of free dyes.
3.2 Theoretical criteria to evaluate efficiency
In order to establish some criteria to evaluate the relative efficiency of coumarin dyes as DSSC sensitizers, it is interesting to review the working scheme of this type of cells. As can be seen in Fig. 5, a DSSC working scheme is based on several processes in which different energy levels are involved. Thus, we have to consider that the electron injection occurs into a semiconductor conduction band state and that the oxidized dye is regenerated through reduction by an electrolyte. Additionally in a type I cell, the first step is a photoexcitation of the dye to an excited state, and for an efficient sunlight harvesting the dye absorption range should cover the whole visible and some of the near-infrared regions. On comparing two dyes with similar structures, the light harvesting would be more efficient for the dye with a lower absorption threshold and a wider first band in the electronic absorption spectrum. The second step involves the electron injection from the dye excited state to the semiconductor conduction band. The rate constant for electron injection is proportional to the number of accessible states, and the effective density of states in the TiO2 conduction band is considered to increase exponentially with energy. It has been reported that a 0.3 eV shift in relative energy between the dye excited state and the TiO2 conduction band provokes a 10-fold change in injection rate.44,45 Therefore, between two dyes with similar structures, the electron injection would be more efficient for that dye with the higher excited state related to the semiconductor conduction band edge. The composition of an excitation is routinely obtained from the calculations (see Tables 1 and 2 for instance) although it is customary to simplify and regard the excitation as an HOMO–LUMO one. Therefore, the energy of the excited state has been approximated as the LUMO orbital energy. In a previous theoretical work, the energy for the conduction band edge was estimated from experimental measurements.9,44 Nevertheless, in this work, the semiconductor has been included explicitly in the models and consequently the energy levels for the dye and for the semiconductor are calculated simultaneously.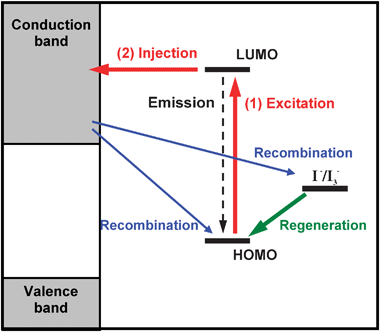 | ||
| Fig. 5 Schematic representation of the main processes involved in a DSSC. | ||
| Transition energy/eV (excitation number) | Oscillator strength | Wave function |
|---|---|---|
| First band | ||
| 1.39 (8) | 0.0427 | H → L + 8 (0.63), H → L + 5 (0.27), H → L + 7 (−0.11) |
| Second band | ||
| 1.73 (14) | 0.4347 | H → L + 13 (0.51), H → L + 5 (0.29), H → L + 12 (−0.21), H → L + 8 (−0.16) |
| 1.84 (18) | 0.4114 | H → L + 13 (0.37), H → L + 14 (0.36), H → L + 5 (−0.25), H → L + 15 (−0.25), H → L + 8 (0.14) |
| 1.87 (19) | 0.1119 | H → L + 14 (0.59), H → L + 15 (0.27), H → L + 13 (−0.14), H → L + 5 (0.13) |
| 1.94 (21) | 0.1800 | H → L + 15 (0.58), H → L + 16 (0.17), H → L + 17 (0.16), H → L + 5 (−0.15), H → L + 13 (0.13), H → L + 20 (0.12) |
| 1.99 (24) | 0.0239 | H → L + 16 (0.66), H → L + 17 (−0.19) |
| 2.04 (25) | 0.0596 | H → L + 17 (0.63), H → L + 20 (−0.19), H → L + 16 (0.12) |
| 2.16 (32) | 0.0811 | H − 1 → L + 8 (0.59), H − 1 → L + 5 (0.33), H − 1 → L + 7 (−0.14) |
| Third band | ||
| 2.31 (34) | 0.1874 | H − 2 → L + 4 (0.50), H → L + 21 (−0.27), H → L + 20 (0.23), H − 1 → L + 8 (0.16), H − 1 → L + 5 (−0.15), H − 1 → L + 9 (0.12) |
| 2.32 (35) | 0.1254 | H − 2 → L + 4 (0.48), H → L + 21 (0.38), H → L + 20 (−0.21), H − 1 → L + 8 (−0.12), H − 1 → L + 5 (0.10) |
| 2.32 (36) | 0.2597 | H → L + 21 (−0.51), H − 1 → L + 5 (0.23), H − 1 → L+8 (−0.21), H → L + 20 (−0.15), H − 1 → L + 9 (−0.14), H − 2 → L + 5 (−0.13), H − 2 → L + 10 (−0.12) |
In summary, we will use the electronic absorption spectra (position and width of the first band and absorption threshold) and the position of the LUMO level related to the conduction band edge to establish criteria in order to evaluate the relative efficiency of coumarin dye derivatives.
3.3 Adsorbed dyes
The simulated absorption spectra from real-time TD-DFT calculations for adsorbed coumarin dyes are shown in Fig. 6. The main effect that can be observed in the spectra after adsorption is a widening of the first band, while the maximum position remains almost unaltered for all dyes (small red-shift of 0.1–0.2 eV). The widening occurs at both sides of the main band; the absorption threshold is red-shifted and consequently it extends into the visible region.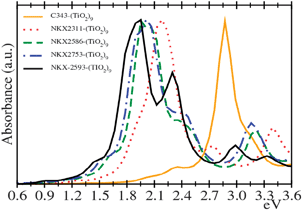
Like in the case of free dyes, the adsorbed C343 main peak is considerably blue-shifted with respect to the other dyes. The absorption maximum position for the rest of dyes is very similar, and there is only a slight red-shift following the sequence: NKX-2311, NKX-2586, NKX-2753 and NKX-2593. Albeit, adsorbed NKX-2593 spectrum covers the widest zone in the visible region and it shows the lowest absorption threshold.
Upon binding the energy levels of dye and cluster are reorganized. We have analyzed the last occupied and the first virtual orbitals for the free and the adsorbed systems to gain insight into the energy levels redistribution. HOMO and LUMO orbitals of free dyes have been described in Section 3.1 and are shown in Fig. 3. We can classify free cluster orbitals into four groups. The inner occupied orbitals (HOMO−6, HOMO−5, HOMO−4 and HOMO−3) are delocalized over the whole cluster. The last occupied orbitals (HOMO−2, HOMO−1 and HOMO) are localized on the cluster region closest to the Ti atom which the molecule will bind to. The first virtual orbitals (LUMO, LUMO+1, LUMO+2 and LUMO+3) are localized on the cluster region farthest to that Ti atom. Finally, next virtual orbitals (from LUMO+4 to LUMO+10) are delocalized over the whole cluster. Fig. 7 illustrates a representative orbital of each group.
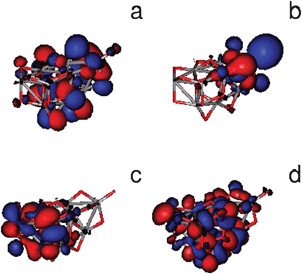 | ||
| Fig. 7 Selected occupied and virtual frontier molecular orbitals of the (TiO2)9 cluster. (a) HOMO−5, (b) HOMO, (c) LUMO and (d) LUMO+8. | ||
Adsorbed system orbitals follow the same scheme for all the dyes except for C343. The main feature of these orbitals is that there is very little mixing between cluster and dye orbitals. In general, orbitals are either completely localized on the cluster or on the dye. The last occupied orbitals (HOMO−3, HOMO−2, HOMO−1 and HOMO) are very similar to the last free dye occupied orbitals. In particular, HOMOs of adsorbed systems are almost identical to the same orbitals for free dyes and they have no contribution from the cluster. In contrast, first virtual orbitals correspond to the same orbitals of free cluster, and they are localized on the cluster region farthest to the dye molecule. The LUMO orbital of free dyes corresponds to LUMO+4 for adsorbed NKX-2586 and LUMO+5 for NKX-2311, NKX-2753 and NKX-2593. These orbitals have been named LUMO* (or L*) for each system. Finally, the free dye LUMO+1 orbital corresponds to LUMO+20 for adsorbed dyes. Some orbitals of adsorbed NKX-2593 have been represented in Fig. 8.
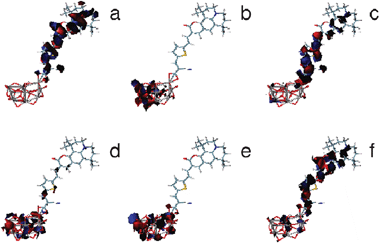 | ||
| Fig. 8 Selected occupied and virtual frontier molecular orbitals of adsorbed NKX-2593: (a) HOMO, (b) LUMO, (c) LUMO+5 or LUMO*, (d) LUMO+8, (e) LUMO+17, (f) LUMO+20. | ||
To better visualize the orbital scheme of the different systems, in Fig. 9 the computed Kohn–Sham orbital energies corresponding to free and adsorbed NKX-2593 and (TiO2)9 cluster have been represented. The energies have been shifted to align the free dye HOMO energy with the adsorbed system HOMO energy and the free cluster LUMO energy with the adsorbed system LUMO energy. The free dye HOMO orbital is located within the cluster band gap and free dye LUMO (and consequently LUMO*) orbitals are located within the semiconductor conduction band.
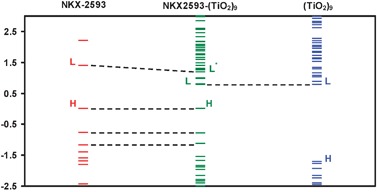 | ||
| Fig. 9 Molecular orbital energies for the ground state. From left to right free NKX-2593, adsorbed NKX-2593 and free (TiO2)9 cluster. | ||
Adsorbed C343 presents a different orbital scheme. We observe more mixing between cluster and dye orbitals and every orbital has some cluster contribution. LUMO* corresponds to LUMO+13 for free dye and now, it is delocalized over the whole system. C343 LUMO is more unstable than the LUMO orbital for the other dyes arising in a region where there are much more cluster orbitals, and for this reason the mixing becomes more notorious.
Similarly to the case for free dyes, we have analyzed the main contributions to the bands in the spectra for adsorbed dyes. Important similarities for all dyes can be observed. In all spectra there is a triad of bands in which the main one is the most intense and it is in the middle. The first band corresponds to excitations from the HOMO orbital to virtual orbitals localized on the cluster. Obviously, these excitations do not appear in the spectrum of free dyes and they are responsible for the widening of the first band of the spectra in the low energy zone after adsorption. For some dyes, in this band there are minor contributions from the LUMO* orbital. The presence of this band indicates a direct charge transfer from the dye to the semiconductor, which is unexpected since coumarin dyes are considered type I sensitizers. The presence of both injection mechanisms could lead to the observation of different injection rates (as it has been observed for alizarin dye).46
Next, the main band is contributed from several excitations from the HOMO orbital to different orbitals delocalized over the cluster. Nevertheless, all excitations with considerable intensity involve LUMO+5 (LUMO*) which is an orbital completely localized on the molecule. Finally, the third band comprises excitations from HOMO−1 and HOMO−2 (both localized on the dye) to orbitals of different nature but with LUMO* contribution. The assignments of the main electronic excitations for adsorbed NKX-2593 are shown in Table 2.
To complete the analysis the HOMO and LUMO* energies for the five adsorbed dyes have been represented in Fig. 10. The energy gap between these two orbitals is related to the position of the absorption maximum in the spectra. In this representation the LUMO orbital energy has been set to zero. As it has been said above, the LUMO orbital is very similar to the same orbital for free cluster. In consequence, the zero value corresponds to the lower edge of the semiconductor conduction band. This figure allows us to combine the different criteria to evaluate efficiency described in Section 3.2.
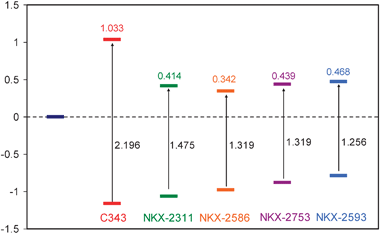 | ||
| Fig. 10 HOMO and LUMO* energies for adsorbed dyes. Energies have been shifted to set the zero energy to the lower edge of the semiconductor conduction band. | ||
HOMO and LUMO* energy gaps are slightly smaller than HOMO and LUMO gaps for free dyes (differences ranged from 0.10–0.15 eV). This is due to a stabilization of the LUMO orbital after mixing with cluster orbitals. HOMO and LUMO* energy gap of adsorbed C343 dye is rather larger than those for the rest of dyes. In consequence, its first band in absorption spectra is blue-shifted in the series and C343 is the dye with the lowest efficiency as sensitizer. Differences between NKX-2311, NKX-2586, NKX-2753 and NKX-2593 are small. Regarding the HOMO–LUMO* energy gap we would predict the following efficiency ordering: C343 < NKX-2311 < NKX-2586 = NKX-2753 < NKX-2593.
On the other hand, the higher the LUMO* orbital is located related to the lower edge of the semiconductor conduction band, the more efficient the electron injection is. Adsorbed C343 is the system with the highest-lying LUMO*. Nevertheless, C343 would not be efficient as a sensitizer because it has almost no absorption in the visible region. For the rest of dyes, the gap between LUMO* and the conduction band edge increases following the sequence: NKX-2586, NKX-2311, NKX-2753 and NKX-2593.
When combining these criteria C343 would be the dye with a lowest efficiency because of its negligible absorption in the visible region. This prediction agrees with experimental data.9 It could also be stated that the efficiency increases following the sequence: NKX-2311, NKX-2753 and NKX-2593. In this series the π electron system increases, the main band becomes wider, the maximum position of the main band and the absorption threshold slightly red-shift, the energy gap between HOMO and LUMO* becomes smaller and the LUMO* is located higher related to the lower edge of the semiconductor conduction band. This efficiency ordering also agrees well with experimental data.9,11
NKX-2586 and NKX-2753 present similar properties and we would expect similar efficiencies for both dyes as sensitizers. Nevertheless for cells sensitized with NKX-2586 the efficiency is almost 50% of the efficiency obtained in cells based on NKX-2753. The reason for this discrepancy is that we are not considering all the factors affecting the global efficiency. As we mentioned in the Introduction, coumarin dyes are liable to experience π-stacked aggregation. Dyes containing three methine units like NKX-2586 are especially prone to suffer dye aggregation. This effect leads to inefficient electron injection because of intermolecular energy transfer between the dyes, resulting thus in a much lower cell efficiency than for NKX-2753.
4. Conclusions
In this work we have investigated five coumarin based dyes: C343, NKX-2311, NKX-2586, NKX-2753 and NKX-2593. TD-DFT calculations have been performed to study their electronic structure and optical properties. First, optimized geometries of the free organic dyes as well as adsorbed on a TiO2 cluster have been obtained in the electronic ground state. Next, two different TD-DFT implementations (time and frequency domains) were employed to explore the optical response of those structures.From the results, some parameters such as the position and width of the first band in the electronic absorption spectra, the absorption threshold and the LUMO energy with respect to the conduction band edge have been chosen to establish useful criteria to evaluate the efficiency of these molecules as sensitizers in DSSCs.
On comparing similar sensitizers, the light harvesting process is more favorable when the absorption threshold is lower and the first band in the spectrum becomes wider extending into the visible region. Secondly, the electron injection in the semiconductor is more efficient when the excited state is located higher in the conduction band.
Thus, we predict, in good agreement with experimental evidences, the following efficiency sequence: C343 ≪ NKX-2311 < NKX-2753 < NKX-2593. However, our calculations indicate that NKX-2586 and NKX-2753 would exhibit similar efficiencies in contrast with the experimental results which show a much higher efficiency for NKX-2753. The reason lies on the tendency of NKX-2586 to suffer π-stacked aggregation phenomena resulting in an inefficient electron injection.
In this work we have established useful theoretical criteria to evaluate the efficiency of organic molecules as sensitizers in DSSCs. These criteria will be used in future works to design more efficient DSSC sensitizers, reducing economical cost and synthesis effort.
Acknowledgements
This work was funded by the Spanish Ministerio de Ciencia e Innovación, MICINN, projects MAT2008-4918 and CSD2008-0023. RSA thanks the Junta de Andalucía for a predoctoral grant (P08-FQM-3661 and EXC/2005/FQM-1126). Part of the calculations has been carried out at the Andalusia Supercomputing Center–Servicio de Supercomputación del Centro Informático Científico de Andalucía (C.I.C.A).References
- M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS.
- W. R. Duncan and O. V. Prezhdo, Annu. Rev. Phys. Chem., 2007, 58, 143 CrossRef CAS.
- Z. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170 RSC.
- A. Mishra, M. K. R. Fischer and P. Bäulere, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS.
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS.
- Z. Chen, F. Li and C. Huang, Curr. Org. Chem., 2007, 11, 1241 CrossRef CAS.
- D. Jacquemin, J. Preat, J. E. A. Perpete and C. Adamo, Int. J. Quantum Chem., 2010, 110, 2121 Search PubMed.
- K. Hara, K. Sayama, Y. Ohga, A. Shinpo, S. Suga and H. Arakawa, Chem. Commun., 2001, 569 RSC.
- K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597 CrossRef CAS.
- K. Hara, Y. Tachibana, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 77, 89 CrossRef CAS.
- K. Hara, M. Kurashige, Y. Dan-Oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC.
- Z. Wang, Y. Cui, Y. Dan-Oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17017 Search PubMed.
- K. Hara, Y. Dan-Oh, C. Kasada, Y. Ohga, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Langmuir, 2004, 20, 4205 CrossRef CAS.
- Z. Wang, K. Hara, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, H. Arakawa and H. Sugihara, J. Phys. Chem. B, 2005, 109, 3907 CrossRef CAS.
- J. M. Rhem, G. L. McLendon, Y. Nagasawa, K. Yoshihara, J. Moser and M. Grätzel, J. Phys. Chem., 1996, 100, 9577 CrossRef.
- H. N. Ghosh, J. B. Asbury and T. Lian, J. Phys. Chem. B, 1998, 102, 6482 CrossRef CAS.
- R. R. Frontiera, J. Dasgupta and A. Mathies, J. Am. Chem. Soc., 2009, 131, 15630 CrossRef CAS.
- K. Deuk Seo, H. Min Song, M. Jun Lee, M. Pastore, C. Anselmi, F. De Angelis, M. K. Nazeeruddin, M. Gräetzel and H. Kyu Kim, Dyes Pigm., 2011, 90, 304 Search PubMed.
- R. Impronta, V. Barone and F. Santoro, Angew. Chem., Int. Ed., 2007, 46, 405 CrossRef CAS.
- R. Impronta, V. Barone, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2006, 125, 05410 Search PubMed.
- D. Jacquemin, E. A. Perpète, G. Scalmani, M. J. Frisch, X. Assfeld, I. Ciofini and C. Adamo, J. Chem. Phys., 2006, 125, 164324 CrossRef.
- I. Georgieva, N. Trendafilova, A. Aquino and H. Lischka, J. Phys. Chem. A, 2005, 109, 11860 CrossRef CAS.
- M. Sulpizi, P. Carloni, J. Hutter and U. Rothlisberger, Phys. Chem. Chem. Phys., 2003, 5, 4798 RSC.
- Y. Kurashige, T. Nakajima, S. Kurashige and K. Hirao, J. Phys. Chem. A, 2007, 111, 5544 CrossRef CAS.
- B. M. Wong and J. G. Cordaro, J. Chem. Phys., 2008, 129, 214703 CrossRef.
- T. Stein, L. Kronik and R. Baer, J. Chem. Phys., 2009, 131, 244119 CrossRef.
- X. Zhang, J. Zhang and Y. Xia, J. Photochem. Photobiol., A, 2008, 194, 167 CrossRef CAS.
- R. Sánchez-de-Armas, J. Oviedo, M. A. San Miguel, P. Ordejón, M. Pruneda and J. F. Sanz, J. Chem. Theory Comput., 2010, 6, 2856 CrossRef CAS.
- R. Sánchez-de-Armas, M. A. San-Miguel, J. Oviedo, A. Márquez and J. F. Sanz, Phys. Chem. Chem. Phys., 2011, 13, 1506 RSC.
- R. Sánchez-de-Armas, J. Oviedo, M. A. San Miguel and J. F. Sanz, J. Phys. Chem. C, 2011, 115, 11293 CAS.
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter, 1991, 43, 1993 CrossRef CAS.
- L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 1982, 48, 1425 CrossRef CAS.
- E. Artacho, D. Sánchez-Portal, P. Ordejón, A. García and J. M. Soler, Int. J. Quantum Chem. Status Solidi B, 1999, 215, 809 Search PubMed.
- J. Junquera, O. Paz, D. Sanchez-Portal and E. Artacho, Phys. Rev. B: Condens. Matter, 2001, 64, 235111 CrossRef.
- E. Anglada, J. M. Soler, J. Junquera and E. Artacho, Phys. Rev. B: Condens. Matter, 2002, 66, 205101 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- D. Sánchez-Portal, P. Ordejón, E. Artacho and J. M. Soler, Int. J. Quantum Chem., 1997, 65, 453 CrossRef.
- A. Tsolakidis, D. Sánchez-Portal and R. M. Martin, Phys. Rev. B: Condens. Matter, 2002, 66, 235416 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Rob, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef.
- Z. Qu and G. Kroes, J. Phys. Chem. B, 2006, 110, 8998 CrossRef CAS.
- S. Hamad, C. R. A. Catlow, S. M. Woodley, S. Lago and J. A. Mejías, J. Phys. Chem. B, 2005, 109, 15741 CrossRef CAS.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808 CrossRef CAS.
- S. E. Koops, P. R. F. Barnes, B. C. O'Regan and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 8054 CrossRef CAS.
- S. Kaniyankandy, S. Verma, J. A. Mondal, D. K. Palit and H. N. Ghosh, J. Phys. Chem. C, 2009, 113, 3593 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cp22058f |
| This journal is © the Owner Societies 2012 |