Fluorescence detection of single guest molecules in ultrasmall droplets of nonpolar solvent
Masakazu
Yasuda
a,
Atsushi
Iida
a,
Syoji
Ito
*ab and
Hiroshi
Miyasaka
*ac
aDivision of Frontier Materials Science, Graduate School of Engineering Science and Center for Quantum Materials Science under Extreme Conditions, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: sito@chem.es.osaka-u.ac.jp; miyasaka@chem.es.osaka-u.ac.jp; Fax: +81-6-6850-6244; Tel: +81-6-6850-6243
bPRESTO, Japan Science and Technology Agency (JST), Kawaguchi, Saitama 332-0012, Japan
cCREST, Japan Science and Technology Agency (JST), Kawaguchi, Saitama 332-0012, Japan
First published on 15th November 2011
Abstract
We have investigated emissive behaviours of individual perylenebisimide derivatives, N,N′-dipropyl-1,6,7,12-tetrakis(4-tert-butylphenoxy)-3,4,9,10-perylenetetra-carboxydiimide (BP-PDI), in single ultrasmall droplets of n-octane at room temperature by using confocal and wide-field microscopic techniques. Single BP-PDIs in the small droplets show no distinguishable blinking in the time courses of fluorescence intensity. This is attributed to small probabilities of the formation of the long-lived ionized state leading to the off-state of the fluorescence. Temporal change in the degree of polarization of fluorescence and wide-field fluorescence images indicated short-time adsorption of the fluorescent molecules at the interfaces between n-octane and watery environments. Fluorescence correlation spectroscopy revealed that the adsorption/desorption processes took place at least in two different time scales, probably due to the difference in the adsorption geometry and/or in the interaction, such as van der Waals interaction and hydrogen bonding, between the dye and the interface.
Introduction
Since the pioneering works of Moerner and Kador1 in 1989 and Oritt and Bernard2 in 1990, the optical detection of individual molecules has been applied to the investigation of various chemical processes in a variety of fields of science, yielding detailed information that is not easily accessible by the traditional ensemble measurements.3–5 In these years, much attention has been devoted to realizing single-photon emitting sources6 potentially applicable to quantum information processing. A number of systems have been reported, such as single fluorescent molecules,7–9 single quantum dots,10–13 trapped single ions14 and atoms.15 In addition to these single quantum systems, detailed investigations on organic multi-chromophoric systems revealed that emitted photons from dendrimers,16,17nanocrystals18 and conjugated polymers19,20 show antibunching behaviours owing to rapid energy migration and effective singlet–singlet annihilation among chromophores in the case where sizes of the systems are well controlled and the conditions of photoexcitation are optimized.Definitive emission, room temperature operation, small blinking probability, and long-term durability seem most important conditions required for actual applications of organic single-photon emitting sources. In general, organic dyes with large fluorescence quantum yields have fluorescent lifetimes of several ns. This short excited state lifetime contributes to the definitive photon emission by pulsed excitation. However, fluorescence dyes usually show photo-blinking when embedded in glassy matrices or immobilized on surfaces of solids. That is, these dyes show random interconversion between a bright emissive (on) and a dark (off) state,4,21,22 which can be utilized for a super resolution (localization) fluorescence microscopy.23
The dark state in these systems has been attributed to several processes;24,25 triplet state formation, occasional quenching by oxygen and photoionization that is probably induced by the stepwise two-photon absorption via the long-lived intermediate species such as a triplet state.25,26 The lifetime of triplet states at room temperature is at most in the order of several ms. Hence the triplet state itself does not seem to result in the long-lived off-state formation, although it plays a significant role under cryogenic conditions.8 The oxygen molecule generally could contribute to the long off-state formation under the conditions where the residence time of oxygen around fluorescent molecules is sufficiently long owing to small diffusion coefficients in the host environments. Experimental results17 on the effect of oxygen on the off-time appearance in PMMA and Zeonex, however, did not show a remarkable difference, although the diffusion constant of O2 is different from each other in these two polymers. This result indicates that oxygen does not effectively contribute to the formation of the long off-state.
In photoionization, it is worth noting that extensive investigations were performed26,27 for the conventional ensemble systems by using laser multiphoton excitation. These studies revealed that the charge recombination between a parent cation and an electron ejected via the two-photon ionization in polymer matrices28 and on surfaces of porous materials29 takes place in the order of ms to several hours even at room temperature. On the other hand, the recombination time of the cation–electron pair produced via the multiphoton ionization is at most several tens of ps in nonpolar alkane solutions26,27,30 because of the large mobility of the electron and/or the intrinsic fast charge recombination process.31,32 Hence, we can expect that the blinking through the photoionization is suppressed in the case where fluorescent molecules are confined in small droplets of nonpolar solvents. Along this line, in order to confirm the contribution of photoionization to the blinking phenomena, we have investigated dynamic behaviours of individual organic molecules encapsulated in micro/nano-droplets of octane solution by means of single molecule detection methods. In addition, the present approach at the single molecule level could provide information on the dynamic aspects of molecules in tiny droplets which are widely used, such as in spatially controllable small reaction cages, micro-electrophoresis,33 high throughput analysis34 of chemical35 and biological systems,36 and efficient chemical synthesis in microfluidic channels.37,38 In the following, we will show a sample preparation procedure to realize single molecule detection in small droplets as well as our experimental setup and discuss the effect of a liquid–liquid interface and droplet size on the fluorescent properties of single guest molecules.
Experimental
Sample preparation
N,N′-Dipropyl-1,6,7,12-tetrakis(4-tert-butylphenoxy)-3,4,9,10-perylenetetra-carboxydiimide (BP-PDI, Yamada Chemical) was used as received. Water was purified by a Milli-Q system (Direct-Q, Millipore). n-Octane (GR grade, Wako) was used as received. The mixture of water and n-octane solution of BP-PDI (ca. 2.0 × 10−10 M) was sonicated for 300 seconds to prepare the droplets of n-octane whose size is smaller than several μm. Diameters of the individual droplets were determined by the optical microscopy. Low concentration of BP-PDI in the initial octane solution resulted in low probability of the inclusion of BP-PDIs in the droplets as will be discussed in later sections. This emulsion was added to an aqueous solution of agarose (LM-200, Dojindo, 2.0 wt%) at 343 K, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 volume ratio between the emulsion and the agarose solutions. This mixed solution (0.02 mL) was sandwiched with two well-cleaned cover slips and was kept at 283 K for several hours. The structure of the droplet with BP-PDI molecules was schematically illustrated in Fig. 1(a).
∶1 volume ratio between the emulsion and the agarose solutions. This mixed solution (0.02 mL) was sandwiched with two well-cleaned cover slips and was kept at 283 K for several hours. The structure of the droplet with BP-PDI molecules was schematically illustrated in Fig. 1(a).
 | ||
| Fig. 1 (a) A schematic illustration of single dye molecules encapsulated in micro/nano-droplets immobilized in the hydrogel. (b) Confocal and wide-field microscopic systems used in the present study (see the text). | ||
Apparatus for single molecule detection
Fig. 1(b) shows a schematic diagram of a single molecule detection system used in the present work. A confocal detection setup with a pinhole of 50 μm diameter was installed in an inverted optical microscope (IX-71, Olympus) to measure time trajectories of fluorescence intensity, fluorescence lifetimes, fluorescence correlation curves and photon correlation histograms (photon antibunching). A piezoelectric stage (P-517. 2CL, PI) was employed for the spatial scanning. A second harmonic (532 nm) of a mode-locked Nd:YAG laser (DPM-1000&SBR-5080-FAP, coherent, pulse duration of ca. 30 ps) was used as an excitation light source. The repetition rate was adjusted to 8 MHz by using an electro-optical modulator (Model360-80, Conoptics). The laser light with circular polarization was focused onto the sample by an objective (NA 1.30, ×100). To obtain photon correlation histograms on the basis of the Hanbury-Brown and Twiss type photon correlation setup with the pulsed laser excitation,39 fluorescence from the droplets was collected by the objective and guided into a pair of avalanche photodiodes (APD, SPCM-AQR-14, Perkin Elmer) after passing through a half beam splitter. A long-pass filter (Semrock, LP01-532RU) was placed to block the scattered light of 532 nm from the sampling volume. A time-correlated single photon counting (TCSPC) module (PicoHarp300, PicoQuant) was used to record the arrival time of all the photons detected, of which results were analyzed by using homemade software coded on LabView (National Instruments).For wide-field fluorescence imaging, the collimated laser light at 532 nm was used for the irradiation of the sample plane by focusing it on the back aperture of the objective.40–42 In the measurement, the repetition rate of the laser was set to 80 MHz. Fluorescence from the sample was collected by the objective and detected by an EM-CCD camera (Cascade II, Roper Scientific) as sequential images at the time intervals of 534.8 ms. The long-pass filter was placed in the optical path to block the excitation light at 532 nm.
Results and discussion
Formation and immobilization of droplets with single molecules
Fig. 2(a) shows an optical transmission image of the specimen prepared by the procedure as explained in the experimental section. Droplets ranging from 200 nm to several μm are observed in the agarose gel. It was confirmed that these droplets were unmoved in the gel at least during the observation time (more than several minutes), ensuring that the droplet was kept tightly for the single molecule measurement by the stage-scanning. Fig. 2(b) shows a fluorescence image of the same area, where the bright spot is confirmed in one of the droplets. This result indicates that some of the droplets actually include fluorescent molecules while many of the droplets do not. The small number of droplets with the fluorescent dye is due to the initial concentration of the fluorescent molecules, as explained in the experimental section. | ||
| Fig. 2 (a) Optical transmission image of n-octane droplets immobilized in the agarose hydrogel. (b) Fluorescence image of the same area, measured with the EM-CCD. (c) Size distribution of the n-octane droplets, obtained from the optical transmission images of 204 droplets. | ||
As shown in Fig. 2(a), the size of the droplet was estimated to be in the range of 200 nm to several μm. It should be noted, however, that this estimation was based on the optical transmission images. In other words, the droplets with the size < ca. 200 nm in diameter were not clearly detected in this measurement. Although the size of the estimation is restricted, the average diameter for 204 droplets detected by optical transmission images was estimated to be ca. 400 nm as shown in Fig. 2(c).
Detection of fluorescence from single molecules in single nanodroplets
Fig. 3(a) shows a typical fluorescence intensity trajectory of a single BP-PDI in a n-octane droplet smaller than 300 nm in diameter. The time course of the fluorescence lifetime for the BP-PDI is also plotted in the figure. Although the large fluctuation of the fluorescence intensity is not so remarkable, a few downward spikes are observed. As will be discussed in later sections, these fluorescence spikes are ascribable to transient adsorption of the dye at the interface. The fluorescence lifetime of the BP-PDI also shows no significant change in the time range from 3 to 25 s. This result is consistent with the small fluctuation in the fluorescence intensity trajectory. At 25 s, the fluorescence intensity suddenly decreased to the background level, as frequently observed in trajectories of fluorescence intensity of single molecules in many systems.3–5,21 Such a one-step photo-bleaching was observed for 144 droplets among 213 emissive droplets with the diameter of ca. 300 nm. On the other hand, as will be shown in the following section, two-step bleaching was observed for 39 droplets and 32 droplets exhibited multi-step (≥3) bleaching. | ||
| Fig. 3 (a) A typical fluorescence intensity trajectory (solid line) and the time-course of fluorescence lifetime (solid rectangles) for a single BP-PDI in a nanodroplet of n-octane and (b) its photon correlation histogram. (c) The fluorescence intensity trajectory (solid line) and the time-course of fluorescence lifetime (solid rectangles) for a nanodroplet including two BP-PDIs. (d) The photon correlation histograms corresponding to the time trajectory data (Fig. 3c) in the time range from 1 to 7 s (solid line) and from 7 to 21 s (dashed line). | ||
Fig. 3(b) shows the second-order photon correlation histogram obtained by the analysis of the data in Fig. 3(a). The signal around the time origin (0 ns), NC, corresponds to the case where more than one photon were detected at a single excitation event, while signals appearing with the time interval of the excitation pulse (125 ns), NL, are originated from the excitation by other laser pulses at different times. The ratio of NC to the average number of photons in other lateral peaks, NL, is related to the number of fluorescent dyes or light-emitting sources in the observation area. Obviously, the signal at the time origin, NC, is zero (antibunching) in the case where fluorescence photons are emanated from a single-photon emitting source. On the other hand, NC/NL values are 0.5, 0.67 and 0.75, respectively, for the cases where 2, 3 and 4 photons were detected at single excitation events.16,17 As shown in Fig. 3(b), the value of NC is almost zero, indicating that a single fluorescent molecule is involved in the droplet. This photon antibunching behaviour is consistent with the one-step photo-bleaching in Fig. 3(a).
As stated in the previous section, more than one-step photo-bleaching was observed for 33% of the droplets. Fig. 3(c) shows a typical example where two-step photo-bleaching was clearly exhibited. Fig. 3(d) shows the photon correlation histogram for the trajectory in the time range from 1 to 7 s in Fig. 3(c). The NC/NL value was rather close to 0.5, which is consistent with the two-step photo-bleaching in Fig. 3(c). That is, two BP-PDIs simultaneously emitted photons in this time range. On the other hand, the NC/NL value for the trajectory in the time range from 7 to 21 s is almost zero (noise level) as shown in Fig. 3(d). This result indicates that the two BP-PDIs underwent a stepwise photo-bleaching in the droplet.
It is worth noting that the fluorescence trajectories of single BP-PDI molecules in the n-octane droplets did not exhibit apparent blinking which has been generally observed in fluorescence intensity trajectories of single molecules immobilized in solids.3–5,21 As a typical example of the polymer system, we show a fluorescence intensity trajectory of a single BP-PDI embedded in PMMA in Fig. 4. Off-state intervals ranging from <1 s to a few tens of seconds frequently appear in the trajectory of the fluorescence intensity. On the other hand, among the 200 droplets of octane solution with the diameter of ≤200–600 nm, the blinking behaviour was observed only for 12 droplets and most of these droplets (10 droplets) had the diameter of ≤200 nm.
 | ||
| Fig. 4 (a) A typical fluorescence intensity trajectory of a single BP-PDI embedded in PMMA and (b) its photon correlation histogram. | ||
As briefly mentioned in the introductory section, several mechanisms have been proposed for the blinking behaviour of single molecules,24,25 such as triplet state formation, quenching by oxygen located in the vicinity of the fluorescent molecule and photoionization.3–5,21,25 Among these, photoionization through the two-photon absorption, probably due to the stepwise two-photon absorption via the triplet state under the weak excitation conditions, has been considered to contribute to the long-time off-state formation. In general, an electron ejected from a molecule via photo-ionization undergoes recombination in the case where no permanent chemical reaction involving the parent cation and/or the ejected electron takes place. The time constant of recombination is strongly dependent on the environment.26,27 In polymer matrices such as PMMA28 and adsorbed systems,29 the recombination takes place in the time range of ms to several hours even at room temperature. This is because the ejected electron is trapped and stabilized in a trap site in these materials and the long-distance between the parent cation and the electron is fixed. In the case where the detrapping process cannot take place owing to a high activation barrier, a long distance electron tunnelling is a major pathway for the charge recombination. Although the time constant of recombination via the long distance electron tunnelling is strongly dependent on the distance between the two oppositely charged species,43 it is in the range of ms to hours28,29 for the electron–cation pair with a typical pair distance26–30 of ca. 30–40 Å. On the other hand, in nonpolar solvents such as alkanes, the time scale of the recombination after the photoionization is less than a few tens of picoseconds26,27,29–31 because of the large mobility of the electron and/or the intrinsic fast charge recombination rate. Accordingly, the photoionization in nonpolar solutions does not lead to the long-time off-state formation. Moreover, because the distance of 30–40 Å is much shorter than the size of droplets in the present system, most of the ejected electrons via the photoionization rapidly come back to the parent molecules via geminate recombination without the trapping process at the interface. The experimental results that the small droplets showed blinking in the present study could also suggest the effect of the interface acting as a trapping site of electrons. From these results and discussion, we could conclude that the blinking behaviours observed in solid systems are suppressed in the nonpolar droplet probably due to the small possibility to produce long-lived charge separated states via the photoionization. In other words, single droplets of nonpolar solutions serve as emissive nano-systems without blinking. In addition, the present result supports the idea that the photoionization is an important mechanism of the blinking behaviour in the immobilized environments.25
Fluorescence polarization of single BP-PDI in nanodroplets
In usual fluid solutions, rotational diffusion time constants of solute molecules are at most a few hundreds of ps at room temperature.44 Because these rotational processes are much faster than the bin time of the measurement (ms time scale), fluorescence polarization is averaged for BP-PDI located in the solution phase. However, the fluorescence intensity could show the anisotropy while BP-PDI is adsorbed at the interface and immobilized. We could thus expect that the polarization of the fluorescence provides information on the adsorption/desorption dynamics at the interface. For this purpose, the fluorescence was divided into two orthogonal polarization components and each component of the fluorescence was detected by the two different APDs (APD1 and APD2 in Fig. 1(b)).Fig. 5(a) shows fluorescence intensity trajectories with orthogonally polarized two components together with the summation of the two. In Fig. 5(b) where the trajectories in the initial 8 s are plotted, both the trajectories fluctuate overall in opposite signs. The two orthogonal contributions show different intensities (oppositely in signs) in the time range from the beginning of photoexcitation at 532 nm (0.7 s) to 1.7 s, while the summation keeps almost the constant value. In order to more quantitatively analyze the dynamic behaviours in the fluorescence polarization, we employed the degree of polarization, P, defined by eqn (1).
 | (1) |
 | ||
| Fig. 5 (a) Fluorescence intensity trajectories of a BP-PDI in the droplet of n-octane with a diameter of ca. 200 nm, measured with two APDs to detect orthogonally polarized components. Black and gray lines, respectively, show trajectories of the fluorescence whose polarizations are orthogonal to each other. The summation of the two intensities is shown as a black dotted line. (b) Fluorescence intensity trajectories in the initial 8 s. (c) Time dependence of the fluorescence polarization. | ||
Fig. 6(a) shows another example of the fluorescence intensity trajectories of a droplet with the size of ca. 300 nm. In the same manner as Fig. 5(a), two orthogonally polarized fluorescence intensities are, respectively, plotted together with the summation of the two. Fig. 6(b) shows the time profile of P for the trajectories shown in Fig. 6(a). In the time range from 0.5 to 20 s, almost equivalent contributions from the orthogonal polarizations were observed, although some fluctuations with short time intervals as comparable with the bin time appear during this time range (typically at 4.5 s). We will discuss this short time fluctuation in the last section. At 20 s, the fluorescence intensity drastically decreases and the fluctuation of P is pronounced. This decrease of the fluorescence intensity might be due to the molecule adsorbed in such a manner that the absorption transition moment is tilted to the optical axis of the excitation laser light, leading to the small absorption probability. Interestingly, the averaged value of P shows almost zero in the time range of 20–24 s, although the fluctuation is enhanced. This averaged value of P in this time range suggests that the movement of the molecule is restricted in one direction (Z axis) but the molecule still could move in other two directions (X and Y axes) rather freely. It is worth noting again for the result shown in Fig. 5(b) that the summation of the two orthogonally polarized fluorescence intensities was kept almost constant with the non-zero P value. Although the apparent results are different between the two cases (Fig. 5 and 6), it is suggested also for the molecule in Fig. 5 that the movement is restricted in one direction (X or Y axes) but the molecule still could move in other two directions rather freely. That is, motions of the two directions among the three dimensional motions could rather freely take place. These behaviours might be related to the molecular geometry of the adsorption. In the case where one of the two ends of the long axis of BP-PDI is attached to the interface, the molecule still could rotate to some extent although the motions might be restricted. It should be noted that the degree of polarization, P, is in principle obtained through the reduction of the 3-dimensional motion into the two-dimensional one. Hence, it is better to employ the technique of defocus imaging4 in order to acquire the detailed information on the molecular motion. However, it is rather difficult to apply the defocus imaging to the present system because the translational movement of the guest molecules is too fast to be tracked by the imaging method. Although the information from the P value is not sufficient to derive a clear conclusion on the structure and the motion in the adsorbed state, the present result shows that the BP-PDI is temporarily adsorbed at the interface which is followed by the desorption.
 | ||
| Fig. 6 Orthogonally polarized fluorescence intensity trajectories of a BP-PDI that can rotate fast in a nanodroplet of n-octane. (a) Time courses of fluorescence intensities with orthogonal polarizations (black and gray lines) and the sum intensity of them (black dotted line). (b) The time course of the fluorescence polarization calculated from the data shown in Fig. 6(a). | ||
Dynamic motions of single molecules in large droplets
As shown in Fig. 2, 3 and 5, fluorescence trajectories of BP-PDI in droplets with ca. 300 nm diameters did not show apparent blinking. This is due to the small probability of the formation of the long-lived off-state, as was discussed in the previous sections. To obtain the trajectory without apparent blinking, however, it is also important to detect droplets with diameters smaller than the confocal volume of the excitation laser light. That is, the translational diffusion of BP-PDI may induce the apparent off-time in fluorescence trajectories in the case where the size of a droplet is larger than that of the confocal volume. In other words, the fluorescence trajectories for the dyes in larger droplets can provide information on translational/rotational diffusions related to adsorption/desorption dynamics at the interface.Prior to the quantitative analysis of the dynamics of the adsorption/desorption, we show time evolution of fluorescence images of a micrometre-sized droplet in Fig. 7 where the exposure time for each of the images was 500 ms. At t = 0, the fluorescence was observed in the entire area of the droplet, indicating that the fluorescent dye moves in the droplet with a large translational diffusion coefficient compared to the exposure time. On the other hand, the image at 0.54 s shows a bright spot at the interface of the droplet, clearly demonstrating that the dye locates at the interface due to the short-time adsorption. Because the fluorescence emission is observed also in other areas of the droplet at 0.54 s, the time interval of the adsorption is much shorter than the exposure time. At 1.08 s, the image shows the fluorescence emission from the entire area of the droplet. These images clearly show that the fluorescent dyes are adsorbed at the interface for a while.
 | ||
| Fig. 7 Sequential fluorescence images of a n-octane microdroplet including a single BP-PDI. | ||
Fig. 8(a) shows a fluorescence intensity trajectory of BP-PDI in a single micro-droplet with a diameter of 1 μm. This trajectory shows a quite different behaviour from those observed in droplets with smaller sizes than the focusing spot of the excitation laser (Fig. 3, 5 and 6). That is, the fluorescence intensity trajectory of BP-PDI in this droplet exhibits a large fluctuation of the intensity that can be divided into two types of photon bursts, such as spike-like one with a short duration comparable to the bin time and intensity change with a time scale much longer than the bin time. So to precisely elucidate the origins of these behaviours, we calculated an autocorrelation function (ACF) curve of the fluorescence intensity fluctuation and analyzed it by a method for fluorescence correlation spectroscopy (FCS).
 | ||
| Fig. 8 (a) A fluorescence intensity trajectory of BP-PDI in a single micrometre sized droplet of n-octane. (b) Open circles show the fluorescence autocorrelation curve of the BP-PDI in the single droplet of which fluorescence intensity trajectory is shown in (a). Closed circles show a typical fluorescence autocorrelation curve of BP-PDIs in a bulk n-octane solution. Solid and dashed lines are, respectively, the results of the analyses using eqn (2). | ||
The ACF curve of BP-PDI in the single droplet thus obtained is shown in Fig. 8(b) by open circles, together with the ACF curve of multiple BP-PDIs in n-octane bulk solution by closed circles for comparison. The ACF curve of the bulk solution was obtained through a usual FCS measurement for the n-octane solution of BP-PDI with a concentration of ∼10−9 M. The ACF of BP-PDI in the micro-droplet (open circles in Fig. 8(b)) exhibits obviously slower decay, compared to that of the bulk solution. To quantitatively analyze the ACF curves, we employed the following equation (eqn (2)).
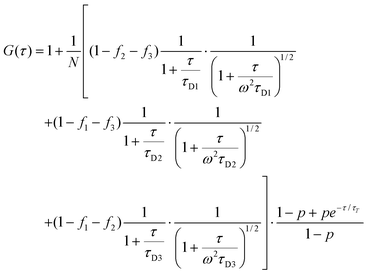 | (2) |
| f 1 | τ D1/ms | f 2 | τ D2/ms | f 3 | τ D3/ms | |
|---|---|---|---|---|---|---|
| Bulk n-octane | 1 | 0.023 | 0 | 0 | ||
| Droplet (φ ≈ 600 nm) | 0.21 | 0.029 | 0.22 | 0.87 | 0.57 | 26 |
| Droplet (φ ≈ 800 nm) | 0.37 | 0.015 | 0.23 | 2.2 | 0.40 | 12 |
| Droplet (φ ≈ 1000 nm) | 0.36 | 0.023 | 0.37 | 2.9 | 0.27 | 49 |
Fastest time constants in Table 1 can be assigned to the free translational diffusion because these time constants are similar to those of BP-PDI in the bulk solution of n-octane. The second and third ones could be attributed to diffusion dynamics affected by the adsorption and desorption at the interface of the droplets. This adsorption/desorption dynamics involves the interaction between the molecules and the interface, as well as the size effect of the diffusive area. Indeed, these parameters were dependent on the size of droplets as shown in Table 1; the contribution of the slowest diffusion, τD3, increased with a decrease in the size of droplets. On the origin of the diffusion times, τD2, it is rather difficult to derive a clear conclusion at the present stage of the investigation. It might be, however, related to the quick desorption owing to the difference in the adsorbed structure and/or the interaction. Some burst-like signals with short time intervals as comparable as the bin time shown in Fig. 3–6 might be related to the component of τD2.
Conclusions
Fluorescence behaviours of individual BP-PDIs in single ultrasmall droplets were investigated by using confocal and wide-field microscopic techniques. Fluorescence intensity trajectories for the individual dyes detected by the confocal microscopy showed discrete photo-bleaching that is a typical behaviour for single molecules. Antibunching behaviours in correlation histograms of the fluorescence photons confirmed the successful detection of individual guest dyes in the droplets. Interestingly, no apparent blinking was observed in the time courses of the fluorescence intensity. This behaviour was attributed to small possibilities for the formation of the long-lived off-state due to photoionization. The small probability of the off-state formation serves as an emissive nano-system without blinking.The polarization degree of the emission indicated the temporary immobilization of the fluorescent molecules, which was detectable with a bin time of ca. 25 ms, due to the adsorption at the interface, while the fluorescent molecules could undergo a rapid rotational diffusion in solution phase. The fluorescence images obtained by the wide-field microscope clearly visualized this temporary adsorption. Fluorescence correlation spectroscopy was applied to the quantitative analysis of the adsorption/desorption processes, which revealed that three components of diffusion velocities were involved in the diffusion process of BP-PDI in the droplets. The fastest diffusion velocity was comparable to that in the bulk solution and was assigned to the lateral motion of the dyes in the solution region of the droplets where the microscopic environment is similar to that in the bulk n-octane. On the other hand, the contributions from the middle and the slowest diffusion velocities were affected by desorption following the adsorption at the interface. Although the origin of the two different desorption processes was not completely elucidated at the present stage of the investigation, the adsorption geometry and/or the difference in the interaction, such as van der Waals interaction and hydrogen bonding between the dye and the interface, was suggested.
Acknowledgements
The present work was partly supported by Grant-in-Aid for Scientific Research (B) (20350009) to HM and Grant-in-Aid for Young Scientists (A) (23681023) to SI from the Japan Society for the Promotion of Science (JSPS).Notes and references
- W. E. Moerner and L. Kador, Phys. Rev. Lett., 1989, 62, 2535–2538 CrossRef CAS.
- M. Orrit and J. Bernard, Phys. Rev. Lett., 1990, 65, 2716–2719 CrossRef CAS.
- Single Molecule Spectroscopy in Chemistry, Physics and Biology, ed. A. Gräslund, R. Rigler and J. Widengren, Springer-Verlag, Berlin, 2001 Search PubMed.
- D. Wöll, E. Braeken, A. Deres, F. C. De Schryver, H. Uji-i and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- Single Molecule Spectroscopy, ed. R. Rigler, M. Oritt and T. Basché, Springer, Berlin, 2010 Search PubMed.
- B. Lounis and M. Orrit, Rep. Prog. Phys., 2005, 68, 1129–1179 CrossRef CAS.
- T. Basché, W. E. Moerner, M. Orrit and H. Talon, Phys. Rev. Lett., 1992, 69, 1516–1519 CrossRef.
- T. Basché, S. Kummer and C. Bräuchle, Nature, 1995, 373, 132–134 CrossRef.
- B. Lounis and W. E. Moerner, Nature, 2000, 407, 491–493 CrossRef CAS.
- P. Michler, A. Imamoğlu, M. D. Mason, P. J. Carson, G. F. Strouse and S. K. Buratto, Nature, 2000, 406, 968–970 CrossRef CAS.
- C. Santori, D. Fattal, J. Vučković, G. S. Solomon and Y. Yamamoto, Nature, 2002, 419, 594–597 CrossRef CAS.
- S. Kako, C. Santori, K. Hoshino, S. Götzinger, Y. Yamamoto and Y. Arakawa, Nat. Mater., 2006, 5, 887–892 CrossRef CAS.
- S. Masuo, H. Naiki, S. Machida and A. Itaya, Appl. Phys. Lett., 2009, 95, 193106 CrossRef.
- B. T. H. Varcoe, S. Brattke, M. Weidinger and H. Walther, Nature, 2000, 403, 743–746 CrossRef CAS.
- M. Keller, B. Lange, K. Hayasaka, W. Lange and H. Walther, Nature, 2004, 431, 1075–1078 CrossRef CAS.
- P. Tinnefeld, K. D. Weston, T. Vosch, M. Cotlet, T. Weil, J. Hofkens, K. Müllen, F. C. De Schryver and M. Sauer, J. Am. Chem. Soc., 2002, 124, 14310–14311 CrossRef CAS.
- S. Masuo, T. Vosch, M. Cotlet, P. Tinnefeld, S. Habuchi, T. D. M. Bell, I. Oesterling, D. Beljonne, B. Champagne, K. Müllen, M. Sauer, J. Hofkens and F. C. De Schryver, J. Phys. Chem. B, 2004, 108, 16686–16696 CrossRef CAS.
- S. Masuo, A. Masuhara, T. Akashi, M. Muranushi, S. Machida, H. Kasai, H. Nakanishi, H. Oikawa and A. Itaya, Jpn. J. Appl. Phys., Part 2, 2007, 46, L268–L270 CrossRef CAS.
- P. F. Barbara, A. J. Gesquiere, S.-J. Park and Y. J. Lee, Acc. Chem. Res., 2005, 38, 602–610 CrossRef CAS.
- S. Masuo, T. Tanaka, S. Machida and A. Itaya, Appl. Phys. Lett., 2008, 92, 233114 CrossRef.
- J. P. Hoogenboom, J. Hernando, E. M. H. P. van Dijk, N. F. van Hulst and M. F. García-Parajo, ChemPhysChem, 2007, 8, 823–833 CrossRef CAS.
- (a) P. Tinnefeld, J. Hofkens, D.-P. Herten, S. Masuo, T. Vosch, M. Cotlet, S. Habuchi, K. Müllen, F. C. De Schryver and M. Sauer, ChemPhysChem, 2004, 5, 1786–1790 CrossRef CAS; (b) J. N. Clifford, T. D. M. Bell, P. Tinnefeld, M. Heilemann, S. M. Melnikov, J. Hotta, M. Sliwa, P. Dedecker, M. Sauer, J. Hofkens and E. K. L. Yeow, J. Phys. Chem. B, 2007, 111, 6987–6991 CrossRef CAS; (c) J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, Angew. Chem., Int. Ed., 2008, 47, 5465–5469 CrossRef CAS.
- (a) M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS; (b) M. Heilemann, S. van de Linde, A. Mukherjee and M. Sauer, Angew. Chem., Int. Ed., 2009, 48, 6903–6908 CrossRef CAS; (c) S. van de Linde, I. Krstić, T. Prisner, S. Doose, a. M. Heilemannc and M. Sauer, Photochem. Photobiol. Sci., 2011, 10, 499–506 RSC.
- F. Kulzer, F. Koberling, T. Christ, A. Mews and T. Bashé, Chem. Phys., 1999, 247, 23–34 CrossRef CAS.
- M. Haase, C. G. Hübner, F. Nolde, K. Müllen and T. Basché, Phys. Chem. Chem. Phys., 2011, 13, 1776–1785 RSC.
- M. Orrit, Photochem. Photobiol. Sci., 2010, 9, 637–642 CAS.
- H. Miyasaka and N. Mataga, Pulse Radiolysis, ed. Y. Tabata, CRC Press, Boca Raton, 1991, p.173 Search PubMed.
- Y. Hirata and N. Mataga, Prog. React. Kinet., 1993, 18, 273–308 CAS.
- A. Tsuchida, W. Sakai, M. Nakano, M. Ysohida and M. Yamamoto, Chem. Phys. Lett., 1992, 188, 254–258 CrossRef CAS.
- H. Miyasaka, S. Kotani, T. Ota and A. Itaya, Chem. Phys. Lett., 2001, 335, 496–502 CrossRef CAS.
- (a) H. Miyasaka and N. Mataga, Chem. Phys. Lett., 1986, 126, 219–224 CrossRef CAS; (b) H. Miyasaka and N. Mataga, Chem. Phys. Lett., 1987, 134, 480–484 CrossRef CAS.
- T. Zhang, Y. J. Lee, T. W. Kee and P. F. Barbara, Chem. Phys. Lett., 2005, 403, 257–261 CrossRef CAS.
- D. R. Link, E. Grasland-Mongrain, A. Duri, F. Sarrazin, Z. Cheng, G. Cristobal, M. Marquez and D. A. Weitz, Angew. Chem., Int. Ed., 2006, 45, 2556–2560 CrossRef CAS.
- D. T. Chiu, R. M. Lorenz and G. D. M. Jeffries, Anal. Chem., 2009, 81, 5111–5118 CrossRef CAS.
- C. J. Gerdts, D. E. Sharoyan and R. F. Ismagilov, J. Am. Chem. Soc., 2004, 126, 6327–6331 CrossRef CAS.
- J. Shendure, G. J. Porreca, N. B. Reppas, X. Lin, J. P. McCutcheon, A. M. Rosenbaum, M. D. Wang, K. Zhang, R. D. Mitra and G. M. Church, Science, 2005, 309, 1728–1732 CrossRef CAS.
- D. T. Chiu, C. F. Wilson, F. Ryttsén, A. Strömberg, C. Farre, A. Karlsson, S. Nordholm, A. Gaggar, B. P. Modi, A. Moscho, R. A. Garza-López, O. Orwar and R. N. Zare, Science, 1999, 283, 1892–1895 CrossRef CAS.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS.
- R. Hanbury-Brown and R. Twiss, Nature, 1956, 177, 27–29 CrossRef.
- T. Kaji, S. Ito, S. Iwai and H. Miyasaka, J. Phys. Chem. B, 2009, 113, 13917–13925 CrossRef CAS.
- S. Ito, T. Kusumi, S. Takei and H. Miyasaka, Chem. Commun., 6165–6167 Search PubMed.
- S. Ito, S. Fukuya, T. Kusumi, Y. Ishibashi, H. Miyasaka, Y. Goto, M. Ikai, T. Tani and S. Inagaki, J. Phys. Chem. C, 2009, 113, 11884–11891 CAS.
- (a) R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CAS; (b) G. R. Fleming, Chemical Applications of Ultrafast Spectroscopy, Oxford University Press, Oxford, 1986, p.137 Search PubMed.
- T. Bosch, M. Cotlet, J. Hofkens, K. Van Der Biest, M. Lor, K. Weston, P. Tinnefeld, M. sauer, L. Latterini, K. Müllen and F. C. De Schryver, J. Phys. Chem. A, 2003, 107, 6920–6931 CrossRef.
| This journal is © the Owner Societies 2012 |