Mechanism of benzene hydroxylation by high-valent bare FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O2+: explicit electronic structure analysis
O2+: explicit electronic structure analysis
Ji-Lai
Li
*a,
Xiang
Zhang
b and
Xu-Ri
Huang
a
aState Key Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People's Republic of China. E-mail: jilai@jlu.edu.cn; huangxr@jlu.edu.cn; Fax: (+)86-431/88499721
bSchool of Chemistry and Materials Science, Shanxi Normal University, Linfen 041004, People's Republic of China
First published on 9th November 2011
Abstract
The conversion of benzene to phenol by high-valent bare FeO2+ was comprehensively explored using a density functional theory method. The conductor-like screen model (COSMO) was used to mimic the role of solvent effect with acetonitrile chosen as the solvent. Two radical mechanisms and one oxygen insertion mechanism were tested for this conversion. The first radical mechanism can also be named as the concerted mechanism in which the hydrogen-atom abstraction process is accomplished via a four-centered transition state. The second radical mechanism is initiated by a direct hydrogen-atom abstraction with a collinear C–H–O transition structure. It is actually the same as the well-accepted rebound mechanism for the C–H bond activation by heme and nonheme iron-oxo catalysts. The third is an oxygen insertion mechanism which is essentially an aromatic electrophilic attack leading to an arenium σ-complex intermediate. The formation of a precomplex with an η4 coordinate environment in the first radical mechanism is energetically more favorable. However, the relatively lower activation energy barrier of the oxygen insertion mechanism compared to the radical ones makes it highly competitive if the Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2+ collides with benzene in the proper orientation. The detailed potential energy surfaces also indicate that the second radical mechanism, i.e., the benzene C–H bond activation through the rebound mechanism, is less favorable. This thorough theoretical study, especially the electronic structure analysis, may offer very important clues for understanding and studying C–H bond activation by iron-based catalysts and enzymatic reactions in protein active pockets.
O2+ collides with benzene in the proper orientation. The detailed potential energy surfaces also indicate that the second radical mechanism, i.e., the benzene C–H bond activation through the rebound mechanism, is less favorable. This thorough theoretical study, especially the electronic structure analysis, may offer very important clues for understanding and studying C–H bond activation by iron-based catalysts and enzymatic reactions in protein active pockets.
1. Introduction
In nature, oxygen-activating enzymes are amazing substances which can effectively accomplish C–H hydroxylation under physiological conditions. Contrary to the facile functionalization of the C–H bond in nature, the effective C–H bond activation under mild conditions in catalytic chemistry has been a long-standing goal and challenge for human beings. Ever since the focused studies on iron(IV)-oxo complexes, which are assumed to be the key intermediates in the catalytic cycles of iron-containing metalloenzymes or model systems responsible for highly efficient and selective oxidation, high-valent iron chemistry has aroused widespread interest in both biological processes and laboratory researches.1–7 Extensive evidence has been accumulated by experimental methods as well as theoretical studies and the reaction mechanism for this kind of reaction has almost reached a consensus.1–3,8–41 Compared to the relatively simple reaction mechanism, i.e. rebound mechanism, for alkane hydroxylation by iron-oxo complexes which is characterized by an initial H-atom abstraction and then an OH− rebound process, aromatic hydroxylation still poses tantalizing questions concerning both the reaction mechanism and the toxicity of some of the metabolites.42There were three potential reaction mechanisms reported for the aromatic hydroxylation. The first reaction mechanism is generally thought to proceed via formation of arene oxides which rearrange nonenzymically to phenols by the classical NIH shift.43,44 However, as the experimental evidence accumulated, the arene oxide path has been questioned as a dominant metabolic path for the hydroxylation of several arenes.45,46 The second one is proposed to proceed through the generation of a tetrahedral radical or cationic σ-complex.47–49 Based on the density functional theory (DFT) calculations, the reactivity is an interplay of electrophilic and radical pathways, which involve an initial attack on the π-system of the benzene to produce a σ-complex.40 The third one is the direct H-atom abstraction of the C–H bond of arenes. This is essentially the same as the rebound mechanism found in the alkane hydroxylation by iron-oxo complexes. In sharp contrast to the alkane hydroxylation in which the H-atom abstraction is the critical step for the whole reaction, the rebound mechanism for aromatic hydroxylation has been ruled out on the basis of experimental and calculated results; a small kinetic isotope effect (KIE) value, for example.19,22,40,50,51 Besides the different general mechanisms of alkane hydroxylation, the aromatic hydroxylation presents new salient features. A proton-shuttle mechanism that reshuttles a proton from the non-innocent ligand to the oxo group competes with the nonenzymatic conversion of benzene oxide to phenol.40,51
Similar to the well-coordinated iron-oxo catalysts, studies on the gas phase reactions of bare transition metal ions and hydrocarbons have also been extensively conducted.19,35,52–64 These studies provided a wealth of insight concerning the intrinsic interactions between the active sites of catalysts and organic substrates. With respect to biological relevance, the gas phase C–H bond activations of small hydrocarbons by various transition metal-oxides were investigated in Schwarz's and Freiser’s laboratories twenty years ago.65 All these catalysts are totally exposed to their substrates and therefore many intrinsic interactions between the active site of catalysts and organic substrates may exist. A salient feature of this kind of reaction is characterized by a new reaction mechanism beyond the classical ones mentioned above, i.e., the direct and effective C–H bond hydroxylation can be completed through the concerted mechanism via a four-centered transition state. There are also some examples for arene hydroxylation by the bare iron-oxo catalysts. Experimentally, Schwarz and coworkers demonstrated that the gas phase reaction of bare FeO+ and benzene could generate diverse products such as Fe(C6H4)+/H2O, Fe(C5H6)+/CO, Fe(C5H5)+/CO/H, and Fe+/C6H5OH at collision rate of k = 1.3 × 10−9 cm3 molecule−1 s−1 and the major product of Fe+/C6H5OH is further confirmed by the neutralization-reionization (NR) experiment.46 Very recently, experimental combined with theoretical researches on vibrational spectroscopy of intermediates in benzene to phenol conversion by FeO+ have been conducted by Metz and co-workers.66 This prototypical arene oxidation reaction has also been thoroughly studied by pure theoretical methods. Yoshizawa and coworkers reported the reaction pathways and energetics for the conversion of benzene to phenol by FeO+ in the gas phase.19 In addition to the oxygen-insertion mechanism (electrophilic attack) and radical mechanism (rebound mechanism) existing in alkane hydroxylation by FeO+, there is a third mechanism which is named as the concerted mechanism (nonradical mechanism) via a four-centered transition state in the C–H bond activation. Furthermore, Kwapien and coworkers examined the substituent influence on the reaction of oxygen insertion into aromatic C–H bonds.58 They found that the insertion process could be facilitated by stabilizing the intermediate σ-complex.
FeO2+, first known to be formed from the reaction of Fe(II) with ozone and then proposed as the reactive species formed in the reaction of Fe(II) with hydrogen peroxide, is of importance for evaluating the role of iron in the chemistry of the aqueous atmosphere and as has biological relevance for the active center of heme and nonheme iron(IV)-oxo complexes, cytochrome P450 and bleomycin,3,4,29,67 for example. The kinetics of the reaction of bare FeO2+ with phenol, nitrobenzene and m-, o-, and p-nitrophenol have been investigated by the stopped-flow technique.68 Other detailed experimental and theoretical studies have also been contributed to the bare FeO2+-catalyzed reactions as well as the well-coordinated iron(IV)-oxo catalysts.3,4,56,63,67,69 However, among all these studies, no detailed theoretical research has been reported so far on the reaction of bare iron oxide di-cations with benzene despite its outstanding importance. Clearly, the catalysis of benzene hydroxylation by bare FeIVO2+ offers a rich and complex mechanistic puzzle. This reaction represents a paradigmatic, challenging test case for electronic structure approaches to transition metal catalysis. Quantum mechanical calculations can probe subtle mechanistic details and thereby complement the experimental information. Therefore, a thorough theoretical study on this reaction can qualitatively offer the simple concept that is required to elucidate the manifold reactivities of the transition metal-oxide cations. In this article, although we have not considered important ligands effects which often play an essential role in the reactivities of the transition metal complex, detailed electronic structure studies of this simple case can be especially useful in interpreting the reaction profiles and understanding the reactivity and selectivity of these systems and a thorough study on this fundamental reaction can provide important clues on the behavior of larger condensed-phased metal-oxo systems. In addition, the reactions of benzene C–H bond activation by FeOn+ cations with various ligands are also under investigation in our group.
2. Computational methods
All electronic structure calculations were performed with the ORCA program package developed by the Neese group, Bonn University.70 For geometry optimizations, the hybrid B3LYP density functional71,72 in combination with triple-ζ quality TZVP basis set73 were employed throughout the study. The RIJCOSX approximation74 was used to accelerate the calculations using the auxiliary basis set TZV/J. All the geometries were fully optimized without symmetry constraints and the convergence criteria was set to tight SCF convergence in order to obtain fully converged stationary points on the minimum energy pathways.70 For transition state (TS) searching, a relaxed surface scan calculation was performed first to obtan a good estimation of the TS structure and initial Hessian, and then TS optimizations were performed to look for first order saddle points. Harmonic vibrational frequencies were computed to verify the nature of the stationary points. The minimum structures reported in this paper possess only positive eigenvalues of the Hessian matrix, whereas the transition states (TSs) have only one negative eigenvalue. The zero-point energies, thermal corrections and entropy terms for the optimized geometries were obtained from frequency calculations. For some stationary points involved in the reaction processes, for example, 5RC_c, multiconfigurational SCF method complete active space self consistent field (CASSCF(12,9)) calculations were performed to confirm their electronic structures.Final energy refinement calculations were first performed with the hybrid B3LYP density functional using the new default basis sets of triple-ζ quality including high angular momentum polarization functions def2-TZVPP for all elements. CCSD(T) calculations for all reaction process were then employed at the previous B3LYP geometries using def2-TZVP basis sets.75 Both single energy calculations were accelerated by using the density fitting and chain of sphere (RIJCOSX) approximation. The T1 magnitudes, often taken as a diagnostic measure of multi-reference character, lie well within the range of 0.007–0.023, which indicates reliable CCSD(T) results. To take account into the role of solvent effect, the conductor-like screen model (COSMO) was used for all calculations and acetonitrile was chosen as the solvent.
3. Results and discussion
Due to the several accessible spin states for the metal-containing active site, the reaction of benzene with FeO2+ is explored on both the triplet and quintet states. Based on our calculations, the reaction pathways on the quintet state surface are energetically more favorable compared to those on the triplet state one. We therefore concentrate our electronic structure discussion mainly on the quintet state surface. In the present study we considered three reaction pathways indicated in Scheme 1: two radical mechanisms, which both involve a H-atom abstraction process, and an oxygen insertion mechanism. (1) The first radical mechanism is also considered as the concerted mechanism with the hydrogen-atom abstraction going through a four-centered transition state; (2) The second radical mechanism is actually the same as the well-accepted rebound mechanism for the C–H bond activation by heme and nonheme iron-oxo catalysts; (3) The oxygen insertion mechanism is essentially an electrophilic attack mechanism and leads to an arenium cation σ-complex intermediate. The remainder of this article is going to address these three mechanisms in detail.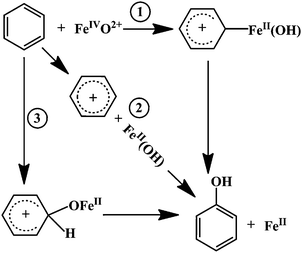 | ||
| Scheme 1 | ||
3.1 Radical mechanism–1
The first radical mechanism, i.e., the concerted mechanism is initiated by forming a η4benzene complex (RC_c) and then followed by a subsequent hydrogen-atom abstraction via a four-centered transition state (TSH_c). During this process, a Fe–C bond is concomitantly formed and finally results in the formation of a hydroxyl intermediate (I_c). The last step of the reaction is accomplished by dissociation of the Fe–C bond and formation of phenol complex P_c, Fe2+(C6H5OH). The potential energy surface of the first radical reaction pathways for the benzene C–H activation by FeO2+ in acetonitrile is shown in Fig. 1, while the corresponding geometry data for all the key point along the reaction pathway as well as the imaginary frequencies for the transition states are presented in Fig. 2. From DFT calculations, the quintet state is the ground state and the triplet state lies ∼8 kcal mol−1 above it. The computed activation energies of hydrogen-atom abstraction relative to the reaction pre-complexes for quintet and triplet states are 25.5 and 30.3 kcal mol−1, respectively. Compared with the higher gas phase energy barrier of the FeO+/benzene reaction,19 we propose a relatively higher reactivity of the title reaction. The main reason lies in the solvent effect which can reduce the field strength of the electric field surrounding a charged particle immersed in it. Therefore, the two positively charged FeO2+ complexes should have higher electrophilicity in the gas phase compared to that in the solvent environment and hence an even higher reactivity is expected in gas phase. In 5TSH_c, the FeO bond is elongated to 1.69 Å which is slightly longer than that of 1.61 Å in the equilibrium structure. The bond distances of C–H and O–H bonds involved in the reaction are calculated to be 1.30 Å and 1.34 Å respectively, which are reasonable for a transition state structure responsible for the cleavage of a C–H bond and the formation of an O–H bond. The newly formed Fe–C bond in the four-centered structure is calculated to be 2.14 Å, which clearly shows the interactions between the iron center and the carbon atom that is involved in the C–H bond cleavage. The computed geometry structures for the triplet transition state, 3TSH_c, closely resemble those found for the quintet state, only with slightly shorter FeO (1.66 Å) and Fe–C (2.11 Å) bonds. The rebound steps for triplet and quintet states encounter significantly different energy barriers. The quintet state has a comparably smooth energy barrier (3.7 kcal mol−1) while the rebound step for triplet state is high in energy, 13.7 kcal mol−1.
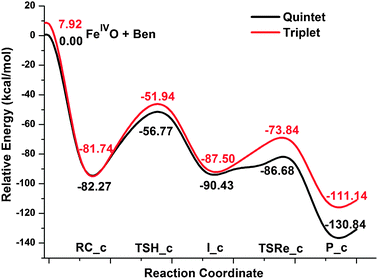 | ||
| Fig. 1 Schematic Gibbs free energy surface (ΔG/kcal mol−1) for benzene hydroxylation in acetonitrile along the radical mechanism (concerted mechanism) at the B3LYP theory level. | ||
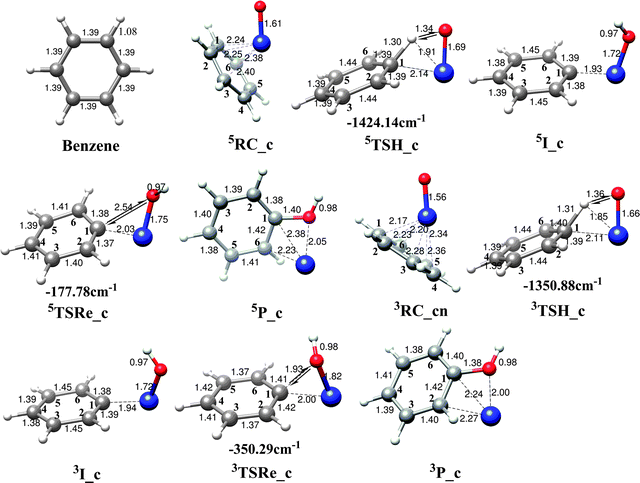 | ||
| Fig. 2 Optimized geometries of the isolated benzene, reactant complexes, transition states, intermediates and products for the benzene hydroxylation by FeO2+ in acetonitrile along the radical mechanism (concerted mechanism). Imaginary frequencies of transition states are shown by double arrows and values are also given. | ||
Fig. 3 shows the schematic frontier molecular orbital (FMO) of the critical points along the reaction pathways. Detailed electronic structure analysis may provide key clues for interpreting the different reactive behaviors of triplet and quintet state surfaces. As shown in Fig. 3, the electronic structure of 5RC_c is better described as the FeIVO center weakly bound to the benzene substrate. The four singly-α-occupied Fe-3d-based orbitals lead to the high spin (HS) iron center and indicate the formal Fe–O bond order of 2. The electronic structure of the reactant 5RC_c is also confirmed by the CASSCF(12,9) calculations. The electronic structure of 5TSH_c has changed greatly compared to 5RC_c and it is better described as the HS–Fe(II) center antiferromagnetically coupled to the oxyl radical. Clearly, two electrons have been transferred to the iron center during this process: (1) Significant electron transfer from the benzene molecule to the FeO2+ complex. Apparently, as the C–H bond of benzene gradual approaches the FeO unit center, the weakly bounded η4 mode species (5RC_c) is changed to the singly-coordinated complex and the newly formed Fe–C bond is mainly attributed to the C–H σ bond electron donation to the iron center. During this process, one β electron of the delocalized benzene π bond orbital is shifted to the Fe-3dxy-based orbital, leading to the doubly-occupied Fe dxy orbital and singly-occupied benzene π bond orbital; (2) The electron redistribution of the FeO center as the FeO bond elongates. The changes in the coordination environment happen concomitantly with the elongation of FeO bond. As already established by previous studies,19 the elongation of the FeO bond results in the electron redistribution of the active center. In this case, a broken symmetry solution with four singly-occupied Fe-3d-based MOs and a β-spin SOMO, which is essentially an Opz orbital, are formed. This process closely resembles the preparatory step found on the quintet state surface of hydrogen-atom abstraction by nonheme iron(IV)-oxo complexes.7,76 From 5TSH_c to 5I_c, one α-spin electron of the C–H bond is shifted to the oxyl radical, finally leading to the HS Fe(II) center antiferromagnetically coupled to the benzene cation radical. Contrary to the weakly bound alkyl radical R˙ and hydroxyl unit in the intermediate species of other studies,7,76,77 the benzene cation radical is strongly bound to the iron center, characterized by the large overlap (0.92) between the Fe dz2 and benzene cation SOMO orbital. The Fe–C bond length in 5I_c is 1.98 Å which also demonstrates the enhanced interaction of the benzene cation radical and iron center.
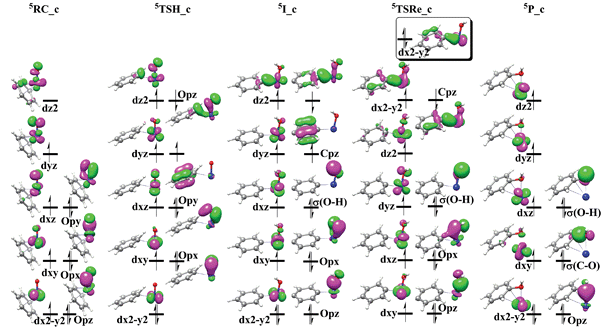 | ||
| Fig. 3 Schematic FMO diagram of 5RC_c, 5TSH_c, 5I_c, 5TSRe_c, 5P_c for the radical mechanism (concerted mechanism). | ||
To ascertain the nature of the intermediate, 5I_c, we recharacterized its electronic structure by using spin density analysis. The calculated positive spin density for the four carbon atoms at ortho and meta positions clearly indicates the first β electron transfer from the benzene ring to the iron center, while the negative spin density on the carbon atom (−0.3) which is directly involved in the C–H bond activation confirms the second α-electron transfer from the σC–H bond and therefore it can be viewed as the radical mechanism.
The last step of the reaction is responsible for the formation of the phenol complex via5TSRe_c, which is a transition state for the dissociation of a Fe–C bond and the formation of a C–O bond. As shown in Fig. 3, the electronic structure of 5TSRe_c is better interpreted as HS Fe(III) center antiferromagnetically coupled to the phenyl radical. Compared to the HS Fe(II) center in 5I_c, the electron evolution during the dissociation of the Fe–C bond is also very complicated. Two electron transfers may be involved in this process: (1) The β-spin electron of Fe dxy orbital is back-transferred to the delocalized π bond of the benzene cation radical, leading to the left α-spin electron of dxy orbital antiferromagnetically coupled to the phenyl radical; the slightly shortened CC bond lengths in benzene confirm the electronic structure analysis discussed above. (2) Close inspection of the strong spin-coupled pair orbitals (overlap of 0.96) indicates the second electron transfer in this process. Fig. 3 presents the localized spin-coupled pair orbitals (dx2−y2 orbital) in the box. Apparently, the doubly-occupied Fe dx2−y2 orbital and empty C p orbital indicates the electron flows from the phenyl radical to the iron center, leading to the HS Fe(II) center and benzene C+. Therefore, the C–O bond formation is better interpreted by the electrophilic attack of the phenyl C+ moiety towards the hydroxyl unit.
3.2 Radical mechanism–2
As we mentioned above, the second radical mechanism for the benzene to phenol conversion catalyzed by bare FeO2+ is actually the same as the well-accepted rebound mechanism for the C–H bond activation by heme and nonheme iron-oxo catalysts.3,4,76,77 This mechanism first requires a direct attack on the hydrogen atom of the benzene to form the iron-hydroxyl intermediate weakly coupled to the phenyl radical. The following step is finished by the recombination of these species and results in the final formation of alcohol. Fig. 4 shows the calculated potential energy surface diagram along the entire reaction coordinate in which both triplet and quintet states are considered. A new initial benzene complex, RC_r, that corresponds to a local minimum is situated as the starting point. The relative energy of this complex on triplet state is calculated to be 10.12 kcal mol−1 above that on the quintet state surface. Like the radical mechanism for the direct benzene to phenol conversion by the FeO+ catalyst which contains the spin crossing of two potential energy surfaces,19 the second radical mechanism might also start with a quintet state reactant and then proceed along the energetically low-lying triplet state reaction pathway. Except for these similarities, there are notable differences for the two catalysts on the quintet state surface. That is, the process catalyzed by the FeO+ catalyst can be partitioned into the first H-atom abstraction step and then the second rebound step, while the process for the FeO2+ catalyst stops immediately after the H-atom abstraction and cannot lead to the final phenol product. Fig. 5 shows the geometric structures of key points along the radical mechanism for both spin state surfaces and the corresponding imaginary frequencies for transition states. The key geometric parameters of the two transition states (3TSH_r and 5TSH_r) that correspond to the hydrogen-atom abstraction closely resemble those found in the π-mechanism of hydrogen-atom abstraction by various FeO2+–L based catalysts, that is, a nearly collinear C–H–O moiety and significantly bent Fe–O–H angle. The calculated O–H and C–H bonds which are going to be formed and broken have reasonable lengths for both spin states. On the quintet state surface, the reduced stretch of the O–H bond compared to that of C–H bond indicates a “late” transition state, while the comparable O–H and C–H bond lengths on triplet state case suggests a little earlier transition state compared to the quintet one. This is also in good agreement with the lower activation energy on the triplet surface. The geometric structures of the intermediates for these two spin state surfaces are dramatically different. The intermediate on the triplet state surface is very similar to that found in the hydrogen-atom abstraction step studied before, i.e., the newly formed iron-hydroxyl species weakly bound to a C6H5+hydrocarbon species which is ready for the following rebound step. However, the two species in 5P_r fall apart from each other and the benzene cation ring has been strongly deformed with two extremely short CC bonds (1.32 Å). The second step of the radical mechanism on the triplet state surface is responsible for the formation of the phenol complex via3TSRe_r, which has a relatively long C–O bond (1.97 Å). The activation energy for the radical rebound step is 4.21 kcal mol−1 which is in accordance with the earlier transition state geometry characters and the weak interaction between the carbon radical center and the O atom of the FeOH moiety.
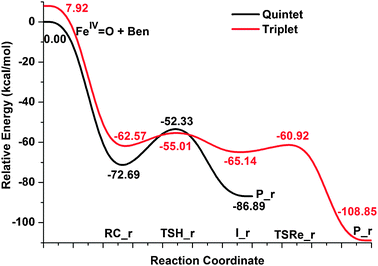 | ||
| Fig. 4 Schematic Gibbs free energy (ΔG/kcal mol−1) surface for benzene hydroxylation in acetonitrile along the radical mechanism (rebound mechanism) at the B3LYP theory level. | ||
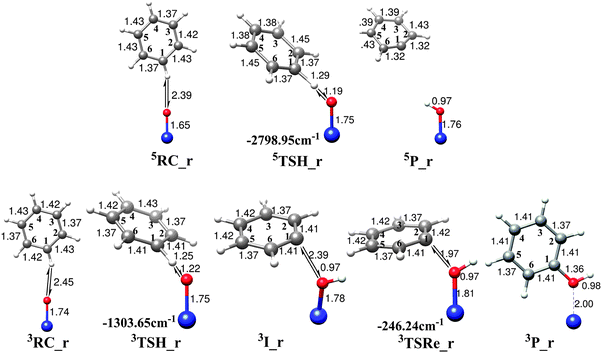 | ||
| Fig. 5 Optimized geometries of the reactant complexes, transition states, intermediates and products for the benzene hydroxylation by FeO2+ in acetonitrile along the radical mechanism (rebound mechanism). Imaginary frequencies of transition states are shown by arrows and values are also given. | ||
Now let us inspect the electronic structure evolution during the whole process. Previous theoretical studies demonstrated that the hydrogen-atom abstraction step on the quintet state surface can happen with either a σ-mechanism or π-mechanism, depending on the interaction of σC–H bond with O-based pz orbital or px/y orbital.8,77 However, in this mechanism, only one π-mechanism is established. The σ-mechanism is ruled out by the initially α occupied Fe based 3dz2 orbital. As shown in Fig. 6, the electronic structure of precomplex 5RC_r is best interpreted as a HS-Fe(III) ion (SFe = 5/2) antiferromagnetically coupled to a phenyl radical. The five single α-spin occupied Fe-based 3d orbitals make it impossible to accept an α electron from the C–H σ bond which is commonly found in the established σ-mechanism. Therefore, the title reaction follows the nonclassical π-mechanism in which an β electron shifts from the C–H σ bond to the low-lying π*xz/yz orbital.76 This mechanism also offers a good explanation for the key geometry parameters, that is, the opposite requirements of optimal orbital overlap and Pauli repulsion lead to bent geometries of 5TSH_r with a relatively small Fe–O–H angle close to 120°. As we mentioned above, the radical mechanism could also happen with the spin flip after the initial precomplex 5RC_r and lead to the crossing of the two potential energy surfaces. Fig. 6 clearly shows the spin inversion of the benzene cation radical on the two spin states. The one α and one β single electron left in the benzene ring in 5I_r makes them react with each other and finally results in the strong deformation of the benzene cation ring with two adjacent CC double bonds. However, the two β single electrons in the benzene cation ring of the triplet state intermediate has no chance to become paired and therefore the reaction could be continued with the radical character of substrate. The electronic structure of 3TSRe_r already demonstrates the benzene cation radical and the FeOH moiety which should be identified as the π mechanism for the rebound step.
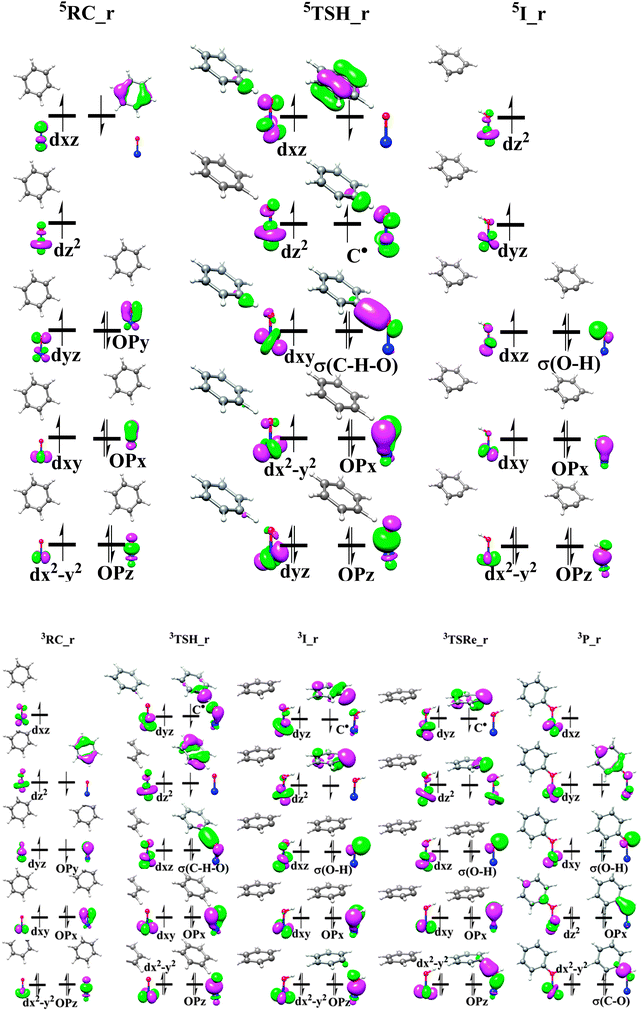 | ||
| Fig. 6 Schematic FMO diagram of quintet and triplet state in the radical mechanism. Top, quintet; Bottom, triplet. | ||
Spin density analysis is also consistent with the electronic structure discussed above. In 5TSH_r, the spin density of the carbon atom that is going to lose a H atom is 0.47, which implicates β-spin electron transfer during the C–H activation. The unchanged negative spin densities on the other C atoms compared to those in the initially formed precomplex indicate that the delocalized benzene cation radical is not involved in the reaction. The zero spin density in 5I_r can be appreciated by the interaction of the β-spin electron of the benzene ring with the α-spin electron generated by the hydrogen-atom abstraction which eventually leads to the distorted C6H5+ species. The total spin density of the benzene ring increases gradually from −1.36 in 3TSH_r to −2.0 in 3I_r which demonstrates that one α-spin electron is transferred during the C–H bond cleavage and more β-spin electrons are accumulated on the phenyl ring.
3.3 Oxygen insertion mechanism
The oxygen insertion mechanism is characteristic of a direct electrophilic attack of the oxygen atom of FeO2+ towards the carbon atom in the benzene ring, leading to formation of a σ-complex intermediate. Subsequently, this intermediate undergoes a rearrangement through a proton transfer transition state to reach the final phenol complex product, Fe2+(C6H5OH), which is the same as that found in the radical mechanism discussed in Section 3.1. Fig. 7 shows the potential energy surface for the oxygen insertion mechanism and Fig. 8 demonstrates the geometry structures of the key points along the reaction including the imaginary frequencies for transition states. The starting point 3RC_i for the triplet state is calculated to be ∼9 kcal mol−1 above that for the quintet state. Therefore, our discussion will mainly focus on the reaction happening on the quintet state surface, and the triplet case is ignored here. As shown in Fig. 7 and 8, first, a precomplex 5RC_i in which the benzene ring is loosely bound to the FeO2+ moiety is initially formed. It is about 10.48 kcal mol−1 above the precomplex (RC_c) in the first radical mechanism. Subsequently, the reaction proceeds through the transition state 5TSO_i to form the energetically low-lying σ-complex intermediate, 5I_i. The calculated energy barrier for this process is only 3.30 kcal mol−1, suggesting that it reacts very quickly. The lower activation energy barrier together with the strong exothermic nature of this process is in good agreement with the geometry character of the transition state. In 5TSO_i, the C–O bond that is going to be formed is calculated to be 2.23 Å, which is far away from the normal C–O bond length of 1.38 Å. Additionally, no significant changes are found for the FeO bond and the geometry parameters of benzene ring. To sum up, all these geometry characters indicate a reactant-like, very early transition state. Starting from 5I_i, the formation of phenol product involves a proton shift in which the hydrogen atom on the oxidized benzene carbon atom migrates as a proton to the oxygen atom. The transition state, 5TSP_i, which corresponds to the breaking of a C–H bond and the formation of an O–H bond is located. The potential energy surface for the proton transfer step is calculated to lie energetically below the first O-atom insertion step. Therefore, the oxygen insertion step is the rate-determining step for the whole oxygen insertion mechanism.
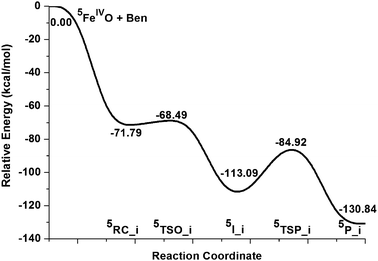 | ||
| Fig. 7 Schematic Gibbs free energy (ΔG/kcal mol−1) surface for benzene hydroxylation in acetonitrile along the oxygen insertion mechanism at the B3LYP theory level. | ||
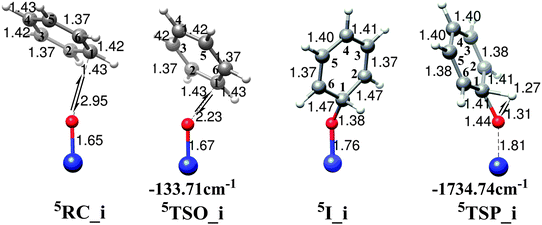 | ||
| Fig. 8 Optimized geometries of the reactant complexes, transition states, intermediates for the benzene hydroxylation by FeO2+ in acetonitrile along the oxygen insertion mechanism. Imaginary frequencies of transition states are shown by arrows and values are also given. | ||
Fig. 9 shows the schematic FMO diagram for the critical points along the reaction pathways in the oxygen insertion mechanism. Clearly, the electronic structure of starting point 5RC_i has the same electronic structures as that found in the precomplex of the second radical mechanism, i.e., a HS Fe(III) ion (SFe = 5/2) antiferromagnetically coupled to a phenyl radical. Compared to the electronic structure of separated FeO2+ (FeIVO2−) and benzene complexes, one electron reduction has occurred at the iron center. The unexpected electronic configuration is better attributed to the high electrophilicity of FeO2+ moiety, which holds two positive charges, and the low energy-lying delocalized benzene π bond. The very early transition state 5TSO_i possesses almost the same electronic structure as 5RC_i, which also has a HS iron center and an antiferromagnetically coupled phenyl radical. The small difference between them is the slightly reduced spin density on phenyl radical in 5TSO_i compared to that in 5RC_i, which indicates the slight second electron transfer from phenyl radical to the iron center. From 5TSO_i to 5I_i, the C–O bond formation leads to ferrous center and an arenium cation which demonstrates that the second electron transfer has been completed. The intermediate 5I_i can be viewed as a σ-complex and the formation of this arenium cation complex causes great deformation of the benzene cation ring, which is characterized by the two elongated C–C single bonds of 1.47 Å. Spin density and total charge analysis also support this view. During this process, the spin density of benzene cation ring has changed from 0.8 to 0 and correspondingly, the atomic charge on the para-position carbon atom is calculated to be +0.28.
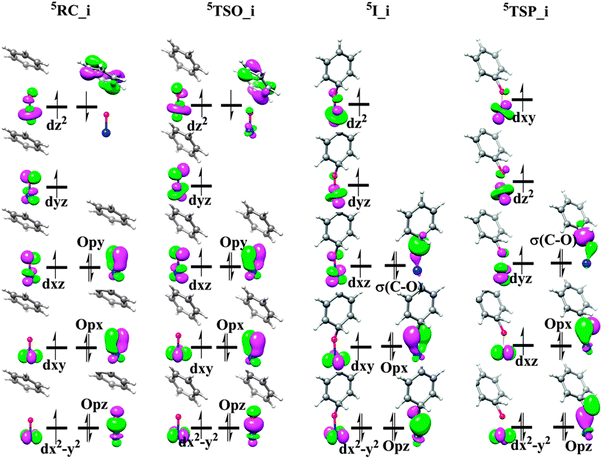 | ||
| Fig. 9 Schematic FMO diagram of 5RC_i, 5TSO_i, 5I_i, 5TSP_i, for the oxygen insertion mechanism. | ||
Starting from the σ-complex 5I_i, the second step of the oxygen insertion undergoes a proton transfer to accomplish the whole reaction process, leading to the phenol product weakly bounded to the Fe2+ ion. As shown in Fig. 7, the rearrangement of the σ-complex 5I_i to the phenol product is connected by a transition state 5TSP_i. In 5TSP_i, it already demonstrates the strong interaction of the proton and one doubly occupied O p-based orbital. As we already know, the proton transfer will break the C–H σ-bond and leave two electrons accumulated at the C atom end. Thereby, accompanying this proton transfer is the electron redistribution around the whole benzene ring. This is also consistent with the geometric changes in the benzene ring in which the slightly elongated C–C bonds in the benzene ring in 5I_i has been converted to the average C–C bonds of 1.40 Å in 5P_c.
3.4 Comparison of the reactivities in different mechanisms
To compare the reactivities of different mechanisms and clarify the most feasible reaction pathway for the benzene C–H bond activation by bare FeO2+, we recalculated the whole potential energy surface using 5RC_c as the reference species. At the B3LYP/def2-TZVPP level, the precomplex 5RC_c in the first radical mechanism (concerted mechanism) is the most stable and the corresponding species for the oxygen insertion and radical mechanisms are 10.48 and 9.58 kcal mol−1 higher, respectively. Among all three reaction mechanisms, the oxygen insertion pathway involves the lowest energy barrier and the other two reaction pathways have comparably similar energy barriers. Considering the small activation energy for the oxygen insertion mechanism, the collision of FeO2+ and benzene in a proper orientation can lead to the oxygen insertion process via5TSO_i. However, the relatively higher energies of precomplexes and transition states in the second radical mechanism (rebound mechanism) makes it the least possible case in the title reaction.
The CCSD(T) level energies based on B3LYP optimized geometries predict a similar reactivity trend for the title reaction. As shown in Table 1, the CCSD(T) results are also in favour of the oxygen insertion mechanism which is characterized by a relatively lower energy barrier. However, close inspection of the energies predicted by B3LYP and CCSD(T) method shows that the B3LYP functional seems to underestimate the energy barriers in both quintet and triplet spin state surfaces for all reaction mechanisms. This is in good agreement with the very recent research by Shaik in which they calibrated many density functional results with high-level ab initio coupled-cluster methods and suggested that the usage of B3LYP in theoretical studies where comparison between quintet and triplet reactivity is important and can be recommended.78
| Entry | ΔG(B3LYP) | ΔG(CCSD(T)) | Entry | ΔG(B3LYP) | ΔG(CCSD(T)) |
|---|---|---|---|---|---|
| 5RC_c | 0.00 | 0.00 | 5I_i | −30.82 | −39.5 |
| 5TSH_c | 25.50 | 39.02 | 5TSP_i | −2.65 | −0.91 |
| 5I_c | −8.15 | 2.01 | 5P_i | −48.57 | — |
| 5TSRe_c | −4.41 | 1.26 | 5RC_r | 9.58 | −8.41 |
| 5P_c | −48.57 | — | 5TSH_r | 29.95 | 27.94 |
| 3RC_c | 0.80 | 0.69 | 5P_r | −4.62 | −20.40 |
| 3TSH_c | 30.33 | 39.15 | 3RC_r | 19.97 | 3.80 |
| 3I_c | −5.23 | 2.75 | 3TSH_r | 27.26 | 22.89 |
| 3TSRe_c | 8.43 | 13.97 | 3I_r | 17.13 | 1.16 |
| 3P_c | −28.87 | — | 3TSRe_r | 21.35 | 20.82 |
| 5RC_i | 10.48 | — | 3P_r | −26.58 | — |
| 5TSO_i | 13.78 | 9.28 |
4. Conclusion
All possible mechanisms for the direct oxidation of benzene by bare FeO2+ catalyst have been discussed extensively through detailed geometric, energetic and electronic structure analyses. Three possible reaction pathways are explored: two radical mechanisms and one oxygen insertion mechanism. The most stable precomplex is formed via a η4 coordination environment for the first radical mechanism and the rate-determining step encounters an energy barrier as high as 25.50 kcal mol−1. The second radical mechanism follows the two-state reactivity pattern. The spin inversion can slightly lower the energy barrier in the radical mechanism and results in a comparable activation energy to that found in the first radical mechanism. The relatively very low activation energy barrier for the oxygen insertion mechanism makes it very competitive if the FeO2+ collides with benzene in the proper orientation. The CCSD(T) calculations predict a similar reactivity trend for these high-valent FeIVO quintet-triplet model systems and indicate the underestimation of the B3LYP reaction barriers.
Due to the two positive charges of the bare FeO2+ catalyst which make it highly electrophilic and the low energy-lying delocalized π bond in the benzene substrate, the title reaction is not experimentally feasible. However, this thorough theoretical study, especially the electronic structure analysis, may offer very important clues for studying the C–H bond activation by iron-based catalysts and enzymatic reactions in protein active pockets. In addition, research by ourselves and some other research groups on the iron-oxo catalysts with various kinds of ligands is under way based on this study. We would like to systematically explain from a computational perspective how the reactivity changes as a function of different ligands coordinated to the FeO2+ core center.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC No. 21073075, 21103064 and 21173097), Research Fund for the Doctoral Program of Higher Education of China (RFDP No. 20100061110046), the Special Funding of State Key Laboratory of Theoretical and Computational Chemistry, Jilin University and Basic Research Fund of Jilin University (No. 421010061439 and 450060445067).References
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS.
- M. Pitie and G. Pratviel, Chem. Rev., 2010, 110, 1018–1059 CrossRef CAS.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS.
- M. T. Green, Curr. Opin. Chem. Biol., 2009, 13, 84–88 CrossRef CAS.
- S. Shaik, S. P. de Visser, F. Ogliaro, H. Schwarz and D. Schroder, Curr. Opin. Chem. Biol., 2002, 6, 556–567 CrossRef CAS.
- S. Ye and F. Neese, Curr. Opin. Chem. Biol., 2009, 13, 89–98 CrossRef CAS.
- M. L. Neidig, A. Decker, O. W. Choroba, F. Huang, M. Kavana, G. R. Moran, J. B. Spencer and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12966–12973 CrossRef CAS.
- X. L. Sun, J. L. Li, X. R. Huang and C. C. Sun, Current Inorganic Chemistry, 2011 Search PubMed.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2010, 110, 575 CrossRef CAS.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS.
- C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS.
- J. Roithova and D. Schroder, Chem. Rev., 2010, 110, 1170–1211 CrossRef CAS.
- D. Schroder, J. Phys. Chem. A, 2008, 112, 13215–13224 CrossRef CAS.
- C. M. Bathelt, A. J. Mulholland and J. N. Harvey, J. Phys. Chem. A, 2008, 112, 13149–13156 CrossRef CAS.
- J. C. Hackett, T. T. Sanan and C. M. Hadad, Biochemistry, 2007, 46, 5924–5940 CrossRef CAS.
- Y. Shiota, K. Suzuki and K. Yoshizawa, Organometallics, 2005, 24, 3532–3538 CrossRef CAS.
- S. P. de Visser, Chem.–Eur. J., 2006, 12, 8168–8177 CrossRef CAS.
- S. P. de Visser, L. Tahsini and W. Nam, Chem.–Eur. J., 2009, 15, 5577–5587 CrossRef CAS.
- M. J. Kang, W. J. Song, A. R. Han, Y. S. Choi, H. G. Jang and W. Nam, J. Org. Chem., 2007, 72, 6301–6304 CrossRef CAS.
- S. P. de Visser, K. Oh, A. R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS.
- J. T. Groves and M. Vanderpuy, J. Am. Chem. Soc., 1976, 98, 5290–5297 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS.
- E. I. Solomon, S. D. Wong, L. V. Liu, A. Decker and M. S. Chow, Curr. Opin. Chem. Biol., 2009, 13, 99–113 CrossRef CAS.
- J. C. Schöneboom, S. Cohen, H. Lin, S. Shaik and W. Thiel, J. Am. Chem. Soc., 2004, 126, 4017–4034 CrossRef.
- H. Hirao, D. Kumar, W. Thiel and S. Shaik, J. Am. Chem. Soc., 2005, 127, 13007–13018 CrossRef CAS.
- H. Hirao, L. Que Jr., W. Nam and S. Shaik, Chem.–Eur. J., 2008, 14, 1740–1756 CrossRef CAS.
- D. Kumar, H. Hirao, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026–8027 CrossRef CAS.
- P. E. M. Siegbahn and T. Borowski, Acc. Chem. Res., 2006, 39, 729–738 CrossRef CAS.
- L. Bernasconi, M. J. Louwerse and E. J. Baerends, Eur. J. Inorg. Chem., 2007, 3023–3033 CrossRef CAS.
- C. Michel and E. J. Baerends, Inorg. Chem., 2009, 48, 3628–3638 CrossRef CAS.
- S. P. de Visser, J. Am. Chem. Soc., 2006, 128, 9813–9824 CrossRef CAS.
- S. P. de Visser, J. Am. Chem. Soc., 2006, 128, 15809–15818 CrossRef CAS.
- S. P. de Visser, L. Tahsini and W. Nam, Chem.–Eur. J., 2009, 15, 5577–5587 CrossRef CAS.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS.
- S. P. de Visser and S. Shaik, J. Am. Chem. Soc., 2003, 125, 7413–7424 CrossRef CAS.
- C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS.
- G. Guroff, J. W. Daly, D. M. Jerina, J. Renson, B. Witkop and S. Udenfrie, Science, 1967, 157, 1524–1530 CrossRef CAS.
- D. M. Jerina and J. W. Daly, Science, 1974, 185, 573–582 CAS.
- R. P. Hanzlik, K. Hogberg and C. M. Judson, Biochemistry, 1984, 23, 3048–3055 CrossRef.
- D. Schroder and H. Schwarz, Helv. Chim. Acta, 1992, 75, 1281–1287 CrossRef.
- T. Vannelli and A. B. Hooper, Biochemistry, 1995, 34, 11743–11749 CrossRef.
- K. R. Korzekwa, D. C. Swinney and W. F. Trager, Biochemistry, 1989, 28, 9019–9027 CrossRef CAS.
- L. T. Burka, T. M. Plucinski and T. L. Macdonald, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6680–6684 CAS.
- R. P. Hanzlik and K. H. J. Ling, J. Am. Chem. Soc., 1993, 115, 9363–9370 CrossRef CAS.
- S. P. de Visser, K. Oh, A. R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS.
- A. Fiedler, D. Schroder, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 1994, 116, 10734–10741 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, Chem.–Eur. J., 1997, 3, 1160–1169 CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, J. Am. Chem. Soc., 1998, 120, 564–572 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, Organometallics, 1998, 17, 2825–2831 CrossRef CAS.
- T. Yumura and K. Yoshizawa, Organometallics, 2001, 20, 1397–1407 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- K. Kwapien and E. Broclawik, Int. J. Quantum Chem., 2008, 108, 2016–2022 CrossRef CAS.
- Z. C. Liu, W. Y. Guo, L. M. Zhao and H. H. Shan, J. Phys. Chem. A, 2010, 114, 2701–2709 CrossRef CAS.
- G. Altinay, M. Citir and R. B. Metz, J. Phys. Chem. A, 2010, 114, 5104–5112 CrossRef CAS.
- D. Schroder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1973–1995 CrossRef.
- S. Malykhin, I. Zilberberg and G. M. Zhidomirov, Chem. Phys. Lett., 2005, 414, 434–437 CrossRef CAS.
- M. J. Louwerse and E. J. Baerends, Phys. Chem. Chem. Phys., 2007, 9, 156–166 RSC.
- H. J. Kulik and N. Marzari, J. Chem. Phys., 2008, 129, 134314 CrossRef.
- D. Schroder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1990, 29, 1433–1434 CrossRef.
- G. Altinay and R. B. Metz, J. Am. Soc. Mass Spectrom., 2010, 21, 750–757 CrossRef CAS.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS.
- D. O. Mártire, P. Caregnato, J. Furlong, P. Allegretti and M. C. Gonzalez, Int. J. Chem. Kinet., 2002, 34, 488–494 CrossRef CAS.
- M. J. Louwerse, P. Vadivelu and E. J. Baerends, J. Phys. Chem. A, 2008, 112, 1000–1012 CrossRef CAS.
- F. Neese, ORCA-an ab initio, density functional and semiempirical program package 2010, Version 2.8, Bonn University, Germany, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, M. Haser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- C. Y. Geng, S. F. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS.
- A. Decker, J. Rohde, E. J. Klinker, S. D. Wong, L. Que, Jr. and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 15983–15996 CrossRef CAS.
- H. Chen, W. Z. Lai and S. Shaik, J. Phys. Chem. Lett., 2010, 1, 1533–1540 Search PubMed.
| This journal is © the Owner Societies 2012 |