Sub-100 fs charge transfer in a novel donor–acceptor–donor triad organized in a smectic film†
T.
Roland
a,
J.
Léonard
a,
G.
Hernandez Ramirez
a,
S.
Méry
a,
O.
Yurchenko
b,
S.
Ludwigs
bc and
S.
Haacke
*a
aInstitut de Physique et Chimie des Matériaux de Strasbourg, Université de Strasbourg - CNRS, 67034 Strasbourg Cedex 2, France. E-mail: Stefan.Haacke@ipcms.u-strasbg.fr
bFreiburg Material Research Center and Freiburg Institute for Advanced Studies, University of Freiburg, 79104 Freiburg, Germany
cInstitute of Polymer Chemistry, University of Stuttgart, 70569 Stuttgart, Germany
First published on 9th November 2011
Abstract
Ultrafast transient absorption spectroscopy is performed on a novel donor–acceptor–donor triad made of two identical bisthiophene derivatives as electron donors and a central perylenediimide moiety as electron acceptor. The triad is extended at both ends by covalently bound siloxane chains that confer self-organisation into thin smectic films at ambient temperature. When diluted in chloroform, selective excitation of the donor moiety leads to resonance energy transfer within 130 fs to the acceptor moiety, followed by the formation of a charge transfer (CT) state in ∼3 ps. The CT state recombines entirely on a 55 ps time scale. In the liquid crystal films, excitonic intermolecular coupling leads to significant changes in the dynamics. Most remarkably, ultrafast intra- and intermolecular CT state formation occurs in about 60 fs, i.e. on a time scale comparable to electronic coherence times. While the intra-molecular CT states recombine on the same time scale as in solution or even faster, inter-molecular CT states live for about 1 ns. Last, triplet states of the perylenediimide moiety dominate the differential absorption after ∼1 ns. We anticipate that the fast recombination of intra-molecular CT states and the triplet state formation may severely limit the photo-current in these materials.
Introduction
Reduced production costs of organic photovoltaic devices are a major argument for research and development1,2 and the technology based on polymer–fullerene bulk heterojunctions (BHJ) is expected to achieve a relevant efficiency-to-cost ratio in the near future.3,4 In particular, the use of low bandgap polymers results in devices with improved absorption of solar energy at longer wavelengths, higher open-circuit voltages and consequently improved power conversion efficiencies,5,6 approaching 8%.7,8However, one major hindrance to higher power conversion efficiencies of polymer solar cells is intimately related to the three-dimensional morphology of the active layer. It has a critical influence on the charge carrier generation, free carrier mobility, connectivity to the electrodes and hence on the overall solar cell efficiency.9–13 In polymer blends, spontaneous phase separation offers a limited degree of control of the mesoscopic structuration of the material.
Although many research efforts are currently directed towards a better control of phase separation in blends, other alternatives include the control of the mesoscopic structuration into ordered heterojunctions.14 The construction of interdigitated nanostructures of donor and acceptor materials perpendicularly oriented with respect to the electrodes has been proposed as an ideal structure for the active layer in organic solar cells.15 One promising way to achieve such mesoscopic structures relies on the self-assembling of molecules made of covalently bonded donor and acceptor moieties, such as block-copolymers,16–18 or molecular dyads or triads.19–23 In contrast to BHJ materials, where excitons have to diffuse to an interface for efficient CT state formation, excitons are formed directly at the donor–acceptor interface of the (macro)molecule. The large tunability in chemical design allows for the fine tuning of the intramolecular properties of these compounds such as CT state formation efficiency, sometimes competing with resonant energy transfer, and charge recombination.24–30 Structuration may be achieved via the self-organization of donor–acceptor molecules into liquid crystalline (LC) films,20–23 recently showing power conversion efficiencies up to 1.5%.23 In addition, LC organization is shown to enhance significantly the charge carrier mobility.31–35 Static and dynamic disorders govern the charge mobility in organic semi-conductors.36 In LC's, the relatively large mobility results from inherent translational disorder37 which is thought to induce a dynamic self-repairing of stacking defects.
Here, we investigate the photophysical properties of a recently synthesized donor–acceptor–donor (DAD) triad that forms a smectic liquid crystalline film at ambient temperature.38,39 The molecule hereafter called DAD is sketched in Fig. 1a. Two bisthiophene derivatives (D) act as electron donors and a central perylenediimide derivative (A) behaves as the electron acceptor. Of essential relevance, the siloxane chains at both ends of the aromatic triad bring the LC properties, favoring the homo-stacking of the molecules, and the self-organization into alternating donor and acceptor lamellae38,39 (see Fig. 1b). Whereas oligothiophene and perylene-diimide moieties are widely used as electron donor and acceptor, respectively, to form dyads, triads or multiads,40–46 to our knowledge no investigation of the photophysical properties of a LC film showing distinct D and A homostacking has ever been reported. We perform transient absorption spectroscopy from femtoseconds to nanoseconds on this triad both in solution and in smectic films, and compare the results. The aim is to reveal the formation and lifetime of charge transfer states and the influence of molecular interactions in the LC film on these properties.
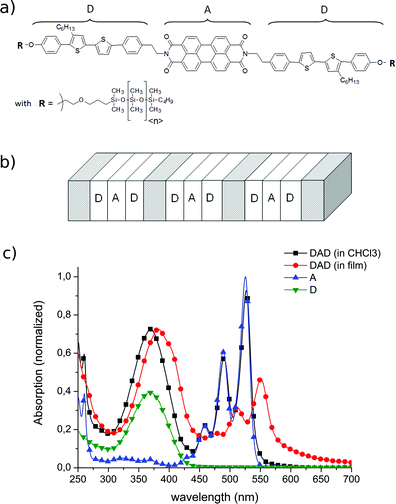 | ||
| Fig. 1 (a) Sketch of the chemical structure of the DAD compound. In the siloxane chains R, 〈n〉 = 8 on average. (b) Sketch of the ordered, bidimensional structuration in LC phase due to the distinct homostacking of the D and A moieties. Grey regions represent lamellae composed of siloxane chains responsible for the LC properties and molecular stacking. (c) Normalized absorption spectra of the two separate components of the triads (D and A in CHCl3), the full triad in solution (CHCl3) and in smectic films. All solutions are dissolved at 0.25 mmol L−1, allowing direct quantitative comparison. The absorbance of DAD in CHCl3 (black) is almost exactly the sum of the spectrum of A (blue), and twice that of D (green). In the film (red), inter-molecular excitonic coupling leads to red-shifts and broadening for both the D and A signatures, and to a relative decrease of the peak absorbance of A with respect to D. The pump pulses at 400 nm selectively excite the D moiety. | ||
Experimental section
The experimental pump–probe setup was previously described in detail.47,48 In short, the femtosecond laser source is a Ti:Sapphire regenerative amplifier laser system (Pulsar, Amplitude Technology) delivering 40 fs long pulses, at 5 kHz, centered at 800 nm. The beam is split into two. One beam is sent through a β-barium borate (BBO) crystal to produce a 400 nm pump beam. The second beam goes to a delay line and through a CaF2 crystal to generate a broad white-light continuum (300–1000 nm). The latter is split into two again: the probe that is sent through the sample, and the reference. Both are sent to a spectrometer and a CCD camera in order to measure the sample absorbance and the white-light power fluctuations respectively. Relative linear polarization between pump and probe beams is set at the magic angle (54.7°) to avoid any signal from anisotropy decay. By chopping the pump beam, the camera records alternatively the absorption spectrum of the unexcited and excited molecules at 220 Hz. This allows us to record differential absorption (ΔA) spectra as a function of the pump–probe time delay, controlled by the delay line. The Instrument Response Function (IRF) is ∼80 fs.47 The pump pulse power density is set to 0.2 mJ cm−2 within the regime of linear dependence of the ground state bleach signals in all experiments reported below. Data are processed to correct the effects of group velocity dispersion (chirp) in the white light continuum. The contributions of the solvent or substrate and the delay-independent background are also subtracted.47 All transient spectra presented in this work are obtained by appending three datasets covering three adjacent spectral windows, from 320 to 750 nm. After chirp correction, the zero time-delay is defined with an error bar of ±20 fs over the entire detection spectral window.For the experiments in solution, DAD is diluted in CHCl3 at 0.25 mmol L−1 and flown through a 0.5 mm quartz capillary thanks to a peristaltic pump. The DAD absorption spectrum is independent of concentration indicating no aggregation at 0.25 mmol L−1. The absorbance is 0.4 at 400 nm. For the experiments on films, these oscillate (∼0.5 Hz) perpendicular to the pump beam under an argon atmosphere to reduce photo-oxidation of the film. Films are obtained by spin coating an ∼300 μL di-chlorobenzene solution of DAD on a quartz substrate. They have an absorbance of 0.15 at 400 nm. The results reported below proved to be reproducible on three identical films prepared independently.
Absorption spectra of the radical anion A˙− and cation D˙+ of the isolated acceptor and donor moieties in solution are recorded as described in ref. 49 with a spectro-electro-chemical setup during electrochemical charging of the sample solution (10−3 M in dichloromethane/0.1 M tetra-n-butylammonium hexafluorophosphate solution, RT, 20 mV s−1). The electrochemical charging was carried out in a thin layer cell with a three-electrode arrangement. Potentials were calibrated against Fc/Fc+. In situUV/Vis spectra were recorded with a Zeiss spectrometer system in reflection mode using the Pt working electrode as mirror.
Results
Steady-state results
Static absorption spectra of D and A in solution (CHCl3) and DAD both in solution and film are displayed in Fig. 1c. D is characterized by a broad absorption band from 300 nm to 425 nm and A by a three-peak absorption structure at 460 nm, 490 nm and 525 nm. It appears that the solution spectrum of DAD (black) is almost the exact sum of the spectrum of A (blue) and twice the spectrum of D (green). This indicates negligible intra-molecular excitonic coupling between D and A in solution. The spectrum of DAD in films (red) mainly shows the same characteristics as in solution, but with a relatively weaker absorption from A, a red shift of both D and A by ∼0.10 eV, and a prominent low-energy shoulder in the A band (575–675 nm). These features indicate significant inter-molecular, excitonic coupling between neighbouring DAD's,50,51 as commonly observed for organic semiconductor thin films. Both in films and in solution, the contribution of D largely dominates the absorption spectra at 400 nm, and hence the pump pulses selectively excite D.DAD in CHCl3
Pump–probe experiments on D and A alone allow us to identify the differential absorption spectra of their excited states A* and D*.52 While in the isolated donor moiety, D* decays on a 220 ps time scale due to inter-system crossing, we will show below that in DAD the excited state D*AD is shortened by energy or charge transfer to A.Selected ΔAspectra for DAD in solution are shown in Fig. 2, at short and long time delays, together with spectra of the relevant species. In Fig. 2a, the zero delay spectrum shows D ground state bleach (negative ΔA) at wavelengths <400 nm, and D* excited state absorption (ESA) at wavelengths >550 nm.52 The 93 fs spectrum (blue) shows increase of the bleach of A with negative peaks at 460, 490 and 525 nm. For longer delays, 240 and 540 fs, the bleach of D below 400 nm partially recovers. Simultaneously, stimulated emission (SE) of A appears, characterized by a new negative peak at 575 nm and an increase of the negative amplitude at 530 nm. The latter is due to the unresolved overlap of the ground state bleach at 525 nm and the SE peak at 535 nm (see Fig. 2c). In addition, the ESA maximum at 705 nm is due to A* as already observed in similar perylene-diimide derivatives.24,53
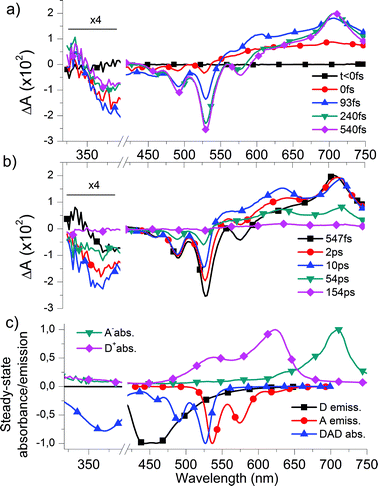 | ||
| Fig. 2 Temporal evolution of the differential absorption (ΔA) spectra of DAD in CHCl3 (a) before 0.5 ps, evidencing RET from D to A, and (b) at longer time delays, showing CT state formation and recombination. Data below 390 nm are magnified by a factor 4. Data from 390 to 420 nm are disregarded, due to pump–pulse related noise. The vertical dashed lines indicate merging of two separate data sets. (c) Normalized absorption spectra of the radical anion A˙− (green) and radical cation D˙+ (pink) in dichloromethane allowing us to identify the signature of the CT state at delay times >10 ps, and normalized absorption spectrum of DAD (blue), and emission spectra of D (black) and A (red) in CHCl3. The latter three are plotted upside-down to facilitate the comparison with the above differential spectra and identify the signatures of the ground state bleach of D and A as well as the SE from A*. | ||
Taken together, these observations give evidence for resonance energy transfer (RET) from D to A: D*AD → DA*D on a 110 fs time scale (see quantitative analysis below). This is not surprising given the large spectral overlap of the fluorescence spectrum of D and the absorption spectrum of A (cf.Fig. 2c).
At longer times (Fig. 2b), ΔAspectra show the decrease of the SE from A* (negative peaks at 530 and 575 nm), while the ground state bleach of A remains constant (negative peaks at 460 and 490 nm). On the same time scale, the apparent redshift of the ESA peak from 705 nm to 712 nm should be interpreted as the ESA signature of A* decaying and giving place to a new positive peak at 712 nm. Simultaneously a second new positive peak appears at 630 nm. Both absorption features are similar to the absorption spectra of the (electrochemically) reduced state of the isolated A moiety (at −1.32 V vs.Fc/Fc+) and oxidized state of the isolated D moiety (at 0.76 V vs.Fc/Fc+), respectively, showing absorption maxima at 709 nm and 623 nm in dichloromethane (Fig. 2c). This indicates the formation of an intra-molecular CT state within a few ps: DA*D → D+A−D. Note that the bleach of D (<400 nm) again increases during the formation of the CT state. Finally, after 154 ps the system has almost completely recovered, due to intra-molecular (geminate) recombination of the CT state.
DAD in films
Representative ΔAspectra for DAD in films are shown in Fig. 3. The top part shows that the bleach of D (at wavelengths <400 nm) is instantaneous and stays at its maximum, in contrast to the sub-ps dynamics observed in solution (Fig. 2a). A weak SE from D* is observed in the 40 fs spectrum (blue) at 420–480 nm. At the same delay, the long wavelength portion is attributed to ESA from D*, characterized by weakly modulated maxima at 620 nm at 710 nm, similar to the 0 fs spectrum in solution (Fig. 2a).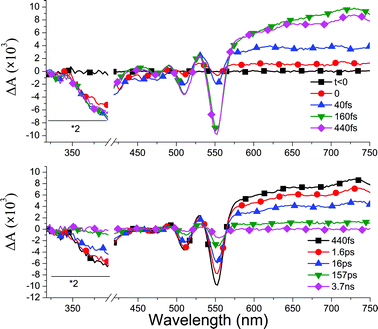 | ||
| Fig. 3 Top: ΔAspectra at selected short times, displaying the crossover from D*AD to D+A−D. Bottom: ΔAspectra at longer delay times. The induced absorption observed at longer wavelengths is attributed to D+A−D. Intra- and intermolecular CT states recombine on different time scales (see text). The incomplete bleach recovery of A (500–560 nm) is due to triplet formation. | ||
Within the next few hundred fs, these two maxima distinctively shift to 640 nm and 730 nm. Simultaneously, the ground state bleach of A, characterized by the negative peaks at 475 nm, 510 nm and 550 nm, rises without any indication for SE of A, unlike in solution. We attribute this spectral evolution to the formation of charge transfer states. Indeed the two weak bands at 640 nm and 730 nm are close to the signatures of D˙+ and A˙− in solution (Fig. 2c). They are broader most probably due to the smaller dielectric constant in the films and possibly larger inhomogeneous broadening. The slight red-shift, with respect to the solution absorption maxima may be due to inter-molecular excitonic coupling, as observed in the steady-state absorption of the film. At longer times (Fig. 3 bottom) we essentially see the decay of the D+A−D CT state. It is slower than in solution, as there is still a significant signal at 157 ps whereas it is almost vanishing at 154 ps in solution (Fig. 2b). Noticeably however, even after 3.7 ns we still observe the bleach of A, and a weak positive ΔA in the 480–600 nm range. This is best seen in Fig. 4, where we show the stationary ΔA signal observed after 2 ns, the ground state bleach of A, and the difference between both. The latter spectrum, as well as ΔA signal are both in good agreement with data published for the triplet absorption of perylene-diimide compounds.54,55 Note that at earlier times, the positive ΔA at 530 nm is most likely not a signature of TA, but rather of D+A−D, since its decay parallels the one of D+A−D at longer wavelengths.
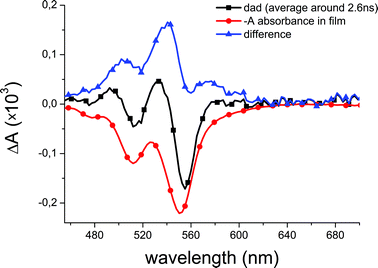 | ||
| Fig. 4 Evidence for formation of the triplet state in A: the ΔAspectrum of DAD in film after 2 ns (black), the absorption spectrum of A in film (red, appropriately scaled), and the difference between both (blue). Comparison with literature data indicates that this long-delay time ΔA is due to triplet formation in the perylene-diimide moiety. | ||
Selected kinetic traces of the transient absorption of DAD in film give a more precise insight on the CT state formation time scale (Fig. 5). The upper trace is the transient signal at 448 ± 5 nm, where the fluorescence of D is maximum. The early negative ΔA is therefore due to stimulated emission from D*. Its duration is shorter than 100 fs, before a transient positive absorption with remarkable non-exponential decay features and oscillations replaces it. Vibrational and dielectric relaxations reduce this absorptive feature and leave the way for a weak bleach signal (A ground state). The 552 nm transient reveals the bleach of A. Its fitting analysis yields a ∼60 fs rise time, which thus corresponds to the formation time of the CT state. Instead the 640 and 730 nm transients show an instantaneous (i.e. IRF-limited) rise, which corroborates our previous assignment of this spectral range to the ESA from D* at early times. At longer times, these transients characterize the CT state as discussed previously. For delays longer than ∼0.5 ps, the kinetic traces associated to A (552 nm), D+ (640 nm), and A− (730 nm) decay monotonously as a consequence of CT recombination. However, while both transient signals at 640 and 730 nm return to zero after 1 ns, the ground state bleach of A (552 nm) does not because of the formation of TA.
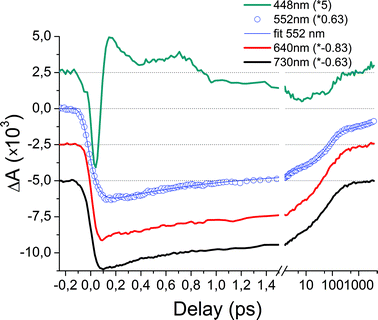 | ||
| Fig. 5 Selected kinetic traces of DAD films. All traces have been scaled (see factors in the graph) and shifted to allow easy comparison. The time scale is linear up to 1.5 ps and then logarithmic. The 730 and 640 nm traces initially characterize the ESA signal from D* and after less than 100 fs the absorption of A− and D+, respectively. The 552 nm trace corresponds to the bleach of A, and the early negative peak of the 448 nm trace is mainly associated with SE from D*. The bleach signal of A (552 nm) rises on a 60 fs time scale which correspond to the lifetime of the SE from D*. After ∼0.5 ps, the bleach recovery of A matches the decay of A− and D+ absorption signals (730 nm and 640 nm), except after a few hundreds ps where the triplet state signature is observed at 552 nm. | ||
Last, we note that the kinetics do not depend on thermal annealing (from 0 up to 24 h, at 150 °C). Since annealing is shown to increase the liquid crystal domain sizes,38,39 this suggests that the charge transfer states are localized on a length scale smaller than the typical domain size.
Solution versus film comparison
The differences between the photophysical processes of DAD in solution and in film are best illustrated by comparing the kinetic traces associated to the ground state bleach of the D moiety in both cases, as displayed in Fig. 6. In solution (black) a partial recovery of D is observed in the first 500 fs, due to resonance energy transfer. This process is absent in the kinetic trace recorded on films (red). While in the latter CT formation occurs on a 60 fs time scale, in solution it takes a few ps, as indicated by the progressive second increase of the D bleach signal. CT recombination is completed within 200 ps in solution, while the full recovery of the D ground state is observed in films for delays >1 ns only.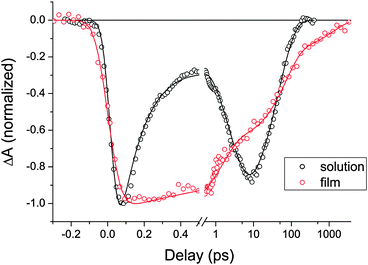 | ||
| Fig. 6 Transient signals observed at 370 ± 20 nm for DAD in film (red circles), in CHCl3 (black circles) and their respective fits by the function given in eqn (1). The scale is linear up to 0.5 ps and logarithmic thereafter. The traces are normalized to the same initial amplitude. In solution, the bleach of D partially recovers after a few hundred fs due to resonance energy transfer, and rises again on a 2.7 ps time scale when the CT state forms. In film instead, the sub-100 fs CT state formation from D* leaves the ground state bleach signal unaffected on short times. Only charge recombination is responsible for the bleach signal recovery on longer time scales. | ||
Data fitting is carried out with a multiple exponential decay convolved with a Gaussian function modeling the IRF, as shown in eqn (1):
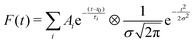 | (1) |
| Solution | Film | |||
|---|---|---|---|---|
| τ | τ | D bleach | A bleach | |
| RET | 130(20) fs | — | — | — |
| CT state formation | 2.7(2) ps | 60(20) fs | — | −100% |
| Rec 1 | — | 1.1(2) ps | 36% | 37% |
| Rec 2 | 55(8) ps | 70(10) ps | 46% | 40% |
| Rec 3 | — | 1.3(3) ns | 18% | 10% |
| Triplet | — | >5 ns | — | 13% |
The fit analysis of the D bleach transient signal in film yields three decay times: 1.1 ps, 70 ps and 1.3 ns, with relative amplitudes reported in Table 1. Analysis of the kinetic traces at 640 nm and 730 nm pertaining to the decay of D+A−D yields the same values, within the experimental uncertainties given in Table 1. Therefore we assign all three time constants to recombination processes of the CT state. Interestingly, as is apparent from Fig. 5, the ground state bleach of A is longer lived. The fit of the 552 nm trace yields the same three time constants and a longer >5 ns component too slow to be accurately determined here. As discussed above, the difference with the D bleach recovery and this long-lived component is attributed to the triplet formation in the A moiety.
Discussion
Remarkable differences are observed between the ultrafast photophysical processes of DAD in liquid crystalline films and in solution. In the latter, RET occurs on a 130 fs time scale and precedes the formation of the CT state on a ∼3 ps time scale. Hence, the charge transfer mechanism is an electron transfer from the HOMO of D to that of A, more stable by ∼1.1 eV in solution.38,39 This is in contrast to the film, where the CT state formation results from an electron transfer taking place on a 60 fs time scale directly from the LUMO of D, to the LUMO of A, more stable by ∼1.5 eV in film.38,39In the film, tight molecular packing may result in geometrical constraints on D and A moieties enhancing the efficiency and speed of the electron transfer from D*, which outperforms the RET process observed in solution. However, we think that in the film excitonic coherence properties must play an additional role in accelerating the charge transfer. Indeed, as compared to the case of DAD in solution, the ground state absorption spectrum of DAD in film indicates significant intermolecular excitonic coupling, induced by the short intra-molecular distances and pairwise side-by-side alignment of D's and A's.38,39 This means that the excitation is delocalized over a few neighboring D's. Remarkably, the 60 fs electron transfer process observed here is as fast or even faster than the typical electronic coherence times observed in conjugated polymers or biomolecular aggregates.56–59 Hence, the electron transfer occurs before the excitonic wavefunction localizes on one individual D, leading to the formation of a CT state itself delocalized over neighboring DAD molecules. The electronic coherence offers several parallel channels for CT state formation, leading to a faster and more efficient formation of delocalized CT states. Since the binding energies of CT states are smaller than those of excitons, the electronic coherence of the former is more sensitive to environment fluctuations and is expected to be shorter-lived. As a result, the delocalized CT states are expected to form immediately intra- and intermolecular localized CT states. Note that intermolecular CT states involve side-by-side aligned DAD moieties, i.e. they are almost perpendicular to the siloxane chains.
The thin film data show that about 80% of CT state recombination occurs within 70 ps, as for the isolated DAD molecules in solution, indicating that most CT states remain intramolecular in the film. Conversion of intra- to intermolecular CT states may occur, but as both species cannot be disentangled spectroscopically in the present study, this process—if present—goes unnoticed. As the longer 1.3 ns decay component is not observed in solution, we attribute it to the lifetime of minority inter-molecular CT states. The shorter 1.1 ps bleach and D+A−D decay component observed in the film are not present in solution. It may be attributed to a fast intra-molecular charge recombination channel which would result from the same geometrical constraints allowing an ultrafast CT state formation. Alternatively, it may be due to bimolecular recombination of CT states.60 Further experiments performed at even lower excitation densities are needed to ascertain this second hypothesis.
Finally, the >5 ns component observed in the film characterizes the bleach recovery of A, but not that of D. We showed above that this is the signature of charge recombination process leading to the formation of neutral D and neutral TA. The formation kinetics of TA is not readily extracted from the data since there is no specific time scale associated to it. However, since the 70 ps and 1.3 ns time scales have a smaller amplitude in the bleach recovery of A as compared to D, the formation of TA most likely occurs on an intermediate time scale ranging from ∼100 ps to ∼1 ns.
In the light of the above, we conclude that two main factors limit the use of these DAD triads for photovoltaic devices. Even when auto-organized in smectic liquid crystals, the formation of inter-molecular CT states remains of secondary importance. Significant intra-molecular CT states recombination occurs on a 1 ps time scale, that is before significant inter-chain diffusion of charges takes place. As already pointed out for other material systems,61 another main limitation comes from the CT relaxation into triplet states, here localized on the perylene-diimide acceptor moiety.
Conclusion
We studied the ultrafast photophysics of a novel DAD molecule both in solution and in a smectic liquid crystalline thin film. The pronounced intermolecular coupling in the LC phase leads to formation of both intra- and intermolecular CT states in less than 100 fs. This ultrafast charge transfer outperforms resonance energy transfer, which was in contrast found to be the primary process for isolated DAD's in solution. Recombination of intramolecular CT states occurs on a 60 ps time scale both in film and solution. In addition, the film shows two other recombination processes. A faster one occurs on a ∼1 ps time scale and corresponds to an unclear mechanism (geminate intramolecular or bimolecular). A slower one occurs on a 1 ns time scale and is associated to the recombination of a minority population (20%) of intermolecular CT states. Their lifetime seems to be limited by the formation of triplet states of the acceptor moiety. We anticipate that the very efficient CT state quenching will seriously limit the short-circuit current and the open circuit voltage of devices made from these materials.62,63 Future work and chemical design of liquid crystal forming DAD's will aim at reducing the intra-molecular CT recombination rate and triplet state formation.Acknowledgements
The authors are grateful to T. Heiser and N. Leclerc for many insightful discussions, and thank O. Crégut, G. Dekyndt, J.-P. Vola and D. Freytag for expert technical assistance. We acknowledge J. Heinze for letting us use his spectrochemical setup. Funding came from CNRS and the University of Strasbourg. T.R. holds a joint PhD grant from CNRS and “Région Alsace”.References
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS.
- A. L. Roes, E. A. Alsema, K. Blok and M. K. Patel, Progr. Photovolt.: Res. Appl., 2009, 17, 372–393 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- L. Huo, J. Hou, S. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., 2010, 122, 1542–1545 CrossRef.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS.
- S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841–843 CrossRef CAS.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- A. Geiser, B. Fan, H. Benmansour, F. Castro, J. Heier, B. Keller, K. E. Mayerhofer, F. Nüesch and R. Hany, Sol. Energy Mater. Sol. Cells, 2008, 92, 464–473 CrossRef CAS.
- M. M. Oosterhout, S. D. Wienk, S. S. van Bavel, R. Thiedmann, L. Jan Anton Koster, J. Gilot, J. Loos, V. Schmidt and R. A. J. Janssen, Nat. Mater., 2009, 8, 818–824 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS.
- K. Tajima and K. Hashimoto, Chem. Rec., 2011, 11, 8–17 CrossRef CAS.
- K. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- S. Barrau, T. Heiser, F. Richard, C. Brochon, C. Ngov, K. van de Wetering, G. Hadziioannou, D. V. Anokhin and D. A. Ivanov, Macromolecules, 2008, 41, 2701–2710 CrossRef CAS.
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS.
- M. Sommer, S. Huettner and M. Thelakkat, J. Mater. Chem., 2010, 20, 10788–10797 RSC.
- A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2002, 15, 3353–3361 CrossRef.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H.-W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 417, 384–387 CrossRef.
- T. Nishizawa, K. Tajima and K. Hashimoto, J. Mater. Chem., 2007, 17, 2440–2445 RSC.
- R. Deschenaux, B. Donnio and D. Guillon, New J. Chem., 2007, 31, 1064–1073 RSC.
- L. Bu, X. Guo, B. Yu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, J. Am. Chem. Soc., 2009, 131, 13242–13243 CrossRef CAS.
- E. Fron, M. Lor, R. Pilot, G. Schweitzer, H. Dincalp, S. D. Feyter, J. Cremer, P. Bäuerle, K. Mullen, M. V. der Auweraer and F. C. D. Schryver, Photochem. Photobiol. Sci., 2005, 4, 61–68 CAS.
- A. M. Ramos, E. H. A. Beckers, T. Offermans, S. C. J. Mesker and R. A. J. Janssen, J. Phys. Chem. A, 2004, 108, 8201–8211 CrossRef CAS.
- P. A. van Hal, R. A. J. Janssen, G. Lanzani, G. Cerullo, M. Zavelani-Rossi and S. D. Silvestri, Chem. Phys. Lett., 2001, 345, 33–38 CrossRef CAS.
- J.-F. Nierengarten, Sol. Energy Mater. Sol. Cells, 2004, 83, 187–199 CrossRef CAS.
- P. A. van Hal, S. C. J. Meskers and R. A. J. Janssen, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 41–46 CrossRef CAS.
- B. P. Karsten, R. K. M. Bouwer, J. C. Hummelen, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. B, 2010, 114, 14149–14156 CrossRef CAS.
- A. Gégout, J.-F. Nierengarten, B. Delavaux-Nicot, C. Duhayon, A. Saquet, A. Listorti, A. Belbakra, C. Chiorboli and N. Armaroli, Chem.–Eur. J., 2009, 15, 8825–8833 CrossRef.
- Y. Shimizu, K. Oikawa, K. Nakayama and D. Guillon, J. Mater. Chem., 2007, 17, 4223–4229 RSC.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- M. Funahashi, Polym. J., 2009, 41, 459–469 CrossRef CAS.
- W. Pisula, M. Zorn, J. Y. Chang, K. Müllen and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1179–1202 CrossRef CAS.
- M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef CAS.
- D. P. McMahon and A. Troisi, ChemPhysChem, 2010, 11, 2067–2074 CrossRef CAS.
- Y. Olivier, L. Muccioli, V. Lemaur, Y. H. Geerts, C. Zannoni and J. Cornil, J. Phys. Chem. B, 2009, 113, 14102–14111 CrossRef CAS.
- G. Hernandez Ramirez, PhD thesis, Université de Strasbourg, 2010, http://tel.archives-ouvertes.fr/tel-00579777/fr/ Search PubMed.
- G. Hernandez Ramirez, B. Heinrich, C. Bourgogne and S. Méry, to be published.
- A. Petrella, J. Cremer, L. D. Cola, P. Bäuerle and R. M. Williams, J. Phys. Chem. A, 2005, 109, 11687–11695 CrossRef CAS.
- J. Cremer, E. Mena-Osteritz, N. G. Pschierer, K. Müllen and P. Bäuerle, Org. Biomol. Chem., 2005, 3, 985–995 CAS.
- L. X. Chen, S. Xiao and L. Yu, J. Phys. Chem. B, 2006, 110, 11730–11738 CrossRef CAS.
- C.-C. You, P. Espindola, C. Hippius, J. Heinze and F. Würthner, Adv. Funct. Mater., 2007, 17, 3764–3772 CrossRef CAS.
- M. K. R. Fischer, T. E. Kaiser, F. Würthner and P. Bäuerle, J. Mater. Chem., 2009, 19, 1129–1141 RSC.
- H. Wonneberger, C.-Q. Ma, M. A. Gatys, C. Li, P. Bäuerle and K. Müllen, J. Phys. Chem. B, 2010, 114, 14343–14347 CrossRef CAS.
- G. Balaji, T. S. Kale, A. Keerthi, A. M. D. Pelle, S. Thayumanavan and S. Valiyaveettil, Org. Lett., 2011, 13, 18–21 CrossRef CAS.
- J. Briand, O. Bräm, J. Réhault, J. Léonard, A. Cannizzo, M. Chergui, V. Zanirato, M. Olivucci, J. Helbing and S. Haacke, Phys. Chem. Chem. Phys., 2010, 12, 3178–3187 RSC.
- A. Sinicropi, E. Martin, M. Ryazantsev, J. Helbingd, J. Briand, D. Sharma, J. Léonard, S. Haacke, A. Cannizzo, M. Chergui, V. Zanirato, S. Fusi, F. Santoro, R. Basosi, N. Ferre and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17642–17647 CrossRef CAS.
- C. Geskes and J. Heinze, J. Electroanal. Chem., 1996, 418, 167–173 CrossRef CAS.
- M. Kasha, H. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- R. Stomphorst, T. Schaafsma and G. van der Zwan, J. Phys. Chem. A, 2001, 105, 4226–4234 CrossRef CAS.
- T. Roland, G. Hernandez Ramirez, J. Léonard, S. Méry and S. Haacke, J. Phys.: Conf. Ser., 2011, 276, 012006 CrossRef.
- Y. Meyer and P. Plaza, Chem. Phys., 1995, 200, 235–243 CrossRef CAS.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- T. Kircher and H.-G. Löhmannsröben, Phys. Chem. Chem. Phys., 1999, 1, 3987–3992 RSC.
- E. Collini and G. D. Scholes, Science, 2009, 323, 369–373 CrossRef CAS.
- Q.-H. Xu, G. D. Scholes, M. Yang and G. R. Fleming, J. Phys. Chem., 1999, 103, 10348–10358 CAS.
- T. Brixner, J. Stenger, H. M. Vaswani, M. Cho, R. E. Blankenship and G. R. Fleming, Nature, 2005, 434, 625–628 CrossRef CAS.
- G. S. Engel, T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mančal, Y.-C. Cheng, R. E. Blankenship and G. R. Fleming, Nature, 2007, 446, 782–786 CrossRef CAS.
- V. Gulbinas, I. Minevičiūtė, D. Hertel, R. Wellander, A. Yartsev and V. Sundström, J. Chem. Phys., 2007, 127, 144907 CrossRef CAS.
- D. Veldman, O. Ipek, S. C. J. Meskers, J. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kroon, S. S. van Bavel, J. Loos and R. A. J. Janssen, J. Phys. Chem. A, 2008, 130, 7721–7735 CAS.
- C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef CAS.
- M. Hallermann, E. D. Como, J. Feldmann, M. Izquierdo, S. Filippone, N. Martin, S. Juchter and E. von Hauff, Appl. Phys. Lett., 2010, 97, 023301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cp22122a |
| This journal is © the Owner Societies 2012 |