A multiscale description of molecular adsorption on gold nanoparticles by nonlinear optical spectroscopy†
C.
Humbert
*a,
O.
Pluchery
b,
E.
Lacaze
b,
A.
Tadjeddine
a and
B.
Busson
a
aUniv Paris-Sud, Laboratoire de Chimie Physique, CNRS, Bâtiment 201 Porte 2, 91405 Orsay, France. E-mail: christophe.humbert@u-psud.fr; Fax: +33 169153328; Tel: +33 16915 3290
bInstitut des Nanosciences de Paris (INSP), CNRS-UMR 7588, UPMC Univ Paris 6, 4 place Jussieu, 75252 Paris, France
First published on 14th November 2011
Abstract
Nonlinear optical Sum and Difference-Frequency spectroscopies are used to probe and model the surface of thiophenol-functionalised gold nanoparticles grafted on a Si(100) substrate through two different silanization procedures. By scanning the [980–1100 cm−1] infrared spectral range with the CLIO Free Electron Laser, ring deformation vibrations of adsorbed thiophenol are investigated. Quantitative data analysis addresses three levels of organization: microscopic, nanoscopic and molecular. Grafting with p-aminophenyl-trimethoxysilane shows an increase of around 40% in surface density of nanoparticles (Ns) as compared to 3-aminopropyl-triethoxysilane. The relative amplitudes of the resonant and nonresonant contributions to the SFG and DFG spectra are discussed in terms of Ns, Fresnel reflectivity factors and local amplification of the nonlinear signals by coupling to the surface plasmon of the particles. They are shown to quantitatively scale with Ns, as measured by atomic force microscopy. Vibration mode assignment is performed through a critical analysis of literature data on IR and Raman spectroscopies coupled to DFT calculations, for which a methodology specific to molecules adsorbed on gold atoms is discussed.
Introduction
Nonlinear optical infrared-visible Sum-Frequency Generation (SFG) spectroscopy is attracting a growing interest for the study of gold nanomaterials as demonstrated in various and recent works.1–3 Since we recently showed that a molecular SFG signal can be strongly enhanced when molecules are grafted on gold nanoparticles (AuNps) in the presence of plasmonic exaltation,3 a study by SFG of adsorbed molecules becomes possible even at very low coverage. SFG, an intrinsic vibrational spectroscopy of interfaces, offers promising perspectives on such nanostructured materials. On one hand, such investigations performed by SFG spectroscopy are most of the time limited to the characterisation of methyl and methylene moieties of organic layers composing the probed interface due to the difficulty in maintaining widely tunable tabletop infrared (IR) sources (Optical Parametric Oscillators (OPO), Optical Parametric Amplifiers) outside the 2.5–4 μm spectral range. Some exceptions relate to the studies of solid/air and liquid/air organic interfaces;4–7 electrochemical or catalytic analyses of cyanide, carbon monoxide and C60fullerene monolayers8 up to 9 μm; amide I modes of proteins at various interfaces9 and more recently to the case of surface phonons of quartz at 12 μm.10 No work was performed so far on nanomaterials functionalized by organic components in the mid-IR spectral range, which however contains the fingerprint of organic molecules. On the other hand, it should be mentioned that a SFG study was performed around 10 μm on the coupling of surface states of Ag nanoclusters embedded in a Si3N4 matrix by tuning the visible excitation wavelength to the surface plasmon resonance (SPR) of the particles.11 This exemplifies the way often used to overcome the IR range limitation by coupling the SFG spectrometer to a Free Electron Laser (FEL).12,13 The latter offers a wide IR domain ranging from 5 to 150 μm, with higher power than any tunable IR source based on a tabletop laser at this date.14In this paper, the coupling of nonlinear optics to the surface optical properties of nanomaterials is investigated with a unique Sum and Difference-Frequency Generation (SFG/DFG) setup coupled to the CLIO FEL. We present SFG and DFG measurements performed on two kinds of samples made of gold spherical nanoparticles grafted on silicon with either p-aminophenyl-trimethoxysilane (APHS) or 3-aminopropyl-triethoxysilane (APTES) to monitor the surface coverage (Scheme 1). AuNps are functionalized with thiophenol (C6H5SH) molecules and we show that reliable information on both the molecules and the particles can be obtained even for a volume density as low as 4%. To reach that purpose, we perform a detailed three scale description (Fig. 1) of the various contributions to the SFG/DFG spectra, which allows to evidence specific chemical and physical properties of the nanostructured samples at all scales: microscopic, where their optical properties are described according to the 3-layer model, with the help of atomic force microscopy (AFM); nanoscopic, where the individual nanoparticles are taken into account to enlighten the differences induced by the grafting molecules and the enhancement effects due to a coupling to the SPR; molecular, where the vibrational response of individual thiophenol molecules is analysed by DFT to understand the nature and activities of the three observed vibration modes, addressing controversy in this rich spectral range.
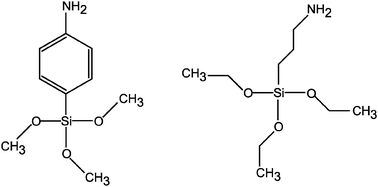 | ||
| Scheme 1 Molecules of p-aminophenyl-trimethoxysilane (APHS, left) and 3-aminopropyl-triethoxysilane (APTES, right) used to graft the gold nanoparticles on the silicon substrate. | ||
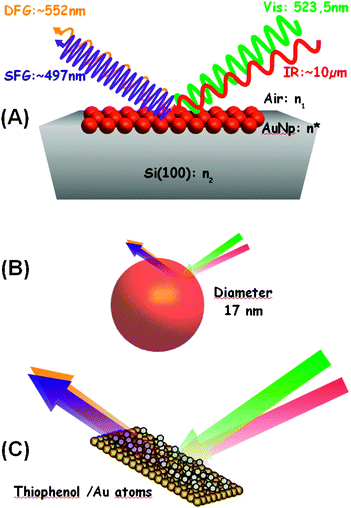 | ||
| Fig. 1 Sketch of the SFG and DFG processes on the surface of AuNps at different scales: (A) microscopic; (B) nanoscopic; (C) molecular. | ||
Results and discussion
Vibrational fingerprint in nonlinear optical spectroscopy
SFG and DFG experimental data of thiophenol adsorbed on AuNps are presented in Fig. 2 for the two grafting silane molecules, i.e. APHS (2A and 2B) and APTES (2C and 2D), respectively. Three observations can be made from these measurements. Firstly, all SFG/DFG spectra display three vibration modes (labelled 1, 2 and 4 with reference to the discussion below) related to the phenyl ring of thiophenol, located at 1000 ± 2, 1023 ± 2 and 1071 ± 2 cm−1, interfering with a constant background generated by the AuNps. Secondly, the interference induces different shapes in SFG and DFG spectra. In the SFG case, the interference pattern produces a peak for the 1000 cm−1 mode, a dip for the 1071 cm−1 mode and a Fano profile for the intermediate 1023 cm−1 mode. In contrast, for the DFG case, all modes appear as peaks. This is related to the change in sign of the damping constant (Γn) of the Lorentzian oscillators between SFG and DFG, detailed below (see eqn (1)–(2) as well as ref. 15). The third interesting feature relates to the silane used to graft the AuNps on the silicon substrate. We observe that SFG/DFG intensities of the AuNps and the 3 vibration modes contributions are stronger in the case of APHS silanisation. This is an indication of a higher coverage of the silicon substrate with APHS, as will be proved further.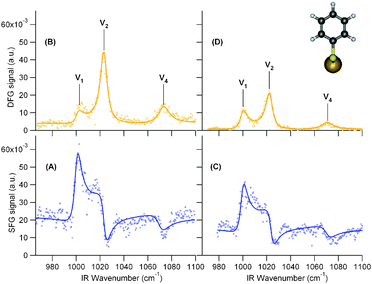 | ||
| Fig. 2 Left: SFG (A) and DFG (B) spectra of thiophenol molecules (insert) adsorbed at the AuNps/APHS/Si(100) interface. Right: SFG (C) and DFG (D) spectra of thiophenol molecules adsorbed at the AuNps/APTES/Si(100) interface. Lines are fits to the data based on eqn (1) and (2). SFG and DFG scales are the same to ease the comparison between the spectra. | ||
To extract quantitative information from these measurements, we develop hereafter an analysis of the various parameters contributing to the SFG/DFG intensity.
SFG/DFG spectroscopy at the microscale
The intensity I(ωFG), where FG stands for either SFG (ωSFG = ωvis + ωIR) or DFG (ωDFG = ωvis − ωIR), of interfaces is modelled by:16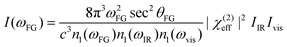 | (1) |
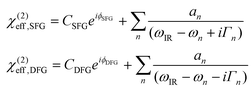 | (2) |
Eqn (2) are used to fit the four experimental spectra in Fig. 2. Prior to the fits, spectra were corrected from the stray light background, account was taken of the actual visible and IR powers (eqn (1)) and spectra were normalized to a common reference sample (ZnS crystal). There are several non-equivalent sets of parameters which may produce a correct fit, even if they do not correspond to a global minimum of the fitting algorithm. In order to fully explore the sets of possible fit parameters, we used the procedure already applied to thiophenol in ref. 18 by fitting the data with complex Lorentzian amplitudes, getting the eight alternate sets of parameters and analysing them to reach the best fits with real amplitudes. In order to choose the correct fit, we have therefore imposed constraints on the parameters. First, we have looked for coherent signs for the amplitudes of the vibrations. The only possibility showed opposite signs for mode 1 on one side and modes 2 and 4 on the other side. This is coherent with the SFG literature on these modes.14,18 A plausible explanation is that the IR transition moment of mode 1 has an opposite sign upon adsorption on AuNps with respect to the two others as shown in Table 1. Fitting the curves in this condition leads to an NR phase (ϕSFG and ϕDFG) of the AuNps contribution analogous to ref. 3 and 18 on silver and AuNps. In order to rationalize the comparison between the curves, we fixed Γn to their mean values, respectively, 3.5, 3.5 and 5.5 cm−1. We note that the larger Γ4 was already observed in ref. 12 and 18. Finally, the most stringent constraint lies on the amplitudes. Considering that any external effect on the amplitudes of the modes (Fresnel factor, local field, coupling to the SPR) must affect the three modes in the same way, the ratios of the three amplitudes must remain constant, as displayed in Table 1. We remark that the mandatory theoretical coherence between vibrational SFG and DFG spectra recorded on the same sample imposed severe constraints on our fits, leading to a unique solution for the parameters which in fact differs from the global minimum found by the fitting program. The usefulness to record DFG in parallel to SFG is evidenced in this case as it allows to calibrate the fits of SFG data.15,18
| SFG (APHS) | DFG (APHS) | SFG (APTES) | DFG (APTES) | ||
|---|---|---|---|---|---|
| AuNp | C | 0.147 | 0.0630 | 0.124 | 0.0261 |
| ϕ (°) | 137.57 | 77.7342 | 143.345 | 81.161 | |
| Mode 1 | ω 1/cm−1 | 1000.2 | 1001.3 | 999.5 | 999.9 |
| a 1/a.u. | −0.339 | −0.584 | −0.289 | −0.461 | |
| Γ 1/cm−1 | 3.5 | 3.5 | 3.5 | 3.5 | |
| Mode 2 | ω 2/cm−1 | 1023.6 | 1023.3 | 1023.8 | 1022.4 |
| |a2/a1| | 0.91 | 0.91 | 0.91 | 0.91 | |
| Γ 2/cm−1 | 3.5 | 3.5 | 3.5 | 3.5 | |
| Mode 4 | ω 4/cm−1 | 1070.4 | 1072.9 | 1070.8 | 1070.1 |
| |a4/a1| | 0.50 | 0.50 | 0.50 | 0.50 | |
| Γ 4/cm−1 | 5.5 | 5.5 | 5.5 | 5.5 | |
The following conclusions may be drawn from the fit parameters. Firstly, the values for the NR background of AuNps remain coherent with the FG response of bulk gold.15 This effect explains well the interference patterns observed in the FG experimental spectra and relates to the excitation of interband transitions in the AuNps, resonant with the FG energies.3 Actually, the NR amplitude (CSFG, CDFG) is higher for APHS than for APTES, with ratios 1.19 for SFG and 2.41 for DFG, respectively. In addition, the NR phases (ϕ) are coherent between APHS and APTES. Mean values lie around 80° for DFG and 140° for SFG, the latter being a bit higher than usual for analogous materials. It means that the nonlinear signal from AuNps is not far from phase-quadrature with respect to the vibration modes of the thiophenol molecules, and follows the trends observed for molecules adsorbed on bulk gold.15 This hints at analogous contributions from AuNps, implying in turn analogous local environments between both samples. This conclusion is confirmed by the observation that the overall positions of the three peaks remain remarkably constant within experimental uncertainties, even when the molecular adlayer is changed from APHS to APTES. Secondly, it is possible to compare the absolute resonant amplitudes from the values of fit parameters a1. For a given sample, the ratio DFG/SFG gives an estimate of the local amplification due to a resonance phenomenon with the surface plasmon of the particles at DFG and SFG frequencies, respectively. This ratio is 1.72 for APHS and 1.60 for APTES. The resonant and NR amplitudes ratios (APHS/APTES) will be discussed in more detail further.
Optical and morphological factors at the nanoscale
As mentioned above, in order to extract the chemical and physical information from our FG spectroscopic measurements, we need to detail the role of the various contributions to the FG intensity defined in eqn (1) and (2). The χ(2)eff third rank complex tensor can be expressed in ppp polarisation combination as:χ(2)eff,ppp = −Fx(ωFG)Fx(ωvis)Fz(ωIR)cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θFG cosθvis sinθIRχ(2)xxz − Fx(ωFG)Fz(ωvis)Fx(ωIR)cosθFG sinθvis cosθIRχ(2)xzx + Fz(ωFG)Fx(ωvis)Fx(ωIR)sinθFG cosθvis cosθIRχ(2)zxx + Fz(ωFG)Fz(ωvis)Fz(ωIR)sinθFG sinθvis sinθIRχ(2)zzz θFG cosθvis sinθIRχ(2)xxz − Fx(ωFG)Fz(ωvis)Fx(ωIR)cosθFG sinθvis cosθIRχ(2)xzx + Fz(ωFG)Fx(ωvis)Fx(ωIR)sinθFG cosθvis cosθIRχ(2)zxx + Fz(ωFG)Fz(ωvis)Fz(ωIR)sinθFG sinθvis sinθIRχ(2)zzz | (3) |
| χ(2)eff,ppp = −Fxxzχ(2)xxz − Fxzxχ(2)xzx + Fzxxχ(2)zxx + Fzzzχ(2)zzz | (4) |
The Fresnel factors Fi (i = x,y,z) describe the surface reflectivity, and we calculate them in a three-layer model: Air/AuNps/Silicon, with respective refractive indices n1, n* and n2, as depicted in Fig. 1(A). In fact, the silane monolayer and the thiophenol submonolayer do not affect significantly the sample reflectivity in the probed spectral range with respect to the Si substrate or the AuNps layer, whose contributions largely dominate in our samples. For an isotropic surface, the explicit complex contribution of the Fresnel factors can be calculated as16
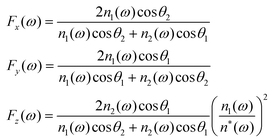 | (5) |
θ1 = n2(ω)sinθ2. Fi should be calculated for each optical frequency contributing to the FG process, i.e. for ω = ωIR, ωvis, ωSFG and ωDFG. The index of air is taken as unity and, for the n2 refractive index of silicon, we used a double exponential fit of the literature data.19 The difficulty lies in the derivation of an expression for the refractive index of the AuNp layer (n*) in the desired spectral ranges. To address this point, we evaluate the dielectric constant ε* = (n*)2 by considering a layer of nanoparticles embedded in a host matrix (air in our case, with εh = 1). We choose here the Maxwell–Garnett formalism20 for the effective dielectric constant: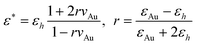 | (6) |
In order to determine the AuNp density (hence the volume ratio), we performed atomic force microscopy (AFM) measurements for the two systems, i.e.nanoparticles attached to silicon through APHS and APTES as displayed in Fig. 3. In a first step, at large scale (10 μm), we always obtained images similar to the ones shown in Fig. 3a for APHS and Fig. 3b for APTES. These AFM images allowed us to establish the proportions of the AuNp monolayer on the substrate. In a second step, we zoomed on the monolayers (see insets in Fig. 3a and b).
 | ||
| Fig. 3 AFM images of AuNps grafted on Si(100) through (a) APHS and (b) APTES silanization at different scales (10 × 10 μm2 and 500 × 500 nm2). | ||
The local densities were thus obtained as an average on six different areas. Combining this value to the monolayer proportion finally allowed us to establish the average AuNp surface density (Ns), leading to a value of 3.12 × 1010 cm−2 for APHS and 2.28 × 1010 cm−2 for APTES. For particles with a 17 ± 2 nm mean diameter, this led to volume densities within the intermediate layer vAu = 4.72% and 3.45% for APHS and APTES grafting, respectively.
The resulting Fresnel contributions to the SFG and DFG signals (i.e. related to χ(2)eff,ppp) appear in Table 2 for AuNps grafted on silicon through APTES or APHS, with the dominant components underlined. For the sake of comparison, the same quantities are calculated for the Air/Thiophenol/Au(111) 3-layer model interface within the same spectral range. In this second case, the organic layer cannot be dismissed because the nonlinear process occurs inside this layer. We used n1 = 1, 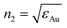 from ref. 19 and 21 for Au(111) and n* = 1.2 as calculated from the Lorentz-Lorentz model of an organic monolayer.16 It is worth noting that the |Fzzz| contribution largely dominates in the SFG/DFG intensity of the AuNps with respect to the other components. This is mainly due to the high refractive index of silicon at all wavelengths, which favours the Fz component. In contrast, for Au(111), the main effect stands in the high refractive index of gold in the IR range, which only favours the Fz(ωIR) component. Another interesting feature is that the dominant Fresnel factors of the AuNps are of the same order of magnitude as the Au(111) ones, as underlined in Table 2. This result shows that a direct comparison between intensities of SFG/DFG spectra can be performed for thiophenol grafted on Au(111) and AuNps. This validates the comparison performed in ref. 3 which evidenced an enhancement as high as 25 due to plasmonic exaltation.
from ref. 19 and 21 for Au(111) and n* = 1.2 as calculated from the Lorentz-Lorentz model of an organic monolayer.16 It is worth noting that the |Fzzz| contribution largely dominates in the SFG/DFG intensity of the AuNps with respect to the other components. This is mainly due to the high refractive index of silicon at all wavelengths, which favours the Fz component. In contrast, for Au(111), the main effect stands in the high refractive index of gold in the IR range, which only favours the Fz(ωIR) component. Another interesting feature is that the dominant Fresnel factors of the AuNps are of the same order of magnitude as the Au(111) ones, as underlined in Table 2. This result shows that a direct comparison between intensities of SFG/DFG spectra can be performed for thiophenol grafted on Au(111) and AuNps. This validates the comparison performed in ref. 3 which evidenced an enhancement as high as 25 due to plasmonic exaltation.
| SFG | APTES | APHS | Au(111) |
|---|---|---|---|
| |Fxxz| | 0.106 | 0.102 | ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) 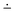 ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) |
| |Fxzx| | 0.108 | 0.102 | 1.655 × 10−2 |
| |Fzxx| | 0.118 | 0.113 | 1.616 × 10−2 |
| |Fzzz| | |
![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) |
![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) |
| DFG | APTES | APHS | Au(111) |
|---|---|---|---|
| |Fxxz| | 0.111 | 0.107 | ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) |
| |Fxzx| | 0.113 | 0.107 | 1.723 × 10−2 |
| |Fzxx| | 0.109 | 0.103 | 1.979 × 10−2 |
| |Fzzz| | ![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) |
|
|
From the comparison between the APTES and APHS samples in Table 2, the differences follow the values of the surface densities. The Fresnel contribution decreases when vAu increases. It is in fact very sensitive to the surface densities of particles through the (n*)2 factor in eqn (5). We also observe that, for a visible wavelength set at 523.5 nm, the Fresnel contribution to the SFG signal is slightly stronger than the DFG one as a consequence of the optical properties of gold in the visible range.
As a consequence of the above analysis, the tensor decomposition of the ppp contribution defined in eqn (4) simplifies into:
| χ(2)eff,ppp ≈ Fzzzχ(2)zzz | (7) |
This proves that only components of the molecular response perpendicular to the surface, i.e.IR transition moments and Raman polarizabilities, contribute to the signal, even for spherical AuNps for which one could have expected equal contributions of all directions. The presence of the silicon surface beneath the AuNps leads to this selection rules, which is an important result of the present calculations.
Consistency between microscale and nanoscale
In order to validate the FG data analysis at the microscale and nanoscale used so far, we have to check whether the whole set of data is consistent. We focus on the comparison between APTES and APHS samples, which in principle only differ by the surface density of adsorbates. The ratios of the fit parameters for APHS and APTES grafting are displayed in Table 3, together with the χ(2)zzz ratios deduced for the resonant and NR contributions after correction of Fresnel effects (eqn (7)).The quantity χ(2)zzz is in principle proportional to the surface density of FG sources on the sample,16 in our case thiophenol molecules for the resonant part and AuNps for the NR part. Considering that the surface density of molecules scales as the surface density of particles, the χ(2)zzz ratio should be directly proportional to the ratio of Ns quantities, provided that local field amplification is constant for a given spectroscopy. We can check in Table 3 that it is the case indeed for SFG (resonant and NR) and for the DFG resonant signals within 8% error, which remains compatible with error amplitudes in the experimental measurements and fit parameter estimations. Only the NR DFG value deviates from the three others. This is due to the fact that the NR backgrounds in DFG suffer from their low absolute values which hamper their quantitative analysis. As for the comparison between DFG and SFG for both samples, it includes a different local field amplification factor related to frequency dependence of the plasmon resonances. The coupling between FG and SPR processes has not yet been fully addressed by experimental methods, but it seems reasonable to consider that the visible laser excites the SPR, inducing an enhancement equal for both SFG and DFG signals. In addition, the SFG and DFG beams may also couple to the SPR, generating an additional factor to the amplification. This FG factor depends on the SPR properties at the FG frequencies, accounting for the differences in amplitude between SFG and DFG responses. As already evidenced in a previous study,3 the SPR of isolated particles (around 520 nm) should favour the SFG response. However, gold nanoparticles tend to aggregate upon adsorption onto the substrate (Fig. 3), leading to the appearance of a second red-shifted plasmon resonance, which eventually boosts the DFG response. This has already been shown for a similar deposition procedure of AuNps on silicon substrates.3 This explains why, in the present study, the local amplification favours DFG rather than SFG. We estimate the DFG/SFG amplification ratio as 1.89 for APHS and 1.73 for APTES when corrected for Fresnel effects, i.e. calculated from χ(2)zzz ratios.
As a summary of the analysis developed so far for the FG intensity coupled to AFM measurements and to calculations of the Fresnel contributions, we may draw the four following conclusions: (1) the four sets of SFG and DFG data are consistent, involving the contributions of three identical vibration modes in the direction normal to the silicon surface; (2) the amplitude ratios of the three modes remain constant whatever the nonlinear vibrational spectroscopy and the silane, hinting at analogous molecular geometries on the particles and local environments; (3) the differences between absolute values of the amplitudes (resonant and non resonant), after correction of Fresnel reflectivity effects, are due to the surface density of nanoparticles, which differs as a function of the silane used to graft them on the silicon; (4) an additional enhancement factor, related to SPR specific for each sample, accounts for the difference between absolute SFG and DFG amplitudes. The great sensitivity of the technique to the surface density finally illustrates the potential role of such spectroscopies in the quantitative comparison of analogous composite materials.
Thiophenol IR and Raman properties at the molecular scale
After the investigation of the FG intensity at the microscale and nanoscale, the last step in such an analysis, when done on a flat surface, would be to relate the microscopic χ(2) to the molecular response, provided that at least two polarization contributions could be measured (usually ppp and ssp). This is routinely done under the conditions that the surface is isotropic and all molecules share the same tilt (even if dual angle distributions appear necessary for complex cases22). An additional approximation on the structure of the molecule (i.e. cylindrical symmetry around the direction of the IR transition moment of a given vibration) is commonly added.16 As a consequence, a multiscale description relates molecular IR and Raman activities of the vibration modes to the microscopic SFG response, giving access to unknown parameters, commonly the molecular tilt angle. A complete simulation of an SFG spectrum, including several vibration modes, must take into account the relative IR and Raman activities and their full decomposition in the molecular frame. It requires usually a molecular DFT calculation.6,7The situation in the case of AuNps is more complicated than the flat surface archetype, as further effects blur the molecular response building up the microscopic FG signal and its analysis. Orientation averaging represents a challenge indeed when the molecules decorate gold nanoparticles. Molecular orientation on one particle is not constant but rather distributed along a sphere. In addition, the density of the molecular packing at the surface of the particle is unknown, as is therefore the degree of order at this scale. As for the particles themselves, their distributions of shapes and sizes complicate the analysis. In addition, the distribution functions of the molecular tilt and twist angles are unknown. It would seem rather hazardous at this stage to draw hypotheses on these parameters. Finally, for SFG and DFG spectroscopies, Fresnel factors involving a polarization of light parallel to the surface (i.e. ssp contribution) are very unfavourable owing to the high refractive index of silicon, as calculated in Table 2. As a consequence, we consider that it is not possible, without additional data on the structure and organization of the molecules at the interface, to use FG data for a detailed orientation analysis. Nevertheless, we demonstrate that DFT analysis of the geometry and vibration modes of the thiophenol molecule, both free and bonded to gold, leads to important information when linked to the SFG and DFG data.
Several publications have focussed on the analysis of thiophenol vibration modes by IR,23,24 Raman24–27 and SFG5,12 spectroscopies. A comparison evidences that there are still some discrepancies in their interpretations. In the early literature, thiophenol was described in the convenient C2v geometry as an extension of the analysis of monosubstituted benzenes to make the analytical calculations easier.24 This means that the H atom was considered negligible and merged into a SH pseudo-atom. For the free thiophenol molecule, experimental data show that there are four active modes in the 1000–1100 cm−1 range, for which qualitative IR and Raman activities are shown in Table 4. The positions and activities of the modes are taken from ref. 24 and 25, even if these values change slightly within the literature and with the experimental tool. In the same way, thiophenol linked to a metal atom, either silver- or gold-benzenethiolate, has been investigated by IR, Raman, SERS and SFG. A summary of the main bands of interest is also shown in Table 4, from ref. 25. Only three modes survive upon adsorption on the metal, but their origin and symmetries are debated. Within the C2v approximation, in-plane modes may be symmetric/a1 or antisymmetric/b2 with respect to the plane perpendicular to the phenyl ring containing the S atom. Ref. 24 and 25 on one side and ref. 26 on the other side disagree on the attribution of modes 3 and 4. For the latter, mode 3 is symmetric and 4 antisymmetric, vice versa for the former. The origin of the SERS and silver-phenylthiolate Raman activity at 1075 cm−1 is also debated, as ref. 26 attributes it to an increase of the activity of peak 3 whereas ref. 25 and 27 talk about a strong shift of the position of the Raman-active mode 4. Finally, ref. 25 reports a fourth mode at 1065 cm−1, which is only IR-active. Such uncertainties on the actual active modes have rather strong impact on the interpretation of SFG data, either for absolute Raman and IR activities or for geometry and orientation purposes.
DFT analysis of free and adsorbed thiophenol molecule
To clarify the vibrational properties of thiophenol, we have calculated by DFT its molecular geometry and vibration modes in the 1000–1100 cm−1 region. We first made use of the most usual functional (B3LYP) and basis sets (6-311++G(d,p)) for such molecular systems to account for our data. In order to validate the calculations, we first tried to reproduce the IR and Raman data before analysing the SFG intensities. As will be illustrated below, we faced two problems to account for the literature results. Firstly, Table 1 and the discussion above show that adsorption on gold plays a great role in the energies of the vibrations in this region, which implies that it has to be taken into account in the calculations. In order to simulate adsorption onto the gold nanocrystal on the geometric and vibrational parameters, calculations have been performed both on free thiophenol and on a model thiophenol molecule linked to one gold atom (gold-benzenethiolate, GBT) as illustrated in Scheme 2. Such a procedure was used for the simulation of electronic properties of CO on platinum28 and CN− on transition metals,29 although more accurate predictions require the use of metal clusters or periodic slabs and heavier calculation methods.30 It appeared that B3LYP functional produces inconsistent mode assignment with GBT (see below and ESI†). In order to produce consistent results, functional PBE0 was successfully used on both molecules. However, such basic DFT results were not satisfactory enough to reproduce experimental data, even with a correction factor. Therefore, a second-order perturbation anharmonicity step was added to frequency calculations,31 which resulted in a very nice accuracy on the predicted frequencies (we note that this method does not provide anharmonic corrections for the IR and Raman activities. Details of the DFT calculations of the present paper are given in the ESI†).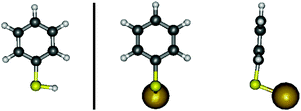 | ||
| Scheme 2 Molecules of thiophenol (left, called planar in the text) and gold-benzenethiolate (right, two different views of the same geometry, called perpendicular in the text) in their lowest energy conformations. | ||
As illustrated in Scheme 2, the ground state geometries differ appreciably. Thiophenol has a planar configuration whereas GBT converges to a perpendicular geometry, where the S–Au bond rotates by 90° to reach the symmetry plane perpendicular to the phenyl ring. As for the latter, it is highly probable that the actual rotation angle around the C–S bond in a rather densely packed zone lies between the two extreme values as a consequence of molecular packing and low free rotation barriers. For this system, additional steric and molecular packing effects at the surface of the gold nanoparticle are not taken into account here and further restrict the free rotation. Both geometries possess Cs symmetry.
All calculations confirm the presence of four vibration modes between 1000 and 1150 cm−1 (Table 5). The harmonic approximation for the vibration energies is, as expected, too high (see ESI†). We could check that a uniform scaling factor, even for as few as these four modes, does not satisfactorily account for the experimental frequencies. In contrast, a mere anharmonic calculation limited to the four modes under study gives very satisfactory results without any scaling factor for B3LYP, and with a uniform additional scaling factor 0.992 for PBE0 (Table 5). For GBT, it appears that B3LYP is unable to handle the metal atom in a correct way within our calculation scheme, leading to inconsistent results for the assignment and position of the modes. PBE0 proves more robust in that case, giving very satisfactory results after anharmonic correction and with the same 0.992 scaling factor as for thiophenol. From Tables 4 and 5, we may draw the following conclusions: (i) modes 1, 2 and 4 are SFG-active for GBT; (ii) mode 3 is SFG-inactive as a consequence of its vanishing Raman activity in GBT; (iii) upon adsorption on gold, mode 4 experiences a strong red shift in position (experimental value 18 cm−1, calculated value 24 cm−1), which could lead to its confusion with mode 3. The explanation of ref. 27 is correct, as this mode encompasses a strong contribution of the C–S stretch, which is directly perturbed by the replacement of the terminal H by the Au atom; (iv) DFT predicts very well the positions of the modes and their evolution upon adsorption as long as anharmonicity is considered; (v) the coarse prediction of qualitative IR and Raman activities is correct when compared to experimental values; (vi) DFT lacks accuracy on the fine prediction of actual values for IR and Raman activities, especially for GBT.
| Mode | ω exp/cm−1 | ω anharm/cm−1 B3LYP | ω anharm/cm−1PBE0 | IR (calc.) | Raman (calc.) | |
|---|---|---|---|---|---|---|
| Thiophenol | 1 | 1002 | 1005.2 | 997.6 | w | s |
| 2 | 1026 | 1025.8 | 1029.6 | s | s | |
| 3 | 1072 | 1074.6 | 1072.0 | m | w | |
| 4 | 1093 | 1091.0 | 1099.6 | s | m | |
| GBT | 1 | 1000 | — | 998.0 | m | s |
| 2 | 1025 | — | 1024.9 | m | s | |
| 3 | 1065 | — | 1067.0 | m | 0 | |
| 4 | 1075 | — | 1075.1 | m | vs |
Considering Table 5, the effects of adsorption on the IR and Raman activities inferred from DFT can be summarized as follows. Upon adsorption, the IR activity of mode 1 is enhanced, whereas it decreases for modes 2 and 4 and remains constant for mode 3. In contrast, Raman activity of modes 2 and 4 rises upon adsorption, whereas it vanishes for mode 3. However, a direct comparison remains hindered by the geometry change between thiophenol and GBT. In addition, as seen before, we have to consider at least a partial free rotation around the C–S bond. In order to separate the effects of the bonding to gold and of the rotation around the C–S bond, we also have performed similar calculations on the two complementary conformers of higher ground state energy: thiophenol in a perpendicular configuration and GBT in a planar geometry. The results show that around 2/3 of the energy shift of mode 4 are due to the gold bond and 1/3 due to the conformation. Taking into account the effect of the atom exchange alone leads to an ∼16 cm−1 shift, very close to the experimental value. The ∼5 cm−1 slight shift of mode 3 is mainly due to conformation. As for the IR and Raman intensities, the effects are less clear. It appears that the Raman enhancement of modes 2 and 4 and the damping of mode 3 are mainly due to the gold atom, even if conformation also favours a decrease of the Raman activity for mode 3. The geometric factor is the cause of the decrease in the IR activity of mode 4, whereas both effects participate to increase for mode 1 and mode 2. In all cases, it appears obvious that the adsorption process must be explicitly taken into account when modelling vibrational properties of molecules adsorbed on the metal.
DFT proves very useful for the assignment of the modes and the comparison of their linear optical activities (i.e.IR and Raman activities). The three vibration modes appearing in Fig. 2 are therefore identified with modes 1, 2 and 4 (Fig. 4 below), respectively, their position being in full agreement with the calculated ones (Tables 1 and 5).
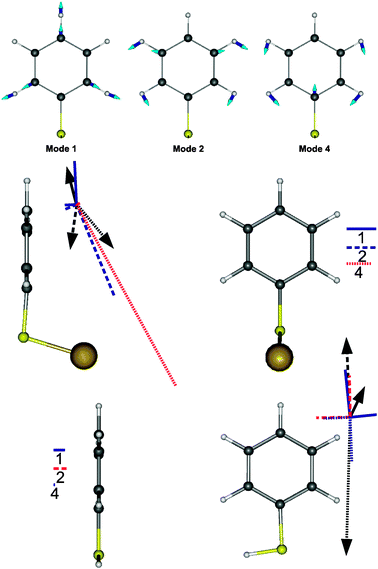 | ||
| Fig. 4 Top: illustration of the vibrational activity of mode 1 (998.0 cm−1), mode 2 (1024.9 cm−1) and mode 4 (1075.1 cm−1) of the GBT system as calculated by DFT. The dominant contributions of the atomic displacements are indicated by arrows in order to display the symmetric character of these particular modes. Middle: IR transition moments (arrows) and Raman polarisabilities of GBT calculated by DFT (blue for positive, red for negative, PBE0 functional). Left panel shows the contributions in the symmetry plane, while the right panel shows the out-of-plane Raman contribution, together with the line code for the modes. Bottom: IR transition moments and Raman polarisabilities of free thiophenol (Right: in the symmetry plane. Left: perpendicular to it). The scales are common to both molecules. | ||
Considering the plane of symmetry of the GBT molecule, they are all totally symmetric (inactive mode 3 is antisymmetric). Their IR transition dipole moments have therefore two components, one dominant along the C1–C4 axis and the other perpendicular to the ring plane. The Raman tensor has four independent nonvanishing terms, building up a Raman polarizability ellipsoid with three nonvanishing principal axes. Focussing on GBT, the transition dipole of mode 1 presents a component parallel to the phenyl plane, oriented in the opposite direction with respect to those of modes 2 and 4. As for Raman polarisabilities, mode 4 is opposite to modes 1 and 2. As a consequence, DFT calculations on GBT cannot reproduce correctly the signs of the SFG activities of the three modes (Table 1). The same analysis performed on the free thiophenol molecule accounts for the correct sign alternation (Fig. 4). However, all modes also have a minor contribution perpendicular to the ring, and the Raman polarizability contributions include a total of four non-vanishing terms, which make uncertain the conclusions on the final signs of the resonant hyperpolarizabilities. In addition, as stated above, the various orientations of the molecules at the surface of the particles require caution when correlating the individual molecular properties to the averaged FG response, in particular when one considers that the actual geometry of the molecule on the surface is not necessarily exactly the perpendicular one.
As far as SFG intensities are concerned, we note that their qualitative behaviour cannot be correctly reproduced by DFT. In fact, considering an estimate of SFG activity from the product of IR and Raman activities as a first order approximation, it appears that the GBT molecule overestimates the contribution of mode 4, whereas in the free molecule mode 1 is largely underestimated (which is coherent with the dramatic changes induced by adsorption). In fact, neither the free molecule nor the gold-bonded one leads to a molecular SFG activity of mode 1 bigger than that of mode 2 in the molecular frame. Of course, orientation averaging will lead to partial cancellation of some hyperpolarizability contributions to the microscopic FG activity as a consequence of symmetry, so that the latter cannot be inferred directly from the product of IR and Raman activities. Nevertheless, considering that (1) DFT already encounters troubles to account for the IR and Raman data from the literature; (2) a calculation of the effects of orientation averaging seems out of reach as a consequence of the lack of knowledge on the actual orientation distributions of the molecules on the nanoparticles; (3) discrepancies between experimental data and DFT simulations (e.g. mode 1 vs. mode 4) are too strong to be due only to orientation effects; (4) a careful analysis of individual components of the IR dipole moments and Raman polarizabilities in the molecular frame shows that they are roughly identically weighted for the three modes and that, even by linear combination of components, it is not possible to recover the activity ratios (for example, all Raman polarizability components of mode 2 are basically twice those of mode 1); we consider that, even with a complete description of the structure at the nanoscale, the present DFT results may not quantitatively describe the FG spectroscopy results. The reasons lie in the amount of approximations done during the calculations: choice of the exchange–correlation functionals (B3LYP and PBE0 might not be the most appropriate for the Au atom), modelling of the Au surface by only one atom, no frequency dispersion. The fact that we did not take into account the effects of local electric fields (either from the laser beams or from the SPR) may also partially account for this result. Surprisingly, we note that good estimates of the SFG intensity ratios can be obtained by combining the IR activity of the GBT molecule to the Raman activity of the free molecule. In particular, it becomes possible to recover the leading SFG intensity of mode 1. However, at this stage, no theoretical background justifies such a methodology.
The literature confirms that an accurate calculation of the 1000–1100 cm−1 region of phenyl ring vibrations by DFT is in general not a straightforward task. This zone seems rather impervious to standard DFT analysis. Adsorption on the metal further induces strong modifications of the vibrational parameters (energy position, IR and Raman activities), at least for vibration modes impacted by the S-metal link. Such discrepancies between DFT calculations and experimental SFG data have already been pointed out in this zone in the past.7 In that paper, the authors even extended their comment to the whole fingerprint mid-infrared region [1000–2000 cm−1]. In a general way, the few published attempts to reproduce SFG results by DFT give contrasting results: an overall agreement may sometimes be found6 although accuracy remains out of reach. Again, this is probably due to the inadequacy of the exchange–correlation parts of the functionals used in the literature for such analyses (mainly B3LYP). However, contrary to the mid-infrared situation, it seems possible to account rather nicely for the high energy range [2000–3000 cm−1].7,32
To summarize, we have shown that the free molecule alone could not account for all the experimental properties of the system. Only a comparison between the calculations performed on thiophenol and GBT could elucidate most of the properties. More precisely, mode assignment and energies were successfully reached for GBT through the addition of anharmonic corrections. The free molecule leads to better relative signs of the SFG activities, whereas none accounted satisfactorily for the SFG relative amplitudes. Considering the rather high amount of work necessary to account for the essential vibrational properties of adsorbed thiophenol, coupled to the difficulty encountered to quantitatively predict SFG intensities from the single molecule, caution seems to be recommended when using DFT results as an input for SFG predictions, especially when conclusions are drawn on molecular orientation of adsorbates on a surface.
Experimental
All chemicals were purchased from Aldrich, unless otherwise mentioned. AuNps are synthesized from 1.7 mg of gold salts (HAuCl4·3H2O 99.999%) dissolved in 20 ml Millipore water following the Turkevich method. The solution was then heated up until boiling point and stirred vigorously. At that point, 0.8 mL of 8.5 × 10−4 M trisodium citrate (Na3C6H5O7) was quickly added at one time while heating and stirring for 30 minutes. The resulting solution turned successively from light yellow to deep gray and finally dark red after a few minutes. The corresponding UV-visible absorbance spectrum displayed the expected absorption peak at 524 nm usually for AuNps in water.3 Wafers of ultrasonically cleaned n-doped silicon (1 × 1 cm2, Siltronix) were silanized in an absolute methanol solution. After being grafted on such a wafer in methanol, AuNps were functionalized with a 10−3 M thiophenol (C6H5SH) solution dissolved in dichloromethane (CH2Cl2). The use of two different silanes, differing by their chain length and nature (p-aminophenyl-trimethoxysilane (APHS), H2N(C6H4)Si(OCH3)3 or 3-aminopropyl-triethoxysilane (APTES), H2N(CH2)3Si(OC2H5)3), aimed at monitoring and comparing the different surface coverages by the AuNps.
SFG and DFG measurements were performed in a ppp-polarisation scheme for the SFG/DFG, visible and IR beams, respectively. The sketch of the experiments is depicted in Fig. 1. It is clear that the only change between SFG and DFG experimental setups resided in the detection angle and the measured frequency of each nonlinear signal, both related to the SFG/DFG rules of momentum and energy conservations (kSFG/DFG = kVis ± kIR and ωSFG/DFG = ωVis ± ωIR), as detailed in ref. 3. The SFG/DFG setup coupled to the CLIO Free Electron Laser (FEL) European Facility is described in detail elsewhere.14 Briefly, its dedicated pulsed and intense IR laser beam (∼1 ps, ∼10 μJ pulse−1 reduced to 2 μJ pulse−1 for SFG/DFG experiments, ∼5 cm−1 resolution) was tuned around 10 μm in order to probe specific vibration modes of ring deformation of thiophenol. The visible laser beam at 523.5 nm (∼5 ps, 4 μJ pulse−1, ∼6 cm−1 resolution) originated from the frequency doubling of a pulsed and amplified Nd∶YLF laser source at 1047 nm through a nonlinear BBO crystal. The laser sources had the same repetition rate for the macropulses (25 Hz) and micropulses (62.5 MHz) fixed by the CLIO FEL clock, in order to synchronize temporally the IR and visible beams at the same point of the probed samples. Under these conditions, SFG and DFG coherent nonlinear processes were always generated simultaneously, provided that spatial and temporal overlap of the input beams was achieved at the interface. The angles of incidence of the visible and IR beams were 55° and 65° with respect to the surface normal, respectively. The visible beam fixed at 523.5 nm was near the maximum of the SPR of the 17 ± 2 nm diameter AuNps. SFG/DFG signals were recorded by photomultipliers after spatial and spectral filtering through a monochromator. Data were normalized by the SFG/DFG signal of a ZnS reference crystal to take account of all laser fluctuations and absorptions on the laser paths.
The samples surface density was analyzed with AFM (Digital Instrument, DI3100) in a tapping mode. The silicon tips used in our measurements have 130 kHz working frequency and their curvature radius at the apex is around 10 nm. This latter does not allow us to obtain sufficient lateral resolution to image correctly isolated nanoparticles but it allows us to identify each gold nanoparticle and give their mean diameter (17 ± 2 nm). Finally AFM allows us to determine the number of nanoparticles grafted on the APHS and APTES surface in order to deduce the AuNps surface density used in the calculations of the Fresnel factors in the SFG and DFG intensities.
Conclusions
We have presented IR-vis SFG/DFG measurements on thiophenol-functionalized AuNps grafted on a silicon substrate through two different silanes. This is the first experimental evidence, to the best of the authors' knowledge, of a vibrational nonlinear activity in the fingerprint spectral range observed on the surface of spherical metallic nanoparticles (17 ± 2 nm diameter). This has been made possible thanks to the sensitivity of the vibrational FG technique, based on a setup coupled to the CLIO FEL. However, the levels of FG intensities measured above make us confident that analogous measurements should be possible even using tabletop laser sources in the [1000–2000 cm−1] range. For an extension towards lower energies, the FEL remains mandatory on such materials.A careful analysis of experimental results shows that FG spectroscopy can actually become quantitative as a function of the surface density, as long as a proper data analysis is carried out: calibration of absolute FG intensities, validation of the fitting procedure by imposing compatibility between SFG and DFG, full account of Fresnel reflectivity effects. As a result, the essential proportionality of the nonlinear (resonant and nonresonant) susceptibilities to surface density of nanoparticles is shown. Provided that an absolute calibration is achieved and the method extended towards more commonly accessible frequency regions (e.g. 2500–3500 cm−1), it becomes an interesting tool for the analysis of such composite materials, complementary to microscopy (AFM, STM) and linear optical techniques (IR, Raman). This opens the door to more routine analysis of the surface chemistry of nanoparticles using nonlinear optical spectroscopies. In addition, derived methods which lower the detection threshold, like heterodyne SFG or doubly resonant SFG,33 should prove helpful for that purpose.
In the investigated samples, we have shown that grafting gold nanoparticles on silicon with APHS is more efficient than with APTES, with an increase of the surface density by around 40%, confirming results of ref. 2. Apart from this parameter, which impacts the absolute FG intensities, both grafting silanes lead to very similar structures at the molecular and nanoscopic scale, since the SFG activities of the molecular vibrations and gold nanoparticles share the same properties.
We intend to continue this systematic work of understanding the optical and chemical properties of complex materials based on metallic nanoparticles. There are still further steps to undertake, concerning an accurate description of the interplay between the surface plasmon of the particles and the molecular vibrations mediated by nonlinear optical spectroscopies (SFG/DFG). Improving theoretical and computational methodology is mandatory to bridge the gap between molecular properties deduced from DFT calculations and a description at the nanoscale of the orientation distribution of molecules at the surface of the particles. In the future, applications including more complex molecular systems adsorbed on AuNps and related, for instance, to heterogeneous catalysis and biosensors could be accurately described at the nanoscale with SFG/DFG spectroscopy. In fact, we believe that the experimental and theoretical procedures developed in this work could be used to systematically monitor the surface chemical properties of such nanomaterials, whatever their sizes, shapes or organization on a given substrate.
Acknowledgements
The authors acknowledge C. Six, A. Gayral, J.-P. Berthet and J.-M. Ortega for their scientific and technical support for the SFG optical setup and the CLIO Free Electron Laser; P. Archirel for his help in DFT calculations. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 226716.Notes and references
- T. Kawai, D. J. Neivandt and P. B. Davies, J. Am. Chem. Soc., 2000, 122, 12301 CrossRef CAS; C. Humbert, B. Busson, J.-P. Abid, C. Six, H. H. Girault and A. Tadjeddine, Electrochim. Acta, 2005, 50, 3101 CrossRef; C. Weeraman, A. K. Yatawara, A. N. Bordenyuk and A. V. Benderskii, J. Am. Chem. Soc., 2006, 128, 14244 CrossRef; A. N. Bordenyuk, C. Weeraman, A. Yatawara, H. D. Jayathilake, I. Stiopkin, Y. Liu and A. V. Benderskii, J. Phys. Chem. C, 2007, 111, 8925 Search PubMed.
- G. Tourillon, L. Dreesen, C. Volcke, Y. Sartenaer, P. A. Thiry and A. Peremans, Nanotechnology, 2007, 18, 41530 CrossRef.
- O. Pluchery, C. Humbert, E. Lacaze and B. Busson, Phys. Chem. Chem. Phys., 2009, 11, 7729 RSC.
- B. Busson and A. Tadjeddine, J. Phys. Chem. C, 2008, 112, 11813 CAS.
- A. A. Mani, Z. D. Schultz, Y. Caudano, B. Champagne, C. Humbert, L. Dreesen, A. A. Gewirth, J. O. White, P. A. Thiry and A. Peremans, J. Phys. Chem. B, 2004, 108, 16135 CrossRef CAS.
- J. Guthmuller, F. Cecchet, D. Lis, Y. Caudano, A. A. Mani, P. A. Thiry, A. Peremans and B. Champagne, ChemPhysChem, 2009, 10, 2132 CrossRef CAS; F. Cecchet, D. Lis, J. Guthmuller, B. Champagne, G. Fonder, Z. Mekhalif, Y. Caudano, A. A. Mani, P. A. Thiry and A. Peremans, J. Phys. Chem. C, 2010, 114, 4106 Search PubMed; F. Cecchet, D. Lis, J. Guthmuller, B. Champagne, Y. Caudano, C. Silien, A. A. Mani, P. A. Thiry and A. Peremans, ChemPhysChem, 2010, 11, 607 CrossRef; D. Lis, J. Guthmuller, B. Champagne, C. Humbert, B. Busson, A. Tadjeddine, A. Peremans and F. Cecchet, Chem. Phys. Lett., 2010, 489, 12 CrossRef.
- L. J. Richter, C. S. C. Yang, P. T. Wilson, C. A. Hacker, R. D. van Zee, J. J. Stapleton, D. L. Allara, Y. Yao and J. M. Tour, J. Phys. Chem. B, 2004, 108, 12547 CrossRef CAS.
- A. Peremans, Y. Caudano, P. A. Thiry, P. Dumas, W. Q. Zheng, A. Le Rille and A. Tadjeddine, Phys. Rev. Lett., 1997, 78, 2999 CrossRef CAS; G. A. Somorjai and G. Rupprechter, J. Phys. Chem. B, 1999, 103, 1623 CrossRef; T. Dellwig, G. Rupprechter, H. Unterhalt and H. J. Freund, Phys. Rev. Lett., 2000, 85, 776 CrossRef; F. Vidal, B. Busson, A. Tadjeddine and A. Peremans, J. Chem. Phys., 2003, 119, 12492 CrossRef; F. Vidal, B. Busson and A. Tadjeddine, Chem. Phys. Lett., 2005, 403, 324 CrossRef; B. Bozzini, B. Busson, G. P. de Gaudenzi, C. Mele and A. Tadjeddine, J. Solid State Electrochem., 2008, 12, 303 CrossRef.
- S. Ye, K. T. Nguyen, S. V. Le Clair and Z. Chen, J. Struct. Biol., 2009, 168, 61 CrossRef CAS.
- W. T. Liu and Y. R. Shen, Phys. Rev. Lett., 2008, 101, 016101 CrossRef.
- A. Traverse, C. Humbert, C. Six, A. Gayral and B. Busson, Europhys. Lett., 2008, 83, 64004 CrossRef.
- R. Braun, B. D. Casson, C. D. Bain, E. W. M. van der Ham, Q. H. F. Vrehen, E. R. Eliel, A. M. Briggs and P. B. Davies, J. Chem. Phys., 1999, 110, 4634 CrossRef CAS.
- B. Bozzini, L. D'Urzo, C. Mele, B. Busson and A. Tadjeddine, J. Phys. Chem. C, 2008, 112, 11791 CAS.
- C. Humbert, B. Busson, C. Six, A. Gayral, M. Gruselle, F. Villain and A. Tadjeddine, J. Electroanal. Chem., 2008, 621, 314 CrossRef CAS.
- A. Le Rille and A. Tadjeddine, J. Electroanal. Chem., 1999, 467, 238 CrossRef CAS; A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103 CrossRef.
- X. Zhuang, P. B. Miranda, D. Kim and Y. R. Shen, Phys. Rev. B: Condens. Matter, 1999, 59, 12632 CrossRef CAS.
- S. H. Lin and A. A. Villaeys, Phys. Rev. A, 1994, 50, 5134 CrossRef CAS.
- B. Busson and A. Tadjeddine, J. Phys. Chem. C, 2009, 113, 21895 CAS.
- E. D. Palik, in Handbook of Optical Constants of Solids, Academic, New York, 1985 Search PubMed.
- D. E. Aspnes, J. B. Theeten and F. Hottier, Phys. Rev. B: Condens. Matter, 1979, 20, 3292 CrossRef CAS.
- P. G. Etchegoin, E. C. Le Ru and M. Meyer, J. Chem. Phys., 2006, 125, 164705 CrossRef CAS.
- X. Chen, J. Wang, A. P. Boughton, C. B. Kristalyn and Z. Chen, J. Am. Chem. Soc., 2007, 129, 1420 CrossRef CAS.
- Coblentz Society, Inc., “Evaluated Infrared Reference Spectra” in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P.J. Linstrom and W.G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899.
- D. W. Scott, J. P. McCullough, W. N. Hubbard, J. F. Messerly, I. A. Hossenlopp, F. R. Frow and G. Waddington, J. Am. Chem. Soc., 1956, 78, 5463 CrossRef CAS.
- H. Joo, M. S. Kim and K. Kim, J. Raman Spectrosc., 1987, 18, 57 CrossRef.
- K. T. Carron and L. G. Hurley, J. Phys. Chem., 1991, 95, 9979 CrossRef CAS.
- M. A. Bryant, S. L. Joa and J. E. Pemberton, Langmuir, 1992, 8, 753 CrossRef CAS.
- H. Nakatsuji, H. Morita, H. Nakai, Y. Murata and K. Fukutani, J. Chem. Phys., 1996, 104, 714 CrossRef CAS.
- M. Tadjeddine and J. P. Flament, Chem. Phys., 1999, 240, 39 CrossRef CAS.
- M. Tadjeddine and J. P. Flament, Chem. Phys., 2001, 265, 27 CrossRef CAS; O. Pluchery, M. Tadjeddine, J. P. Flament and A. Tadjeddine, Phys. Chem. Chem. Phys., 2001, 3, 3343 RSC; G. L. Beltramo, T. E. Shubina, S. J. Mitchell and M. T. M. Koper, J. Electroanal. Chem., 2004, 563, 111 CrossRef.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef.
- M. Buck, Phys. Chem. Chem. Phys., 2003, 5, 18 RSC.
- I. V. Stiopkin, H. D. Jayathilake, A. N. Bordenyuk and A. V. Benderskii, J. Am. Chem. Soc., 2008, 130, 2271 CrossRef CAS; M. B. Raschke, M. Hayashi, S. H. Lin and Y. R. Shen, Chem. Phys. Lett., 2002, 359, 367 CrossRef; L. Dreesen, C. Humbert, Y. Sartenaer, Y. Caudano, C. Volcke, A. A. Mani, A. Peremans and P. A. Thiry, Langmuir, 2004, 20, 7201 CrossRef; B. Bozzini, L. D'Urzo, C. Mele, B. Busson, C. Humbert and A. Tadjeddine, J. Phys. Chem. C, 2008, 112, 11791 Search PubMed; T. Miyamae, Y. Miyata and H. Kataura, J. Phys. Chem. C, 2009, 113, 15314 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of all DFT calculations. See DOI: 10.1039/c1cp21091b |
| This journal is © the Owner Societies 2012 |