The synthesis, structures and reactions of zinc and cobalt metal–organic frameworks incorporating an alkyne-based dicarboxylate linker†
Andrew D.
Burrows
*a,
Laura C.
Fisher
ab,
David
Hodgson
a,
Mary F.
Mahon
a,
Naomi F.
Cessford
c,
Tina
Düren
c,
Christopher
Richardson
ad and
Sean P.
Rigby
be
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cInstitute for Materials and Processes, School of Engineering, The University of Edinburgh, Edinburgh, EH9 3JL
dSchool of Chemistry, University of Wollongong, Wollongong, NSW 2522, Australia
eDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 20th October 2011
Abstract
The reaction of zinc(II) nitrate and 4,4′-ethynylenedibenzoic acid (H2edb) in DMF at 80 °C gave the metal–organic framework material [Zn4O(edb)3(H2O)2]·6DMF 1 in which edb ligands connect Zn4O centres into a doubly-interpenetrated cubic network with a similar topology to observed with other linear dicarboxylates in the IRMOF series. Analysis of the nitrogen isotherm revealed the material to have a BET surface area of 1088 m2 g−1, which is approximately one-third of the value calculated from GCMC simulations, suggesting incomplete activation or pore blocking in the activated material. The reaction of cobalt(II) nitrate and H2edb in DMF gave [Co3(edb)3(DMF)4]·2.6DMF 2. The structure of 2 is based on Co3(O2CR)6 linear secondary building units that are linked by the edb ligands into a two-dimensional network. When 2 was placed under vacuum, a colour change from pale pink to deep blue was observed, which is consistent with loss of the coordinated DMF molecules. When treated with [Co2(CO)8], crystals of 1 turned dark red, and IR analysis is consistent with coordination of Co2(CO)6 fragments to the alkyne groups. However, the colour change was restricted to the external crystal surfaces. This is a likely consequence of partial framework collapse, which occurs following coordination of Co2(CO)6 to the alkyne groups. Coordination changes the preferred angle between carboxylate groups in the edb ligand, which in turn introduces strain into the network.
Introduction
Since the demonstration that coordination network structures can exhibit permanent porosity,1 there has been an enormous surge of interest in this class of compound.2–6 Much of the interest in these materials, often referred to as metal–organic frameworks (MOFs), has related to their hydrogen adsorption and storage properties.7 More recently, however, areas such as catalysis,8carbon capture9 and drug delivery10 have become increasingly important. As the field matures, there has also been increasing interest in MOFs based on linkers that contain additional functionalities. As part of a broader programme on functionalised MOFs,11,12 we were attracted to the ligand 4,4′-ethynylenedibenzoate (edb) because of the potential for further reaction of the carbon-carbon triple bond after the ligand had been incorporated into a MOF structure.
There are two previous reports of the use of the edb ligand to prepare MOFs. In 2008, Hupp, Nguyen and co-workers reacted H2edb with zinc(II) in the presence of 4,4′-bipyridyl (bipy) to give [Zn(edb)(bipy)], which exhibits a four-fold interpenetrated structure containing distorted tetrahedral zinc(II) centres that are inter-linked by edb and bipy ligands.13 Introducing substituents onto the 3- and 3′- positions of the benzene rings on the edb ligands changes the nature of the network. The resultant structures are based on ‘paddle wheel’ secondary building units (SBUs) that are connected by the linkers into three-dimensional networks, with the degree of interpenetration dependent on the size of the substituent. Also in 2008, Song and co-workers reported the syntheses and structures of [Zn2(edb)2(DMSO)2]·1.6H2O and [Eu2(edb)3(DMSO)2(MeOH)2]·2DMSO·3H2O.14 The zinc compound has a two-dimensional network structure in which paddle wheel SBUs are linked by the edb ligands, with DMSO ligands in the axial positions preventing the network from extending in the third dimension. The structure of the europium compound consists of Eu2(O2CR)6 SBUs that are linked into a three-dimensional network.
There were two main aims of the research described in this paper. Firstly, we wanted to determine whether this ligand could form networks that are isoreticular to known series of MOFs, thereby broadening the range of networks based on edb. Secondly, we wanted to establish whether the carbon-carbon triple bond could be modified post-synthetically15 by coordination to an organometallic centre. Such post-synthetic modifications through coordination of a metal fragment to a MOF are relatively rare. Coordination of a Cr(CO)3 fragment to the benzene rings of a 1,4-benzenedicarboxylate linker has been reported by Kaye and Long,16 whereas Lin and co-workers modified a cadmium MOF by coordinating a titanium centre to give an enantioselective catalyst.17 Coordination of catalytic metal centres to frameworks in which an amine or alcohol has been post-synthetically converted to a polydentate ligand has also been reported.18–23
Results and discussion
With the first of these aims in mind, we initially targeted the well-established IRMOF series of MOFs, developed by Yaghi and co-workers.24 These materials, when activated to remove included solvent molecules, have the general formula [Zn4O(dicarboxylate)3] and form cubic networks. We used similar reaction conditions to those we had previously employed for preparing zinc MOFs with 2-substituted 4,4′-biphenyldicarboxylate ligands.11 Thus, the reaction between Zn(NO3)2·6H2O and H2edb in DMF was carried out at 80 °C to produce cream-coloured cubic crystals. These were shown by single crystal X-ray crystallography to be [Zn4O(edb)3(H2O)2]·6DMF 1. Compound 1 has a structure based on Zn4O(O2CR)6 SBUs that are connected by linear linkers into a cubic network structure, which is doubly interpenetrated. This establishes that 1 is isoreticular to the IRMOFs.A closer look at the Zn4O(O2CR)6 SBU (Fig. 1a) reveals the presence of two water molecules coordinated to one of the zinc(II) centres, giving this metal centre [Zn(3)] distorted octahedral geometry. The cis-oxygen atoms are related by angles at Zn(3) that range from 69.7(2) to 86.3(4)°, while the trans-oxygen atoms are related by angles of between 167.8(3) and 172.4(4)°. This compares with angles between 99.85(14)° and 118.6(3)° for the distorted tetrahedral zinc centres [Zn(1) and Zn(2)]. The water molecules project into the pores of the interpenetrated structure. The change in metal ion geometry from tetrahedral to octahedral does not significantly affect the geometry of the SBU, nor compromise the formation of the cubic network. Similar observations have been made in the structures of isoreticular MOFs containing 4,4′-biphenyldicarboxylate ligands functionalised as the 2-position with an aldehyde,11hydrazone11 or sulfide group.12
![The structure of [Zn4O(edb)3(H2O)2]·6DMF 1, showing (a) the Zn4O(O2CR)6(OH2)2 SBU, with the primed atom generated by the symmetry operation x, –y, z, (b) the cubic network structure, and (c) the interpenetration, with the red spheres representing the largest sphere that can fit into the pores between the networks following removal of the included solvent molecules.](/image/article/2012/CE/c1ce06044a/c1ce06044a-f1.gif) | ||
| Fig. 1 The structure of [Zn4O(edb)3(H2O)2]·6DMF 1, showing (a) the Zn4O(O2CR)6(OH2)2 SBU, with the primed atom generated by the symmetry operation x, –y, z, (b) the cubic network structure, and (c) the interpenetration, with the red spheres representing the largest sphere that can fit into the pores between the networks following removal of the included solvent molecules. | ||
As seen in Fig. 1b, the eight edb ligands on two opposite sides of each cube in the network contain benzene rings that are almost co-planar (the angle between the two mean ring planes is 10°), whereas the four edb ligands linking these faces together contain benzene rings that are perpendicular to each other (90° imposed by crystallographic symmetry). Despite its interpenetrated structure, 1 has significant pore volume in the form of channels that are approximately 12.0 Å in width. These contain diffuse solvent molecules, estimated by the PLATON SQUEEZE routine as six DMF molecules per formula unit. Fig. 1c shows the structure viewed down the a-axis. The red spheres represent the largest van der Waals sphere (diameter 16.0 Å) that could occupy the pore space without touching the framework itself.
The thermogravimetric analysis (TGA) of 1 revealed a mass loss between 150 °C and 200 °C of 37%, representing removal of the included solvent molecules. This mass loss is consistent with eight DMF molecules per formula unit (calc. 36.7%), and is larger than that expected on the basis of the crystal structure. This difference could reflect inaccuracies in the estimation of the quantity of highly disordered solvent molecules in pores of this size. After solvent loss the MOF is stable until 300 °C, when it decomposes.
Prior to recording N2 adsorption measurements, 1 was heated at 250 °C for 8 h. An X-ray powder diffraction pattern recorded after this pre-treatment confirmed it had not lost crystallinity. Fig. 2a shows the N2 isotherm for the activated sample of 1 in comparison with the simulated adsorption based on a GCMC protocol. The BET surface area for 1, calculated using the Rouquerol method,25 gives a value of 1088 m2 g−1, which is approximately one-third the value suggested by the simulations (3156 m2 g−1). This difference may be related to either incomplete activation or partial framework collapse on activation. Interestingly, as shown in the log plot in Fig. 2b, there is initially higher experimental uptake than anticipated from the simulations. This means that the solid-fluid energy is higher in the experiments than in the simulations, which in turn implies that something is creating stronger adsorptions sites than would be expected from the crystallographic data. Although the nature of this is unknown, the presence of something in the pores facilitating the increased initial uptake is consistent with the lower than expected total nitrogen uptake.26
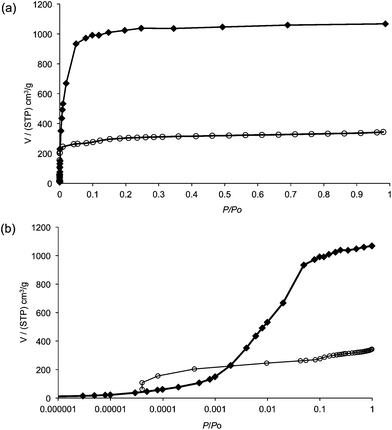 | ||
| Fig. 2 (a) The N2 isotherm for an activated sample of 1 (o) compared with the simulated data (◆), (b) the same data shown in a logarithmic plot to emphasise the initial uptake. | ||
The structure of 1 can be compared with those of [Zn4O(bpdc)3] (IRMOF-9/10, bpdc = 4,4′-biphenyldicarboxylate) and [Zn4O(tpdc)3] (IRMOF-15/16, tpdc = 1,4′,4′′-terphenyldicarboxylate).24 In terms of ligand length, edb (13.8 Å) lies between bpdc (11.2 Å) and tpdc (15.5 Å). Thus, it might be expected that the pore sizes present in the structure of 1 would lie between those in IRMOF-9 and IRMOF-15, both of which are also doubly-interpenetrated. As noted above, the diameter of the largest van der Waals sphere that could occupy the pore space without touching the framework is 16.0 Å, which is greater than the equivalent value for IRMOF-9 (14.5 Å). The equivalent value for IRMOF-15 (12.8 Å) is lower than expected since the two interpenetrated networks are maximally displaced from each other in this structure, in contrast to the interpenetration observed in 1 and IRMOF-9, in which the two networks lie relatively close to each other.
IRMOF-10 and IRMOF-16 are non-interpenetrated isomers of IRMOF-9 and IRMOF-15, respectively, and were crystallised from more dilute solutions. Although an analogous non-interpenetrated network has not been unambiguously established for [Zn4O(edb)3], carrying out the reaction under dilute conditions led to the observation of a small number of crystals with the space group Pm–3m and unit cell length a = 19.549 Å. Given the similarity to the structure of IRMOF-16 (Pm–3m, a = 21.4903(13) Å), these crystals can be tentatively identified as the non-interpenetrated version of 1. Unfortunately, these crystals did not diffract well, thwarting a full analysis by single crystal methods.
The second system targeted was based on cobalt(II). While an extended isoreticular series is not known for cobalt dicarboxylate MOFs, there are a number of structures that contain Co3(O2CR)6 SBUs, in which the six carboxylates radiate at approximately 60° angles from a Co3 hub.27–30 Such networks are not unique to cobalt, and have also been observed for other metals including zinc,31–34cadmium35 and magnesium.36,37 The reaction between Co(NO3)2·6H2O and H2edb in DMF yielded bright pink needle-shaped crystals. These were analysed by single crystal X-ray crystallography and shown to be [Co3(edb)3(DMF)4]·2.6DMF 2. The structure of 2 is based on SBUs that each contain a chain of three Co atoms bridged by dicarboxylate ligands and capped at each end by DMF ligands, as shown in Fig. 3a. These SBUs are linked together by dicarboxylate ligands to form two dimensional sheets, as illustrated in Fig. 3b.
![The structure of [Co3(edb)3(DMF)4]·2.6DMF 2, showing (a) the Cd3(O2CR)6(DMF)4 SBU, with the primed atom generated by the symmetry operation 2 – x, –y, –z, and (b) the two-dimensional network structure. Hydrogen atoms have been omitted for clarity.](/image/article/2012/CE/c1ce06044a/c1ce06044a-f3.gif) | ||
| Fig. 3 The structure of [Co3(edb)3(DMF)4]·2.6DMF 2, showing (a) the Cd3(O2CR)6(DMF)4 SBU, with the primed atom generated by the symmetry operation 2 – x, –y, –z, and (b) the two-dimensional network structure. Hydrogen atoms have been omitted for clarity. | ||
There are two types of cobalt environments in the MOF. The two terminal cobalt atoms in each Co3(O2CR)6 SBU [Co(1)] exhibit distorted octahedral geometries, forming four bonds to edb oxygen atoms and, in addition, two bonds to DMF oxygen atoms. The central cobalt atom [Co(2)] forms six bonds to edb oxygen atoms. Four of the six carboxylate groups in the SBU are in the bridging bidentate coordination mode, with the other two chelating/bridging bidentate. This latter coordination mode enforces a narrow bond angle at Co(1) [O(4)′–Co(1)–Co(3)′ 60.51(13)°], which is the biggest contributor to the distortion from a regular octahedral geometry at this metal centre. The sheets in the structure stack in an ABA fashion, with the DMF ligands of one sheet projecting into the triangular pores present in the neighbouring sheets. The structure of 2 is related to those previously reported for the bpdc compounds [Co3(bpdc)3(DMA)2]·DMA·EtOH28 and [Zn2Co(bpdc)3(DMF)2]·4DMF27 which both have similar two-dimensional sheets, and for [NH2Me2]2[Co3(bpdc)4]·2MeOH·1.5DMF29 in which analogous sheets are pillared by further bpdc linkers to form an interpenetrated three-dimensional network.
The X-ray powder diffraction pattern for 2 matches that simulated from the crystal structure, revealing that the bulk material is homogeneous. However, crystals of 2 rapidly lose solvent, with an accompanying loss of crystallinity, suggesting that 2 is not a good candidate for gas adsorption studies. When crystals of 2 were placed under vacuum, they changed colour from pink to a deep blue. This is consistent with loss of the coordinated DMF molecules from the terminal cobalt centres, leading to a change in metal geometry. Similar observations were seen when the pink crystals of 2 were soaked in non-coordinating solvents such as CHCl3, though the material lost crystallinity during this process, so hampering analysis of the material by X-ray techniques.
The presence of the alkyne group on the edb ligand and the relatively large, accessible pore space suggested that 1 might be an appropriate candidate for our second goal of post-synthetic modification. The possibility of coordinating the alkyne group to a metal centre was investigated by soaking a sample of the MOF in a CH2Cl2 solution of [Co2(CO)8] under nitrogen for several days. This led to a colour change of the crystals, but on observation under a microscope it was found that this colour change was limited to the external surfaces of the crystals, with the interiors unaltered. The X-ray powder diffraction patterns remained unaltered, confirming that no change in the gross structure had occurred. Infrared spectroscopy revealed the presence of additional peaks at 2027, 2055 and 2092 cm−1 following the reaction, all of which compare well to the expected values for ν(CO) in a complex of the type [Co2(CO)6(PhC≡CPh)] (2025, 2050 and 2090 cm−1).38 Notably, the bridging carbonyl band for [Co2(CO)8] (1895 cm−1) is absent, confirming that the observed carbonyl stretches are not simply due to adsorbed starting material.
Why is the post-synthetic modification restricted to the external crystal surfaces? The pore windows of the MOF are approximately 12.0 × 12.0 Å whereas [Co2(CO)8] has approximate dimensions 7.4 × 7.5 × 8.8 Å, so the carbonyl complex is small enough to enter into the MOF. When a metal fragment coordinates to an alkyne, the carbon-carbon bond order is reduced, and as a consequence the substituents are normally observed to bend away from the metal centre(s). For example, in the crystal structure of [Co2(CO)6(PhC≡CPh)], each phenyl ring has rotated by 40° from the linearity observed in PhC≡CPh.39 In a MOF, the resultant change in the preferred angle between the two coordinating carboxylate groups in the linker will introduce strains into the network. Such strains might lead to partial collapse at the surface of the framework, thus preventing further reaction by blocking entrances to the crystal interior.
In conclusion, we have demonstrated that edb can form MOFs that are isoreticular with those formed by other dicarboxylates such as the shorter bpdc, and that the pore volumes of the resultant MOFs are consistent with the length of the linkers. Post-synthetic modification reactions with metal carbonyls reveal that the carbon-carbon triple bond can coordinate to a metal fragment, though reactions to date have been limited to modifications of the external crystal surfaces. We are currently working to circumvent this limitation with a view to extending the range of post-synthetic modification reactions possible with MOFs containing the edb linker.
The EPSRC are thanked for financial support
Experimental
General synthetic details and the synthesis of 4,4′-ethynylenedibenzoic acid (H2edb) are described in the ESI.Synthesis of [Zn4O(edb)3(H2O)2]·6DMF 1
Zn(NO3)2·6H2O (0.160 g, 0.54 mmol) and H2edb (0.049 g, 0.16 mmol) were placed in a pressure tube with DMF (10 cm3). The reaction mixture was heated to 80 °C in an oil bath until the solids had dissolved, then stirred for 10 min. The reaction mixture was then transferred to a programmable oven where it was heated at 80 °C for 5 days, before cooling slowly to room temperature. The resultant cream crystals of 1 were separated by filtration. Yield 0.050 g (61%). Samples of 1 were activated to remove the DMF by heating at 250 °C for 8 h, and this activated sample was used for the microanalysis. Found: C, 51.7; H, 2.55%. Calc. for C48H28O15Zn4 (1 – 6DMF) C, 52.1; H, 2.55%.Synthesis of [Co3(edb)3(DMF)4]·2.6DMF 2
Co(NO3)2·6H2O (0.079 g, 0.27 mmol) and H2edb (0.080 g, 0.27 mmol) were placed in a pressure tube with DMF (10 cm3) and heated at 80 °C for 5 days, before being cooled slowly to room temperature. Pink crystals were formed which were kept under fresh DMF until required for characterisation. Yield 0.048 g (52%). These crystals rapidly decomposed on removal from DMF, preventing an accurate microanalysis from being obtained. However, the identity of the bulk material was confirmed by comparing the X-ray powder diffraction pattern with that simulated from the crystal structure (Fig. S3).Solvatochromism was observed by placing the crystals in a small amount of solvent in a round bottom flask, which was then placed under vacuum. After the solvent evaporated the crystals slowly changed colour to a deep blue. The blue crystals were seen to break down in air, so were kept under nitrogen. On addition of DMF they returned to their original pink colour but appeared to have lost some crystallinity as they had become opaque. The loss of crystallinity was confirmed by X-ray powder diffraction.
The reaction of 1 with [Co2(CO)8]
[Co2(CO)8] (0.010 g, 0.029 mmol) was placed in a Schlenk tube, and dissolved in dried, deoxygenated dichloromethane (10 cm3). This solution was transferred under nitrogen to a second Schlenk tube containing 1 (0.020 g, 0.013 mmol), and the reaction mixture was left for 5 days at room temperature. After this time, the resultant dark red crystals were separated by filtration and examined under a microscope before IR analysis.Crystallography
The data reported for 1 were collected at 200 K, as data collections at lower temperatures were hampered by a possible phase transition. The crystals decomposed in the X-ray beam, so low sigmas on the intensity data were sacrificed in order to get a ‘complete’ data set before degradation of the sample. This adversely affected R(int), such that data were truncated to 22°. The asymmetric unit for 1 consists of one full zinc atom, one full edb ligand, two half-occupancy zinc atoms (located at special positions coincident with a crystallographic mirror plane in the space group symmetry), one half of a edb ligand (wherein O6, O7, O8, C18–26, C29, C30, O8 – and associated hydrogens where relevant also lie on the mirror plane), two half water molecules (O9, O10 – also on the mirror symmetry element), and approximately three DMF molecules. The water hydrogen atoms could not be located reliably, and hence were omitted from the final least squares refinement. The DMF was very diffuse/disordered. Hence the data were subjected to the PLATON SQUEEZE routine, from which the estimate of three molecules per asymmetric unit arose. This solvent was accounted for in the unit cell contents during the final least squares cycles.The asymmetric unit for 2 consists of one and a half cobalt centres, one full edb ligand, one half of an edb ligand, one DMF ligand and two areas of free solvent. The latter comprises of one DMF fragment with 50% occupancy plus one DMF fragment with 80%. The partial solvent was refined subject to similarity restraints for the bond distances and ADPs therein. There was clearly some disorder in these two areas, which is not unsurprising, but this could not be readily modelled. The largest difference peak in the electron density map is associated with the free solvent.
References
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- R. Robson, Dalton Trans., 2008, 5113 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284 RSC.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482 CrossRef CAS.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Chem. Commun., 2009, 4218 RSC.
- T. Gadzikwa, B. S. Zeng, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 3672 RSC.
- B. T. N. Pham, L. M. Lund and D. Song, Inorg. Chem., 2008, 47, 6329 CrossRef CAS.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498 RSC.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2008, 130, 806 CrossRef CAS.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS.
- M. J. Ingleson, J. Perez Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680 RSC.
- X. Zhang, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2009, 265, 155 CrossRef CAS.
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492 CrossRef CAS.
- T. Gadzikwa, O. K. Farha, K. L. Mulfort, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2009, 3720 RSC.
- K. K. Tanabe and S. M. Cohen, Angew. Chem., Int. Ed., 2009, 48, 7424 CrossRef CAS.
- S. Bhattacharjee, D.-A. Yang and W.-S. Ahn, Chem. Commun., 2011, 47, 3637 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552 CrossRef CAS.
- M. T. Luebbers, T. Wu, L. Shen and R. I. Masel, Langmuir, 2010, 26, 11319 CrossRef CAS.
- Y. Wang, B. Bredenkötter, B. Rieger and D. Volkmer, Dalton Trans., 2007, 689 RSC.
- S. H. Kim, H. S. Huh and S. W. Lee, J. Mol. Struct., 2007, 841, 78 CrossRef CAS.
- F. Luo, Y.-X. Che and J.-M. Zheng, Cryst. Growth Des., 2009, 9, 1066 CAS.
- Y. Du, A. L. Thompson, N. Russell and D. O'Hare, Dalton Trans., 2010, 39, 3384 RSC.
- M. Edgar, R. Mitchell, A. M. Z. Slawin, P. Lightfoot and P. A. Wright, Chem.–Eur. J., 2001, 7, 5168 CrossRef CAS.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC.
- H. Kim, G. Park and K. Kim, CrystEngComm, 2008, 10, 954 RSC.
- S. M. Hawxwell, H. Adams and L. Brammer, Acta Crystallogr., Sect. B: Struct. Sci., 2006, B62, 808 CAS.
- A. D. Burrows, K. Cassar, T. Düren, R. M. W. Friend, M. F. Mahon, S. P. Rigby and T. L. Savarese, Dalton Trans., 2008, 2465 RSC.
- R. P. Davies, R. J. Less, P. D. Lickiss and A. J. P. White, Dalton Trans., 2007, 2528 RSC.
- C. A. Williams, A. J. Blake, C. Wilson, P. Hubberstey and M. Schröder, Cryst. Growth Des., 2008, 8, 911 CAS.
- H. Sternberg, H. Greenfield, R. A. Friedel, J. Wotiz, R. Markby and I. Wender, J. Am. Chem. Soc., 1954, 76, 1457 CrossRef CAS.
- D. Gregson and J. A. K. Howard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1024 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 839201 and 839202. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce06044a |
| This journal is © The Royal Society of Chemistry 2012 |