DOI:
10.1039/C1CE06015E
(Paper)
CrystEngComm, 2012,
14, 160-168
In situ syntheses, crystal structures and magnetic properties of CuII and MnII coordination assemblies based on a novel heteroalicyclic dicarboxylate tecton and N-donor co-ligands†
Received
6th August 2011
, Accepted 27th September 2011
First published on 10th November 2011
Abstract
This work focuses on coordination chemistry of a heteroalicyclic dicarboxylate ligand 5,6-dihydro-1,4-dithiin-2,3-dicarboxylate (L) with transition metals in the presence of N-donor co-ligands. A series of five CuII and MnII coordination complexes with L building blocks and different chelating/bridging co-ligands have been prepared, namely [Cu(L)(phen)]n (1), [Cu(L)(4bpy)(H2O)]n (2), {[Cu2(L)2(μ3-OH)](H2dabco)0.5(H2O)}n (3), {[Mn2(L)(phen)4](H2O)2(ClO4)2}2 (4), and {[Mn2(L)(2bpy)4](2bpy)0.5(ClO4)2}2 (5) (phen = 1,10-phenanthroline, 4bpy = 4,4′-bipyridine, dabco = 1,4-diazabicyclo[2,2,2]octane, 2bpy = 2,2′-bipyridine), in which the ligand L is obtained by an in situhydrolysis reaction of 5,6-dihydro-1,4-dithiin-2,3-dicarboxylic anhydride in the presence of lithium hydroxide. Single-crystal X-ray diffraction reveals that these complexes display a variety of coordination motifs, from the discrete tetranuclear species (4 and 5) to infinite 1-D arrays (1 and 3) and 2-D → 3-D polycatenated architecture (2), which are regulated by the multiple coordination modes of L. Furthermore, the introduction of auxiliary N-heterocyclic ligands plays a critical role in extending the dimensionality of these metallosupramolecular systems via coordination and/or secondary interactions such as hydrogen bonds and aromatic stacking. The magnetic properties and thermal stability of these complexes have also been investigated and discussed in detail.
Introduction
As a flourishing research field, the rational design and construction of metallosupramolecular materials, especially coordination polymers, has attracted intensive attention both experimentally and theoretically, because of not only their intriguing and often complicated crystalline architectures,1 but also their potential applications as functional materials in several aspects such as photoluminescence,2 catalysis,3 ion exchange,4 gas storage,5 magnetism,6 and so forth.7 Though numerous coordination systems have been documented so far, to accurately predict and control the resulting crystalline lattices still represents a great challenge in synthetic chemistry owing to the unpredictability of the assembly process.8 For example, the in situ metal–ligand reaction may occur during the coordination assemblies, which is currently regarded as a feasible and effective strategy to obtain novel coordination solids that are inaccessible or not easily achieved by the conventional methods.9
Currently, ligands containing the carboxylate group have been mostly used in the construction of various coordination architectures, in virtue of its versatile coordination modes, such as unidentate, chelating, and bridging in syn-syn, syn-anti, or anti-anti configuration,10 which are also closely related to the magnetic properties of the resulting coordination systems. In this regard, polycarboxylate ligands, especially those based on rigid aromatic backbones (benzene-, naphthalene-, anthracene-, and pyrene-based derivatives) have been applied to successfully prepare numerous polymeric coordination complexes with interesting structures and properties, where benzenedicarboxylate,111,3,5-benzenetricarboxylate,12naphthalenedicarboxylate,139,10-anthracene-dicarboxylate,14 and 2,7-pyrenedicarboxylate15 represent the most typical examples. To the contrary, only limited studies on coordination assemblies based on alicyclic or heteroalicyclic multicarboxylate ligands have been known.16 Furthermore, the carboxylate-bridged CuII and MnII species have been usually acknowledged as potential molecule-based magnetic materials.17
On the other hand, the introduction of auxiliary neutral N-donor ligands can significantly influence the coordination assembled systems with multicarboxylate ligands. For instance, the exo-bidentate rod-like ligands such as 4,4′-bipyridine (4bpy)18 and 1,4-diazabicyclo[2,2,2]octane (dabco),19 normally serve as ideal connectors between the metal ions for the propagation of coordination networks from 1-D to 3-D due to their two potential divergent coordination sites. Notably, the ligand dabco can display different forms (neutral dabco,19a,b monoprotonated Hdabco+,19c,d and diprotonated H2dabco2+19e) depending on the pH conditions. Meanwhile, the terminal chelating ligands such as 2,2′-bipyridine (2bpy)20 and 1,10-phenanthroline (phen),20c,21 favor the formation of low-dimensional patterns, but have the ability to construct complicated supramolecular networks through aromatic interactions.
Recently, we have reported a novel 3-D AgI coordination polymer based on a dicarboxylic acid with a heteroalicyclic skeleton, namely 5,6-dihydro-1,4-dithiin-2,3-dicarboxylate (L), which was formed in situ from the hydrolysis of 5,6-dihydro-1,4-dithiin-2,3-dicarboxylic anhydride (dhca, see Scheme 1).22 In this case, the ligand L displays multiple coordination modes using all of the potential coordination sites. Herein, we will present the in situ synthesis, crystal structures, thermal stability, and magnetic properties of a series of CuII and MnII complexes with L as the primary building block and different N-donor auxiliary co-ligands.
 |
| | Scheme 1 | |
Experimental section
Materials and general methods
All the starting reagents and solvents for synthesis were commercially available and used as received without further purification. Elemental analyses of C, H, and N were performed on a Vario EL III Elementar analyzer. IR spectra were recorded in the range of 4000–400 cm−1 on a Bruker Tensor 27 OPUS FT-IR spectrometer with KBr pellets. Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker D8 Advance diffractometer (Cu-Kα, λ = 1.54056 Å) at 40 kV and 100 mA, by using a Cu-target tube and a graphite monochromator. The powder samples were prepared by crushing the single crystal products and the intensity data were recorded by continuous scan in a 2θ/θ mode from 3° to 80° with a step size of 0.02° and a scan speed of 8° min−1. Simulation of the PXRD patterns was taken by the single-crystal data and diffraction-crystal module of Mercury (Hg). Thermogravimetric and differential thermal analysis (TG-DTA) experiments were carried out on a Perkin-Elmer Diamond SII thermal analyzer from room temperature to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
Preparation of complexes 1–5
[Cu(L)(phen)]n (1).
To an aqueous solution (10 mL) of dhca (0.05 mmol, 9.4 mg) and phen (0.05 mmol, 9.0 mg), LiOH (0.1 mmol, 2.4 mg) was added with vigorous stirring and heating. Then, Cu(ClO4)2·6H2O (0.05 mmol, 18.5 mg) was mixed with the above clear solution. After stirring for ca. 5 min, the resultant solution was filtered and left to stand under ambient conditions. Blue block crystals of 1 were obtained after a period of one week by slow solvent evaporation. Yield: ca. 30% (6.7 mg, based on phen). Anal. Calc. for C18H12CuN2O4S2: C, 48.26; H, 2.70; N, 6.25%. Found: C, 48.35; H, 2.78; N, 6.39%. IR (cm−1): 1576s, 1521w, 1432m, 1377m, 1327s, 1148w, 1082w, 1050w, 862m, 785w, 748m, 721m, 468w.
[Cu(L)(4bpy)(H2O)]n (2).
A mixture of dhca (0.05 mmol, 9.4 mg), 4bpy (0.05 mmol, 7.8 mg) and LiOH (0.1 mmol, 2.4 mg) was added to water (5 mL). The suspension was stirred and heated until the solids were dissolved. After cooling, the solution was filtered into the bottom of a straight glass tube, above which a CH3OH/H2O buffer (10 mL, v/v = 1/1) was placed. A methanol solution (5 mL) of Cu(ClO4)2·6H2O (0.05 mmol, 18.5 mg) was carefully layered onto the buffer. Upon evaporation of the solvents, blue block crystals of 2 were observed on the tube wall after ca. 2 weeks. Yield: ca. 50% (11.0 mg, based on 4bpy). Anal. Calc. for C16H14CuN2O5S2: C, 43.48; H, 3.19; N, 6.34%. Found: C, 43.59; H, 3.25; N, 6.49%. IR (cm−1): 3385bm, 1603vs, 1549m, 1490w, 1462w, 1414m, 1325s, 1222m, 1074m, 920w, 822m, 752w, 726w, 698m, 644w, 474w.
{[Cu2(L)2(μ3-OH)](H2dabco)0.5(H2O)}n (3).
The same synthetic procedure as that for 1 was used except that phen was replaced by dabco (0.05 mmol, 5.6 mg), affording dark-blue block crystals of 3 after three days upon slow evaporation of solvent. Yield: ca. 40% (6.3 mg, based on dhca). Anal. Calc. for C15H18Cu2NO10S4: C, 28.76; H, 2.90; N, 2.24%. Found: C, 28.59; H, 2.78; N, 2.46%. IR (cm−1): 3487bm, 2894bm, 1601s, 1571vs, 1466w, 1380vs, 1083m, 989w, 927w, 787w, 749m, 706m, 468w.
{[Mn2(L)(phen)4](H2O)2(ClO4)2}2 (4).
The same synthetic procedure as that for 1 was used except that the metal salt was replaced by Mn(ClO4)2·6H2O (0.05 mmol, 18.1 mg), producing yellow block crystals of 4 after ca. one week. Yield: ca. 20% (3.2 mg, based on phen). Anal. Calc. for C54H40Cl2Mn2N8O14S2: C, 51.08; H, 3.17; N, 8.82%. Found: C, 51.15; H, 3.16; N, 8.85%. IR (cm−1): 3414bm, 1597s, 1517m, 1425s, 1373w, 1342w, 1144w, 1100vs, 848m, 775w, 727m, 624m.
{[Mn2(L)(2bpy)4](2bpy)0.5(ClO4)2}2 (5).
The same synthetic method as that for 1 was used except that the metal salt was replaced by Mn(ClO4)2·6H2O (0.05 mmol, 18.1 mg) and phen was replaced by 2bpy (0.05 mmol, 7.8 mg), producing yellow block crystals of 5 after ca. one week. Yield: ca. 16% (2.1 mg, based on 2bpy). Anal. Calc. for C51H40Cl2Mn2N9O12S2: C, 50.41; H, 3.32; N, 10.38%. Found: C, 50.29; H, 3.38; N, 10.46%. IR (cm−1): 1599s, 1490w, 1474m, 1439s, 1373m, 1316w, 1246w, 1157w, 1099vs, 1015m, 767m, 738w, 707w, 649w, 623m, 413w.
Caution! Metal perchlorate complexes in the presence of organic ligands are potentially explosive. Only a small amount of material should be handled with great care.
X-ray data collection and structure determination
Single-crystal X-ray diffraction data for all complexes were collected on a Bruker Smart 1000 CCD area-detector diffractometer at room temperature with Mo-Kα radiation (λ = 0.71073 Å) by ω scan mode. The program SAINT23 was used for integration of the diffraction profiles and a semi-empirical absorption correction was applied using the SADABS program.24 All the structures were solved by direct methods using the SHELXS program of the SHELXTL package and refined by full-matrix least-squares methods with SHELXL.25 Metal ions in all the complexes were located from the E-maps, and the other non-H atoms were located in successive difference Fourier syntheses and refined with anisotropic thermal parameters on F2. Hydrogen atoms except for those of water were generated theoretically and refined with isotropic thermal parameters riding on the parent atoms. Crystallographic data and structural refinement details for 1–5 are summarized in Table 1. Selected bond parameters for 1–5 are listed in Table S1, ESI.†
| |
1
|
2
|
3
|
4
|
5
|
| Empirical formula |
C18H12CuN2O4S2 |
C16H14CuN2O5S2 |
C15H18Cu2NO10S4 |
C108H80Cl4Mn4N16O28S4 |
C102H80Cl4Mn4N18O24S4 |
| Formula weight |
447.96 |
441.95 |
627.62 |
2539.68 |
2431.64 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P21/c |
C
2
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
P
|
|
a/Å |
7.9092(5) |
15.308(3) |
7.7845(5) |
13.9737(7) |
14.4203(8) |
|
b/Å |
11.2366(6) |
16.129(2) |
12.2006(6) |
13.9846(6) |
14.8169(7) |
|
c/Å |
19.2416(10) |
7.999(2) |
12.2707(6) |
16.1404(8) |
15.5464(7) |
|
α/° |
90 |
90 |
109.260(4) |
112.092(4) |
89.536(4) |
|
β/° |
95.413(6) |
110.94(3) |
94.393(5) |
98.552(4) |
62.541(5) |
|
γ/° |
90 |
90 |
99.606(4) |
104.588(4) |
65.838(5) |
| V/A3 |
1702.42(17) |
1844.4(7) |
1073.86(10) |
2722.1(2) |
2621.5(2) |
|
Z
|
4 |
4 |
2 |
1 |
1 |
| D/g cm−3 |
1.748 |
1.592 |
1.941 |
1.549 |
1.540 |
|
μ/mm−1 |
1.557 |
1.440 |
2.424 |
0.714 |
0.735 |
|
R
int
|
0.0264 |
0.0631 |
0.0181 |
0.0452 |
0.0218 |
| GOF |
0.994 |
1.023 |
0.996 |
0.981 |
1.011 |
|
R
1, wR2 [I > 2s(I)] |
0.0303/0.0532 |
0.0585/0.1231 |
0.0376/0.1112 |
0.0511/0.0896 |
0.0448/0.1066 |
|
R
1, wR2 (all data) |
0.0547/0.0553 |
0.0845/0.1292 |
0.0532/0.1153 |
0.1294/0.0966 |
0.0681/0.1198 |
Magnetic studies
Magnetic measurements of 1–4 were carried out in the Unitat de Mesures Magnètiques (Universitat de Barcelona) on polycrystalline samples (ca. 30 mg) with a Quantum Design SQUID MPMS-XL magnetometer equipped with a 5 T magnet. Diamagnetic corrections were calculated using Pascal's constants and an experimental correction for the sample holder was applied. Magnetic susceptibility data were collected for crushed crystalline samples of 1–4 in the 2 to 300 K temperature range at an applied field of 0.3 T for 4 and 1 T for the other three samples. Additional data were collected at an applied field of 200 G in the 2–30 K temperature range and no field-dependence of the low temperature data was observed for any of the samples.
Results and discussion
Synthesis and characterization
All complexes were obtained by in situ reaction of dhca with the corresponding metal perchlorate in the presence of N-donor co-ligands and LiOH. According to our experimental results, when a direct combination of different CuII or MnII salts with relevant ligands was carried out in solution, single crystals of complexes 1 and 3–5 suitable for X-ray diffraction could be obtained by solvent evaporation, with the pH value of the reaction solution ca. 6–7. As for 2, a direct solution reaction affords either no evidence of solid formation or a mass of uncharacterized yellow precipitate. It is well known that properly lowering the reaction rate may facilitate the growth of well-shaped single crystals. Considering this point, we have sequentially achieved the isolation of 2 using the solution diffusion method. In each synthetic case, an approximately equimolar amount of reagents was used for the starting materials, which however, was independent of the composition of the products. Significantly, the use of LiOH to adjust the pH value of the reaction systems is a key point for the crystallization of 1–5, and when other basic reagents such as NaOH, KOH, and 2,6-dimethylpyridine were used instead, crystalline products suitable for X-ray diffraction could not be obtained under similar conditions. All complexes are air-stable and insoluble in common organic solvents and water, due to their polymeric and neutral nature. The compositions of complexes 1–5 were completely characterized by IR, elemental analysis, and X-ray single-crystal diffraction techniques. The phase purities of the bulk samples were further identified by powder X-ray diffraction (PXRD) patterns, which display essential similarity to those of the calculated ones (see Fig. S1, ESI†). For the IR spectra of 1–5, the absence of characteristic absorption bands of anhydride (ca. 1750 and 1800 cm−1 for antisymmetric and symmetric stretching vibrations of two carbonyl groups) confirms the hydrolysis of dhca. As a consequence, the antisymmetric and symmetric stretching vibrations of carboxylate groups appear in the range of 1571–1603 and 1380–1439 cm−1 for 1–5, respectively. In addition, the strong and moderate absorption bands observed at ∼1100 and ∼620 cm−1 reveal the presence of perchlorate anions in 4 and 5.
Structural description for complexes 1–5
[Cu(L)(phen)]n (1).
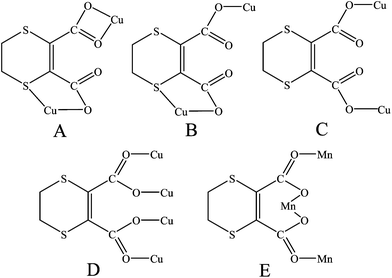
X-ray single crystal diffraction indicates that complex 1 consists of infinite helical coordination arrays with the intrachain Cu⋯Cu separation of 6.7691(5) Å. The asymmetric unit has one CuII ion, one L, and one phen ligand. In 1, the CuII center is six-coordinated by one chelating phen ligand, one sulfur and three carboxylate oxygen atoms from two different L ligands (see Fig. 1a), showing a typical Jahn–Teller distortion with significantly longer axial Cu–S and Cu–O2 distances of 2.7690(8) and 2.678(2) Å (see Table S1, ESI† for detailed bond distances). The L ligand takes a tetradentate mode, which utilizes three oxygen atoms of two carboxylate groups and one sulfur atom, and is chelated to two metal ions (see Scheme 2, A). Thus, the CuII centers are extended via the L ligands to form alternate left- and right-handed helical chains with the dangling phen arms, running along the [010] direction (see Fig. 1b) with a pitch of 11.2366(9) Å (equal to b-axis). The phen entities, located on both sides of the coordination chains, possess a large π-system and promote the formation of interchain π–π stacking interactions between the benzene rings (centroid-to-centroid and centroid-to-plane distances: 3.76 and 3.42 Å, see black dashed lines in Fig. S2, ESI†), as well as between benzene and pyridyl rings (centroid-to-centroid and centroid-to-plane distances: 3.55 and 3.43 Å, see red dashed lines in Fig. 1c). As a result, a 2-D achiral layered motif along the (011) plane is generated with the nearest interchain Cu⋯Cu distance of 7.9092(7) Å.
![Crystal structure of 1. (a) A local view showing the coordination environment of CuII (symmetry code: A = −x + 2, y − 1/2, −z + 1/2). (b) View of the right- and left-handed helical arrays along [010].](/image/article/2012/CE/c1ce06015e/c1ce06015e-f1.gif) |
| | Fig. 1 Crystal structure of 1. (a) A local view showing the coordination environment of CuII (symmetry code: A = −x + 2, y − 1/2, −z + 1/2). (b) View of the right- and left-handed helical arrays along [010]. | |
 |
| | Scheme 2 | |
[Cu(L)(4bpy)(H2O)]n (2).
Compared with phen, the rod-like bidentate 4bpy tecton can not only be involved in aromatic stacking contacts, but also be effective to extend the lower-dimensional motifs into higher-dimensional frameworks via coordination bonds. In this context, we used 4bpy instead of phen as an auxiliary ligand, forming a novel 2-D → 3-D polycatenated architecture. Complex 2 crystallizes in the acentric space groupC2 with the Flack parameter of 0.08(3). The asymmetric unit contains one CuII ion, one L, one 4bpy, and one water ligand. As shown in Fig. 2a, each octahedral CuII ion is surrounded by two carboxylate oxygen atoms from one L and two pyridyl nitrogen donors of 4pby in the equatorial plane, as well as one sulfur atom of L and one aqua molecule occupying the axial positions. As expected, the coordination geometry shows Jahn–Teller elongation with Cu–S and Cu–Oaqua bond distances [2.756(3) and 2.8070(78) Å respectively] being significantly longer than those of the other Cu–Ocarboxylate and Cu–N (see Table S1, ESI† for detailed bond distances). In this case, each L connects two CuII centers using two unidentate carboxylate donors and one sulfur donor (see Scheme 2, B). This binding mode leads to the generation of a 1-D coordination pattern along the [001] direction. Then, such 1-D [CuL]n chains are linked by the 4bpy bridges in two nearly vertical directions of [110] and [10], which result in a 2-D (4,4) network (see Fig. 2b). Each CuII ion acts as a 4-connected vertex of the layer, which shows nearly parallelogram meshes with the dimensions of 8.00 × 11.12 Å2, being large enough that it can be threaded by one equivalent grid from another layer. That is, each 4pby of one layer is inserted into one grid of another vertical layer, and vice versa, each grid is penetrated by one 4bpy from the vertical direction. As a consequence, these 2-D arrays are entangled into an unusual 3-D polycatenated architecture (2-D → 3-D, see Fig. 2c). In addition, an intramolecular O–H⋯O bond between water and the uncoordinated carboxylate oxygen is observed (see Table S2, ESI† for details), and the potential solvent-accessible area is 142.6 Å3 (7.7% of the unit cell volume), as calculated by the PLATON software.26
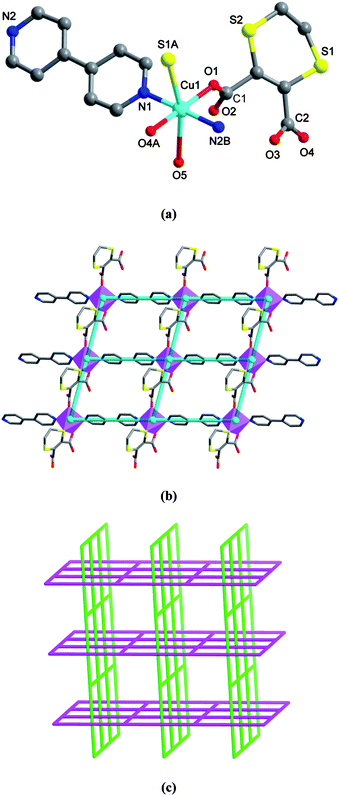 |
| | Fig. 2 Crystal structure of 2. (a) A local view showing the coordination environment of CuII (symmetry codes: A = x, y, z − 1; B = x − 1/2, y − 1/2, z). (b) 2-D (4,4) coordination network, in which the turquoise balls represent the 4-connected CuII centers. (c) Schematic representation of the 2-D → 3-D polycatenated supramolecular architecture. | |
{[Cu2(L)2(μ3-OH)](H2dabco)0.5(H2O)}n (3).
In complex 3, the cyclic aliphatic diamine dabco is introduced to act as the counter cation in its protonated form. There are two independent CuII ions, two L ligands, one hydroxyl group as well as half a diprotonated H2dabco, and one lattice water molecule in each asymmetric unit. As shown in Fig. 3a, the Cu1 atom is four-coordinated by three carboxylate oxygen atoms from distinct L ligands and one hydroxyl in μ3-mode, to complete a nearly ideal square planar geometry. Differently, Cu2 adopts a square-pyramidal coordination sphere with the τ27 parameter of 0.05, where the basal plane is composed of three carboxylate oxygen donors and one μ3-OH group, and the apical position is occupied by another hydroxyl ion. Detailed bond parameters for both CuII coordination spheres are listed in Table S1, ESI†. The two unique L ligands in 3 show different coordination modes, acting as 2-connected (see Scheme 2, C) or 4-connected bridges (see Scheme 2, D). As a result, two pairs of such L ligands and two μ3-OH groups join four CuII centers to generate a tetranuclear subunit, and these subunits are further propagated to form a 1-D array along [100] (see Fig. 3b). Within the subunit, the neighboring Cu1⋯Cu2 separations are 3.1936(5) and 3.3192(8) Å, and the Cu2⋯Cu2 distance of 2.8888(5) Å is significantly shorter. Additionally, the lattice water molecules are also involved in hydrogen bonds with the coordination chains to further stabilize the structure (see Table S2, ESI† for details).
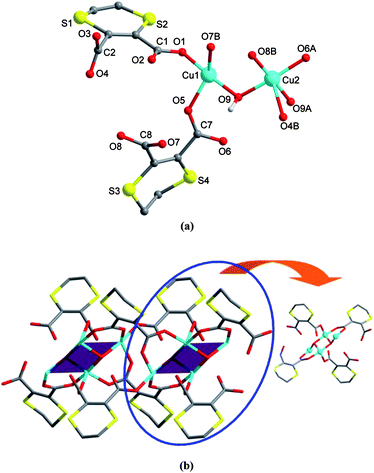 |
| | Fig. 3 Crystal structure of 3. (a) A local view showing the coordination environments of CuII centers (symmetry codes: A = −x + 2, −y, −z; B = −x + 3, −y, −z). (b) A perspective of the 1-D chain, highlighting the tetranuclear subunit. | |
{[Mn2(L)(phen)4](H2O)2(ClO4)2}2 (4) and {[Mn2(L)(2bpy)4](2bpy)0.5(ClO4)2}2 (5).
Complexes 4 and 5 show similar coordination structures except for the nature of the auxiliary chelating ligands (phen for 4 and 2bpy for 5) and the lattice guests (water for 4 and 2bpy for 5). Thus, only the crystal structure of 4 is described herein in detail. X-ray single crystal analysis of 4 indicates a discrete centrosymmetric tetranuclear complex. The asymmetric unit consists of two crystallographically unique MnII centers, one ligand L, four chelating phen ligands, as well as two lattice water and two perchlorate anions. Interestingly, both Mn1 and Mn2 have the coordination geometry of distorted octahedron with the N4O2 coordination environment, formed by four nitrogen donors from two phen ligands and two carboxylate oxygen atoms from L (see Fig. 4a). The difference in the two MnII spheres is that the two oxygen atoms bound to Mn1 are from two L ligands, but those to Mn2 are from the same L ligand. The Mn–N and Mn–O bond distances lie in the range of 2.232(4)–2.337(4) Å and 2.099(3)–2.138(4) Å, being comparable to those found for similar complexes in the literature (see Table S1, ESI† for detailed bond distances). For each L ligand, two carboxylate groups take the same bridging coordination mode to join three MnII centers (see Scheme 2, E).
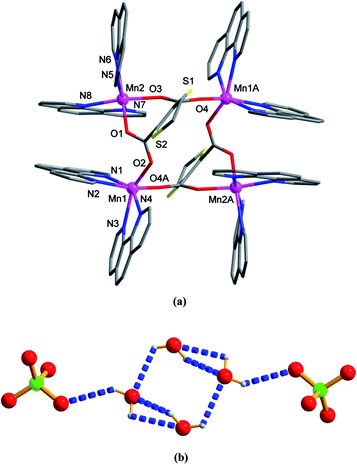 |
| | Fig. 4 Crystal structure of 4. (a) Local coordination environments of MnII (symmetry code: A = −x, y + 2, −z). (b) View of the discrete hexamer linked by hydrogen bonds. | |
Notably, the lattice perchlorate anions and water molecules in 4 are linked to form a [(H2O)4(ClO4)2]2−clustervia O–H⋯O hydrogen bonding (see Table S2, ESI† for details). This hexamer is composed of a water tetramer adopting rectangular geometry with two additional perchlorate pendants at its diagonally opposite ends (see Fig. 4b). In addition, the neighboring tetranuclear coordination units are extended into a 2-D layer running parallel to the (101) plane by numerous intra- and intermolecular π–π stacking interactions (see black dashed lines in Fig. S3, ESI†) between pyridyl and benzene rings of phen in a face-to-face fashion, with the centroid-to-centroid separations ranging from 3.48 to 3.82 Å. These 2-D arrays are arranged in a parallel mode, between which the [(H2O)4(ClO4)2]2−clusters are accommodated (see Fig. S4, ESI†). A calculation of the guest-accessible area reveals the values of 47.1 and 606.9 Å3 before and after excluding the perchlorate and water guests (1.7% and 22.3% per unit cell volume).
Because of the different stacking nature of the auxiliary ligands, no typical stacking interactions exist in 5 (see Fig. S5, ESI†). The final 3-D crystalline lattice is constructed from the tetranuclear units by multiple weak hydrogen bonds, where the 2bpy and perchlorate guests act as the bridges. Examination of this structure with the PLATON program reveals that there are no guest-accessible voids.
Thermal stability of 1–5
Complexes 1–5 are air stable and their thermal stability was studied by TG-DTA experiments (see Fig. S6, ESI† for the TG-DTA curves). The TGA trace reveals that complex 1 remains intact until heating to ca. 165 °C, and then suffers two consecutive sharp weight losses. Accordingly, two intensive peaks are observed at 180 and 256 °C in the DTA curve. The final residue has an 18.26% weight of the total sample and seems to be CuO (calculated: 17.86%). The TGA curve of 2 indicates that the first weight loss of 4.31% occurring between ca. 45 and 85 °C (calculated: 4.07%) may correspond to the elimination of one lattice aqua molecule (a peak at 71 °C in DTA). Then, the residual framework starts to quickly decompose beyond ca. 130 °C with consecutive weight loss (four peaks observed at 147, 189, 208, and 269 °C in DTA) and does not end until heating to 800 °C. As for 3, the original weight loss of 2.12% (calculated: 2.87%) in the range 60–170 °C corresponds to a gradual removal of one lattice water molecule. And this dehydration process is closely followed by a sharp (peaking at 203 and 245 °C in the DTA curve) and then a slow and consecutive weight loss ending at ca. 700 °C. According to the remaining weight of 26.47%, the final residue may be CuO (calculated: 25.35%). With regard to 4, the release of free water molecules is found from ca. 25 to 65 °C (experimental and calculated: 2.41% and 2.84%) with a peak at 53 °C in DTA. The residual component remains largely unchanged until the decomposition starts at ca. 255 °C, upon which a series of consecutive weight losses occur (peaking at 307 and 465 °C in DTA) and do not stop until the heating ends at 800 °C. Complex 5 is thermally stable upon heating to ca. 230 °C, followed by three stages of weight loss peaking at 295, 317, and 360 °C. The weight loss of 75.18% observed from ca. 230 to 450 °C is very close to the calculated value of 74.17%, corresponding to the exclusion of perchlorate as well as lattice and coordinated 2bpy ligands. Then, a slow weight loss occurs until heating to 800 °C (peaking at 711 °C), and the residue holds a weight of 15.15% that corresponds to that of MnO2 (calculated: 14.30%).
Magnetic properties of 1–4
[Cu(L)(phen)]n (1).
The χT product for 1 has a value of 0.43 cm3 K mol−1 at 300 K, in good agreement with that expected for a free CuII ion (S = 1/2, g = 2.0, χT = 0.375 cm3 K mol−1 at 300 K). As the temperature decreases, the χT product remains nearly constant until below 50 K when it drops sharply. A Curie plot is shown in Fig. S7, ESI† in which the solid line is the best fitting to the Curie–Weiss law with the fitting parameters C = 0.43 cm3 K mol−1 and Θ = −1.69 K. The negative value of the Weiss constant indicates weak antiferromagnetic coupling through the L ligand that acts as a tetradentate unit using its four oxygen atoms to bridge the CuII ions. The magnetization vs. field plot for 1 follows the Brillouin law for an isolated CuII ion with g = 2.0, which indicates that even at 2 K the thermal energy is sufficient to overcome the weak antiferromagnetic coupling observed (see Fig. S8, ESI†).
[Cu(L)(4bpy)(H2O)]n (2).
The χT product for 2 has a value of 0.37 cm3 K mol−1 at 300 K, in good agreement with that expected for a free CuII ion (S = 1/2, g = 2.0, χT = 0.375 cm3 K mol−1 at 300 K). As the temperature decreases, the χT product remains nearly constant until below 50 K when it drops sharply. A Curie plot is shown in Fig. S9, ESI†, where the solid line is the best fitting to the Curie–Weiss law with the fitting parameters C = 0.37 cm3 K mol−1 and Θ = −1.16 K. The negative value of the Weiss constant indicates weak antiferromagnetic coupling. This is not unexpected when examining the crystal structure of 2, which consists of interlocked 2-D nets where the CuII ions are linked by 4bpy and the dicarboxylato ligand L. The ligand 4bpy is known to facilitate weak (of about 1–2 K) antiferromagnetic coupling as seen for 2. The magnetization vs. field plot for 2 follows the Brillouin law for an isolated CuII ion with g = 2.0, which indicates that even at 2 K the thermal energy is sufficient to overcome the weak antiferromagnetic coupling observed (see Fig. S10, ESI†).
{[Cu2(L)2(μ3-OH)](H2daco)0.5(H2O)}n (3).
Complex 3 is a coordination polymer where each repeating unit is a tetranuclear CuII cluster. The χT product per repeating [Cu4] unit has a value of 1.87 cm3 K mol−1 at 300 K, in good agreement with that expected for four non-interacting CuII ions (S = 1/2, g = 2.2, χT = 1.81 cm3 K mol−1 at 300 K). As the temperature decreases, the χT product remains nearly constant until below 50 K it drops sharply. Using the spin Hamiltonian Ĥ = −2Jbb[S2·S2′] − 2Jbt[S1·S2 + S1·S2′ + S1′·S2 + S1′·S2′] − 2Jtt[S1·S1′], where Si refers to the spin of the Cu1, Cu1′, Cu2 and Cu2′ atoms (see Scheme 3), and using the equivalent operator approach one can write a Van Vleck equation that can be used to model the experimental data.28 The fitting parameters were g and Jbb and Jbt, Jtt was fixed to a value of zero due to the long distance between Cu1 and Cu1′. The best fitting is shown in Fig. 5 as a solid line, with the fitting parameters g = 2.16, Jbb = 84.4 cm−1 and Jbt = −2.8 cm−1. The combination of ferromagnetic and weak antiferromagnetic coupling leads to a spin ground state of S = 2 for 3, with an excited states S* = 1 and S** = 0 within 15 cm−1 above the ground state, for each tetranuclear unit.
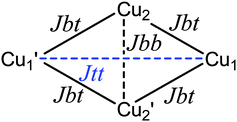 |
| | Scheme 3 | |
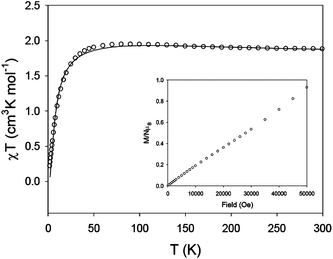 |
| | Fig. 5
χT
vs. T plot for 3. The solid line is the best fit to the experimental data (see text for fitting parameters). Inset: Magnetization vs. field plot at 2 K. | |
The energy states of the polymer of Cu4 units will be in a narrower energy range. The magnetization vs. field plot at 2 K, shown in Fig. 5 inset, confirms the presence of low-lying excited states as well as the non-magnetic isolation of the Cu4 units. In fact, the Cu4 units should be antiferromagnetically coupled, as the bridging mode of the ligand in 3 is similar to that found in 1 and 2. These antiferromagnetic interactions between Cu4 units have not been taken into account in the Van Vleck model for 3, since they are not important in the high temperature region, but they cannot be ignored in the magnetization, measured at 2 K. The antiferromagnetic coupling between Cu4 units would lead to S = 0 for complex 3, with low lying excited states.
{[Mn2(L)(phen)4](H2O)2(ClO4)2}2 (4).
Complexes 4 and 5 show similar structures and thus only the magnetic properties of 4 are studied herein. The χT product has a value of 17.8 cm3 K mol−1 at 300 K, in good agreement with that expected for four non-interacting MnII ions (S = 5/2, g = 2.0, χT = 17.5 cm3 K mol−1 for four MnII ions at 300 K). As the temperature decreases the χT product remains nearly constant until below 50 K when the χT product sharply drops to a value of 2.5 cm3 K mol−1 at 2 K (see Fig. 6). This drop reflects the Boltzman population of the S = 0 spin ground state of 4. The magnetization vs. field behaviour for 4 is shown in Fig. 6 inset. At 2 K the magnetization shows a linear increase with the field, in agreement with an S = 0 spin ground state with accessible excited states of larger S values. In 4, the four MnII ions are arranged in the corners of a square, linked by syn-anticarboxylato groups from the two ligands, which leads to weak antiferromagnetic coupling for MnII. The magnetic data was simulated with the software package MAGPACK29 with the exchange Hamiltonian Ĥ = −2J[Ŝ1Ŝ2] + ΣDŜiz2. For MnIID = 0. The best agreement between experimental and simulated data (solid line in Fig. 6) was obtained for g = 2.05, J = −0.5 cm−1, resulting in a spin ground state of ST = 0 for the tetranuclear MnII complex 4.
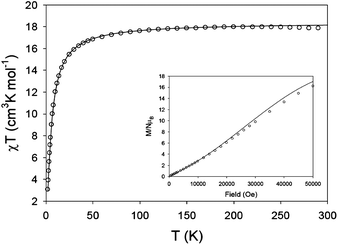 |
| | Fig. 6
χT
vs. T plot for 4. The solid line is a simulation calculated using MAGPACK. Inset: Magnetization vs. field plot at 2 K. | |
Conclusion
We have successfully prepared five new CuII and MnII coordination complexes showing discrete tetranuclear motifs as well as infinite 1-D and 2-D coordination arrays with an in situ generated dicarboxylate ligand L bearing a heteroalicyclic skeleton. Through a structural comparison of these complexes, it is apparent that the chosen metal ions and the ancillary co-ligands exert a significant influence on the resulting polymeric architectures. Moreover, the L ligand displays diverse bridging fashions, which could be regarded as a potential connector to assemble novel supramolecular complexes. Variable-temperature magnetic susceptibility measurements of 1–4 indicate the different magnetic interactions between the metal centers arising from diverse exchange pathways. The current results will encourage us to perform further systematic research on coordination assemblies of such heteroalicyclic dicarboxylate tectons, which may enrich the building-block methodology and structural database of coordination architectures and also extend their potential applications.
Acknowledgements
This work was supported by the National Natural Science Fund of China (Nos. 20801049 and 21071129), Plan for Scientific Innovation Talent of Henan Province (No. 114100510017), Program for New Century Excellent Talents in University (No. NCET-10-0143), Tianjin Normal University, and the Spanish Government (CTQ2009-06959 and Ramón y Cajal contract).
References
-
(a) R. Robson, Dalton Trans., 2008, 5113 RSC;
(b) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC;
(c) B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545 CrossRef CAS;
(d) Q.-M. Wang and T. C. W. Mak, J. Am. Chem. Soc., 2001, 123, 7594 CrossRef CAS;
(e) X.-H. Bu, M.-L. Tong, H.-C. Chang, S. Kitagawa and S. R. Batten, Angew. Chem., Int. Ed., 2004, 43, 192 CrossRef CAS;
(f) S. Horiuchi, Y. Nishioka, T. Murase and M. Fujita, Chem. Commun., 2010, 46, 3460 RSC;
(g) J.-Z. Hou, M. Li, Z. Li, S.-Z. Zhan, X.-C. Huang and D. Li, Angew. Chem., Int. Ed., 2008, 47, 1711 CrossRef CAS;
(h) N. W. Ockwig, O. Delgado-Friederichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS;
(i) V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC.
-
(a) R.-Q. Zou, R.-Q. Zhong, M. Du, D. S. Pandey and Q. Xu, Cryst. Growth Des., 2008, 8, 452 CrossRef CAS;
(b) W. T. Chen and S. Fukuzumi, Inorg. Chem., 2009, 48, 3800 CrossRef CAS;
(c) J.-J. Jang, L. Li, T. Yang, D.-B. Kuang, W. Wang and C.-Y. Su, Chem. Commun., 2009, 2387 RSC;
(d) R. Cao, J. Lü and S. R. Batten, CrystEngComm, 2008, 10, 784 RSC;
(e) Y. Ikawa, M. Takeda, M. Suzuki, A. Osuka and H. Furuta, Chem. Commun., 2010, 46, 5689 RSC;
(f) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC;
(g) X. Hu, W. Dou, C. Xu, X.-L. Tang, J.-R. Zheng and W.-S. Liu, Dalton Trans., 2011, 40, 3412 RSC.
-
(a) Y. Fujii, J. Terao and N. Kambe, Chem. Commun., 2009, 1115 RSC;
(b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC;
(c) J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094 CrossRef CAS;
(d) C. E. Willans, K. M. Anderson, P. C. Junk, L. J. Barbour and J. W. Steed, Chem. Commun., 2007, 3634 RSC;
(e) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440 CrossRef CAS.
-
(a) S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC;
(b) J. Zhao, L. Mi, J. Hu, H. Hou and Y. Fan, J. Am. Chem. Soc., 2008, 130, 15222 CrossRef CAS;
(c) Z.-J. Zhang, W. Shi, Z. Niu, H.-H. Li, B. Zhao, P. Cheng, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2011, 47, 6425 RSC;
(d) L. Mi, H. Hou, Z. Song, H. Han and Y. Fan, Chem.–Eur. J., 2008, 14, 1814 CrossRef CAS.
-
(a) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490 CrossRef CAS;
(b) B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130, 6411 CrossRef CAS;
(c) J. Luo, H. Xu, Y. Liu, Y. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626 CrossRef CAS;
(d) C.-R. Tan, S.-H. Yang, N. R. Champness, X. Lin, A. J. Blake, W. Lewis and M. Schröder, Chem. Commun., 2011, 47, 4487 RSC.
-
(a) Y.-F. Zeng, X. Hu, F.-C. Liu and X.-H. Bu, Chem. Soc. Rev., 2009, 38, 469 RSC;
(b) H.-L. Sun, Z.-M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081 CrossRef CAS;
(c) S. Paul and A. Misra, Inorg. Chem., 2011, 50, 3234 CrossRef CAS;
(d) E. Coronado and K. R. Dunbar, Inorg. Chem., 2009, 48, 5047 CrossRef CAS.
-
(a) E. Cariati, R. Macchi, D. Roberto, R. Ugo, S. Galli, N. Masciocchi and A. Sironi, Chem. Mater., 2007, 19, 3704 CrossRef CAS;
(b) Q.-X. Yao, W.-M. Xuan, H. Zhang, C.-Y. Tu and J. Zhang, Chem. Commun., 2009, 59 RSC;
(c) M. R. Kishan, J. Tian, P. K. Thallapally, C. A. Fernandez, S. J. Dalgarno, J. E. Warren, B. P. McGrail and J. L. Atwood, Chem. Commun., 2010, 46, 538 RSC.
- S. R. Batten, J. Solid State Chem., 2005, 178, 2475 CrossRef CAS.
-
(a) H.-B. Zhu and S.-H. Gou, Coord. Chem. Rev., 2011, 255, 318 CrossRef CAS;
(b) X.-D. Chen, H.-F. Wu and M. Du, Chem. Commun., 2008, 1296 RSC;
(c) H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC;
(d) X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2007, 40, 162 CrossRef CAS.
-
(a) M. Du, Z.-H. Zhang, Y.-P. You and X.-J. Zhao, CrystEngComm, 2008, 10, 306 RSC;
(b) H. Arora, F. Lloret and R. Mukherjee, Inorg. Chem., 2009, 48, 1158 CrossRef CAS;
(c) H. Chun and H. Jung, Inorg. Chem., 2009, 48, 417 CrossRef CAS;
(d) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS;
(e) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
-
(a) T.-L. Hu, R.-Q. Zou, J.-R. Li and X.-H. Bu, Dalton Trans., 2008, 1302 RSC;
(b) W. Chen, J.-Y. Wang, C. Chen, Q. Yue, H.-M. Yuan, J.-S. Chen and S.-N. Wang, Inorg. Chem., 2003, 42, 944 CrossRef CAS;
(c) X.-L. Wang, Y.-F. Bi, H.-Y. Lin and G.-C. Liu, Cryst. Growth Des., 2007, 7, 1086 CrossRef CAS;
(d) E. Suresh, K. Boopalan, R. V. Jasra and M. M. Bhadbhade, Inorg. Chem., 2001, 40, 4078 CrossRef CAS;
(e) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS.
-
(a) Q. Fang, G. Zhu, M. Xue, Z. Wang, J. Sun and S. Qiu, Cryst. Growth Des., 2008, 8, 319 CrossRef CAS;
(b) J. Zhang, Y.-B. Chen, S.-M. Chen, Z.-J. Li, J.-K. Cheng and Y.-G. Yao, Inorg. Chem., 2006, 45, 3161 CrossRef CAS;
(c) C. Livage, N. Guillou, J. Marrot and G. Férey, Chem. Mater., 2001, 13, 4387 CrossRef CAS.
-
(a) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS;
(b) J. Yang, Q. Yue, G.-D. Li, J.-J. Cao, G.-H. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 2857 CrossRef CAS;
(c) B. Liu, R. Zou, R. Zhong, S. Han, H. Shioyama, T. Yamada, G. Maruta, S. Takeda and Q. Xu, Microporous Mesoporous Mater., 2008, 111, 470 CrossRef CAS.
-
(a) D. Tanaka, S. Horike, S. Kitagawa, M. Ohba, M. Hasegawa, Y. Ozawa and K. Toriumi, Chem. Commun., 2007, 3142 RSC;
(b) C.-S. Liu, J.-J. Wang, Z. Chang, L.-F. Yan and T.-L. Hu, Z. Anorg. Allg. Chem., 2009, 635, 523 CrossRef CAS;
(c) C.-S. Liu, E. C. Sañudo, J.-J. Wang, Z. Chang, L.-F. Yan and X.-H. Bu, Aust. J. Chem., 2008, 61, 382 CrossRef CAS;
(d) S. Ma, X.-S. Wang, C. D. Collier, E. S. Manis and H.-C. Zhou, Inorg. Chem., 2007, 46, 8499 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS.
-
(a) Z. Lin and M.-L. Tong, Coord. Chem. Rev., 2011, 255, 421 CrossRef CAS;
(b) R.-X. Yao, Z.-M. Hao, C.-H. Guo and X.-M. Zhang, CrystEngComm, 2010, 12, 4416 RSC;
(c) H. Kumagai, M. Akita-Tanaka, K. Inoue, K. Taka-hashi, H. Kobayashi, S. Vilminot and M. Kurmoo, Inorg. Chem., 2007, 46, 5949 CrossRef CAS.
-
(a) L.-F. Ma, B. Liu, L.-Y. Wang, C.-P. Li and M. Du, Dalton Trans., 2010, 39, 2301 RSC;
(b) L.-F. Ma, B. Liu, L.-Y. Wang, J.-L. Hu and M. Du, CrystEngComm, 2010, 12, 1439 RSC;
(c) Z.-H. Ni, J. Tao, W. Wernsdorfer, A.-L. Cui and H.-Z. Kou, Dalton Trans., 2009, 2788 RSC;
(d) R.-Q. Zhong, R.-Q. Zou, M. Du, T. Yamada, G. Maruta, S. Takeda, J. Li and Q. Xu, CrystEngComm, 2010, 12, 677 RSC;
(e) C.-S. Liu, E. C. Sañudo, M. Hu, L.-M. Zhou, L.-Q. Guo, S.-T. Ma, L.-J. Gao and S.-M. Fang, CrystEngComm, 2010, 12, 853 RSC;
(f) S.-M. Fang, Q. Zhang, M. Hu, E. C. Sañudo, M. Du and C.-S. Liu, Inorg. Chem., 2010, 49, 9617 CrossRef CAS.
-
(a) J.-C. Jin, Y.-Y. Wang, W.-H. Zhang, A. S. Lermontov, E. K. Lermontova and Q.-Z. Shi, Dalton Trans., 2009, 10181 RSC;
(b) M. Felloni, A. J. Blake, P. Hubberstey, C. Wilson and M. Schröder, Cryst. Growth Des., 2009, 9, 4685 CrossRef CAS;
(c) D. M. Shin, I. S. Lee and Y. K. Chung, Inorg. Chem., 2003, 42, 8838 CrossRef CAS.
-
(a) M.-H. Bi, G.-H. Li, Y.-C. Zou, Z. Shi and S.-H. Feng, Inorg. Chem., 2007, 46, 604 CrossRef CAS;
(b) M.-H. Bi, G.-H. Li, J. Hua, X.-M. Liu, Y.-W. Hu, Z. Shi and S.-H. Feng, CrystEngComm, 2007, 9, 984 RSC;
(c) J.-H. Yu, X.-M. Wang, L. Ye, Q. Hou, Q.-F. Yang and J.-Q. Xu, CrystEngComm, 2009, 11, 1037 RSC;
(d) L. A. Paton and W. T. A. Harrison, Angew. Chem., Int. Ed., 2010, 49, 7684 CrossRef CAS;
(e) W. Zhang, H.-Y. Ye, H.-L. Cai, J.-Z. Ge, R.-G. Xiong and S. D. Huang, J. Am. Chem. Soc., 2010, 132, 7300 CrossRef CAS;
(f) J.-T. Li, D.-K. Cao, B. Liu, Y.-Z. Li and L.-M. Zheng, Cryst. Growth Des., 2008, 8, 2950 CrossRef CAS.
-
(a) J. Wang, Z.-J. Lin, Y.-C. Qu, N.-L. Yang, Y.-H. Zhang and M.-L. Tong, Inorg. Chem., 2008, 47, 190 CrossRef CAS;
(b) Y.-Q. Guo, B.-P. Yang, J.-L. Song and J.-G. Mao, Cryst. Growth Des., 2008, 8, 600 CrossRef CAS;
(c) Y. Xu, J.-Q. Xu, K.-L. Zhang, Y. Zhang and X.-Z. You, Chem. Commun., 2000, 153 RSC.
-
(a) C. Pettinari, F. Marchetti, A. Cingolani, R. Pettinari, S. I. Troyanov and A. Drozdov, J. Chem. Soc., Dalton Trans., 2000, 831 RSC;
(b) A. Bencini and V. Lippolis, Coord. Chem. Rev., 2010, 254, 2096 CrossRef CAS;
(c) Z.-F. Chen, Z.-L. Zhang, Y.-H. Tan, Y.-Z. Tang, H.-K. Fun, Z.-Y. Zhou, B. F. Abrahams and H. Liang, CrystEngComm, 2008, 10, 217 RSC.
- M. Hu, Q. Zhang, L.-M. Zhou, S.-M. Fang and C.-S. Liu, Inorg. Chem. Commun., 2010, 13, 1548 CrossRef CAS.
-
Bruker AXS, SAINT Software Reference Manual, Madison: WI, 1998 Search PubMed.
-
G. M. Sheldrick, SADABS, Siemens Area Detector Absorption Corrected Software, University of Göttingen: Germany, 1996 Search PubMed.
-
G. M. Sheldrick, SHELXTL NT Version 5.1. Program for Solution and Refinement of Crystal Structures, University of Göttingen: Germany, 1997 Search PubMed.
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University: Utrecht, The Netherlands, 2005 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
C. J. O'Connor, Progress in Inorganic Chemistry, 1982, 29, 203. Ed. S. J. Lippard, John Wiley & Sons Search PubMed.
- J. J. BorrásAlmenar, J. M. Clemente, E. Coronado and B. S. Tsukerblat, Inorg. Chem., 1999, 38, 6081 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns for 1–5 (Fig. S1), additional structural figures for 1, 4 and 5 (Figs. S2–S5), TG-DTA curves for 1–5 (Fig. S6), Curie plots and magnetization vs. field plots for 1 and 2 (Figs. S7–S10), selected bond parameters for 1–5 (Table S1), and H-bonding geometries for 2–4 (Table S2). CCDC reference numbers 838704–838708. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce06015e |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.