DOI:
10.1039/C1CE05639E
(Paper)
CrystEngComm, 2012,
14, 169-177
A series of 1D, 2D and 3D coordination polymers based on a 5-(benzonic-4-ylmethoxy)isophthalic acid: syntheses, structures and photoluminescence†
Received
28th May 2011
, Accepted 8th September 2011
First published on 18th October 2011
Abstract
Seven new coordination polymers, namely, [Zn(HL)(H2O)] (1), [Zn(HL)(phen)]·1.5H2O (2), [Zn(HL)(L1)] (3), [Zn2(HL)2(L2)2]·2H2O (4), [Zn(HL)(L3)0.5] (5), [Zn(HL)(L4)] (6) and [Cu2(L)(OH)(H2O)]·0.5H2O (7) (H3L = 5-(benzonic-4-ylmethoxy) isophthalic acid, phen = 1,10-phenathroline, L1 = 1,2-bis(1,2,4-triazole-1-yl)ethane, L2 = 1,3-bis(1,2,4-triazole-1-yl)propane, L3 = 1,6-bis(1,2,4-triazole-1-yl)hexane and L4 = 4,4′-bis(1,2,4-triazole-1-ylmethyl)biphenyl), have been hydrothermally synthesized and characterized by single-crystal X-ray diffraction. In compounds 1–6, the H3L ligand is partially deprotonated to form HL2−, while it is completely deprotonated in 7. Compound 1 shows a 3D framework with 4-connected (42·7·83)2 topology. Compound 2 displays a 1D ribbon structure. The neighboring ribbons are further linked by hydrogen-bonding interactions to form a 3D supramolecular architecture. Compound 3 exhibits a 2D undulated sheet. The sheets are further penetrated into each other to give rise to a 3D polycatenation structure. Compound 4 displays a 2D supramolecular layer structure. Compound 5 shows a 3D (3,6)-connected (4·8)(4·82·103) net. Compound 6 reveals a 3D four-fold interpenetrating diamondoid architecture. Compound 7 displays a (3,8)-connected (4·62)(44·68·812·104) topology. These compounds have been characterized by powder X-ray diffractions (PXRD) and thermal gravimetric analyses (TGA). In addition, the photoluminescent behaviours of 1–6 have been investigated in detail.
Introduction
In recent years, considerable efforts have been focused on the design and synthesis of novel multidimensional coordination polymers, not only because of their intriguing variety of architectures and topologies but also because of their tremendous potential applications in the fields of ion exchange, gas storage, luminescence and heterogeneous catalysis.1 It is well known that the construction of coordination polymers is mainly dependent on the combination of several factors, such as the organic ligand, solvents, and metal atoms.2 Among them, the anions play an important role in the construction of novel architectures. Among various polycarboxylates, 5-(benzonic-4-ylmethoxy)isophthalic acid (H3L), as a tricarboxylate ligand, is a good candidate for the construction of coordination polymers. The H3L ligand contains a hetero-oxygen atom and a –CH2– spacer at the center of the molecule. Therefore, the two phenylene rings can freely twist to meet the requirements of the coordination geometries of metal atoms in the assembly process. In addition, it can bridge the central metals to give a series of interesting structures through complete or partial deprotonation of its three carboxylate groups. As far as we know, coordination polymers constructed from H3L and auxiliary N-donor ligands have been scarcely investigated so far.3 Besides, neutral N-donor ligands, especially flexible bridging N-donor ligands, also have significant effects on the construction of coordination polymers.4 In this regard, 1-substituted bis(1,2,4-triazole) ligands have been found to be one kind of useful flexible building block in the construction of coordination polymers with versatile topologies.5
In this work, seven compounds based on H3L acids and flexible N-donor ligands have been synthesized under hydrothermal conditions, namely, [Zn(HL)(H2O)] (1), [Zn(HL)(phen)]·1.5H2O (2), [Zn(HL)(L1)] (3), [Zn2(HL)2(L2)2]·2H2O (4), [Zn(HL)(L3)0.5] (5), [Zn(HL)(L4)] (6) and [Cu2(L)(OH)(H2O)]·0.5H2O (7) (phen = 1,10-phenathroline, L1 = 1,2-bis(1,2,4-triazole-1-yl)ethane, L2 = 1,3-bis(1,2,4-triazole-1-yl)propane, L3 = 1,6-bis(1,2,4-triazole-1-yl)hexane and L4 = 4,4′-bis(1,2,4-triazole-1-ylmethyl)biphenyl) (Chart 1). The coordination modes of the H3L ligands on the ultimate structures have been discussed. In addition, the luminescent properties for 1–6 have also been investigated.
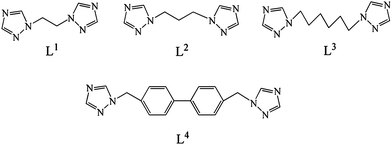 |
| | Chart 1 Structures of the bis(1,2,4-triazole) ligands used in this work. | |
Experimental
Materials and measurements
All reagents and solvents for syntheses were purchased from commercial sources and used as received. The ligand H3L was synthesized as in a previously reported method.6 The C, H and N elemental analysis was conducted on a Perkin–Elmer 240C elemental analyzer. The FT-IR spectra were recorded from KBr pellets in the range 4000–400 cm−1 on a Mattson Alpha-Centauri spectrometer. The phase purities of the bulk samples were identified by X-ray powder diffraction on a Siemens D5005 diffractometer. TGA was performed on a Perkin–Elmer TG-7 analyzer. The photoluminescent properties were measured on a FLSP920 Edinburgh Fluorescence Spectrometer.
Syntheses of L1–L4.
A mixture of 1,2,4-triazole (3.5 g, 50 mmol), NaOH (2.0 g, 50 mmol) and DMF (40 mL) was heated at 60 °C for 1 h, then 1,2-dibromoethane (4.7 g, 25 mmol) was added. After stirring at 60 °C for 2 h, the mixture was evaporated in a water bath to a viscous state. The product was extracted by chloroform three times. The solvent was removed from the filtrate by rotary evaporation, and a white solid of L1 formed immediately. The preparations of L2–L4 were similar to that of L1 except that 1,3-dibromopropane for L2, 1,6-dibromohexamethylene for L3 and 4,4′-bis(chloromethyl)-1,1′-biphenyl for L4 were used instead of 1,2-dibromoethane, respectively.
Synthesis of [Zn(HL)(H2O)] (1).
A mixture of ZnCl2 (0.032 g, 0.23 mmol), H2L (0.050 g, 0.16 mmol), and water (8 mL) was placed in a Teflon reactor, and the pH value was adjusted to 4.0 by addition of 0.25 mol L−1NaOH solution. The mixture was heated at 140 °C for 3 days, and then it was gradually cooled to room temperature. The crystals were obtained in a 58% yield. Anal. Calcd for C16H12ZnO8 (Mr = 397.63): C, 48.33; H, 3.04%. Found: C, 48.64; H, 2.93%. IR data (KBr, cm−1): 3504 (m), 3112 (m), 2912 (m), 2635 (m), 2514 (m), 1682 (s), 1536 (s), 1455 (m), 1386 (s), 1275 (s), 1136 (s), 1058 (s), 1015 (w), 967 (w), 882 (w), 774 (s), 726 (m), 666 (w).
Synthesis of [Zn(HL)(phen)]·1.5H2O (2).
A mixture of ZnCl2 (0.032 g, 0.23 mmol), H2L (0.050 g, 0.16 mmol), phen (0.029 g, 0.16 mmol), 1 mL 0.25 mol L−1NaOH solution, and water (8 mL) was placed in a Teflon reactor. The mixture was heated at 150 °C for 3 days, and then it was gradually cooled to room temperature. Colorless block crystals were obtained in a 60% yield. Anal. Calcd for C28H21ZnN2O8.50 (Mr = 586.84): C, 57.31; H, 3.61; N, 4.77%. Found: C, 57.14; H, 3.67; N, 4.85%. IR data (KBr, cm−1): 3471 (s), 3075 (m), 2874 (m), 1682 (m), 1571 (s), 1378 (s), 1245 (m), 1108 (m), 1037 (m), 846 (m), 779 (m), 727 (m), 618 (w), 577 (w).
Synthesis of [Zn(HL)(L1)] (3).
A mixture of ZnCl2 (0.032 g, 0.23 mmol), H2L (0.050 g, 0.16 mmol), L1 (0.026 g, 0.16 mmol), 1 mL 0.25 mol L−1NaOH solution, ethanol (2 mL) and water (6 mL) was placed in a Teflon reactor. The mixture was heated at 120 °C for 3 days, and then it was gradually cooled to room temperature. Colorless block crystals were obtained in a 47% yield. Anal. Calcd for C22H18ZnN6O7 (Mr = 543.79): C, 48.59; H, 3.33; N, 15.45%. Found: C, 48.74; H, 3.38; N, 15.31%. IR data (KBr, cm−1): 3437 (s), 3148 (m), 3032 (m), 1713 (s), 1606 (s), 1570 (s), 1381 (s), 1265 (s), 1204 (m), 1129 (s), 994 (m), 916 (w), 880 (w), 832 (w), 765 (m), 674 (m), 580 (m).
Synthesis of [Zn2(HL)2(L2)2]·2H2O (4).
The preparation of 4 was similar to that of 2 except that L2 (0.028 g) was used instead of phen. Colorless block crystals were obtained in a 53% yield. Anal. Calcd for C46H44Zn2N12O16 (Mr = 1151.67): C, 47.97; H, 3.85; N, 14.59%. Found: C, 47.76; H, 3.73; N, 14.43%. IR data (KBr, cm−1): 3437 (s), 3121 (m), 2921 (w), 2621 (s), 1686 (s), 1567 (s), 1369 (s), 1264 (s), 1174 (w), 1136 (m), 998 (m), 897 (m), 777 (m), 736 (m), 673 (m), 646 (m).
Synthesis of [Zn(HL)(L3)0.5] (5).
A mixture of ZnCl2 (0.032 g, 0.23 mmol), H2L (0.050 g, 0.16 mmol), L3 (0.035 g, 0.16 mmol), 1 mL 0.25 mol L−1NaOH solution, and water (8 mL) was placed in a Teflon reactor. The mixture was heated at 160 °C for 3 days, and then it was gradually cooled to room temperature. Colorless block crystals were obtained in a 43% yield. Anal. Calcd for C84H72Zn4N12O28 (Mr = 1959.02): C, 51.50; H, 3.70; N, 8.58%. Found: C, 51.39; H, 3.73; N, 8.61%. IR data (KBr, cm−1): 3133 (m), 3045 (w), 2948 (s), 2865 (s), 2488 (w), 1714 (s), 1544 (s), 1410 (m), 1280 (s), 1275 (m), 1130 (s), 1001 (s), 892 (m), 858 (m), 771 (m), 673 (m), 641 (m).
Synthesis of [Zn(HL)(L4)] (6).
The preparation of 6 was similar to that of 5 except that L4 (0.035 g) was used instead of L3. Colorless block crystals were obtained in a 35% yield. Anal. Calcd for C34H26ZnN6O7 (Mr = 695.79): C, 58.69; H, 3.77; N, 12.07%. Found: C, 58.79; H, 3.70; N, 12.01%. IR data (KBr, cm−1): 3549 (m), 3478 (s), 3414 (s), 3137 (s), 3053 (s), 2979 (m), 2851 (w), 1705 (s), 1621 (s), 1558 (s), 1502 (m), 1405 (s), 1320 (m), 1268 (m), 1240 (m), 1132 (s), 1056 (m), 999 (m), 965 (w), 940 (w), 888 (w), 782 (s), 734 (m), 674 (m), 647 (m).
Synthesis of [Cu2(L)(OH)(H2O)]·0.5H2O (7).
The preparation of 7 was similar to that of 1 except that CuCl2·2H2O (0.057 g) was used instead of ZnCl2, and the temperature was changed into 130 °C. Blue crystals were obtained in a 52% yield. Anal. Calcd for C32H26Cu4O19 (Mr = 968.69): C, 39.68; H, 2.70%. Found: C, 39.74; H, 2.66%. IR data (KBr, cm−1): 3386 (s), 2501 (m), 1679 (m), 1544 (s), 1400 (s), 1294 (s), 1238 (m), 1138 (m), 1044 (m), 962 (w), 912 (w), 765 (s), 692 (w), 599 (w).
Crystallographic diffraction data for compounds 1–7 were recorded on a Oxford Diffraction Gemini R CCD with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K. Absorption corrections were applied using multi-scan technique. All the structures were solved by Direct Method of SHELXS-97 and refined by full-matrix least-squares techniques using the SHELXL-97 program within WINGX.7Non-hydrogen atoms were refined with anisotropic temperature parameters. The hydrogen atoms attached to carbons were generated geometrically. The hydrogen atoms of hydroxyl and water molecules were located from difference Fourier maps and refined with isotropic displacement parameters. The detailed crystallographic data and structure refinement parameters for 1–7 are summarized in Table 1. Selected bond distances and angles and hydrogen bonds for the compounds are given in the ESI (Tables S1–S2)†.
Table 1
Crystal data and structure refinements for compounds 1–7
| |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
|
R
1 = Σ||Fo| − |Fc‖/Σ|Fo|.
wR
2 = |Σw(|Fo|2−|Fc|2)|/Σ|w(Fo2)2|1/2.
|
| Formula |
C16H12ZnO8 |
C28H21ZnN2O8.50 |
C22H18ZnN6O7 |
C46H44Zn2N12O16 |
C84H72Zn4N12O28 |
C34H26ZnN6O7 |
C32H26Cu4O19 |
|
fw
|
397.63 |
586.84 |
543.79 |
1151.67 |
1959.02 |
695.79 |
968.69 |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
P
|
P21/c |
P21212
|
P
|
|
a/Å |
11.9530(7) |
9.955(5) |
8.6590(3) |
11.8109(6) |
15.1240(7) |
14.9780(5) |
6.8167(4) |
|
b/Å |
7.2717(3) |
10.550(4) |
9.9594(4) |
13.7859(5) |
13.8252(5) |
32.6149(11) |
10.8308(5) |
|
c/Å |
17.7746(8) |
14.073(3) |
12.8686(6) |
16.9387(6) |
9.5104(4) |
6.3668(2) |
12.0167(7) |
|
α (deg.) |
90 |
88.346(2) |
94.713(4) |
67.887(4) |
90 |
90 |
86.596(4) |
|
β (deg.) |
104.482(5) |
69.302(3) |
101.475(3) |
87.405(4) |
102.674(4) |
90 |
83.997(5) |
|
γ (deg.) |
90 |
64.914(5) |
92.540(3) |
71.034(4) |
90 |
90 |
73.499(4) |
|
V/Å3 |
1495.85(13) |
1234.7(8) |
1081.82(8) |
2407.07(20) |
1940.10(14) |
3110.22(18) |
845.58(8) |
|
Z
|
4 |
2 |
2 |
2 |
1 |
4 |
1 |
|
D
c/g cm−3 |
1.766 |
1.578 |
1.669 |
1.589 |
1.677 |
1.486 |
1.902 |
|
F(000) |
808 |
602 |
556 |
1184 |
1004 |
1432 |
486 |
| Observed reflection/unique |
6486/3455 |
9086/5617 |
8204/4916 |
17725/10961 |
8435/4491 |
14173/6632 |
6155/3847 |
|
R(int) |
0.0262 |
0.0124 |
0.0202 |
0.0264 |
0.0227 |
0.0400 |
0.0200 |
| GOF on F2 |
0.902 |
1.042 |
0.880 |
0.871 |
0.854 |
0.748 |
0.924 |
|
R
1
a[I > 2σ(I)] |
0.0335 |
0.0279 |
0.0347 |
0.0347 |
0.0285 |
0.0336 |
0.0281 |
|
wR
2
b
|
0.0698 |
0.0745 |
0.0724 |
0.0594 |
0.0563 |
0.0315 |
0.0637 |
Results and discussion
Structure of [Zn(HL)(H2O)] (1)
As illustrated in Fig. 1a, the Zn(II) ion is five-coordinated by four carboxylate oxygen atoms from four HL anions and one water molecule (Zn–O = 1.928(2)–2.459(2) Å). Each HL anion coordinates to four Zn(II) atoms in a μ4-η1:η1:η0:η1:η0:η1 coordination mode (Chart 2a). In this way, the Zn(II) centers are connected by HL anions to yield a 3D framework (Fig. 1b). Actually, in order to simplify the intricate structure of 1, both the Zn(II) atom and HL anion can be considered as 4-connected nodes, respectively (Fig. S1†). From a topological perspective, the 3D framework of 1 can be described as a 4-connected net with a Schläfli symbol of (42·7·83)2 (Fig. 1c).8 In addition, there are intermolecular hydrogen-bonding interactions between the water molecule and the carboxylate O atoms. These hydrogen bonds further stabilize the structure of the 3D framework (Table S2†).
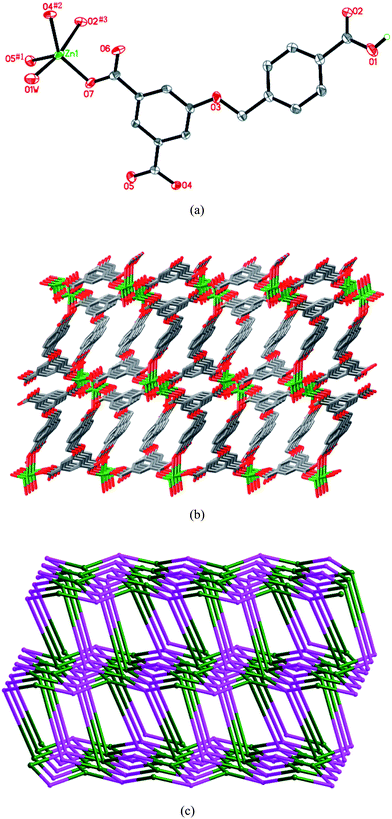 |
| | Fig. 1 (a) ORTEP diagram showing the coordination environment of the Zn(II) atom in 1. Symmetry codes: #1 −x + 2, −y, −z + 2; #2x, −y − 1/2, z − 1/2; #3 −x + 1, y + 3/2, −z + 3/2. (b) View of the 3D structure. (c) Schematic diagram (OLEX) showing the 3D (42·7·83)2 net of 1. | |
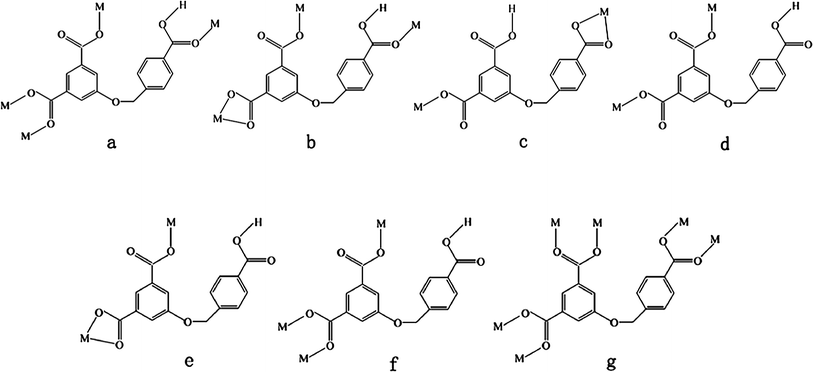 |
| | Chart 2 Coordination modes of the HL2− and L3− ligands in compounds 1–7. | |
Structure of [Zn(HL)(phen)]·1.5H2O (2)
When phen was introduced into the reaction system of 1, a quite different ribbon structure of 2 was obtained. The Zn(II) ion of 2 is six-coordinated by four oxygen atoms from three HL anions (Zn–O = 1.991(1)–2.304(2) Å) and two nitrogen atoms (Zn–N = 2.095(1)–2.143(2) Å) from one phen ligand (Fig. 2a). The HL anion exhibits a μ3-η1:η1:η0:η1:η0:η1 coordination mode (Chart 2b). The Zn(II) atoms are linked viaHL anions to form a 1D ribbon structure (Fig. 2b). The chelating phen ligands are located on both rims of the ribbon. Adjacent ribbons are further connected by face-to-face π–π interactions between phen ligands (with average face-to-face distance of 3.34 Å and centroid-to-centroid distance of 3.53 Å), resulting in a 3D supramolecular architecture (Fig. 2c). Moreover, there exist hydrogen-binding interactions among adjacent ribbons (Table S2†). The lattice water molecules and the uncoordinated carboxylic group of the HL anion act as hydrogen donors (O7⋯O3#5 = 2.5806(18) Å, O1W⋯O1 = 2.968(3) Å, O1W⋯O1#6 = 3.017(3) Å, O2W⋯O2#4 = 2.932(6) Å, #4 −x + 1, −y + 1, −z + 1; #5x − 1, y + 1, z + 1; #6 −x, −y + 1, −z + 2). These hydrogen bonds further stabilize the 3D supramolecular architecture of 2.
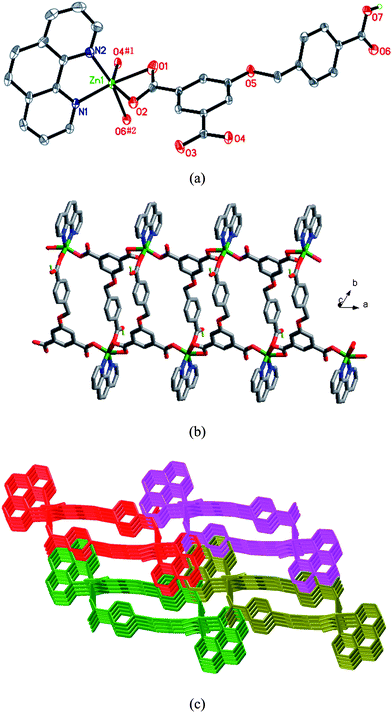 |
| | Fig. 2 (a) ORTEP diagram showing the coordination environment of the Zn(II) atom in 2. Symmetry codes: #1x − 1, y, z; #2 −x, −y + 2, −z + 2. (b) View of the 1D chain of 2. (c) View of the 3D supramolecular architecture of 2 formed by π–π stacking interactions of neighboring chains. | |
Structure of [Zn(HL)(L1)] (3)
When the flexible bis(1,2,4-triazole) ligand L1 was utilized in place of the chelating phen ligand of 2, a 2D → 3D polycatenated framework of 3 was obtained. The Zn(II) atom is five-coordinated by three carboxylate oxygen atoms from two HL anions, and two nitrogen atoms from two L1 ligands (Fig. 3a). The HL anion shows a μ2-η0:η1:η0:η0:η1:η1 coordination mode (Chart 2c). Two crystallographically unique L1 ligands lie about inversion centers. The Zn(II) atoms are linked by HL anions and L1 ligands to generate a deeply corrugated 2D (4,4) sheet (Fig. 3b). The open space within each sheet provides the chance for the parallel polycatenation of the (4,4) sheets (Fig. 3c). Each sheet is penetrated by two others (one above and one below) which have parallel but not coincident mean planes, leading to an overall 3D entanglement (Fig. 3d). As far as we know, only a few examples of 2D → 3D structures entangled in parallel mode have been observed up to now.9
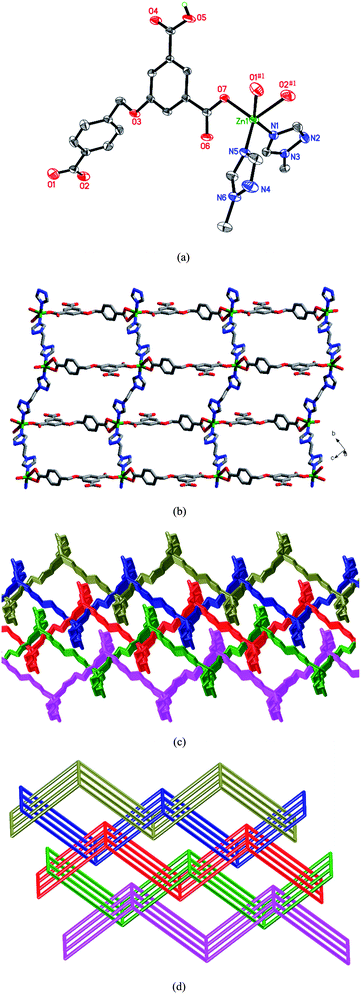 |
| | Fig. 3 (a) Coordination environment of the Zn(II) atom in 7. Symmetry codes: #1x, y + 1, z + 1. (b) View of the 2D undulated layer. (c) The 2D → 3D parallel interpenetration of the layers in 3. (d) Schematic representation of the 2D → 3D polycatenation of different (4,4) motifs. | |
Structure of [Zn2(HL)2(L2)2]·2H2O (4)
When L1 was replaced by L2, a completely different compound 4 was obtained. As shown in Fig. 4a, there are two kinds of crystallographically unique Zn(II) centers in the structure. Each Zn(II) cation is coordinated by two oxygen atoms from two HL anions, and two nitrogen atoms from two L2 ligands in a slightly distorted tetrahedral geometry. Two HL ligands show μ2-η0:η1:η0:η1:η0:η0 and μ2-η1:η1:η0:η1:η0:η0 coordination modes, respectively (Chart 2d and 2e). The L2 ligands bridge adjacent Zn(II) atoms to give a dimeric unit [Zn(L2)2Zn] with the Zn⋯Zn distance of 9.956 Å. As shown in Fig. 4b, the [Zn(L2)2Zn] dimers are further connected by HL anions to give a 1D ribbon structure. The dangling 4-carboxybenzyloxy groups of HL anions locate on both rims of the ribbon structure. The 1D ribbon structures of 4 are further stacked by the face-to-face π–π interactions between the phenyl rings of adjacent HL anions (with face-to-face and centroid-to-centroid distances of 3.32 and 3.66 Å, respectively) to generate a 2D supramolecular network (Fig. 4c). In addition, the intermolecular hydrogen-bonding interactions among adjacent chains further stabilize the 2D supramolecular network of 4.
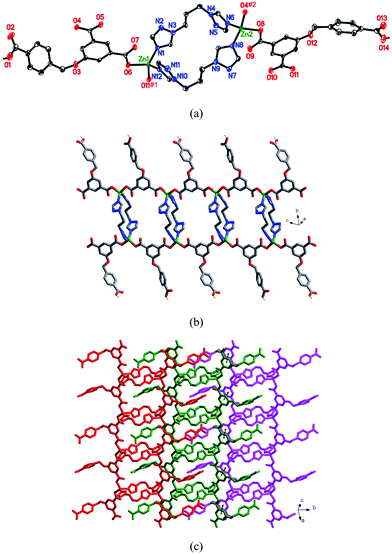 |
| | Fig. 4 (a) Coordination environments of the Zn(II) atoms in 4. Symmetry codes: #1 −x + 1, −y + 1, −z + 1; #2 −x + 2, −y + 1, −z. (b) View of the 1D chain structure of 4. (c) View of the 2D supramolecular layer connected by face-to-face π–π interactions. | |
Structure of [Zn(HL)(L3)0.5] (5)
When the L2 anion of 4 was replaced by L3, a different 3D structure of 5 was obtained. As shown in Fig. 5a, the Zn(II) cation is coordinated by three oxygen atoms from three HL anions, and one nitrogen atom from one L3 ligand in a slightly distorted tetrahedral geometry. The L3 ligand lies about an inversion center. The HL anion is partly deprotonated, and displays a μ3-η1:η1:η0:η1:η0:η0 coordination mode (Chart 2f). Neighboring Zn(II) atoms are linked by HL anions to generate a 2D layer. The 4-carboxybenzyloxy groups of HL anions, as the dangling arms, locate at both sides of the layer (Fig. 5b). The layers are further connected by L3 ligands to give a 3D framework. If Zn(II) atoms and HL anions can be considered as four-connected and three-connected nodes, respectively, the structure of 5 can be symbolized as a 3D (4·8)(4·82·103) net with two kinds of nonequivalent points (Fig. 5c).
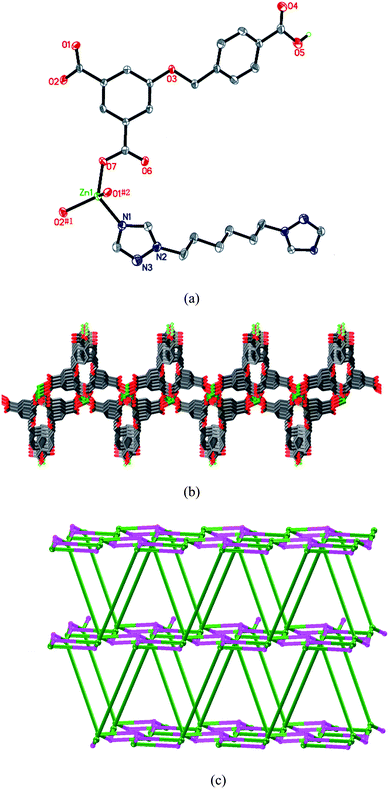 |
| | Fig. 5 (a) Coordination environment of the Zn(II) atom in 5. Symmetry codes: #1 −x + 2, y + 1/2, −z + 3/2; #2 −x + 2, −y, −z + 2. (b) Representation of the 2D Zn-HL layer. (c) Schematic diagram (OLEX) of the (4·8)(4·82·103) network of 5. | |
Structure of [Zn(HL)(L4)] (6)
Each Zn(II) center in 6 is five-coordinated by three oxygen atoms from two HL anions, and two nitrogen atoms from two L4 ligands (Fig. 6a). The HL anion shows a μ2-η1:η1:η0:η1:η0:η0 coordination mode (Chart 2e). The Zn⋯Zn separations bridged by the HL anion and L4 ligand are 9.82 and 18.14 Å, respectively. Zn(II) ions are bridged by HL anions and L4 ligands to give a 3D diamond framework (Fig. 6b). Because of the spacious nature of the single network, the potential voids are filled via mutual interpenetration of identical 3D frameworks, generating a four-fold interpenetrating architecture (Fig. 6c). According to topology classification, this diamondoid architecture belongs to Class Ia (all the independent nets are related by a single vector and the whole interpenetrating arrays are generated by translating a single net 3 times),10 with the translating vector of [001] 6.37 Å.
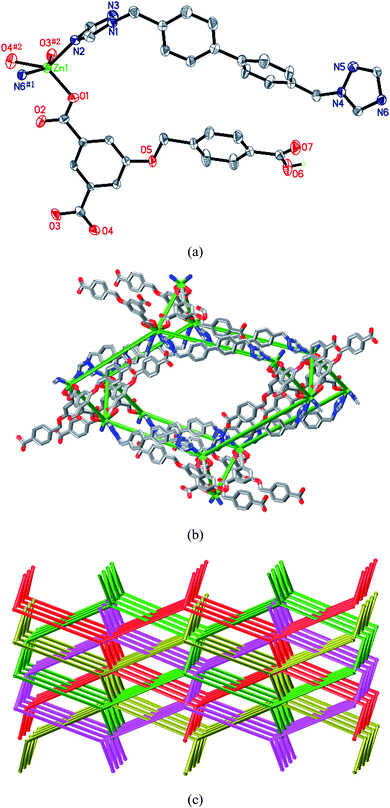 |
| | Fig. 6 (a) Coordination environment of the Zn(II) atom in 6. Symmetry codes: #1 −x + 3/2, y + 1/2, −z + 1; #2x + 1/2, −y + 1/2, −z − 1. (b) View of a single diamond structure of 6. (c) View of the four-fold interpenetrating diamond diagram (OLEX) of 6. | |
Structure of [Cu2(L)(OH)(H2O)]·0.5H2O (7)
As shown in Fig. 7a, the asymmetric unit contains two crystallographic Cu(II) atoms. The Cu1 cation is coordinated by three oxygen atoms from three L anions, one μ3–OH and one water molecule. Cu2 is coordinated by three oxygen atoms from three L anions and two μ3–OH. Cu1 and Cu2 ions have distorted square pyramidal environments, in agreement with the structural index τ values of 0.383 for Cu1 and 0.263 for Cu2, respectively (τ = 0 and 1 for perfect square-pyramidal and trigonal-bipyramidal geometries, respectively).11 Due to the Jahn–Teller effect, the Cu–O distance at the axial site (Cu1–O1W 2.287(3) and Cu2–O7 2.374(2) Å) is much longer than those of equatorial planes (1.9156(15)–1.9935(15) Å). All of the coordination bonds show typical values for the Cu(II) ion. As shown in Fig. 7b, two μ3–OH groups link four Cu(II) ions to form a rhombus-like tetranuclear unit. The separation of Cu2⋯Cu2#4 ions is 3.027 Å, indicating the existence of a weak Cu⋯Cu interaction.12 Each L anion bridges three tetranuclear units in a μ6-η1:η1:η1:η1:η1:η1 coordination mode (Chart 2g). In this way, L anions linked the neighboring tetranuclear [Cu4O10] units to generate a 3D framework. If the tetranuclear [Cu4O10] unit was considered as an eight-connected node, and the L anion was considered as a three-connected node (Fig. S2†), the topology of this 3D framework can thus be described as a (4·62)(44·68·812·104) net (Fig. 7c).
![(a) Coordination environments of the Cu(ii) atoms in 7. Symmetry codes: #1 −x + 4, −y + 1, −z + 3; #2x, y − 1, z; #3 −x + 2, −y + 2, −z + 2; #4 −x + 2, −y + 1, −z + 2; #5x − 1, y, z − 1. (b) View of the [Cu4O10] unit. (c) View of the 3D (3,8)-connected framework with (4·62)(44·68·812·104) topology.](/image/article/2012/CE/c1ce05639e/c1ce05639e-f7.gif) |
| | Fig. 7 (a) Coordination environments of the Cu(II) atoms in 7. Symmetry codes: #1 −x + 4, −y + 1, −z + 3; #2x, y − 1, z; #3 −x + 2, −y + 2, −z + 2; #4 −x + 2, −y + 1, −z + 2; #5x − 1, y, z − 1. (b) View of the [Cu4O10] unit. (c) View of the 3D (3,8)-connected framework with (4·62)(44·68·812·104) topology. | |
Effect of organic ligands on the complex structure
From the structure description above, we can see that the neutral ligand has a significant effect on the complex structure. Compound 1 displays a 3D framework. In comparison with 1, the introduction of neutral N-donor ligands (phen, L1, L2, L3 and L4) in 2–6 not only replaces the coordinated water molecule of the Zn(II) ion, but also changes the coordination modes of the HL anions. When the secondary N-donor ligand phen was utilized, a structurally different compound 2 was obtained. In 2, phen occupies the coordinate sites of Zn(II) ion, and generates a 1D chain structure.13 The structural differences of compounds 3–6 are closely related to the spacers of bis(1,2,4-triazole) ligands. Due to the presence of different spacers, –(CH2)2–, –(CH2)3–, –(CH2)6– and –CH2–(phenyl)2–CH2– in L1, L2, L3 and L4, respectively, the neutral N-donor ligands display different lengths, geometries and flexibilities. As a result, these different properties of the N-donor ligands result in the differences of their complex frameworks.14 For example, in 3, 5 and 6, Zn(II) centers are bridged by L1, L3 and L4 ligands to form chains, whereas two Zn(II) ions are bridged by L2 ligands to generate a [Zn2(L2)2] loop in 4.
On the other hand, the structures of the compounds are strongly related to the coordination modes of H3L ligands (Chart 2). In compound 1, the μ4-HL anions are partly deprotonated, and link Zn(II) ions to give a 3D framework. In compounds 2–4 and 6, the HL anions bridge Zn(II) atoms to generate 1D chains in different bidentate coordination modes. In 5, μ4-HL anions connect Zn(II) atoms to give a 2D layer. In 7, the L anion coordinates to six Cu(II) centers to form a (3,8)-connected (4·62)(44·68·812·104) topology.
In the IR spectrum of compounds 1–6, the vibration bands at 1682 cm−1 for 1, 1682 cm−1 for 2, 1713 cm−1 for 3, 1686 cm−1 for 4, 1714 cm−1 for 5 and 1705 cm−1 for 6 were observed, which indicates the incomplete deprotonation of the H3L ligand.3a The peaks around 1536 and 1386 cm−1 for 1, 1571 and 1378 cm−1 for 2, 1570 and 1381 cm−1 for 3, 1567 and 1369 cm−1 for 4, 1544 and 1410 cm−1 for 5, 1558 and 1405 cm−1 for 6, and 1544 and 1400 cm−1 for 7 can be attributed to the asymmetric and symmetric vibrations of the carboxylate groups. The broad bands centered around 3504 cm−1 for 1, 3471 cm−1 for 2, 3437 cm−1 for 4 and 3386 cm−1 for 7 are attributable to the O–H stretching frequency of the water molecules.4d
Thermal analysis
To characterize the compounds more fully in terms of thermal stability, the thermal behaviors of 1–7 were examined by TGA. The experiments were performed on samples consisting of numerous single crystals with a heating rate of 10 °C min−1 (Fig. S3†).
For compound 1, the weight loss corresponding to the release of a lattice water molecule is observed from 147 to 233 °C (obsd 5.10%, calcd 4.53%). The anhydrous compound begins to decompose at 362 °C. The remaining weight corresponds to the formation of ZnO (obsd 20.2%, calcd 20.47%). For 2, the first weight loss corresponding to the release of a water molecule is observed before 115 °C (obsd 2.4%, calcd 4.60%), and the departure of organic components occurs from 296 °C. For 3–6, the decomposition of organic components occurs at 331–565, 276–550, 236–645 and 298–570 °C, respectively. Compound 7 releases its water molecule from room temperature to 160 °C (obsd 3.7%, calcd 5.57%), and the anhydrous compound begins to decompose at 284 °C.
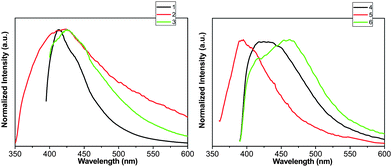
Luminescent properties
Solid-state luminescent studies were carried out for compounds 1–6 and free ligands H3L, phen, L1, L2, L3 and L4 (Fig. 8, Fig. S4† and Table 2). The emission bands for the free ligands are probably attributable to the π* → n or π* → π transitions.15
Table 2 Wavelengths of the emission maximums and excitation (nm), and the luminescent lifetimes (ns) of 1–6 and ligands
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
|
|
λ
em (nm) |
413 |
424 |
426 |
423 |
395 |
462 |
|
|
λ
ex (nm) |
358 |
372 |
372 |
352 |
353 |
353 |
|
|
τ (ns) |
0.92 |
1.91,10.14 |
3.60 |
2.01 |
1.93 |
1.29,6.73 |
|
| ligand |
|
phen
|
L1 |
L2 |
L3 |
L4 |
H3L
|
|
λ
em (nm) |
|
380 |
437 |
408 |
414 |
443 |
392,412 |
|
λ
ex (nm) |
|
325 |
326 |
320 |
312 |
305 |
364 |
|
τ (ns) |
|
1.39 |
|
|
|
0.88,6.78 |
1.22,3.78 |
The emission spectra of compounds exhibit emissions at about 413 nm (λex = 358 nm) for 1, 424 nm (λex = 372 nm) for 2, 426 nm (λex = 372 nm) for 3, 423 nm (λex = 352 nm) for 4, 395 nm (λex = 353 nm) for 5 and 462 nm (λex = 353 nm) for 6. The luminescent decay curves are well fitted into a single-exponential function with τ = 0.92 ns for 1, 3.60 ns for 3, 2.01 ns for 4 and 1.93 ns for 5. The decay curves can be fitted with a double-exponential decay function with τ1 = 1.91 ns (61.72%) and τ2 = 10.14 ns (38.28%) for 2; τ1 = 1.29 ns (34.76%) and τ2 = 6.73 ns (65.24%) for 6. The emissions of compounds 1–6 are mainly based on the organic ligands.
The emissions of compounds 1, 3–5 may originate from the corresponding HL anions, because similar emissions are observed for H3L ligand (λem = 392 and 412 nm, λex = 364 nm). The luminescent lifetimes of these compounds are very close to the ones of the free H3L ligand [τ1 = 1.22 ns (65.6%) and τ2 = 3.78 ns (34.4%)].16 The broad peaks for 2 and 6 exhibit a red-shift with respect to the free H3L, phen (380 nm, λex = 325 nm, τ = 1.39 ns) and L4 ligands [443 nm, λex = 305 nm, τ1 = 0.88 ns (88.6%) and τ2 = 6.78 ns (11.35%)], respectively. On the basis of the emission peaks and the luminescent lifetimes, the N-donor ligands and O-donor anions may show contributions to the broad fluorescent emissions of 2 and 6 simultaneously.17
Conclusion
In summary, seven coordination polymers based on 5-(benzonic-4-ylmethoxy)isophthalic acid have been prepared and characterized through single-crystal X-ray diffraction analyses. These compounds show fascinating 2D and 3D frameworks with various topologies. The structural diversities of the complexes indicate that this flexible tricarboxylate is a good candidate for the construction of coordination polymers with fascinating motifs. It is anticipated that other new coordination polymers with intriguing structures as well as physical properties may also be synthesized through using this ligand.
Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21071028, 21001023), the Science Foundation of Jilin Province (20090137, 20100109), the Fundamental Research Funds for the Central Universities, the Specialized Research Fund for the Doctoral Program of Higher Education, the Training Fund of NENU's Scientific Innovation Project and the Analysis and Testing Foundation of Northeast Normal University for support.
References
-
(a) A. Erxleben, Coord. Chem. Rev., 2003, 246, 203 CrossRef CAS;
(b) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS;
(c) S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC;
(d) Q. Wang, R. Yang, C.-F. Zhuang, J.-Y. Zhang, B.-S. Kang and C.-Y. Su, Eur. J. Inorg. Chem., 2008, 1702 CrossRef CAS.
-
(a) J. Yang, J.-F. Ma, Y.-Y. Liu, J.-C. Ma and S. R. Batten, Cryst. Growth Des., 2008, 8, 4383 CrossRef CAS;
(b) H.-Y. Bai, J.-F. Ma, J. Yang, Y.-Y. Liu, H. Wu and J.-C. Ma, Cryst. Growth Des., 2010, 10, 995 CrossRef CAS;
(c) L.-P. Zhang, J.-F. Ma, J. Yang, Y.-Y. Pang and J.-C. Ma, Inorg. Chem., 2010, 49, 1535 CrossRef CAS;
(d) B. Li, G. Li, D. Liu, Y. Peng, X. Zhou, J. Hua, Z. Shi and S. Feng, CrystEngComm, 2011, 13, 1291 RSC;
(e) B. Li, Y. Zhang, G. Li, D. Liu, Y. Chen, W. Hu, Z. Shi and S. Feng, CrystEngComm, 2011, 13, 2457 RSC.
-
(a) L. Chen, G.-J. Xu, K.-Z. Shao, Y.-H. Zhao, G.-S. Yang, Y.-Q. Lan, X.-L. Wang, H.-B. Xu and Z.-M. Su, CrystEngComm, 2010, 12, 2157 RSC;
(b) X. Zhang, J.-X. Yang, J. Zhang, J.-K. Cheng, M.-L. Sun and Y.-G. Yao, Inorg. Chem. Commun., 2011, 14, 986 CrossRef CAS.
-
(a) M.-H. Hu, G.-L. Shen, J.-X. Zhang, Y.-G. Yin and D. Li, Cryst. Growth Des., 2009, 9, 4533 CrossRef CAS;
(b) L.-L. Wen, F. Wang, J. Feng, K.-L. Lv, C.-G. Wang and D.-F. Li, Cryst. Growth Des., 2009, 9, 3581 CrossRef CAS;
(c) H.-Y. Bai, J.-F. Ma, J. Yang, L.-P. Zhang, J.-C. Ma and Y.-Y. Liu, Cryst. Growth Des., 2010, 10, 1946 CrossRef CAS;
(d) W.-Q. Kan, J. Yang, Y.-Y. Liu and J.-F. Ma, Polyhedron, 2011, 30, 2113 CrossRef.
-
(a) L. Yi, X. Yang, T. Lu and P. Cheng, Cryst. Growth Des., 2005, 5, 1215 CrossRef CAS;
(b) D.-Z. Wang, C.-S. Liu, J.-R. Li, L. Li, Y.-F. Zeng and X.-H. Bu, CrystEngComm, 2007, 9, 289 RSC;
(c) Y.-Y. Liu, L. Yi, B. Ding, Y.-Q. Huang and P. Cheng, Inorg. Chem. Commun., 2007, 10, 517 CrossRef CAS;
(d) B. Ding, Y.-Y. Liu, Y.-Q. Huang, W. Shi, P. Cheng, D.-Z. Liao and S.-P. Yan, Cryst. Growth Des., 2009, 9, 593 CrossRef CAS;
(e) X.-L. Tong, D.-Z. Wang, T.-L. Hu, W.-C. Song, Y. Tao and X.-H. Bu, Cryst. Growth Des., 2009, 9, 2280 CrossRef CAS;
(f) N. Liang, J. Wang, D. Yuan, B. Li and H. Li, Inorg. Chem. Commun., 2010, 13, 844 CrossRef CAS;
(g) P. I. M. List, S. J.-L. Oldenbroek, E. N. Lugthart, G. A. Albada, P. Gamez, J. G. Haasnoot, S. J. Teat, O. Roubeau, I. Mutikainen and J. Reedijk, Inorg. Chim. Acta, 2011, 370, 164 CrossRef;
(h) X. He, J.-J. Liu, H.-M. Guo, M. Shao and M.-X. Liu, Polyhedron, 2010, 29, 1062 CrossRef CAS;
(i) K. Liu, W. Shi and P. Cheng, Dalton Trans., 2011, 40, 8475 RSC.
- Y. H. He, Y. L. Feng, Y. Z. Lan and Y. H. Wen, Cryst. Growth Des., 2008, 8, 3586 CAS.
-
(a)
G. M. Sheldrick, SHELXS-97, Programs for X-ray Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997 Search PubMed;
(c)
L. J. Farrugia, WINGX, A Windows Program for Crystal Structure Analysis, University of Glasgow, Glasgow, UK, 1988 Search PubMed.
-
(a)
A. F. Wells, Three-dimensional Nets and Polyhedra; Wiley-Interscience: New York, 1977 Search PubMed;
(b)
A. F. Wells, Further Studies of Three-Dimensional Nets; ACA Monograph 8; American Crystallographic Association: New York, 1979 Search PubMed;
(c) O. V. Dolomanov, A. J. Blake, N. R. Champness and M. Schröder, J. Appl. Crystallogr., 2003, 36, 1283 CrossRef CAS.
- Y.-Y. Liu, Z.-H. Wang, J. Yang, B. Liu, Y.-Y. Liu and J.-F. Ma, CrystEngComm, 2011, 13, 3811 RSC and refs. therein.
- Y.-Y. Liu, J.-F. Ma, J. Yang, J.-C. Ma and G.-J. Ping, CrystEngComm, 2008, 10, 565 RSC.
- L. Rodríguez, E. Labisbal, A. Sousa-Pedrares, J. A. García-Vázquez, J. Romero, M. L. Durán, J. A. Real and A. Sousa, Inorg. Chem., 2006, 45, 7903 CrossRef.
- X. Li, D. Cheng, J. Lin, Z. Li and Y. Zheng, Cryst. Growth Des., 2008, 8, 2853 CAS.
- Y.-Y. Liu, J.-F. Ma, J. Yang, J.-C. Ma and Z.-M. Su, CrystEngComm, 2008, 10, 894 RSC.
-
(a) L. Carlucci, G. Ciani and D. M. Proserpio, Chem. Commun., 2004, 380 RSC;
(b) J. Fan, G. T. Yee, G. B. Wang and B. E. Hanson, Inorg. Chem., 2006, 45, 599 CrossRef CAS.
-
(a) X. Shi, G. Zhu, Q. Fang, G. Wu, G. Tian, R. Wang, D. Zhang, M. Xue and S. Qiu, Eur. J. Inorg. Chem., 2004, 185 CrossRef;
(b) G. Tian, G. S. Zhu, Q. R. Fang, X. D. Guo, M. Xue, J. Y. Sun and S. L. Qiu, J. Mol. Struct., 2006, 787, 45 CrossRef CAS.
-
(a) L.-Y. Zhang, G.-F. Liu, S.-L. Zheng, B.-H. Ye, X.-M. Zhang and X.-M. Chen, Eur. J. Inorg. Chem., 2003, 2965 CrossRef CAS;
(b) J. Fan, T. Okamura, Z.-H. Zhang, G.-X. Liu, K.-B. Yu, W.-Y. Sun and N. Ueyama, Inorg. Chem., 2006, 45, 3941 CrossRef;
(c) H. Wu, H.-Y. Liu, J. Yang, B. Liu, J.-F. Ma, Y.-Y. Liu and Y.-Y. Liu, Cryst. Growth Des., 2011, 11 DOI:10.1021/cg200005q.
- Y.-Y. Liu, J.-F. Ma, J. Yang and Z.-M. Su, Inorg. Chem., 2007, 46, 3027 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond distances and angles; structure illustrations for compounds 1 and 7; TGA, luminescence decay and PXRD curves. CCDC reference numbers 828044–828050. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05639e |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.