In situ formation of new organic ligands to construct two novel self-charge-transfer Pb(II)-based frameworks†
Yi
Liu
a,
Melvin Jun
Wei Tan
a,
Fengxia
Wei
a,
Yufeng
Tian
b,
Tom
Wu
b,
Christian
Kloc
a,
Fengwei
Huo
a,
Qingyu
Yan
a,
Huey Hoon
Hng
a,
Jan
Ma
a and
Qichun
Zhang
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore. E-mail: qczhang@ntu.edu.sg
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore, 637371, Singapore
First published on 7th November 2011
Abstract
Employing in situ-formed new organic ligands, two Pb(II)-based metal–organic frameworks were synthesized. Compound 1 possesses a 3D framework while compound 2 has a layered structure. Solid state UV-visible absorptions indicate that the two compounds are semiconductors with band gaps of 1.70 and 1.78 eV. Magnetic properties reveal that they are temperature-independent diamagnetic.
Metal–organic framework (MOF) is a classical system coming from the marriage between metal ions and organic ligands.1 The variety of organic ligands (e.g. shapes, sizes and lengths) together with the choice of metal centers (e.g. coordination number) and reaction conditions (e.g.solvent, temperature, pH value) can create enormous structural diversity and multifunctional properties (e.g. porous for storage, chiral, magnetic, separation, sensing, catalytic, drug-delivering, bio-imaging).2–9 To frustrate us, the metal centers and the organic ligands in most metal–organic frameworks have no “communicating” (electron interactions) although electron-transfer (partially or completely) is a common phenomenon in individual metal–organic complex systems. The charge-transfer (or communicating) between metal centers and organic ligands in one framework could switch MOFs from insulating to semiconducting, and even conducting states. New applications in electronic or semiconductor devices could be made possible with such switching.
On the other hand, most of the research on MOF so far has focused on d-block transition MOFs, and main group MOFs have been relatively less explored. Compared with the transition metal atoms, the main group metal lead atom presents a larger radius and adopts [Xe]4f125d106s26p2 electronic configurations. When the lead atom is oxidised, it is easy to get the Pb(II) cation, which has a [Xe]4f125d106s2 electronic structure. Such an electronic configuration will give the Pb(II) cation variable coordination numbers (from 2 to 10), which provide unique opportunities for the construction of fascinating network topologies and some interesting physical properties such as luminescence,10,11 ion exchange,12 and nonlinear optics.13 Thus, lead-based structural chemistry has recently been receiving increasing attention by virtue of its fantastic architectures and good physical properties.14–16 As a functional center, Pb(II) cation prefer to coordinate with organic ligands containing O, N, and S donor atoms or their combination thereof.17–19 Among all the organic ligands, carboxylate ligands are often employed to assemble various Pb(II) complexes with interesting structures because of its excellent coordinating ability, versatile coordination fashion, and large conjugated systems. However, Pb(II) coordination polymers, in which Pb(II) atoms bind to oxygen coming from rigid benzoquinone ligands, are rarely reported as yet.
In this work, we successfully synthesized a family of Pb(II) MOFs, C9H7Cl2NO5Pb (1) and C16H12N2O5SPb (2), in which two novel rigid organic ligands were in situ formed from the parent compounds tetrachloro-1,4-benzoquinone and 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ).‡ Both Pb(II)-based frameworks were prepared at ambient conditions through diffusion methods. Compound 1 was obtained as dark-red crystals by slow diffusion of dimethylformamide (DMF) solution containing tetrachloro-1,4-benzoquinone into another yellowish DMF solution containing Pb(SC6H5)2 (SPh = benzenethiolate) in 4–6 weeks. The preparation of compound 2 is similar to that of compound 1, except that tetrachloro-1,4-benzoquinone was replaced by DDQ. Although two complexes were synthesized in a similar way, there are great differences in the newly generated organic ligands and coordination modes of the Pb(II) center.
Single crystal X-ray diffraction showed that compound 1 possesses a 3D framework while compound 2 has a layered structure.§ Compound 1 crystallizes in the orthorhombic space groupPnma. The asymmetric unit of 1 consists of one half Pb(II) atom, half coordinated μ2-DMF molecule and half ligand 3. The new ligand 3 was derived from tetrachloro-1,4-benzoquinone, in which two para-position Cl atoms were replaced by O atoms (Scheme 1).20Pb(II) atom exhibits a greatly distorted bicapped trigonal prism, where two oxygen atoms (from two different coordinated DMF molecules) as capped atoms and six other oxygen atoms (from four different ligands 3) as vertex atoms generate the PbO8 geometry. Each bicapped trigonal prism is connected with the other two via edge-sharing and vertex-sharing to form a 1D zig–zag infinite chain along the a-direction (Fig. 1a). These chains are further interconnected together by the in situ formed ligands to construct the final 3D framework with 1D long channels. The approximate dimension of the channels is 14.46 × 10.37 Å2 (based on Pb⋯Pb distances), as shown in Fig. 1b. If the Pb1 center is selected as a single node, and ligand 3 and DMF as linear bridging connectors, respectively, the distance of the adjacent Pb1 nodes in the 1D zig–zag infinite chain is 4.274 Å and that separated by the ligand 3 is 8.848 Å. Thus, a typical diamond-type topology was formed, belonging to rod packing motifs, which is equivalent to the 3D layer structure of 1 (Fig. 1c). PLATON21 calculation showed that the resulting effective free volume, after removal of encapsulated DMF molecules, was 29.4% of the crystal volume (358.9 Å3 out of the 1221.6 Å3 unit cell volume).
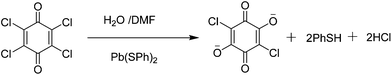 | ||
| Scheme 1 Schematic drawing of formation of ligand 3. | ||
 | ||
| Fig. 1 (a) View of the Pb(II) polyhedra chain in 1; (b) Structure of 3D framework of 1 down the a-axis; (c) Topological view showing the equivalent 3D framework of 1. | ||
Compound 2 belongs to another structure type with the monoclinic system with P21/c space group and features a 2D layered structure. In the asymmetric unit of compound 2, there are one independent Pb(II) cation, one terminal coordinated DMF molecule and one ligand 4. The new ligand 4 originated from the reaction between DDQ and benzenethiolate, meanwhile, two O atoms substituted for one Cl atom and one nitrile group in DDQ (Scheme 2). This new ligand 4 is obviously different from the new ligand 3 because of the incorporation of benzenethiolate ligand. The Pb(II) center is in hexacoordinated mode, and shows an extremely distorted trigonal antiprism geometry composed of eight O atoms from one terminal coordinated DMF molecule and three different ligands 4 (Fig. 2a). Thus, each PbO8 polyhedron shares the edge with another one, constructing a Pb2O10 dimer. Each Pb2O10 dimer is further bridged to four adjacent Pb2O10 dimers through the linkage of four ligands 4 to make up a 2D layered MOF parallel to the bc-plane. Then, the layers are overlapped and separated from each other by the terminal coordinated DMF molecules to form 1D long channels with a size of ∼13.82 × 8.96 Å2 (based on Pb⋯Pb distances), as shown in Fig. 2b, and the benzenethiolate species are located in the channels. There exist N⋯H hydrogen bonds between 2D layers in compound 2, which are shown in Fig. S1.† In both compounds, the Pb–O bond distances vary from 2.420(6) to 3.010(4) Å with an average value of 2.715 Å, which is comparable to those found in other reported lead complexes.10,11 Topological analysis shows that is a typical two-dimensional 44 square grid (Fig. 2c).
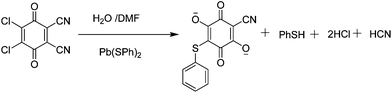 | ||
| Scheme 2 Schematic drawing of formation of ligand 4. | ||
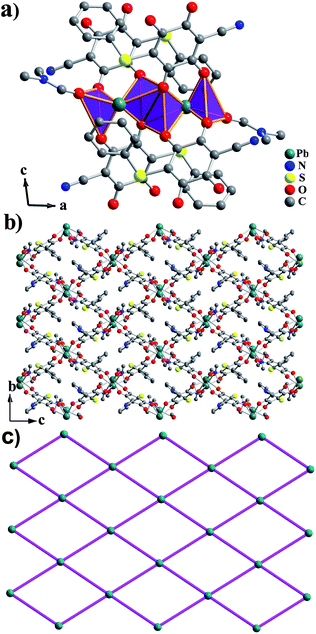 | ||
| Fig. 2 (a) View of the Pb(II) dimers in 2; (b) View of a single layer of 2 down the a-axis; (c) Topological view showing the equivalent 2D framework of 2. | ||
The powder XRD patterns of compounds 1 and 2 are consistent with the simulated ones obtained on the basis of single-crystal structure, indicating that the as-synthesized samples (Fig. S2†) are pure. For the patterns of compound 2, there are slight differences in reflection intensities between the simulated and the experimental one, which are due to the variation in the crystal orientation of the power samples.
The solid state UV-visible absorption for compounds 1 and 2 show that they are semiconductors with band gaps of 1.70 eV and 1.78 eV, which agree with their dark red color (Fig. 3). Such low band gaps suggest that there might be some charge transfer between the Pb(II) centers and organic ligands. This result was furthermore confirmed by the UV-vis absorption of the organic ligands (<350 nm, Fig. S3†). Magnetic properties for compounds 1 and 2 have been studied at an applied field of 5000 Oe in the temperature range of 5–273 K. The molar magnetic susceptibilities (Fig. S4 and S5†) show that both compounds are temperature-independent diamagnetic, and the magnetic properties are dominated by diamagnetic contributions from the Pb(II) cations and the ligands. The diamagnetic properties of both complexes indicated that the charge transfer between Pb(II) metal centers and organic ligands is not complete (0 < q < 1).
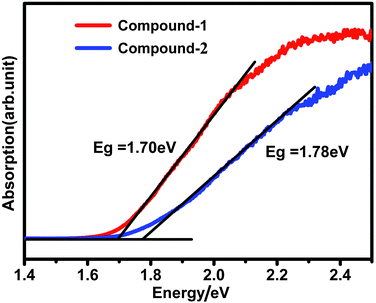 | ||
| Fig. 3 The UV-visible absorbance of compounds 1 and 2. | ||
To study the thermal stability of compounds 1 and 2, the thermogravimetric analysis (TGA) was performed on polycrystalline samples in flowing N2 with a heating rate of 10 °C min−1. TGA curve of compound 1 (Fig. S6†) shows there is no mass change from room temperature to 200 °C. The first step weight loss of 15.55% (calcd. 15.00%) from 200 °C to 274 °C corresponds to the removal of one coordinated μ2-DMF molecule, and is then followed by a long slope which can be attributed to the decomposition of the organic ligands. The TGA curve of compound 2 (Fig. S7†) shows that compound 2 is stable up to 100 °C. The first stage is a weight loss of 12.70% starting at 100 °C, which is due to the loss of one terminal coordinated DMF molecule (calcd: 13.25%). The subsequent weight loss of 2 can be due to the decomposition of the organic ligands. Comparing the TGA curves of compounds 1 and 2, indicates that compound 1 is much more stable than compound 2.
In summary, two novel semiconducting self-charge-transfer Pb(II) polymers have been synthesized and structurally characterized. In situ formation organic ligands (3–4) present in the two compounds are from benzoquinone ligands, to which little attention has been paid so far. This work not only greatly enriches the chemistry of the self-assembly of Pb(II)-based MOFs but also gives impetus to the further exploration of the coordination chemistry of the benzoquinone ligands.
Acknowledgements
The authors thank the support from the NTU start-up grant and Tier 1 fund from MOE.Notes and references
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- (a) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (b) S. T. Zheng, J. T. Bu, Y. F. Li, T. Wu, F. Zuo, P. Y. Feng and X. H. Bu, J. Am. Chem. Soc., 2010, 132, 17062 CrossRef CAS; (c) X. H. Bu, M. L. Tong, H. C. Chang, S. Kitagawa and S. R. Batten, Angew. Chem., Int. Ed., 2004, 43, 192 CrossRef CAS; (d) J. R. Li, Y. Tao, Q. Yu, X. H. Bu, H. Sakamoto, S. Kitagawa and S. R. Batten, Chem.–Eur. J., 2008, 14, 2771 CrossRef CAS.
- (a) D. Zhao, D. J. Timmons, D. Q. Yuan and H. C. Zhou, Acc. Chem. Res., 2011, 44, 123 CrossRef CAS; (b) L. Q. Ma, J. M. Falkowski, C. Abney and W. B. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS; (c) F. J. Song, C. Wang, J. M. Falkowski, L. Q. Ma and W. B. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef; (d) R. E. Morris and X. H. Bu, Nat. Chem., 2010, 2, 353 CrossRef CAS; (e) J. Zhang, S. M. Chen, A. Zingiryan and X. H. Bu, J. Am. Chem. Soc., 2008, 130, 17246 CrossRef CAS; (f) K. Tanaka, S. Oda and M. Shiro, Chem. Commun., 2008, 820 RSC.
- (a) J. S. Hu, L. F. Huang, X. Q. Yao, L. Qin, Y. Z. Li, Z. J. Guo, H. G. Zheng and Z. L. Xue, Inorg. Chem., 2011, 50, 2404 CrossRef CAS; (b) S. Nayak, K. Harms and S. Dehnen, Inorg. Chem., 2011, 50, 2714 Search PubMed; (c) Q. C. Zhang, X. H. Bu, Z. E. Lin, T. Wu and P. Y. Feng, Inorg. Chem., 2008, 47, 9724 CrossRef CAS.
- (a) Z. Y. Gu and X. P. Yan, Angew. Chem., Int. Ed., 2010, 49, 9702 CrossRef CAS; (b) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (c) B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS; (d) Y.-S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135 RSC; (e) B. L. Chen, S. Q. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS.
- (a) B. L. Chen, L. B. Wang, F. Zapata, G. D. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef CAS; (b) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC; (c) G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832 CrossRef CAS; (d) J. R. Li, Y. Tao, Q. Yu and X. H. Bu, Chem. Commun., 2007, 1527 RSC.
- (a) L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (b) J.-Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (c) A. Corma, H. Garcia and F. X. L. I. Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS; (d) C. D. Wu and W. B. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075 CrossRef CAS; (e) D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916 CrossRef CAS.
- (a) P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS; (b) Q. Zhang, X. H. Bu, Z. Lin, T. Wu and P. Feng, Inorg. Chem., 2008, 47, 9724 CrossRef CAS; (c) J. Y. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376 CrossRef CAS; (d) Y. Liu, F. Boey, L. L. Lao, H. Zhang, X. Liu and Q. Zhang, Chem.–Asian J., 2011, 6, 1004 Search PubMed.
- (a) K. M. L. Taylor-Pashow, J. Della Rocca, Z. G. Xie, S. Trans and W. B. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS; (b) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2009, 9, 172.
- (a) L. Zhang, Z. J. Li, Q. P. Lin, Y. Y. Qin, J. Zhang, P. X. Yin, J. K. Cheng and Y. G. Yao, Inorg. Chem., 2009, 48, 6517 CrossRef CAS; (b) J. D. Lin, S. T. Wu, Z. H. Li and S. W. Du, CrystEngComm, 2010, 12, 4252 RSC; (c) E. C. Yang, J. Li, B. Ding, Q. Q. Liang, X. G. Wang and X. J. Zhao, CrystEngComm, 2008, 10, 158 RSC.
- (a) J. Yang, G. D. Li, J. J. Cao, Q. Yue, G. H. Li and J. S. Chen, Chem.–Eur. J., 2007, 13, 3248 CrossRef CAS; (b) K. L. Huang, X. Liu, J. K. Li, Y. W. Ding, X. Chen, M. X. Zhang, X. B. Xu and X. J. Song, Cryst. Growth Des., 2010, 10, 1508 Search PubMed; (c) Y. H. Zhao, H. B. Xu, Y. M. Fu, K. Z. Shao, S. Y. Yang, Z. M. Su, X. R. Hao, D. X. Zhu and E. B. Wang, Cryst. Growth Des., 2008, 8, 3566 CrossRef CAS.
- K. Kavallieratos, J. M. Rosenberg and J. C. Bryan, Inorg. Chem., 2005, 44, 2573 CrossRef CAS.
- X. R. Meng, Y. L. Song, H. W. Hou, Y. T. Fan, G. Li and Y. Zhu, Inorg. Chem., 2003, 42, 1306 CrossRef CAS.
- (a) D. S. Li, Y. P. Wu, P. Zhang, M. Du, J. Zhao, C. P. Li and Y. Y. Wang, Cryst. Growth Des., 2010, 10, 2037 CrossRef CAS; (b) X. L. Wang, Y. Q. Chen, Q. Gao, H. Y. Lin, G. C. Liu, J. X. Zhang and A. X. Tian, Cryst. Growth Des., 2010, 10, 2174 CrossRef CAS; (c) G. P. Yang, L. Hou, Y. Y. Wang, Y. N. Zhang, Q. Z. Shi and S. R. Batten, Cryst. Growth Des., 2011, 11, 936 CrossRef CAS; (d) Z. Y. Du, H. B. Xu, X. L. Li and J. G. Mao, Eur. J. Inorg. Chem., 2007, 4520 CrossRef CAS.
- (a) B. Ding, Y. Y. Liu, X. X. Wu, X. J. Zhao, G. X. Du, E. C. Yang and X. G. Wang, Cryst. Growth Des., 2009, 9, 4176 Search PubMed; (b) J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma and S. R. Batten, Cryst. Growth Des., 2009, 9, 1894 CrossRef CAS; (c) S. H. Li, S. K. Gao, S. X. Liu and Y. N. Guo, Cryst. Growth Des., 2010, 10, 495 CAS.
- (a) T. F. Liu, J. Lü, C. B. Tian, M. N. Cao, Z. J. Lin and R. Cao, Inorg. Chem., 2011, 50, 2264 Search PubMed; (b) A. Thirumurugan, R. A. Sanguramath and C. N. R. Rao, Inorg. Chem., 2008, 47, 823 CrossRef CAS.
- R. L. Davidovicha, V. Stavilab, D. V. Marinina, E. I. Voita and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef CAS.
- (a) K. L. Zhang, Y. Chang, C. T. Hou, G. W. Diao, R. T. Wu and S. W. Ng, CrystEngComm, 2010, 12, 1194 RSC; (b) J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma and S. R. Batten, Inorg. Chem., 2007, 46, 6542 CrossRef CAS; (c) C. Gabriel, C. P. Raptopoulou, V. Psycharis, A. Terzis, M. Zevou, C. Mateescu and A. Salifoglou, Cryst. Growth Des., 2011, 11, 382 Search PubMed.
- R. L. Davidovicha, V. Stavilab and K. H. Whitmire, Coord. Chem. Rev., 2010, 254, 2193 CrossRef CAS.
- (a) J. T. Wrobleski and D. B. Brown, Inorg. Chem., 1979, 18, 498 CrossRef CAS; (b) M. Verdaguer, A. Michalowicz, J. J. Girerd, N. A. Berding and O. Kahn, Inorg. Chem., 1980, 19, 3271 CrossRef CAS; (c) S. Kawata, S. Kitagawa, H. Kumagai, T. Ishiyama, K. Honda, H. Tobita, K. Adachi and M. Katada, Chem. Mater., 1998, 10, 3902 CrossRef CAS.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2001 Search PubMed.
- G. M. Sheldrick, Shelxtl NT, Program for solution and refinement of crystal structures, version 5.1, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Simulated and experimental X-ray powder diffraction pattern, UV-Vis absorption, Magnetic properties, TGA curve and IR spectra. CCDC reference numbers 836172 and 836173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce06006f |
| ‡ Synthesis of C9H7Cl2NO5Pb (1): Pb(SC6H5)2 (0.3 mmol, 60.18 mg) was dissolved in 4.6 mL DMF solution to form a yellow solution. To this solution, a 2.3mL DMF solution containing tetrachloro-1,4-benzoquinone (0.3 mmol, 73.76 mg) was layered on top. After 4–6 weeks, dark-red block crystals were collected in 42% yield (Based on Pb(SC6H5)2). Anal. calc. for C9H7Cl2NO5Pb (1): H, 1.45%; N, 2.87%; C, 22.18%. Found: H, 1.23%; N, 2.72%; C, 22.68%. Synthesis of C16H12N2O5SPb (2): Crystals of 2 were prepared by a similar procedure to that of complex 1, except that tetrachloro-1,4-benzoquinone was replaced by 2,3-dichloro-5,6-dicyanobenzoquinone. After 4–6 weeks, dark-red plate crystals were collected in 57% yield (Based on Pb(SC6H5)2). Anal. calc. for C16H12N2O5SPb (2): H, 2.19%; N, 5.08%; C, 34.84%; S, 5.81%. Found: H, 2.07%; N, 4.93%; C, 35.02%; S, 5.72%. |
| § Crystallographic data for 1: C9H7Cl2NO5Pb; Mr = 487.25; orthorhombic; space groupPnma; a = 8.140(2), b = 14.460(3), c = 10.370(2) Å; V = 1221.6(4) Å3, Z = 4, ρc = 2.649 g cm−3, μ = 14.258 mm−1, F(000) = 896, GOF = 0.984, a total of 8992 reflections were collected, 2553 of which were unique (Rint = 0.0271). R1 = 0.0467 and wR2 = 0.0730 for all data, R1 = 0.0278 and wR2 = 0.0657 for data with I > 2σ(I). Crystallographic data for 2: C16H12N2O5SPb; Mr = 551.53; monoclinic; space groupP21/c; a = 13.550(3), b = 8.9600(18), c = 13.820(3) Å; β = 99.16(3)°; V = 1656.5(6) Å3, Z = 4, ρc = 2.212 g cm−3, μ = 10.341 mm−1, F(000) = 1040, GOF = 1.040, a total of 11739 reflections were collected, 3783 of which were unique (Rint = 0.0310). R1 = 0.0506 and wR2 = 0.1290 for all data, R1 = 0.0323 and wR2 = 0.0873 for data with I > 2σ(I). Single crystal X-ray diffraction data for the two compounds were collected on a Bruker APEX II CCD diffractometer equipped with a graphite-monochromatized Mo-Kα radiation source with a radiation wavelength of 0.71073 Å at 293 K. Empirical absorption was performed, and the structure was solved by direct methods and refined with the aid of a SHELX-TL program package.22 All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were calculated and refined using a riding model. |
| This journal is © The Royal Society of Chemistry 2012 |