α,α′,α′′-Tris(hydroxyimino)-1,3,5-benzenetriacetonitrile: A three-fold symmetric, versatile and practical supramolecular building block†
Christer B.
Aakeröy
*,
Michelle M.
Smith
and
John
Desper
Department of Chemistry, Kansas State University, Manhattan, KS 66506, USA. E-mail: aakeroy@ksu.edu
First published on 3rd November 2011
Abstract
The synthesis of a three-fold symmetric, aromatic molecule containing three co-planar effective hydrogen-bond moieties that can become a versatile and reliable hub for the assembly of extended, porous organic networks is presented.
Trimesic acid has achieved near-classic status as a building block in structural chemistry1 and organic crystal engineering.2 Examples include hexagonal sheets of trimesic acid with space-filling coronene within the channels,3 the hydrogen-bond based complementarity of 1,3,5-tris(4-pyridyl)triazine with trimesic acid leading to a planar architecture clathrated with pyrene,4 and starting with the crystal structure determination of trimesic acid itself over forty years ago.5 This molecule has also been frequently employed in inorganic crystal engineering, and recent interest in metal–organic frameworks6 clearly demonstrates that trimesic acid remains an important and versatile tecton for supramolecular synthesis. The general appeal of trimesic acid7 is undoubtedly a result of its high symmetry that can be promoted into extended networks through three directional hydrogen-bonding (or metal-coordinating) sites. The co-planarity of the binding sites allows for the production of flat layers with large hexagonal channels, catenated and non-catenated structures,8 and ‘extended’ hexagonal networks through the use of symmetric ditopic hydrogen-bond acceptors. However, given the potential structure-building qualities, chemical simplicity, and commercial availability of trimesic acid, there is arguably a disproportionately small number of structure determinations of neutral organic architectures based around trimesic acid.9 The reason for this can undoubtedly be traced first to the poor solubility of trimesic acid. Generally speaking, as most supramolecular synthesis is carried out in solution, it is important that all reactants have comparable solubility if they are to be assembled together into one crystalline heteromeric architecture with desired connectivity and dimensionality. Second, since trimesic acid displays perfect self-complementarity comprising particularly strong intermolecular interactions, hydrogen-bond based acid⋯acid dimers, it is difficult to make trimesic acid ‘abandon’ itself, without having to resort to deprotonation of one or more of the carboxylic acid moieties, (which frequently leads to unpredictable structures and chemical compositions10). Against this background, we wanted to design an alternative to trimesic acid as well as to other recently reported trigonal molecules such as benzene-1,3,5-tri-p-phenylphosphonic acid11 and 1,3,5-cyclohexanetricarboxylic acid.12
Our target, therefore, is an aromatic molecule with three-fold symmetry containing three co-planar effective hydrogen-bond donor moieties that, through less favorable self-complementarity and improved solubility can become a versatile and reliable hub for the assembly of extended, organic networks. A suitable molecule that meets all requirements is α,α′,α′′-tris(hydroxyimino)-1,3,5-benzenetriacetonitrile, 2, Scheme 1, because aromatic cyanooximes are capable hydrogen-bond donors,13cyanooxime catemers and dimers are less stable than carboxylic acid dimers,14 and solubilities of oximes are generally superior to those of their acid analogues. Herein we present the synthesis and characterization of 2 as well as the crystal structure of a co-crystal of 2 and a symmetric ditopic hydrogen-bond acceptor, 1,2-di(4-pyridyl)ethane, 3, Scheme 2, which displays precisely the extended network structure that was desired and postulated.
 | ||
| Scheme 1 | ||
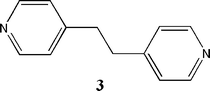 | ||
| Scheme 2 | ||
Synthesis of 1: To a solution of 1,3,5-tris(bromomethyl)benzene (3.02 g, 8.50 mmol) in THF (25 ml) was added sodium bicarbonate (saturated solution, 30 mL) and sodium cyanide (4.17 g, 85.0 mmol), followed by 30 mL water. The solution was left to stir for 48 h after which it was acidified with 1 M HCl, and THF was removed viarotary evaporation to leave behind an off-white solid. Yield: 1.53 g (92%); 1H NMR (200 MHz, CDCl3): δ 3.81 (s, 6H), 7.31 (s, 3H); IR: ν 2253 cm−1 (C≡N); MP: 110–115 °C. Despite repeated attempts we were not successful at growing crystals of 1 suitable for single-crystal X-ray diffraction.
Synthesis of 2: Na metal (0.540 g, 23.5 mmol) was dissolved in 540 mL 2-propanol. 1 (0.80 g, 4.10 mmol) was dissolved in ∼100 mL 2-propanol and added to the sodium solution. Methyl nitrite gas was generated in situ (see Supplementary details for full procedure) and passed through the solution, which caused it to turn yellow. The solution was left to stir for 5 days, after which a precipitate emerged. This solid, the sodium salt of 2, was filtered off and dried. It was then dissolved in water which was acidified to ∼pH 2 whereupon a brown precipitate appeared. In addition, 2-propanol was removed from the first filtrate producing a small amount of solid which was re-dissolved in water, acidified to ∼pH 2, resulting in a brown precipitate. Both solids were combined, dissolved in methylene chloride and a small amount of methanol, and purified by column chromatography. (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexanes/ethyl acetate) to yield a colorless solid. Yield: 0.822 g (71%); 1H NMR (400 MHz, DMSO-d6): δ 8.11 (s, 3H), 14.24 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 110.34, 124.29, 130.49, 132.17; IR: ν 2248 cm−1 (C≡N), ν 1040 cm−1 (N–O), ν 3395 cm−1 (O–H); MP: 240–245 °C (decomp.). m/z: 305.06 (M + Na)+. We were not able to grow crystals of 2 suitable for single-crystal X-ray diffraction.
:1 hexanes/ethyl acetate) to yield a colorless solid. Yield: 0.822 g (71%); 1H NMR (400 MHz, DMSO-d6): δ 8.11 (s, 3H), 14.24 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 110.34, 124.29, 130.49, 132.17; IR: ν 2248 cm−1 (C≡N), ν 1040 cm−1 (N–O), ν 3395 cm−1 (O–H); MP: 240–245 °C (decomp.). m/z: 305.06 (M + Na)+. We were not able to grow crystals of 2 suitable for single-crystal X-ray diffraction.
Co-crystallization of α,α′,α′′-tris(hydroxyimino)-1,3,5-benzenetriacetonitrile 1,2-di(4-pyridyl)ethane, 2:3: 2 (5 mg, 0.0177 mmol) was dissolved in ethyl acetate and added to a solution of 3 in ethyl acetate (5.0 mg, 0.0266 mmol), and the resulting solution was left to evaporate at ambient conditions. This produced an off-white powder that was recrystallized from acetonitrile to yield pale, yellow plates, mp: 246–248 °C.
X-ray data were collected on a Bruker Kappa Apex II four-circle CCD diffractometer at 120 K using a fine-focus molybdenum Kα tube. Data were collected using APEX215 software. Initial cell constants were found by small widely separated “matrix” runs. An entire hemisphere of reciprocal space was collected. Scan speed and scan width were chosen based on scattering power and peak rocking curves.
Initial analysis of the dataset showed that the sample suffered from merohedral twinning. Conversion of the triclinic unit cell using the matrix [−1 0 0][1 −2 0][0 0 1] gave a cell that was nearly monoclinic (C-centered) by metric (a = 89.68°; g = 90.32°) and symmetry (Rmerg = 23.9%) criteria. The existing triclinic HKLF 4 data were converted to an HKLF 5 format, with twin law corresponding to 2-fold rotation down the pseudo-monoclinic unique axis, using locally written software. The ratio of the two twin components could then be refined using the BASF command (which refined to ∼13%). Data were reduced with SHELXTL.16 The structure was solved by direct methods (on the original HKLF 4 format dataset) without incident. All hydrogen atoms were assigned to idealized positions and were allowed to ride. Heavy atoms were refined with anisotropic thermal parameters. Absorption correction was not performed (μ = 0.089 mm−1).
The asymmetric unit contains two tris(cyanooximes), three dipyridylethane molecules, and several solvent molecules. Excepting the solvent molecules, the unit cell contents were divided into two RESIdues, each containing one tris(oxime) and three half dipyridylethane molecules. Two of the three heterocycles exhibited criss-cross disorder of the ethano linkage. Anisotropic thermal parameters for closely spaced atoms on these fragments were pairwise constrained using EADP commands. Relative occupancies of the species were allowed to refine using free variables. Geometries of the molecules were restrained using the SAME command.
Significant residual electron density was present in the difference Fourier map after taking into account the major species. For purposes of refinement, several acetonitrile fragments were constructed using DFIX commands. Occupancy for each species was refined during initial stages of data processing. Since the molecules were close to inversion centers, suppression of bonds between unique and equivalent molecules was achieved by using the PART –n command. (With each molecule having occupancy of 50%, each unique molecule can safely be assumed to be adjacent to a void.) No attempt was made to locate or refine the solvent protons. Twelve of the 20 strongest peaks in the final difference Fourier map, with strongest peak at 0.94 electrons/Å3, are associated with solvent.
The crystal structure determination of 2:317 demonstrates that the two components are present in a 1:1.5 ratio, which reflects the fact that 2 has three hydrogen-bond donors and each dipyridyl molecule contains two hydrogen-bond acceptors. The observed ratio means that the numbers of donors and acceptors are matched perfectly and each three-fold symmetric tecton forms three O–H⋯N hydrogen bonds to three different dipyridyl units, Table 1, Fig.1.
| D–H⋯A | r(D⋯A) Å | <(DHA)° |
|---|---|---|
| a OXX_1 and OXX_2 represent the unique oxygen atoms of the three oximes on the two crystallographically unique tritopic oximes. NXX_1 and NXX_2 represent the hydrogen-bond acceptor sites located on nitrogen atoms located on six unique bipyridine fragments (see ESI for detailed labeling scheme). | ||
| O21_1–H21_1⋯N31_1 | 2.609(2) | 157.8 |
| O23_1–H23_1⋯N41_1 | 2.676(2) | 170.6 |
| O25_1–H25_1⋯N51_1 | 2.718(3) | 161.6 |
| O21_2–H21_2⋯N31_2 | 2.598(2) | 157.7 |
| O23_2–H23_2⋯N41_2 | 2.620(4) | 157.5 |
| O25_2–H25_2⋯N51_2 | 2.591(4) | 165.8 |
 | ||
| Fig. 1 The central motif in the crystal structure of 2:3; each molcule of 2 forms three O–H⋯N hydrogen bonds with neighboring pyridyl-based molecules (disordered atoms removed for clarity). | ||
As each dipyridyl moiety acts as a bridge between two tricyanooximes, the end result is an infinite 2-D assembly constructed from three unique O–H⋯N hydrogen bonds, Fig. 2.
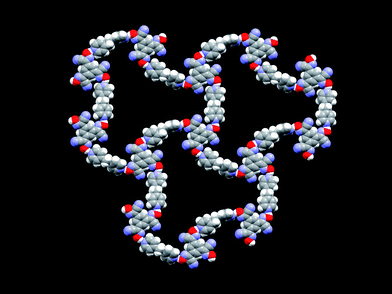 | ||
| Fig. 2 The extended hydrogen-bonded network in the crystal structure of 2:3; disordered solvent molecules (removed for clarity) occupy the space within the host structure. | ||
Each crystallographically unique layer is constructed by three O–H⋯N hydrogen bonds (shown in Table 1, underscore 1 and 2, respectively).
There are no O–H⋯N hydrogen bonds between layers and the layer-layer orientation, which is slightly off-set, is therefore controlled by relatively weak dispersion forces and is non-specific. The overall structure looks deceptively open, but the lattice also contains several disordered solvent molecules. The solvent is lost from the crystal upon standing, which produces a microcrystalline powder. The ease with which lattice solvent is lost can probably be attributed to the fact that the relative orientation of neighbouring 2-D networks leads to a 3-D architecture with continuous channels, Fig. 3. It is worth noting, however, that the intended primary host network by itself is not disrupted by any solute–solvent hydrogen-bonds
 | ||
| Fig. 3 A view down the a- axis shows the channels (occupied by solvent molecules, running through the crystal structure of 2:3. | ||
The hydrogen bonds between pyridyl and cyano-oxime moieties in this structure fall in the range of 2.59–2.72 Å (for the O⋯N distances) and 158–171° (for the O–H⋯N angles). A search of the CSD18 produced 15 hits of co-crystals between a cyano-oxime and a N-heterocyclic hydrogen bond acceptor and the corresponding distances and angles are, 2.58–2.68 Å and 157–179°, respectively. It is of interest to note that in the case where cyano-oximes form homomeric interactions (ten hits in the CSD), the corresponding O⋯N distances are significantly longer at 2.77–2.90 Å. This observation is readily explained by the fact that in the case of oxime-oxime hydrogen bonds the acceptor is either the N-nitrile or the N-oxime both of which are substantially weaker hydrogen-bond acceptors than the pyridyl moiety employed in this present structure (most cyanooxime-based co-crystals in the CSD involve imidazole/benzimidazole acceptors which are also very strong). Finally, over 90% of all co-crystals containing a carboxylic acid-py, O–H⋯N, hydrogen bond have O⋯N distances in the range of 2.54–2.73 Å which further illustrates that the cyano-oxime moiety is capable of forming hydrogen-bonds of comparable strength as those displayed by carboxylic acids.
The desired improvement in solubility vis-a-vis the analogous carboxylic acid was attained; 2 is soluble in a variety of organic solventscf.ethanol, diethylether, ethylacetate and dichloromethane. The synthesis and characterization of 2 as well as the demonstration that this molecule can indeed produce porous crystalline molecular solids indicate that 2 may offer a platform for several research ventures in a variety of areas. For example, previous work has shown that the assembly of porous monolayers/networks of trimesic acid on gold19 and graphite20 surfaces is driven by intermolecular hydrogen bonds, and it is likely that, in addition to the obvious opportunities for cyanooxime-driven organic- and inorganic21 crystal engineering, 2 may also be a useful candidate for nanopatterning and deposition on surfaces as it shares several important chemical/structural characteristics with trimesic acid.22
We gratefully acknowledge financial support from NSF (CHE-0957607) and from the Terry C. Johnson Center for Basic Cancer Research.
Notes and references
- F. H. Herbstein, M. Kapon and G. M. Reisner, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 348 CrossRef.
- (a) J. D. Wuest, Chem. Commun., 2005, 5830–5837 RSC; (b) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS; (c) G. R. Desiraju, Crystal Engineering: The Design of Organic Solids; Elsevier: Amsterdam, 1989 Search PubMed; (d) F. Vögtle, Supramolecular Chemistry; Wiley: Chichester, 1991 Search PubMed; (e) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives; VCH: New York, 1995 Search PubMed; (f) Making Crystals by Design: Methods, Techniques and Applications, ed., Braga, D.; Grepioni, F.Wiley-VCH, Weinheim, 2007 Search PubMed.
- O. Ermer and J. Neudörfl, Helv. Chim. Acta, 2001, 84, 1268–1313 CrossRef CAS.
- B.-Q. Ma and P. Coppens, Chem. Commun., 2003, 2290 RSC.
- D. J. Duchamp and R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 5 CrossRef CAS.
- (a) F. Luo, J.-M. Zheng and S. R. Batten, Chem. Commun., 2007, 3744–3746 RSC; (b) J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239–8247 CrossRef CAS; (c) A. I. Skoulidas, J. Am. Chem. Soc., 2004, 126, 1356–1357 CrossRef; (d) K. M. Ok, J. Sung, G. Hu, R. M. J. Jacobs and D. O'Hare, J. Am. Chem. Soc., 2008, 130, 3762–3763 CrossRef CAS; (e) L. Xu, E.-Y. Choi and Y.-U. Kwon, Inorg. Chem., 2008, 47, 1907–1909 CrossRef CAS; (f) O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Woell, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS; (g) F. A. A. Paz and J. Klinowski, Inorg. Chem., 2004, 43, 3948–3954 CrossRef CAS.
- (a) S. V. Kolotuchin, E. E. Fenlon, S. R. Wilson, C. J. Loweth and S. C. Zimmerman, Angew. Chem., Int. Ed. Engl., 1996, 34, 2654–2657 CrossRef; (b) S. Chatterjee, V. R. Pedireddi, A. Ranganathan and C. N. R. Rao, J. Mol. Struct., 2000, 520, 107–115 CrossRef CAS; (c) S. E. Dale, M. R. J. Elsegood and S. J. Richards, Chem. Commun., 2004, 1278–1279 RSC.
- F. H. Herbstein, M. Kapon and G. M. Reisner, J. Inclusion Phenom., 1987, 5, 211 CrossRef CAS.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef.
- J. Beckmann, R. Rüttinger and T. Schwich, Cryst. Growth Des., 2008, 8, 3271–3276 CrossRef CAS.
- (a) B. R. Bhogala, P. Vishweshwar and A. Nangia, Cryst. Growth Des., 2002, 2, 325 CrossRef CAS; (b) B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2003, 3, 547 CrossRef CAS; (c) N. Shan, A. D. Bond and W. Jones, New J. Chem., 2003, 27, 365 RSC.
- C. B. Aakeröy, J. Desper, D. J. Salmon and M. M. Smith, Cryst. Growth Des., 2006, 6, 1033–1042 CrossRef.
- C. B. Aakeröy, J. Desper and M. M. Smith, Chem. Commun., 2007, 3936–3938 RSC.
- APEX2 v2.2.0 © 2005–2007, Bruker AXS, Madison, WI.
- SHELXTL v6.10, © 2001, Bruker AXS, Madison, WI.
-
Crystal data for 2:3: C65H58.50 N19 O7.50 M = 1225.81, triclinic, space groupP
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.310(2); b = 17.162(3); c = 17.335(3) Å, α = 88.010(10), β = 82.742(3), γ = 70.891(10)°, V = 3153.8(11) Å3Z = 2, Dc = 1.291 g cm−3, μ(Mo-Ka) = 0.089 mm−1, F(000) = 1283, crystal size 0.35 × 0.30 × 0.25 mm., Data were collected at 120(2) K on a Bruker Kappa APEX II diffractometer. A total of 69025 were collected, of which 69025 were unique (Rint = 0.000) and 47925 significant with I > 2σ(I). Structure solution and refinement were carried out with SHELXL-97. Final residuals for I > 2σ(I) were R1 = 0.0816 and w R2 = 0.2282 (GOF = 1.435). Largest diffraction peak and hole = 0.938 and −0.559 eÅ−3.
, a = 11.310(2); b = 17.162(3); c = 17.335(3) Å, α = 88.010(10), β = 82.742(3), γ = 70.891(10)°, V = 3153.8(11) Å3Z = 2, Dc = 1.291 g cm−3, μ(Mo-Ka) = 0.089 mm−1, F(000) = 1283, crystal size 0.35 × 0.30 × 0.25 mm., Data were collected at 120(2) K on a Bruker Kappa APEX II diffractometer. A total of 69025 were collected, of which 69025 were unique (Rint = 0.000) and 47925 significant with I > 2σ(I). Structure solution and refinement were carried out with SHELXL-97. Final residuals for I > 2σ(I) were R1 = 0.0816 and w R2 = 0.2282 (GOF = 1.435). Largest diffraction peak and hole = 0.938 and −0.559 eÅ−3. - F. A. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- Y. Ye, W. Sun, Y. Wang, X. Shao, X. Xu, F. Cheng, J. Li and K. Wu, J. Phys. Chem. C, 2007, 111, 10138 CrossRef CAS.
- L. Kampschulte, M. Lackinger, A.-K. Maier, R. S. K. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829 CrossRef CAS.
- (a) N. Gerasimchuk, T. Maher, P. Durham, K. V. Domasevitch, J. Wilking and A. Mokhir, Inorg. Chem., 2007, 46, 7268 CrossRef CAS; (b) D. Robertson, J. F. Cannon and N. Gerasimchuk, Inorg. Chem., 2005, 44, 8326 CrossRef CAS.
- K. G. Nath, O. Ivasenko, J. A. Miwa, H. Dang, J. D. Wuest, A. Nanci, D. F. Derepichka and F. Rosei, J. Am. Chem. Soc., 2006, 128, 4212–4213 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data for synthesis and labeling scheme. CCDC reference number 708971. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05919j |
| This journal is © The Royal Society of Chemistry 2012 |