DOI:
10.1039/C1CE05968H
(Paper)
CrystEngComm, 2012,
14, 246-250
Synthesis and growth mechanism of hollow microcorn-like CdS crystal
Received
30th July 2011
, Accepted 6th September 2011
First published on 25th October 2011
Abstract
Hollow microcorn-like CdS crystals have been synthesized by a facile solvothermal method. The effects of molar ratio of Cd to S, pH value, and reaction time on the morphologies of CdS crystals were investigated by scanning electron microscopy (SEM). The results of transmission electron microscopy (TEM) and selective-area electron diffraction (SAED) show that the CdS crystals had predominately grown along the [001] direction. A possible growth process in the morphology of the hollow microcorn-like CdS crystals has been suggested.
Introduction
Significant efforts have been made to synthesize various chalcogenide semiconductors of micro- and nano-structures due to their fascinating photoelectric properties, and their potential applications, ranging from micro- and nano-scale electronic devices to light-emitting diodes, solar energy conversion, photocatalysis, and so forth.1 Thus, rational control over the morphology, crystalline structure, and size of materials is very important in controlling their chemical and physical properties.2 Among various metal chalcogenides, CdS has been one of the most studied due to its extensive applications in photoelectric conversion in solar cells and light-emitting diodes in flat panel displays.3 CdS nanoboxes, nanowires, nanobelts, nanospheres and some other particular morphologies have been synthesized by several techniques,4 such as the chemical vapor deposition (CVD) process,5 various templates,6polymer gels,7 micelles,8 and molecular precursor methods.9 Although they are effective methods to prepare uniform products, they usually need some special instruments, or lead to harsh conditions and rather complicated procedures. Among various traditional synthesis approaches of micro- and nano-materials, the solvothermal process is an ideal industrial method for its low cost and adaptability. However, most of them used ethylenediamine (EDA) as the solvent,10 and the organic molecule dithiol glycol (HSCH2–CH2SH) or dodecanethiol as the sulfur source.11,12 Though they may be effective methods to prepare CdS micro- and nano-structures, as we know, hydrogen sulfide (H2S) is a virulent gas, and ethylenediamine and thiol are also pungent. So it is desirable to exploit some facile and environmentally friendly methods.
Herein, we propose a simple solvothermal approach to synthesize hollow microcorn-like CdS crystals without using any special additives such as cysteamine or polyvinylpyrrolidone (PVP).13 Advantages of the approach is that there is no need for post-treatment to get rid of the template materials. In our experiments, the raw materials are economical. No toxic by-products were produced in our experiment. The growth mechanism of the hollow microcorn-like CdS crystals has been studied. To acquire products with better purity and quality, the pH value of the reaction solution, reaction time, and molar ratio of Cd/S have also been investigated.
Results and discussion
Hollow microcorn-like CdS crystals can be successfully synthesized by adjusting the pH value of the mixture solution of CdCl2, sodium thiosulfate, and glycerol with NaOH at the solvothermal condition of 180 °C. X-Ray diffraction (XRD) patterns of the as-prepared products with pH = 7, 14, and >14 showed that all the reflection peaks of the products could be readily indexed as pure hexagonal (wurtzite) phase CdS crystals (Fig. 1), which were in good agreement with the literature values (JCPDS card number 06-0314).
 |
| | Fig. 1
XRD patterns of the CdS crystals synthesized with pH = 7 (a), pH = 14 (b), and pH > 14 (c), (Cd/S = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mol/mol, 180 °C for 48 h). :4 mol/mol, 180 °C for 48 h). | |
Several experimental parameters that were important for the formation of pure hollow microcorn-like CdS crystals were concluded as follows: (1) The molar ratio of Cd to S was important for the formation of the hollow microcorn-like CdS crystals with high purity and uniform morphology. The optimal molar ratio of Cd/S was 1:4. (2) The pH value played another key role in forming the pure hollow microcorn-like CdS crystals. The basic condition was crucial to obtain the perfect morphology of the hollow microcorn-like CdS crystals. In the neutral or alkalescent solution (pH = 7), only nanospheres could be obtained. However, the substantive hollow microcorn-like morphology could be observed under highly basic conditions (pH > 14). (3) The reaction time was also an important factor for the continuous growth and formation of hollow microcorn-like microstructures of the CdS crystals. As described above, it was possible to control and tune the morphologies of CdS crystals by rationally changing Cd/S molar ratio, pH value, and reaction time of the precursor solution. The detailed experiment results were as follows.
(1) The effect of Cd/S molar ratio
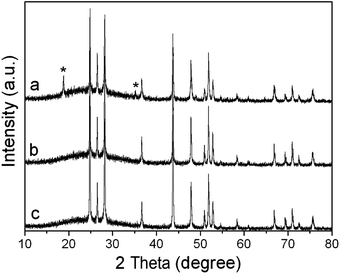
During the experimental process, we found that the Cd/S molar ratio had a great influence on the purity of the hollow microcorn-like CdS crystals. For example, when Cd2+ was constant (1.0 mmol) and the Cd/S molar ratio was kept at 1:2 (2 M NaOH, 180 °C for 48 h), uniform microcorn-like microstructures with the length of about several micrometres were obtained (Fig. 2a). However, the corresponding XRD pattern showed that the product was impure and composed of Cd(OH)2 and CdS (Fig. 3a), which means that the amount of S was deficient. When the Cd/S molar ratio was reduced to 1:4, substantive microcorn-like CdS microstructures appeared (Fig. 2b). The corresponding XRD patterns showed that all the peaks could be indexed as CdS crystals (Fig. 3b). With further decreasing the Cd/S molar ratio to 1:6, the microcorn-like microstructures decreased greatly and many irregular particles appeared (Fig. 2c), though the products were still pure CdS crystals according to the XRD pattern (Fig. 3c). The results indicated that an appropriate Cd/S molar ratio would be necessary to form the well-defined morphology of the CdS crystals. Finally, we fixed the molar ratio of Cd/S at 1:4 in our following experiments.
 |
| | Fig. 2
SEM images of hollow microcorn-like CdS crystals at different Cd/S molar ratios: (a) 1:2, (b) 1:4, (c) 1:6 (2 M NaOH, 180 °C for 48 h). | |
 |
| | Fig. 3
XRD patterns of the hollow microcorn-like CdS crystals at different Cd/S molar ratios: (a) 1:2, (b) 1:4, (c) 1:6 (2 M NaOH, 180 °C for 48 h). * Peaks of Cd(OH)2. | |
(2) The effect of pH value
Further investigation revealed that the pH value played a key role on the morphological transformation. We displayed the SEM images of CdS crystals synthesized at different pH values (7, 13, 14 and >14). As shown in Fig. 4a, CdS crystals synthesized at pH = 7 were sphere-like nanoparticles. Moreover, with increasing the pH value to 13 and 14, mixture of the nanoparticles and the microcorn-like microstructures could be observed, as shown in Fig. 4b and 4c. Obviously, the lower pH value favors the formation of large nanospheres. With the drastic increase of pH value (pH > 14, 2 M NaOH), the morphology of the synthesized CdS crystals varied remarkably. In Fig. 4d, all of the CdS crystals showed the microcorn-like morphology. The above results indicated that the morphology of CdS crystals gradually changed from nanoparticles to microcorn-like microstructures with increasing pH value.
 |
| | Fig. 4
SEM images of CdS crystals at different pH values: (a) pH = 7, (b) pH = 13, (c) pH = 14, (d) pH > 14 (Cd/S = 1:4 mol/mol, 180 °C for 48 h). | |
(3) The effect of time
To investigate the morphology evolution during the growth of the microcorn-like microstructures, we performed the synthesis at 180 °C under different reaction times. A series of SEM images in Fig. 5 showed the morphology evolution of the CdS crystals captured at different reaction stages corresponding to reaction times of 12, 24, and 48 h, which revealed that the morphological evolution of the microcorn-like microstructures was time-dependent. A great deal of colloid suspension formed progressively when NaOH was added to the solution of CdCl2 and sodium thiosulfate. Once the Cd(OH)2 particles had formed, the nucleation and growth of CdS one-dimensional (1D) microstructures would proceed during the next solvothermal process. When the solvothermal process was extended to 12 h, a few star-like morphologies composed of some sphere-like nanoparticles could be observed (Fig. 5a). We also found that some characteristic peaks of the CdS phase with relativly weak intensity and some characteristic peaks of the Cd(OH)2 phase with relatively strong intensity appeared on the XRD pattern (Fig. 6a). After the solvothermal process was extended to 24 h, some microcorn-like crystals were generated (Fig. 5b). The SEM image (Fig. 5b) showed that the products were microcorn-like crystals with the size about 800 nm wide and up to several millimetres long. It was possible that the nascent microcorn-like structure in Fig. 5b was the CdS crystal. This meant that the growth of microcorn-like CdS continuously proceeded through consuming the Cd(OH)2 particles. This tendency could also be interpreted by a contrast in the variation of peak intensity in the XRD patterns (Fig. 6b), in which the intensity of the characteristic peaks of CdS crystals largely intensified, and those of Cd(OH)2 disappeared with an increase in the solvothermal time to 24 h. In classic colloidal models, crystal growth was subject to Ostwald ripening during the synthetic courses.14 During the Ostwald ripening process, larger particles tend to grow even larger at the expense of smaller particles, which means that the smaller particles dissolve into the mother liquor progressively. So after the solvothermal process was extended to 48 h (Fig. 5c), some smaller particles disappeared and the microcorn-like crystals grew bigger. Simultaneously, the intensity of the CdS peaks increased in the XRD pattern (Fig. 6c). As we know, the branching growth has been proposed to be determined by a diffusion process.15 Crystals consume ions or molecules near the surface as they grow, and a concentric diffusion field forms around the crystal, which protrudes further into the region of higher concentration, growing faster than the central parts of the facets. By carefully observation, we could find that the lateral particles of the microcorn-like crystals grew longer with the increase of the solvothermal time. The tubers on the sides of the microcorn-like crystals were close to the mother liquid, and would be in accord to branching growth. The TEM images (Fig. 7) also depicted the hollow microcorn-like structure. The insets in Fig. 7 showed SAED patterns recorded from different positions of a microcorn-like crystal, which indicated that it was single crystalline, and the hollow microcorn-like crystal grew along the [001] direction.
 |
| | Fig. 5
SEM images of CdS crystals at different reaction times: (a) 12 h, (b) 24 h, and (c) 48 h (Cd/S = 1:4 mol/mol, NaOH 2 M, 180 °C). | |
 |
| | Fig. 6
XRD patterns of CdS crystals at different reaction times: (a) 12 h, (b) 24 h, (c) 48 h (Cd/S = 1:4 mol/mol, NaOH 2 M, 180 °C). * Peaks of Cd(OH)2. | |
 |
| | Fig. 7
TEM images of the hollow microcorn-like CdS crystals obtained at Cd/S = 1:4 mol/mol, NaOH 2 M, and 180 °C for 48 h: (a) the tuber of the microcorn, (b) the middle of the microcorn, (c) the tip of the microcorn (inset is the corresponding SAED pattern). | |
Selective preparation of CdS nanostructures can be achieved by controlling the reaction conditions, such as the Cd/S molar ratio, the pH of the solution, the volume ratio of the mixed solventsetc.16 Combining the morphological transformation of the CdS crystals with the change of pH value and reaction time, we could propose the growth mechanism for the hollow microcorn-like CdS crystals as shown in Fig. 8. In principle, a crystal growth process consists of nucleation and growth, which are affected by the intrinsic crystal structure and the external conditions.17 At the beginning, Cd(OH)2 nanoparticles can be prepared by adding sufficient OH− ions in the solution, as long as [Cd2+][OH−]2 is greater than the Ksp value of Cd(OH)2. As one knows, the Ksp of Cd(OH)2 is more than that of CdS, i.e. Ksp[Cd(OH)2] = 5.27 × 10−15 > Ksp(CdS) = 8.0 × 10−27. So CdS can be synthesized from Cd(OH)2 by an ion exchange reaction.18 Subsequently the nanoparticles could coalesce in a way we called oriented attachment.19 The solubility for CdS is very small, which results in a fast nucleation of CdS crystals. These nuclei would preferentially grow along the [001] direction to form 1D micro- or nano-structures. From the crystal structure, 1D CdS described as a number of Cd2+ and S2− are arranged alternately staking along the c-axis. The oppositely charged ions produce positively charged Cd2+ and negatively charged S2− polar surfaces.20 Considering the polar crystal structure of CdS, in general, the +c-axis terminated by Cd2+ is in favor of adsorbing the incoming ions to grow quickly, while growth along the −c-axis terminated by S2− is suppressed. At the higher concentration of OH− (pH > 14.0), OH− may serve as a surface modifier, presumably bound to the Cd2+ polar planes. Thus the dense OH− will prevent the negatively charged S2− polar surface from further reaction, resulting in the formation of microcorn-like structures. In other words, the OH− can directly attach to the positively charged Cd2+ surface and form Cd(OH)2 along the [001] direction. The produced Cd(OH)2 then can be transformed into CdS by ion exchange, which makes this reaction continue. This effect was very prominent at higher concentration of OH−. Due to the Ostwald ripening process, the hollow microcorn-like crystals grew bigger with the increase of reaction time. Meanwhile, some tubers on the sides of the microcorn-like crystal grew longer because of branching growth.15
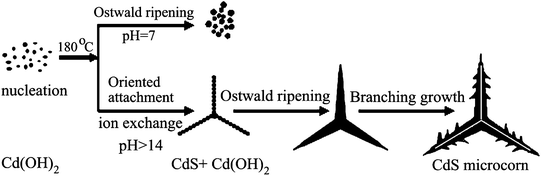 |
| | Fig. 8 Schematic diagram illustrating the morphological change with pH value and reaction time. | |
Experimental section
Synthesis
All of the chemical reagents used in this experiment were of analytical grade and used without further purification. In a typical synthesis, 1.0 mmol of CdCl2 and 4.0 mmol of sodium thiosulfate were dissolved in 40 mL distilled water. Then, 20.0 mL of glycerol were added into the mixed solution and stirred for 5 min. Subsequently, NaOH was added to the above solution to adjust the pH value. After the solution was stirred for a further 10 min, it was transferred to a stainless Teflon-lined autoclave, heated at 180 °C for different hours, and then cooled to room temperature naturally. Then the precipitates were centrifuged, washed several times with deionized water, an ethanol, respectively. Finally, yellow products were dried in a vacuum at 50 °C for 4 h.
Characterizations
Scanning electron microscopy images were obtained on a scanning electron microscope (SEM, JSM-6360LV). The X-ray powder diffraction patterns of the as-prepared samples were recorded by employing a Japan Rigaku Dmax 2000 X-ray diffractometer with Cu-Kα radiation at a scanning rate of 4 ° min−1 in the 2θ range of 10–80°. The selected-area electron diffraction pattern was captured on a Hitachi model H-800 instrument at an accelerating voltage of 200 kV.
Conclusions
We have demonstrated a facile strategy to synthesize CdS crystals with different morphologies including nanospheres, microcorns, and multi-armed microrods. The effects of the concentration of NaOH, the solvothermal time, and the molar ratio of Cd to S on the morphology of the CdS crystals were investigated. This simple synthetic method, which does not use any surfactant, ligand, or template, may be extended to the synthesis of other materials with novel morphologies.
Acknowledgements
This work was financially supported by the Program for Changjiang Scholars and Innovative Research Team in University, the Science Foundation of Jilin Province (20070505), and the Fundamental Research Funds for the Central Universities.
Notes and references
-
(a) O. Hayden, A. B. Greytak and D. C. Bell, Adv. Mater., 2005, 17, 701–704 CrossRef CAS;
(b) P. T. Chin, R. A. Hikmet, M. S. Meskers and R. A. Janssen, Adv. Funct. Mater., 2007, 17, 3829–3835 CrossRef CAS;
(c) S. L. Xiong, B. J. Xi, C. M. Wang, G. C. Xi, X. Y. Liu and Y. T. Qian, Chem.–Eur. J., 2007, 13, 7926–7932 CrossRef CAS;
(d) H. Weiû, A. Fernandez and H. Kisch, Angew. Chem., Int. Ed., 2001, 40, 3825–3827 CrossRef;
(e) J. Tang and A. P. Alivisatos, Nano Lett., 2006, 6, 2701–2706 CrossRef CAS;
(f) X. Fang, Y. Bando, M. Liao, U. K. Gautam, C. Zhi, B. Dierre, B. Liu, T. Zhai, T. Sekiguchi, Y. Koide and D. Golberg, Adv. Mater., 2009, 21, 2034–2039 CrossRef CAS;
(g) X. S. Fang, C. H. Ye, L. D. Zhang, Y. H. Wang and Y. C. Wu, Adv. Funct. Mater., 2005, 15, 63–68 CrossRef CAS;
(h) L. Hu, J. Yan, M. Liao, L. Wu and X. Fang, Small, 2011, 7, 1012–1017 CrossRef CAS.
-
(a) S. H. Choi, E. G. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 2520–2521 CrossRef CAS;
(b) B. Kale, B. Baeg, J. Ook, S. M. Lee, H. Chang, S. J. Moon and C. W. Lee, Adv. Funct. Mater., 2006, 16, 1349–1354 CrossRef CAS;
(c) X. S. Fang, Y. Bando, G. Z. Shen, C. H. Ye, U. K. Gautam, P. M. Costa, C. Y. Zhi, C. C. Tang and D. Golberg, Adv. Mater., 2007, 19, 2593–2596 CrossRef CAS;
(d) Y. F. Shi, Y. Wan, R. L. Liu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 9522–9531 CrossRef CAS.
-
(a) S. Mandal, D. Rautaray, A. Sanyal and M. Sastry, J. Phys. Chem. B, 2004, 108, 7126–7131 CrossRef CAS;
(b) L. Weinhardt, T. Gleim, O. Fuchs, C. Heske, E. Umbach, M. Bar, H. J. Muffler, C. H. Fischer, M. C. Lux-Steiner, Y. Zubavichus, T. P. Niesen and F. Karg, Appl. Phys. Lett., 2003, 82, 571–573 CrossRef CAS.
-
(a) M. R. Kim and D. J. Jang, Chem. Commun., 2008, 5218–5220 RSC;
(b) Z. Dai, J. Zhang, J. Bao, X. Huang and X. Mo, J. Mater. Chem., 2007, 17, 1087–1093 RSC;
(c) J. Han, H. Su, D. Zhang, J. Chen and Z. Chen, J. Mater. Chem., 2009, 19, 8741–8746 RSC.
-
(a) R. M. Ma, L. Dai and G. G. Qin, Appl. Phys. Lett., 2007, 90, 93109 CrossRef;
(b) G. Z. Shen, J. H. Cho, J. K. Yoo, G. C. Yi and C. J. Lee, J. Phys. Chem. B, 2005, 109, 9294–9298 CrossRef CAS.
-
(a) Y. Lifshitz, O. Konovalov, N. Belman, A. Berman and Y. Golan, Adv. Funct. Mater., 2006, 16, 2398–2404 CrossRef CAS;
(b) J. X. Huang, Y. Xie, B. Li, Y. Liu, Y. T. Qian and S. Y. Zhang, Adv. Mater., 2000, 12, 808–811 CrossRef CAS;
(c) S. Mandal, D. Rautaray, A. Sanyal and M. Sastry, J. Phys. Chem. B, 2004, 108, 7126–7131 CrossRef CAS;
(d) D. Routkevitch, T. Bigioni, M. Moskovits and J. M. Xu, J. Phys. Chem., 1996, 100, 14037–14047 CrossRef CAS.
-
(a) M. F. Zhang, M. Drechsler and A. H. Muüller, Chem. Mater., 2004, 16, 537–543 CrossRef CAS;
(b) V. Maheshwari and R. F. Saraf, Langmuir, 2006, 22, 8623–8626 CrossRef CAS;
(c) H. Du, G. Q. Xu and W. S. Chin, Chem. Mater., 2002, 14, 4473–4479 CrossRef CAS;
(d) T. Di Luccio, A. M. Laera and L. Tapfer, J. Phys. Chem. B, 2006, 110, 12603–12609 CrossRef CAS;
(e) L. Wang, Y. S. Liu, X. Jiang, D. H. Qin and Y. Cao, J. Phys. Chem. C, 2007, 111, 9538–9542 CrossRef CAS;
(f) N. Duxin, F. T. Liu, H. Vali and A. Eisenberg, J. Am. Chem. Soc., 2005, 127, 10063–10069 CrossRef CAS.
-
(a) H. Y. Zhao, E. P. Douglas, B. S. Harrison and K. S. Schanze, Langmuir, 2001, 17, 8428–8433 CrossRef CAS;
(b) B. A. Harruff and C. E. Bunker, Langmuir, 2003, 19, 893–897 CrossRef CAS;
(c) B. A. Simmons, S. Li, V. T. John, G. L. McPherson, A. Bose, W. L. Zhou and J. He, Nano Lett., 2002, 2, 263–268 CrossRef CAS.
-
(a) T. Trindade and Paul. O'Brien, Chem. Mater., 1997, 9, 523–530 CrossRef CAS;
(b) P. Yan, Y. Xie, Y. Qian and X. Liu, Chem. Commun., 1999, 1293–1294 RSC;
(c) M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2001, 13, 913–920 CrossRef CAS;
(d) C. J. Barrelet, Y. Wu, D. C. Bell and C. M. Lieber, J. Am. Chem. Soc., 2003, 125, 11498–11499 CrossRef CAS.
-
(a) F. Gao, Q. Y. Lu, S. H. Xie and D. Y. Zhao, Adv. Funct. Mater., 2007, 17, 3829–3835 CrossRef;
(b) H. B. Chu, X. M. Li, G. D. Chen, W. W. Zhou, Y. Zhang, Z. Jin, J. J. Xu and Y. Li, Cryst. Growth Des., 2005, 5, 1801–1806 CrossRef CAS.
-
(a) T. Hirai, T. Saito and I. Komasawa, J. Phys. Chem. B, 2001, 105, 9711–9714 CrossRef CAS;
(b) J. Aldana, N. Lavelle, Y. J. Wang and X. G. Peng, J. Am. Chem. Soc., 2005, 127, 2496–2504 CrossRef CAS;
(c) D. Xu, Z. P. Liu, J. B. Liang and Y. T. Qian, J. Phys. Chem. B, 2005, 109, 14344–14349 CrossRef CAS;
(d) A. G. Young, D. P. Green and A. J. McQuillan, Langmuir, 2006, 22, 11106–11112 CrossRef CAS;
(e) A. G. Young, D. P. Green and A. J. McQuillan, Langmuir, 2007, 23, 12923–12931 CrossRef CAS.
-
(a) C. A. Fernandez and C. M. Wai, Chem.–Eur. J., 2007, 13, 5838–5844 CrossRef CAS;
(b) T. D. Luccio, A. M. Laera and L. Tapfer, J. Phys. Chem. B, 2006, 110, 12603–12609 CrossRef;
(c) T. H. Jia, G. R. Liu, X. Y. Qia, H. B. Xu and M. Y. Gao, Mater. Res. Bull., 2007, 42, 720–730 CrossRef;
(d) A. Datta, A. Priyam, S. Chatterjee, A. K. Sinha, S. N. Bhattacharyya and A. Saha, Colloids Surf., A, 2007, 301, 239–245 CrossRef CAS.
-
(a) S. L. Xiong, B. J. Xi, C. M. Wang, G. F. Zou, L. F. Fei, W. Z. Wang and Y. T. Qian, Chem.–Eur. J., 2007, 13, 3076–3081 CrossRef CAS;
(b) X. H. Li, J. X. Li, G. D. Li, D. P. Liu and J. S. Chen, Chem.–Eur. J., 2007, 13, 8754–8761 CrossRef CAS.
- M. F. Ye, H. Z. Zhong, W. J. Zheng, R. Li and Y. F. Li, Langmuir, 2007, 23, 9064–9068 CrossRef CAS.
-
(a) L. Ye, W. Guo, Y. Yang, Y. F. Du and Y. Xie, Chem. Mater., 2007, 19, 6331–6337 CrossRef CAS;
(b) M. Epifani, J. Arbiol, E. Pellicer and J. R. Morante, Chem. Mater., 2007, 19, 4919–4924 CrossRef CAS;
(c) G. D. Lilly, J. Lee, K. Sun, Z. Tang, K.-S. Kim and N. A. Kotov, J. Phys. Chem. C, 2008, 112, 370–377 CrossRef CAS.
- Y. Zhu, T. Mei, Y. Wang and Y. Qian, J. Mater. Chem., 2011, 21, 11457–11463 RSC.
-
(a) J. B. Wu, H. Zhang, N. Du, X. Y. Ma and D. Yang, J. Phys. Chem. B, 2006, 110, 1107–1110 CrossRef;
(b) J. Zhou, G. Zhao, B. Song and G. Han, CrystEngComm, 2011, 13, 2294–2302 RSC.
- N. Z. Bao, L. M. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20, 110–117 CrossRef CAS.
- M. F. Ye, H. Z. Zhong, W. J. Zheng, R. Li and Y. F. Li, Langmuir, 2008, 23, 9064–9068 CrossRef.
- A. S. Barnard and H. F. Xu, J. Phys. Chem. C, 2007, 111, 18112–18117 CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.